

Abstract
Colicin M (Cma) is specifically imported into the periplasm of Escherichia coli and kills the cells. Killing depends on the periplasmic peptidyl prolyl cis-trans isomerase/chaperone FkpA. To identify the Cma prolyl bonds targeted by FkpA, we replaced the 15 proline residues individually with alanine. Seven mutant proteins were fully active; Cma(P129A), Cma(P176A), and Cma(P260A) displayed 1%, and Cma(P107A) displayed 10% of the wild-type activity. Cma(P107A), Cma(P129A), and Cma(P260A), but not Cma(P176A), killed cells after entering the periplasm via osmotic shock, indicating that the former mutants were translocation-deficient; Cma(P129A) did not bind to the FhuA outer membrane receptor. The crystal structures of Cma and Cma(P176A) were identical, excluding inactivation of the activity domain located far from Pro-176. In a new peptidyl prolyl cis-trans isomerase assay, FkpA isomerized the Cma prolyl bond in peptide Phe-Pro-176 at a high rate, but Lys-Pro-107 and Leu-Pro-260 isomerized at only <10% of that rate. The four mutant proteins secreted into the periplasm via a fused signal sequence were toxic but much less than wild-type Cma. Wild-type and mutant Cma proteins secreted or translocated across the outer membrane by energy-coupled import or unspecific osmotic shock were only active in the presence of FkpA. We propose that Cma unfolds during transfer across the outer or cytoplasmic membrane and refolds to the active form in the periplasm assisted by FkpA. Weak refolding of Cma(P176A) would explain its low activity in all assays. Of the four proline residues identified as being important for Cma activity, Phe-Pro-176 is most likely targeted by FkpA.
Keywords: Bacterial Toxins, Chaperone Chaperonin, Membrane Proteins, Protein Structure, Protein Translocation, Colicin, FkpA
Introduction
Colicin M (Cma)2 is synthesized by Escherichia coli cells that carry a pColBM plasmid and is unspecifically released to a low extent. It kills sensitive E. coli cells by interfering with lipid carrier recycling, leading to inhibition of murein biosynthesis and cell lysis (1, 2). Specifically, Cma cleaves the phosphate ester bond between the lipid carrier (bactoprenol) and the murein precursor (3). Cma enters the periplasm of sensitive cells by binding to the FhuA outer membrane receptor protein and translocation across the outer membrane by an energy-coupled mechanism through the action of the TonB, ExbB, and ExbD proteins (Ton system) (4–6).
Like all colicins, Cma consists of a central receptor binding domain, an N-terminal translocation domain, and a C-terminal activity domain (7), which can be seen in the crystal structure (8). Only the activity domain, which starts at residue 124, is homologous with colicin-M-like proteins of other bacteria (8, 9).
The first E. coli mutants identified that were resistant to added colicin M were mutated in the fhuA, tonB, exbB, or exbD transport genes. Another type of Cma-resistant mutant eluded genetic and function characterization (10) until recently. We localized the mutation to the fkpA gene, which encodes a periplasmic peptidyl prolyl cis-trans isomerase (PPIase)/chaperone. Among 10 colicins tested, only Cma requires FkpA for toxicity (11).
The function of FkpA for E. coli physiology, however, is not clear as fkpA mutants show no phenotype. FkpA is overexpressed under stress conditions, partially controlled by σE through a σE promoter. Deletion of fkpA stimulates transcription of degP, which encodes a heat shock-inducible periplasmic protease (12). Overproduction of FkpA prevents the formation of periplasmic inclusion bodies of MalE31, a defective folding derivative of the MalE-binding protein (13), and increases the yield of functional engineered antibody fragments produced in E. coli (14).
FkpA displays high PPIase activity, as demonstrated by the refolding of ribonuclease T1 and prolyl cis-trans isomerization of oligopeptides (15). Its activity is inhibited by FK506, an inhibitor of prokaryotic and eukaryotic FKBP-type PPIases (15). FkpA forms a dimer; the monomers consist of an N-proximal helical domain (residues 1–114) and a C-proximal domain (residues 115–245) typical for the FKBP family of PPIases (16). The five periplasmic PPIases/chaperones PpiA, PpiD, SurA, FkpA, and Skp usually act on various substrates and can functionally replace each other (17–19). In contrast, the toxicity of Cma strictly depends on FkpA and not on any other E. coli PPIase/chaperone (11).
The strict dependence of Cma toxicity on FkpA prompted us to attempt to identify the prolyl bond targeted by FkpA. If periplasmic Cma activity depends on the FkpA prolyl cis-trans isomerase function and not, or not only on the FkpA chaperone function, replacement of the proline residue that is cis-trans-isomerized should prevent activation. To test this premise we replaced each of the 15 proline residues in Cma individually with alanine. Here we identified one prolyl bond that could serve as the target of FkpA PPIase catalysis.
EXPERIMENTAL PROCEDURES
Bacterial Strains and Plasmids
The bacterial strains and plasmids used and their sources are listed in Table 1. Isolation of the FkpA313 mutant SIP1275 and the temperature-sensitive FkpA43 mutant K458 was described previously (10, 11, 20).
TABLE 1.
E. coli strains and plasmids used in this study
| Strain/Plasmid | Genotype | Reference |
|---|---|---|
| Strain | ||
| AB2847 | aroB thi tsx malT | (1) |
| BL21(DE3) | F−ompT gal dcm lon hsdB (rB− mB−) λ(DE3) lacI lacUV5-T7 gene 1 | (50) |
| BL21 fhuA | BL21(DE3) fhuA | (11) |
| MB97 | AB2847 ΔfhuA | (54) |
| K458 | AB2847 fkpA43 (tolM-ts) | (20) |
| Mo3 | AB2847 fkpA40 | (10) |
| ES965 | KTu50 pES7 cba cbi on pACYC184 | (51) |
| DH5α | supE44 ΔlacU169 [Φ80lacZΔM15] hsdR17 recA1 endA1 gyrA thi-1 relA1 | (52) |
| SIP1275 | AB2847 fkpA313 | (11) |
| Plasmid | ||
| pMLD237 | cma encoding Cma-Hisa | (3) |
| pSP127/56 | pET25b encoding FkpA313-Hisb | (11) |
| pYH15 | pET25b encoding FkpA-Hisb | (11) |
| pYH16 | pET25b encoding FkpA43-Hisb | (11) |
| pGP1–2 | Gene 1 (T7 polymerase) behind λPL controlled by the temperature-sensitive CI857 λ repressor | (53) |
| pPG773 | pT7–7 pbpB-cmi, ApR encoding PbpB′-′Cmi | (33) |
| pSP130/155 | encodes MalE1–26-Cma1–271 with a C-terminal His-tag controlled by the araBAD promoter, araC, Apr | This study |
a Cma with a N-terminal His6 tag.
b FkpA protein with a C-terminal His6 tag.
Generation of Cma Proline Substitution Mutants
The cma gene on plasmid pMLD237 (generously provided by D. Mengin-Lecreulx, Université Paris-Sud, Orsay, France) encodes Cma with an N-terminal His6 tag. We replaced each of the 15 proline residues in Cma with alanine by site-directed mutagenesis of cma with appropriate primers (sequences available upon request) using the QuickChange® site-directed mutagenesis kit (Stratagene, La Jolla, CA) and the PhusionTM high fidelity DNA polymerase (Finnzymes, Espoo, Finland). Mutagenized cma genes on plasmid pMLD237 were introduced into E. coli DH5α by transformation, and the mutations were verified by sequencing the cma genes isolated from the transformants. Cma activity was determined with transformants of E. coli BL21 fkpA carrying the mutated cma genes on pMLD237. For the assay, crude cell extracts were serially diluted 10-fold, and 10 μl were spotted on LB agar plates seeded with E. coli AB2847. The same procedure was used to isolate the mutant Cma(D226A).
To equip Cma with a signal sequence, cma on pMLD237 was amplified by PCR, ligated to a sequence encoding a C-terminal His-tag, and cloned downstream of malE′ of pMA-RQ MalE′-Cma1–130 synthesized by Geneart (Regensburg, Germany), replacing cma1–130. The resulting plasmid pSP130/155 encodes the signal sequence MalE1–26 (Swissprot P0AEX9), Cma1–271 (Swissprot P05820) and the C-terminal His-tag LAHHHHHH under the control of the araBAD promoter. The plasmid contains araC from pMA-RQ MalE′-Cma(1–130). pSP130/155 was mutagenized by site-directed mutagenesis, resulting in plasmids encoding MalE′-Cma(P107A)-His, MalE′-Cma(P129A)-His, MalE′-Cma(P176A)-His, MalE′-Cma(D226A)-His, and MalE′-Cma(P260A)-His.
Isolation of Cma and Mutant Cma
Cma and the proteins from transformants of E. coli BL21(DE3) fkpA harboring the recombinant plasmids were purified on Ni-NTA-agarose as described previously (11), taking advantage of the His6 tag on the proteins. E. coli BL21(DE3) fkpA is not killed by Cma because of the fkpA mutation.
Isolation of Colicin B and Competition Assays
A crude extract of colicin B was isolated from E. coli ES965 (pES7 cba cbi). Synthesis of colicin B was induced by adding 0.4 μg ml−1 mitomycin C in water to the culture and shaking the culture for 150 min at 37 °C. Cells were harvested and suspended in 0.1 m Tris-HCl, pH 7.5, and disrupted by sonication, and the suspension was centrifuged. The soluble fraction containing colicin B was active up to a dilution of 106 and was used for competition assays with Cma and the nearly inactive Cma proline mutants. For the assays, exponentially growing cells of E. coli Mo3 fkpA40 were supplemented with a Cma sample and incubated for 10 min; colicin B at a final dilution titer of 104 was then added. Growth of the cultures was monitored spectrophotometrically for 5 h.
Isolation of FkpA-His6
FkpA-His6 was isolated from cells of E. coli BL21(DE3) fkpA transformed with pYH15 fkpA and purified on Ni-NTA-agarose as described (11). The eluate of the column was concentrated with Amicon ultra-15 tubes, and FkpA was further purified by chromatography on a 320-ml Superdex 75 column with 35 mm HEPES, pH 7.8. The presence of the desired proteins and impurities in the eluted fractions were analyzed by SDS-PAGE. FkpA313-His and FkpA43-His from cells transformed with plasmids pSP127/56 and pYH16, respectively, were isolated accordingly to electrophoretic homogeneity.
Crystallization, Data Collection, Phasing, and Refinement
Crystallization of initial protein crystals were obtained by sitting-drop crystallization with screens purchased from Qiagen mixing 0.4 μl of protein with 0.4 μl of reservoir solution. Drops were prepared using the Honeybee 961 crystallization robot (Genomic Solutions) and automatically imaged using the Rock Imager 54 imaging system (Formulatrix, Waltham). Crystals of wild-type (wt) and mutant proteins were mounted from the crystallization drops and frozen in liquid nitrogen according to the procedure described in Table 3. Data of wt and mutant crystals were collected either at beamline PXII, Swiss Light Source, or beamline ID29 of the European Synchrotron Radiation Facility. Diffraction images were recorded on the detector types mentioned in Table 3. All diffraction images were processed and scaled with the XDS/XCALE program package (21). The structure of the wt crystals was solved by molecular replacement using the MOLREP program starting from the PDB entry 3DA4 (MOLREP). Structures of the two mutant datasets were solved using the wt structure and the program MOLREP. Model building and refinement of all structures was performed in CCP4i (22) and Coot (23). Protein geometry was analyzed using the program PROCHECK (24), and secondary structures were assigned based on the DSSP algorithm (25). All pictures were prepared using the PYMOL program. Statistics are summarized in Table 3.
TABLE 3.
Data collection and refinement statistics
| Cma(WT) | Cma(P107A) | Cma(P176A) | |
|---|---|---|---|
| Data collection | |||
| Space group | C2221 | P1 | C2221 |
| Cell dimensions | |||
| a, b, c (Å) | 52.75, 115.10, 227.71 | 52.85, 63.11, 189.34 | 52.83, 114.77, 225.40 |
| α, β, γ (°) | 90 | 87.59, 82.17, 65.65 | 90 |
| Resolution (Å) | 48-1.67a (1.71-1.67) | 49-2.31 (2.37-2.30) | 47-2.14 (2.19-2.14) |
| Rsym or Rmerge | 0.08 (0.57) | 0.09 (0.40) | 0.19 (0.82) |
| I/σI | 14.2 (1.9) | 5.4 (1.7) | 9.2 (2.0) |
| Completeness (%) | 97.3 (83.9) | 89.5 (72.9) | 99.0 (94.2) |
| Redundancy | 5.2 (3.2) | 1.7 (1.5) | 6.3 (4.9) |
| Refinement | |||
| Resolution (Å) | 48-1.67 (1.71-1.67) | 49-2.31 (2.37-2.31) | 47-2.14 (2.19-2.14) |
| No. reflections | 73413 (5526) | 85754 (4514) | 36063 (1898) |
| Rwork/Rfree | 0.16/0.20 (0.25/0.30) | 0.24/0.27 (0.31/0.33) | 0.18/0.24 (0.26/0.30) |
| No. atoms (all) | 4,733 | 17,005 | 4,376 |
| Protein (chains/residues) | 2/540 | 8/2160 | 2/540 |
| Ligands (nitrate/glycerol) | 3/4 | 1/0 | |
| Water | 434 | 451 | 319 |
| B-Factors | |||
| Protein | 5.7 | 13 | 20.6 |
| Ligands (nitrate/glycerol) | 20/23 | 34/- | |
| Water | 15.1 | 9.1 | 26.1 |
| Root mean square deviations | |||
| Bond lengths (Å) | 0.027 | 0.016 | 0.023 |
| Bond angles (°) | 2.1 | 1.4 | 2.0 |
| Ramachandran statistics | |||
| Residues in favored regions (%) | 532 (99.3) | 2123 (99) | 531 (99.1) |
| Residues in allowed region (%) | 3 (0.6) | 21 (1) | 5 (0.9) |
| Residues in outlier region (%) | 1 (0.2) | 0 (0) | 0 (0) |
| Crystallization conditions | 20% PEG 3350 | 20% PEG 3350 | 20% PEG 3350 |
| 0.2 m NaNO3 | 0.2 m NaNO3, 0.1 m Bis/Tris, pH 6.5 | 0.2 m NaNO3 | |
| Cryo protectant added | 10% PEG 400 | 10% PEG 400 | 10% PEG 400 |
a Values in parentheses are for highest-resolution shell.
Cma Sensitivity Tests
Sensitivity of E. coli cells to Cma was tested with Cma, and mutant derivatives were purified to electrophoretic homogeneity by Ni-NTA-agarose chromatography or with crude extracts of cells that overexpressed Cma or mutant derivatives. In the latter case the Cma protein was by far the most prominent protein in the cell extract, as revealed by SDS-PAGE. The cma transformants released less than 10% of the Cma produced as they lack a Cma lysis gene, which is also absent on natural pColBM plasmids (26). Cells from an 8-ml culture were harvested by centrifugation and suspended in 0.5 ml of 0.1 m Tris-HCl, pH 7.5. An aliquot of 0.2 ml was disrupted by vigorously shaking with glass beads, and cell debris was removed by centrifugation (16,000 × g, 4 °C). 10-Fold dilutions of Cma were spotted onto 20 ml of LB agar plates seeded with 109 cells of the strains to be tested in 3 ml of soft agar. The results are given as the diameter (in mm) of the clear zone of growth inhibition.
To examine the sensitivity of cells overexpressing FkpA, we used E. coli BL21(DE3) fkpA transformed with both pYH15 fkpA and pGP1-2, which encodes the T7 polymerase gene 1 behind λPL, controlled by the temperature-sensitive CI857 λ repressor. Cells were grown at 30 °C to an A578 nm of 0.3 and then shifted for 150 min to 42 °C. 10-Fold-diluted Cma samples were spotted onto LB plates seeded with temperature-induced E. coli BL21(DE3) fkpA (pYH15 fkpA pGP1-2) and incubated overnight at 37 °C. The diameter of the lysis zone was measured.
Osmotic Shock
Osmotic shock not only releases periplasmic proteins into the shock medium (27) but also transfers added proteins into the periplasm (10, 28, 29). The Cma sensitivity of cells after osmotic shock was tested with purified Cma samples.
E. coli AB2847 in 10 ml of LB medium was grown to an A578 nm of 0.5; cells were harvested by centrifugation and then suspended in 1 ml of 10 mm Tris-HCl, 30 mm NaCl, pH 7 (buffer 1). Cells were pelleted by centrifugation, washed once with buffer 1, and centrifuged again. The washed cells were suspended in 0.5 ml of 33 mm Tris-HCl, pH 7 (buffer 2), and 0.1-ml aliquots were added to four tubes. Sucrose (40% in buffer 2, 0.1 ml) was added to two tubes, and buffer 2 (0.1 ml) was added to the other two tubes. Cells were suspended by vortexing, kept for 10 min on a shaker, then cooled from room temperature to 4 °C and harvested by centrifugation. Cma (10 μg in 10 μl) was added to an Eppendorf tube containing a cell suspension treated with sucrose and a tube containing an untreated cell suspension. Then 0.2 ml of ice-cold 0.5 mm MgCl2 and 30 μl of 1 mm CaCl2 was added to each tube; the in vivo activity of Cma depends on Ca2+ (20). The contents of the tubes were immediately mixed by vortexing. Two additional samples received no Cma but were otherwise treated the same way. The four samples were kept on ice for 15 min and then serially diluted 10-fold in buffer 2. Aliquots of 0.1 ml were suspended in 3 ml of LB soft agar and spread on 20-ml LB agar plates. After overnight incubation at 37 °C, the colonies were counted.
Activity Assay for Secreted Cma
Cells containing wild-type cma and cma mutant genes fused to the MalE signal sequence were grown in LB medium containing ampicillin to the exponential growth phase. Arabinose was added to the final concentrations indicated in Fig. 4. To avoid killing of cells by Cma released into the medium, transformants of the fhuA mutant MB97 were used. In the case of fkpA mutants this precaution was not required as they are resistant to Cma. Cell growth and lysis were monitored by measuring the optical density at 578 nm.
FIGURE 4.
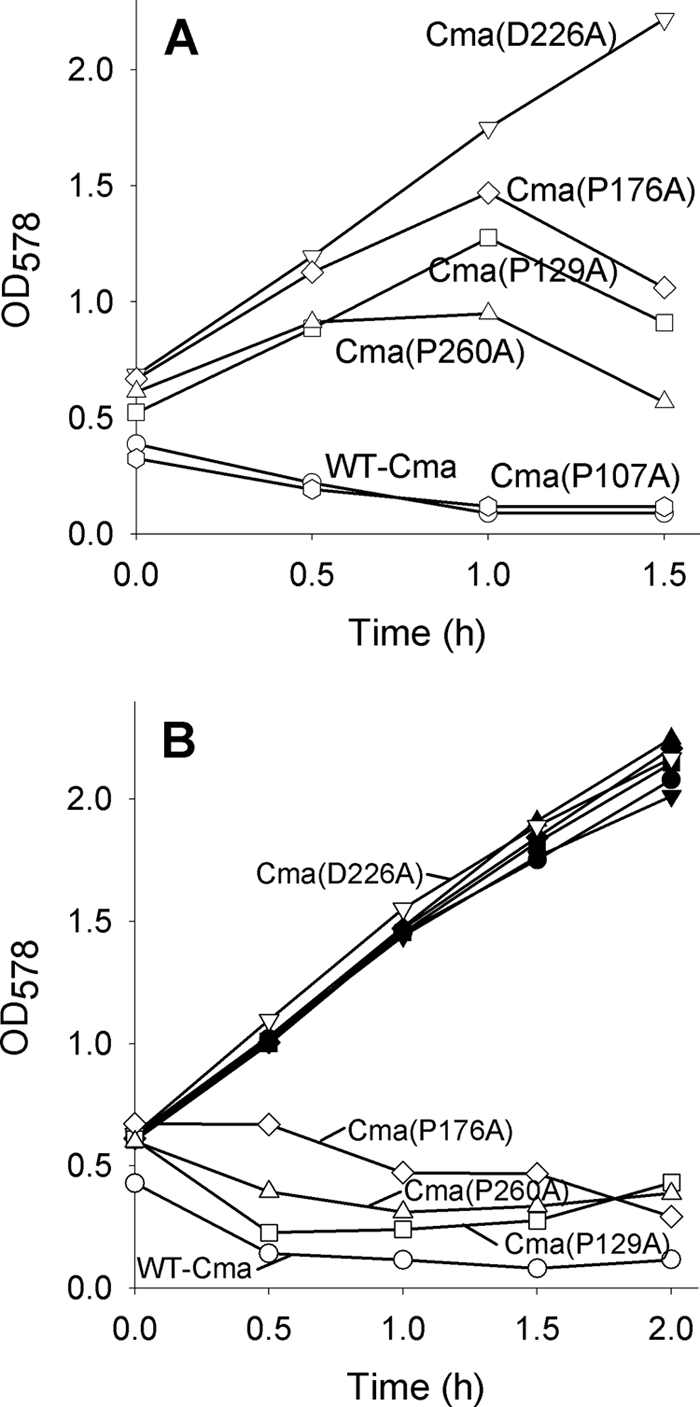
Growth inhibition and lysis of E. coli Mo3 fkpA (filled symbols) and E. coli MB97 fhuA (open symbols) by Cma and its mutant derivatives fused to the MalE signal sequence after induction of Cma synthesis by 0.001% arabinose (A) and 0.01% arabinose (B). Samples were withdrawn at the indicated times, and the optical density (OD) was measured at 578 nm.
Determination of PPIase Activity in Vitro
The cis-trans isomerization of the peptides p-aminobenzoyl-CFPVC-(NO2)Tyr-NH2 (P is Pro-176), p-aminobenzoyl-CKP)AC-(NO2)Tyr-NH2 (P is Pro-107), and p-aminobenzoyl-CLPGC-(NO2)Tyr-NH2 (P is Pro-260) was induced by reducing the disulfide bond with 50 mm DTT in 35 mm HEPES, pH 7.8. The time course of fluorescence of 1 μm concentrations of each peptide at 10 °C was followed at 420 nm after the peptides were transferred from the peptide stock solution in DMSO to the final buffer solution. The excitation wavelength was 320 nm, and the spectral bandwidth was 5 nm. Data were analyzed by single exponential nonlinear regression using a Sigma Plot Scientific Graphing System. The samples contained 31–124 μm of FkpA and its derivatives. kcat/Km was determined by evaluating the linear dependence of kenz from the concentration of FkpA and its derivatives. kenz is the catalyzed first-order rate minus the uncatalyzed rate. kcat/Km was calculated from kenz/Eo, where Eo is the FkpA concentration. Each data point represents the mean of three independent measurements that differed less than 10%.
Cleavage of Cma by Proteinase K
Cma and its derivatives tagged with His6 at the N terminus were purified on Ni-NTA-agarose; 20 μg of each protein was incubated with 0.01 μg of proteinase K for 13 min on ice and then analyzed by SDS-PAGE.
Circular Dichroism (CD)
CD spectra of electrophoretically homogeneous wild-type and mutant Cma proteins were recorded in a Jasco spectral polarimeter model J-810 at 190–240 nm and 20 °C. Temperature-dependent denaturation was measured at 220 nm between 10 and 70 °C.
Denaturation of Cma and Cma Mutants by Urea
Proteins (1 mg ml−1) dissolved in 20 mm potassium phosphate, 0.5 mm MgCl2, 2 mm mercaptoethanol, 150 mm NaCl, or in the same buffer containing 1–8 M urea were incubated for 15 min at 20 °C. The samples were centrifuged, and the fluorescence emission spectrum was measured between 300 and 500 nm (1 nm bandwidth), with excitation at 280 nm (3 nm bandwidth) in a Jasco spectrofluorometer FP-6500. The values given are the average of five determinations.
RESULTS
Isolation of Pro → Ala Colicin M (Cma) Mutants
We previously have shown that FkpA is required for killing E. coli K-12 by Cma (11). If periplasmic Cma activity depends on the FkpA prolyl cis-trans isomerase function and not, or not only on the FkpA chaperone function, replacement of the proline residue that is cis-trans-isomerized should prevent activation. To test this premise we replaced each of the 15 proline residues of Cma individually with alanine. The activities of the Cma mutant proteins encoded on pMLD237 were first tested with crude extracts of the E. coli pMLD237 transformant BL21(DE3) fkpA after IPTG induction of cma transcription. The fkpA mutation prevented the producer cells to be killed by Cma. In the crude extracts the mutant Cma was the dominant protein (supplemental Fig. S1). 10-Fold dilutions of each mutant Cma protein sample were spotted on nutrient agar seeded with Cma-sensitive E. coli AB2847. Four mutants (Cma(P9A), Cma(P11A), Cma(P28A), and Cma(P157A)) showed higher activities than wild-type Cma, and seven mutants had activities similar to that of the wild type (Table 2); these mutants were not studied further. Cma(P129A), Cma(P176A), and Cma(P260A) displayed 1% of the wild-type activity, and Cma(P107A) showed 10% of the wild-type activity (Table 2). Samples of the Cma(P → A) double mutants Cma(P129A,P176A), Cma(P129A,P260A), and Cma(P176A,P260A) were completely inactive (data not shown). The location of the P107A, P129A, P176A, and P260A mutations in the Cma crystal structure is shown in Fig. 1.
TABLE 2.
Activities of the Cma Pro → Ala mutants
Clear zones of growth inhibition are listed.
| Cma | Diameter of the lysis zones |
|||
|---|---|---|---|---|
| 100 dilution | 101 dilution | 102 dilution | 103 dilution | |
| mm | ||||
| Wild-type | 15 | 11 | 8 | 0 |
| P9A | 14 | 11 | 9 | 8 |
| P11A | 15 | 13 | 10 | 9 |
| P16A | 14 | 11 | 9 | 0 |
| P28A | 13 | 10 | 8 | 6 |
| P31A | 12 | 10 | 8 | 0 |
| P35A | 13 | 11 | 8 | 0 |
| P48A | 15 | 11 | 8 | 0 |
| P70A | 13 | 10 | 7 | 0 |
| P94A | 12 | 10 | 8 | 0 |
| P107A | 13 | 11 | 0 | 0 |
| P109A | 13 | 10 | 8 | 0 |
| P129A | 10 | 0 | 0 | 0 |
| P157A | 15 | 12 | 10 | 8 |
| P176A | 10 | 0 | 0 | 0 |
| P260A | 10 | 0 | 0 | 0 |
FIGURE 1.

Location of the mutations in the crystal structure of Cma studied here (8). The predicted structural and functional domains are indicated; orange, N-terminal translocation domain; blue, central receptor binding domain; magenta, C-terminal activity domain; side chains of Pro-107, Pro-129, Pro-176, Pro-260, and Asp-226, which were replaced by alanine in this study are marked green (C-atoms, blue (nitrogen), and red (oxygen).
To examine whether overproduction of FkpA increases the toxicity of the Cma(P → A) mutants, crude extracts of wild-type and the three Cma mutant proteins with the lowest activity were spotted onto nutrient agar plates seeded with E. coli AB2847 carrying in addition to chromosomal fkpA, also fkpA encoded on plasmid pYH15 and the temperature-inducible T7 RNA polymerase gene on plasmid pGP1-2. The toxicity of each Cma protein for the FkpA-overproducing strain and for untransformed strain AB2847 was the same (data not shown), i.e. the amount of chromosomally encoded FkpA was sufficient to confer the respective sensitivity to Cma and its mutant derivatives.
Replacement of Pro-129, Pro-176, and Pro-260 by glycine or serine instead of alanine resulted in mutants with activities similar to those of the P → A mutants (data not shown). The higher flexibility of the glycine bonds did not facilitate restoration of the active Cma conformation after transfer across the outer membrane, and the similarity of serine in the hydrogen-bonded form to proline did not increase Cma activity provided the proline residues were essential for Cma phosphatase activity (see also “Discussion”). In addition, Pro-176 was replaced by leucine, valine, tyrosine, phenylalanine, lysine, and aspartate. The Cma derivatives showed 1% in vivo activity on FkpA wild-type cells, except P176F and P176Y, which displayed 10% of Cma wild-type activity. The activity of all mutant Cma proteins was zero on FkpA mutant cells.
FkpA is not required to produce an active Cma. The activity of wild-type Cma released into the medium or isolated from the cytoplasm is the same when it is produced in wild-type and fkpA mutant cells (11). FkpA is only required after uptake of Cma into the periplasm. Nevertheless, we examined whether the low activity of the four Cma P → A mutants was altered when they were produced in E. coli BL21 FkpA+ cells carrying a fhuA mutation, which confers resistance to Cma. Killing E. coli AB2847 by the Cma mutant proteins synthesized in E. coli BL21 fhuA was the same (1 and 10% activity) as killing by the Cma mutant proteins synthesized in parallel by E. coli BL21 fkpA, i.e. lack of FkpA did not affect the activity of the Cma mutant proteins.
Structural Properties of the Cma P → A Mutant Proteins/Crystal Structures of Cma(P107A) and Cma(P176A)
Because the reduction in toxicity of the four mutant proteins might be related to changes in the structures, we determined the crystal structures of Cma(P107A) and Cma(P176A). We selected these two proteins because Pro-107 forms the only proline cis bond in the Cma structure (8), and as will be shown below, the Pro-176 bond is most likely a target of FkpA. Cma, Cma(P107A), and Cma(P176A) were expressed; the production of similar amounts of the almost inactive Cma derivatives and wild-type Cma indicated that replacement of the proline residues by alanine did not cause gross structural changes that affected the yield and activity of the proteins. We determined an improved structure of wild-type Cma under conditions slightly different from those previously used (8) to 1.67 Å (Fig. 2A). The published structures of Cma (Ref. 8; PDB entries 3DA4 and 3DA3) showed partial disorder, in particular in the flexible N-terminal domain, which is frequently not seen in colicin structures. The new wild-type structure, in contrast, showed an excellent defined structural homogeneity for all main and side-chain atoms (Fig. 2A). We determined the crystal structures of mutants Cma(P107A) and Cma(P176A) to 2.3 Å (eight monomers in the asymmetric unit) and 2.15 Å (two monomers in the asymmetric unit), respectively. Crystallographic data are summarized in Table 3. The root mean square deviation between the mutant (P107A and P176A) and the wild-type structures was 0.3 and 0.2 Å2, respectively, including all Cα-atoms of the molecule.
FIGURE 2.

A, ribbon model representation of the newly determined Cma wt structure in a side view illustration (PDB 2XMX) is shown. The structure consists of three domains, the N-terminal translocation domain (in green; T), the intermediate receptor binding domain (blue; R), and the C-terminal activity domains (orange; R). The secondary structure elements and N (NT) and C termini (CT) of the structure as well as the two residues mutated are marked accordingly (α1-α5, β1-β6, NT, CT, P107, P176). B, superposition of the three structures represented as ribbon models (PDB 2XTR). The wt structure is color-coded in orange, the P107A mutant structure is in blue, and the P176A structure is in red. C, zoom into the region of the extended loop connecting the receptor binding and activity domain is shown. Wt and mutant structures are displayed in the same orientation and color-coded as in B, and adjacent residues of the mutant (P107A) are shown in ball-and-stick representation. D, close-up of the second mutation Pro-176 located in the β1-sheet of the protein is shown. wt and mutant structures are displayed in the same orientation and color coded as in B; adjacent residues of the P176A mutant are shown in a ball-and-stick representation.
Superposition of the Cma wild-type and mutant protein structures revealed identical folding (Fig. 2B). Analysis of the local environment of each mutation revealed that the Lys-Pro cis bond in wild-type Cma was converted to a Lys-Ala trans bond in Cma(P107A), which caused a local distortion of the loop region that connects the receptor binding and the activity domains (Fig. 2C). The structural changes were confined to the region around Pro-107 and elicited no long range structural transitions up to the predicted active center around Asp-226 (Fig. 1) (8, 9, 29). In contrast, the P176A mutation had no visible effect on the local backbone geometry of the β3 strand of Cma, as demonstrated in a section of Cma(P176A) overlaid by wild-type Cma (Fig. 2D). The entire structures were almost identical, which indicates that no conformational change inactivated Cma(P176A).
Structural Properties of the Cma P → A Mutant Proteins in Solution
Although the crystal structure of Cma(P176A) and most likely also of Cma(P107A) excluded structural alterations of the Cma activity domain as the cause of the low toxicity, we examined the structures of the three mutants with 1% wild-type activity in solution using proteinase K degradation, circular dichroism (CD), and tryptophan fluorescence spectroscopy after denaturation with urea. Cma(P107A) was not included in all assays because it displayed a Cma activity higher (10%) than that of the other mutant proteins (1%).
We chose proteinase K cleavage conditions such that Cma was not completely digested; instead, fragments were formed, of which the 24-kDa fragment resulted from Cma truncation at the N terminus (7). By analyzing the formation of this intermediary product instead of analyzing complete digestion, differences in the structure of Cma and its mutant forms should become more apparent. No differences in the cleavage pattern of wild-type Cma and the Cma mutants were observed (supplemental Fig. S2).
Next, we used CD spectroscopy to discern whether the mutant Cma proteins structurally differed from wild-type Cma. The far-UV CD spectra of the four Cma mutants and the Cma wild-type were the same at 20 °C (data not shown). However, at higher temperatures, the CD spectra indicated that Cma(P107A) (Tm = 51.8 °C) and Cma(P129A) (Tm = 48.8 °C) denatured at a lower temperature than Cma (Tm = 55.8 °C), Cma(P176A) (Tm = 55.4 °C), and Cma(P260A) (Tm = 55.0 °C) (supplemental Fig. S3).
We furthermore examined structural changes in the mutant proteins by measuring the fluorescence of urea-denatured proteins. The fluorescence emission maximum shifted to the red from 320 to 355 nm for Cma(P107A) and Cma(P129A) between 4 and 5 m urea and for Cma(P176A), Cma(P260A), and wild-type Cma between 5 and 6 m urea (supplemental Fig. S4). The lower urea stability of Cma(P107A) and Cma(P129A) agrees with their lower stability at higher temperatures.
Translocation of the Cma P → A Mutant Proteins Across the Outer Membrane
We examined whether the Cma P → A mutants were defective in binding to the FhuA receptor and in translocation across the outer membrane. Receptor binding was determined by competition of Cma with the antibiotic albomycin, which binds to the same FhuA receptor as Cma. E. coli Mo3 fkpA40 was used because the fkpA deletion confers resistance to Cma (11). Growth inhibition was, therefore, confined to the action of albomycin. Albomycin inhibited growth of E. coli Mo3 (Fig. 3). Wild-type Cma prevented growth inhibition by albomycin, as did Cma(P107A), Cma(P176A), and Cma(P260A). These proteins probably interfered with binding of albomycin to FhuA. In contrast, Cma(P129A) failed to interfere with albomycin. The low activity of Cma(P129A), therefore, most likely results from poor binding to FhuA.
FIGURE 3.

Binding of wild-type and mutant Cma proteins to the FhuA receptor in a competition assay with albomycin. Exponentially growing E. coli Mo3 fkpA40 in LB medium untreated (■) and treated with 1 μg ml−1 albomycin (▴), treated with 0.1 mg ml−1 wild-type Cma and albomycin (□), with mutant Cma(P107A) and albomycin (♢), with mutant Cma(P129A) and albomycin (●), with mutant Cma(P176A) and albomycin (○), and with mutant Cma(P260A) and albomycin (▵). Growth was monitored spectrophotometrically. The arrows indicate the addition of the Cma samples and albomycin.
Cma and albomycin not only share the common FhuA receptor, but also both use the Ton system for uptake. It is unlikely that albomycin uptake was inhibited because uptake of Cma, and its derivatives used up TonB. TonB can simultaneously interact with several receptors without substantially decreasing their transport rate (30). Reduction of the transport rate of a substrate was only observed after strong overexpression of another TonB-dependent transport system (30). Nevertheless, we determined whether Cma(P176A) and Cma(P260A) interfered with the activity of the Ton system. The competition assay was done with colicin B instead of albomycin; colicin B binds to the FepA receptor and not to FhuA but uses the Ton system for uptake. The Cma P → A mutants did not reduce growth inhibition of the Cma-resistant fkpA mutant Mo3 by colicin B, which indicates that the Cma mutant proteins do not interfere with the Ton system step of albomycin uptake. Interference of Cma with the target site of albomycin is also excluded as albomycin inhibits the seryl-tRNA synthetase; Cma, on the other hand, cleaves the phosphate ester bond between bactoprenol and the murein precursor.
Lack of Translocation Versus Lack of Phosphatase Activity
To differentiate between lack of translocation across the outer membrane and lack of phosphatase activity, we used an osmotic shock procedure. Transferring E. coli cells from a medium of high osmolarity to a medium of low osmolarity renders their outer membrane temporarily permeable to proteins. The procedure was developed and widely used to release periplasmic proteins from cells (27) but can also be used to introduce proteins into cells, as has been shown for colicin E3 (28) and Cma (7, 10, 29). The uptake of Cma by osmotic shock bypasses the requirement for the FhuA outer membrane receptor protein and the energizing Ton system. A variation of this procedure (10) was recently used to differentiate between Cma uptake and activity mutants (9). We purified wild-type and mutant proteins to electrophoretic homogeneity (supplemental Fig. S2) and used the same amounts of protein in the osmotic shock experiments. Osmotic shock treatment rendered cells sensitive to Cma(P107A), Cma(P129A), and Cma(P260A) (0.03 or 0.04% survivors) and to Cma(P176A) but to a much lower extent (0.4% survivors) (Table 4). Killing by Cma(P129A) after osmotic shock agrees with its inability to kill cells under normal conditions because of its failure to bind to FhuA, which is not required to enter osmotically shocked cells. Because Cma(P107A) and Cma(P260A) bound to FhuA but hardly killed cells under normal conditions and were able to kill osmotically shocked cells after being unspecifically translocated into the periplasm, the uptake of these mutant proteins across the outer membrane must be impaired after binding to FhuA. We conclude that Cma(P107A), Cma(P129A), and Cma(P260A) show low activities because they are poorly taken up into the periplasm.
TABLE 4.
Activity of selected Cma mutant proteins introduced into cells by osmotic shock
The percent of surviving cells of AB2847, its fkpA40 derivative, and the pPG773 cmi transformant were determined by plating after treatment with purified Cma wt and Cma mutant proteins transferred into cells by osmotic shock (+ shock) or without shock treatment (− shock). The average percentage of survivors of all experiments after shock treatment without Cma was 6.0.
| Conditions | Surviving cells |
||
|---|---|---|---|
| AB2847 | AB2847 fkpA | AB2847 pPG773 cmi | |
| Percent of input | |||
| Cma wt + shock | 0 | 1.5 | 6.1 |
| Cma wt − shock | 0.2 | 93.3 | 97.4 |
| Cma(P107A) + shock | 0.03 | 1.8 | 8.8 |
| Cma(P107A) − shock | 1.7 | 91.3 | 93.9 |
| Cma(P129A) + shock | 0.04 | 0.8 | 7.0 |
| Cma(P129A) − shock | 1.9 | 95.2 | 95.8 |
| Cma(P176A) + shock | 0.4 | 0.6 | 8.9 |
| Cma(P176A) − shock | 9.4 | 90.4 | 97.3 |
| Cma(P260A) + shock | 0.03 | 0.2 | 5.8 |
| Cma(P260A) − shock | 1.4 | 90.6 | 100 |
Wild-type and mutant Cma proteins were osmotically shocked into an fkpA deletion mutant to test whether FkpA was only required for the energy-coupled transfer of Cma across the outer membrane or also required after unspecific transfer by osmotic shock. The Cma proteins were osmotically shocked into E. coli Mo3 fkpA40 under conditions identical to those for the fkpA wild-type strain. Killing of the fkpA mutant by wild-type Cma and the proline Cma mutant proteins was strongly reduced (Table 4), which shows that regardless of the route across the outer membrane (energy-coupled import or entry by osmotic shock) FkpA was required for wild-type and mutant Cma activity in the periplasm. The results also demonstrated the reliability of the osmotic shock procedure as the Cma activities specifically required FkpA. However, the Cma proteins displayed residual Cma activities; 0.2–1.8% survival compared with 6% survival after osmotic shock in the absence of the Cma proteins (Table 4). After osmotic shock, a fraction of the Cma molecules was active in the absence of FkpA.
This conclusion was further supported by comparison of the surviving fkpA mutant cells with cells that synthesize the Cmi colicin M immunity protein. Cmi renders cells resistant to imported Cma (31). Cmi is located in the periplasm and anchored to the cytoplasmic membrane by its N-terminal hydrophobic sequence (32, 33). Membrane fixation brings Cmi to the site where Cma cleaves its substrate and prevents release of Cmi by osmotic shock. We used the previously constructed plasmid pPG773, in which the translocation and membrane anchor of the penicillin-binding protein PBP3 was fused to the periplasmic activity domain of Cmi (33). Transformants carrying this plasmid confer immunity without induction of T7 polymerase synthesis because it contains an E. coli polymerase promoter. Wild-type Cma and the Cma P → A mutant proteins were completely inactive on plates seeded with E. coli AB2847 pPG773 cmi. After osmotic shock, Cma and the Cma derivatives were inactive (Table 4). The percentage of survivors (average 7%) was in the range of the survivors after osmotic shock treatment without Cma (6%). This shows that the Cma proteins that entered cells by osmotic shock were completely inactivated by Cmi. The fraction of surviving cells into which Cma and the Cma derivatives entered via the normal, energy-coupled route was similar in the fkpA mutant and the cmi transformants (92 and 100%, respectively; Table 4). The almost complete resistance of the fkpA mutant to Cma and its mutant derivatives that enter the cell by energy-coupled uptake (− shock in Table 4) and the partial sensitivity of the fkpA mutant when Cma and its mutant derivatives are osmotically shocked into the cell support the conclusion that a fraction of Cma osmotically shocked into cells is active in the absence of FkpA. This fraction assumes an active conformation without the assistance of FkpA.
Cma(P176A) differed from the other Cma P → A mutants in that its low activity after osmotic shock did not differ in wild-type and fkpA cells (0.4 versus 0.6% survivors). The percentage of survivors of the immune cells treated with Cma(P176A) reached the level obtained with wild-type Cma and the other Cma P → A mutant proteins (Table 4). This suggests that the residual activity of Cma(P176A) in Cmi-deficient cells is Cma-specific.
The number of survivors in the control of the osmotic shock procedure, i.e. without osmotic shock treatment (Table 4), cannot be quantitatively compared with the number of survivors obtained by the plate test (Table 2). In the osmotic shock procedure, test cells are exposed to Cma for 15 min at 4 °C, diluted, and then plated; in the plate test, cells are exposed to Cma overnight at 37 °C.
Killing of Cells by Secreted Wild-type and Cma P → A Mutants
Colicins, including Cma, are not secreted but are rather unspecifically released by partial lysis of the producing cells. Lysis is caused by a lysis protein that is not encoded on pColBM plasmids (26). Therefore, 90% of the Cma stays inside cells. To examine whether Cma translocated across the outer membrane only requires FkpA or whether Cma secreted across the cytoplasmic membrane also requires FkpA, we fused wild-type and mutant Cma proteins to the signal sequence of the periplasmic MalE-binding protein. In a previous paper of our laboratory (34) Cma was fused to the signal peptide of the murein lipoprotein and killed cells of fhuA, tonB, and exbBD uptake mutants but left a tolM, now designated fkpA, mutant unaffected. The Cma constructs chosen in this paper contained the MalE signal peptide for secretion and C-terminal His6 for purification and are designated as secreted Cma (Cmasec). We cloned the fusion gene downstream of the araC arabinose promoter, which tightly controls transcription of downstream genes. This was important for cloning as uninduced transcription might have resulted in cell lysis and failure to obtain transformants. In addition, the level of expression can be modulated over a wide range (up to 1200-fold) depending on the concentration of the arabinose inducer (35). Cmasec transformants grew well in the uninduced state and stopped growing upon induction of cmasec transcription with 0.1% arabinose. Growth inhibition could be caused by the phosphatase activity of Cma or by Cma jamming the secretion pathway by folding in the cytoplasm before secretion, as has for example been shown for the outer membrane LamB protein fused to a large C-terminal fragment of β-galactosidase. In this case cells become sensitive to induction by maltose as enhanced synthesis of the LamB-LacZ hybrid protein impairs secretion of LamB-LacZ and of other proteins (36). Indeed, 0.1% arabinose inhibited not only the growth of cells that synthesized wild-type Cmasec but also of cells that synthesized inactive mutant Cmasec(D226A) (data not shown). Asp-226 is located in the predicted active center of Cma and is essential for Cma activity (8, 9, 29). At an arabinose concentration of 0.001%, Cma(D226A) did not inhibit growth, but wild-type Cma and Cma(P107A), with 10% activity in the plate test, completely inhibited growth and lysed cells (Fig. 4A). Cma(P129A), Cma(P176A), and Cma(P260A) displayed residual activities, with Cma(P176A) having the lowest. To compare growth inhibition of wild-type with an fkpA mutant, we increased the level of the Cma mutant proteins by enhancing transcription with a higher arabinose concentration (0.01%). At this concentration, Cma(D226A) failed to inhibit growth (Fig. 4B), which indicated that growth inhibition by interference with protein secretion played no role. The Cma P → A mutant proteins now inhibited growth, which was abolished in the fkpA strain. Regardless of whether Cma and its mutant derivatives were imported across the outer membrane or secreted across the cytoplasmic membrane, they required FkpA to be active in the periplasm.
Catalysis of Prolyl Cis-Trans Isomerization of the Phe-Pro-176 Bond by FkpA
Cma(P176A) shocked into the periplasm had a 10-fold lower activity than the other Cma P → A mutants. This residual activity was independent of FkpA, which suggested that the replacement of Pro-176 by alanine affected interaction with FkpA. The Phe-Ala-176 bond is not a substrate of FkpA since FkpA-type PPIases do not catalyze cis-trans isomerization of nonprolyl peptide bonds (37). To test whether the Phe-Pro-176 bond serves as a substrate for the FkpA PPIase, we used a novel peptide assay. Disulfide-constrained synthetic pentapeptide derivatives of the amino acid sequence p-aminobenzoyl-Cys-Xaa-Pro-Yaa-Cys-(NO2)Tyr-NH2 exhibit a high content of Xaa-Pro cis conformation (38). Upon opening of the disulfide bridge by reduction, the cis/trans ratio declines to an open-chain cis isomer level of ∼15% (39). Only in the cis conformation is the fluorescence of the p-aminobenzoyl group efficiently quenched by the nitrotyrosine amide. After reduction of the disulfide bridge, fluorescence increases during cis/trans interconversion of the Xaa-Pro bond. This slow reaction is accelerated by PPIases, which use the peptides as substrates.
We compared spontaneous and FkpA-catalyzed cis-trans isomerization of the substrate p-aminobenzoyl-CFPVC-(NO2)Tyr-NH2, which represents the amino acid sequence adjacent to Cma Phe-Pro-176-Val, and used as controls p-aminobenzoyl-CKPAC-(NO2)Tyr-NH2 and p-aminobenzoyl-CLPGC-(NO2)Tyr-NH2, which represent the amino acid sequences adjacent to Cma Lys-Pro-107-Ala and Cma Leu-Pro-260-Gly, respectively. FkpA was purified to electrophoretic homogeneity by Ni-NTA-agarose chromatography and subsequent gel filtration on a Superdex 75 column. FkpA considerably accelerated cis-trans isomerization of the prolyl bond in Phe-Pro-176-Val as compared with uncatalyzed spontaneous cis-trans isomerization (Fig. 5). The kcat/Km value of the catalyzed reaction was similar to the kcat/Km values of FKBP-type PPIases with optimal substrates (40). The kcat/Km value was 14- and 12-fold more efficient than the kcat/Km values of the Lys-Pro-107-Ala and Leu-Pro-260-Gly peptides, respectively (Fig. 6, Table 5). The low value of the Lys-Pro-107-Ala peptide is particularly interesting because it indicates that the only cis prolyl bond in Cma is unlikely to be the site of FkpA cis-trans isomerization.
FIGURE 5.

Time-dependent spontaneous cis-trans isomerization of peptide p-aminobenzoyl-CFPVC-(NO2)Tyr-NH2 (lower curve) and FkpA-catalyzed cis-trans isomerization of peptide p-aminobenzoyl-CFPVC-(NO2)Tyr-NH2 (upper curve) after reduction of the disulfide bond with DDT.
FIGURE 6.

Determination of kcat/Km of FkpA-catalyzed cis-trans isomerization of the Phe-Pro-176 bond in p-aminobenzoyl-CFPVC-(NO2)Tyr-NH2, the Lys-Pro-107 bond in p-aminobenzoyl-CKPAC-(NO2)Tyr-NH2, and the Leu-Pro-260 bond in p-aminobenzoyl-CLPGC-(NO2)Tyr-NH2 by measuring the linear dependence of kenz on the concentration of FkpA.
TABLE 5.
Catalysis of prolyl cis-trans isomerization of Cma peptides by FkpA, FkpA43, and FkpA313
ND, not determined.
| Cma prolyl bond | Kcat/Km FkpA | Kcat/Km FkpA43 | Kcat/Km FkpA313 |
|---|---|---|---|
| m−1s−1 | m−1s−1 | m−1s−1 | |
| Phe-Pro-176 | (1.38 ± 0.12) × 106 | (6.61 ± 0.33) × 105 | (3.45 ± 0.43) × 103 |
| Leu-Pro-260 | (1.15 ± 0.11) × 105 | (1.34 ± 0.17) × 105 | (5.11 ± 1.44) × 103 |
| Lys-Pro-107 | (9.69 ± 0.67) × 104 | ND | ND |
To further support the role of FkpA PPIase activity for Cma activity, we determined the catalytic efficiency toward the Phe-Pro-176-Val and Leu-Pro-260-Gly derivatives using the FkpA313 mutant protein, which contains a G148D replacement in the PPIase domain and does not mediate Cma sensitivity to cells (11). Only a very low in vitro PPIase activity of FkpA313 was observed with both peptides; the activity amounted to 0.4 and 0.2% that of wild-type FkpA (Table 5). The data were further corroborated by the temperature-sensitive mutant FkpA43, which confers 10% activity to Cma at 30 °C and no activity at 42 °C (11). It displayed a 2-fold lower PPIase activity than wild-type FkpA for Phe-Pro-176-Val and a 5-fold lower activity for Leu-Pro-260-Gly than for Phe-Pro-176-Val (measured at 10 °C) (Table 5). The near inactivity of mutant Cma(P176A) in lysing cells and the high efficiency of FkpA in the catalysis of the cis-trans isomerization of Phe-Pro-176-Val peptide suggest that Phe-Pro-176 is the Cma peptide bond that is most likely catalytically cis-trans isomerized by FkpA.
DISCUSSION
Periplasmic folding helper proteins interact with outer membrane proteins, and a considerable degree of redundancy and overlap in their function is observed (41–49). For the periplasmic PPIases FkpA, PpiA, PpiD, and SurA of E. coli, no specificity for a distinct protein has been found, and no defined prolyl bond of a bacterial protein cis-trans isomerized by a PPIase has been identified yet. The strict dependence of Cma toxicity on FkpA allowed us to search for a correlation between Cma activity and FkpA catalysis of cis-trans isomerization of a specific prolyl bond of Cma. Using such an approach, we could identify a critical prolyl bond of Cma.
The 15 Cma proline-to-alanine replacement mutants encompassed all proline residues in Cma. Three mutants showed a strongly reduced (1%) killing of E. coli. A fourth mutant, Cma(P107A), had 10% of Cma wild-type activity in plate assays. This mutant was interesting because Pro-107 forms the only prolyl cis peptide bond in Cma. Strongly increased activity after bypassing the energy-coupled uptake by osmotic shock suggested that three mutant proteins (Cma(P107A), Cma(P129A), and Cma(P260A)) were impaired in uptake. Indeed, Cma(P129A) did not bind to the FhuA receptor. The translocation efficiency of Cma(P107A) was probably affected by the isomerization of the Lys-Pro cis bond to a Lys-Ala trans bond even though this conversion altered the protein crystal structure only locally. Because the molecular mechanism of Cma translocation and in fact of any colicin across the outer membrane is unknown, it is not clear to what extent regions other than the translocation domain (residues 1–35) contribute to uptake across the outer membrane. The same reasoning applies to Cma(P260A), whose mutation is not located in the translocation domain but, rather, in the activity domain.
Cma(P176A) was the only mutant with very low activity (1%) that remained nearly inactive when it was osmotically shocked into cells. It also displayed the lowest activity when it was equipped with a signal sequence, which allowed it to be secreted from the cytoplasm into the periplasm. Because the crystal structure of Cma(P176A) did not differ from the crystal structure of wild-type Cma, it is unlikely that the mutation affects the active center of Cma(P176A). Pro-176 is exposed at the Cma surface (Fig. 1) and its conversion to A176 does not change the Cma structure beyond the mutation site.
Previous results indicate that Pro-176 is not in the active site of Cma (8, 9, 29). Random and site-directed mutagenesis of cma localized the active center around residue D226, a strictly conserved residue in a region in which the Cma-type sequences display the highest level of identity (8, 9). Pro-176 is located far from Asp-226 (Fig. 1). If the P176A mutation affects Cma activity independent of catalysis by FkpA, the mutation must have a long distance structural effect on the active center; such an effect was not observed in the crystal structure. We also found no evidence for a structural alteration of Cma(P176A) in solution; it acted like wild-type Cma with regard to proteinase K digestion, thermal denaturation, and urea denaturation.
FkpA exhibits PPIase and chaperone activities with artificial substrates (13, 15, 16). FkpA reactivates Cma denatured with 5 m guanidine hydrochloride, which is inhibited by FK506 (11), an inhibitor of the PPIase activity. In addition, the G148D mutation in the FkpA PPIase center abolishes Cma renaturation (11) and confers resistance to Cma. Additional mutants in the PPIase domain and only in the PPIase domain, G176D and G180D, were isolated by independent random mutagenesis, which did not confer Cma sensitivity.3 Arié et al. (13) isolated by site-specific mutagenesis FkpA mutants I174S and I174S/G176S and found no or only 5% PPIase activity but unaltered chaperone activity using a standard protease-coupled peptide assay. These mutants strongly favor the PPIase activity in Cma activation. In this study Phe-Pro-176 is the only bond left for cis-trans isomerization. The high catalytic efficiency of FkpA toward the Phe-Pro-176 peptide substrate, which was 14- and 12-fold higher than toward the Lys-Pro-107 and Leu-Pro-260 peptide substrates agrees with this proposal.
Pro-176 is exposed at the surface of Cma (Fig. 1) and can, thus, be approached by FkpA. Pro-107 and -260 are also close to the surface but are less exposed than Pro-176. Pro-129 is buried inside Cma and cannot be reached by FkpA unless strong structural alterations are induced by binding of Cma to FkpA before catalysis of cis-trans isomerization. The location of the Phe-Pro-176 bond makes it the most likely candidate for cis-trans isomerization.
Despite strong evidence for the PPIase activity of FkpA in Cma activation, its chaperone activity should also be considered. Cma(P176A) transferred into the periplasm by osmotic shock did not respond to FkpA as the residual activity was not altered in the fkpA deletion mutant (Table 4). However, Cma(P176A) secreted from the cytoplasm into the periplasm by a signal sequence fused to Cma(P176A) stopped growth of the fkpA wild-type cells and failed to stop growth of the fkpA mutant. Although this activity was weak and was only observed after increased synthesis of Cma(P176A), it nevertheless demonstrates some response of Cma(P176A) to FkpA. We assign this activity to the chaperone function of FkpA in the periplasm. However, an exclusive FkpA chaperone activity for activation of Cma would not show such a high site specificity as found for the inactive point mutants in the PPIase domain.
Cma must unfold at least partially during translocation across the outer membrane and, when fused to a signal sequence, during translocation across the cytoplasmic membrane. Unfolding may convert the Phe-Pro-176 trans bond into a cis bond. Trans to cis isomerization also occurred in Cma mutant proteins in which Pro-176 was replaced by Ala, Gly, Leu, Val, Thr, Lys, and Asp. These derivatives were all inactive or nearly inactive because the lack of Pro prevented FkpA-catalized cis to trans isomerization during refolding in the periplasm. The conversion involves the PPIase, but the chaperone function of FkpA also contributes to Cma activation. Direct determination of the cis to trans conversion is hampered by the few molecules of Cma in the periplasm, which are sufficient to kill cells (20) and the much larger unspecific adsorption of Cma to the cells. In addition, we found no suitable wavelength at which fluorescence changes reflected structural changes during in vitro refolding of denatured Cma by FkpA (11). Cma refolding may contribute energy for translocation. In addition, FkpA may act as a chaperone that binds imported Cma and prevents proteolytic degradation of the unfolded protein. Multiple functions of FkpA (Cma refolding, binding of Cma in the periplasm, and positioning of Cma to the substrate site) best explain the complete dependence of Cma activity on FkpA.
Supplementary Material
Acknowledgments
We thank Dr. Reinhard Albrecht and Kerstin Bär for valuable help in protein crystallization, Dr. Andrei Lupas for generous support, and Karen A. Brune for critically reading the manuscript. We cordially thank the beamline staff from the Max Planck beamline PX10 at the Swiss Light Source for great help and beamline maintenance.
This work was supported by the Max Planck Society, the German Science Foundation (BR 30/25-1), and the Fonds der Chemischen Industrie.

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1–S4.
The atomic coordinates and structure factors (codes 2XMX and 2XTR) have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ (http://www.rcsb.org/).
S. I. Patzer, unpublished results.
- Cma
- colicin M
- PPIase
- peptidyl prolyl cis-trans isomerase
- Ni-NTA
- nickel-nitrilotriacetic acid
- Bis/Tris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
- FKBP
- FK506-binding protein.
REFERENCES
- 1. Schaller K., Höltje J. V., Braun V. (1982) J. Bacteriol. 152, 994–1000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Harkness R. E., Braun V. (1989) J. Biol. Chem. 264, 6177–6182 [PubMed] [Google Scholar]
- 3. El Ghachi M., Bouhss A., Barreteau H., Touzé T., Auger G., Blanot D., Mengin-Lecreulx D. (2006) J. Biol. Chem. 281, 22761–22772 [DOI] [PubMed] [Google Scholar]
- 4. Braun V. (1995) FEMS Microbiol. Rev. 16, 295–307 [DOI] [PubMed] [Google Scholar]
- 5. Braun V., Patzer S. I., Hantke K. (2002) Biochimie 84, 365–380 [DOI] [PubMed] [Google Scholar]
- 6. Postle K., Larsen R. A. (2007) Biometals 20, 453–465 [DOI] [PubMed] [Google Scholar]
- 7. Dreher R., Braun V., Wittmann-Liebold B. (1985) Arch. Microbiol. 140, 343–346 [DOI] [PubMed] [Google Scholar]
- 8. Zeth K., Römer C., Patzer S. I., Braun V. (2008) J. Biol. Chem. 283, 25324–25331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Barreteau H., Bouhss A., Gérard F., Duché D., Boussaid B., Blanot D., Lloubès R., Mengin-Lecreulx D., Touzé T. (2010) J. Biol. Chem. 285, 12378–12389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Braun V., Frenz J., Hantke K., Schaller K. (1980) J. Bacteriol. 142, 162–168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hullmann J., Patzer S. I., Römer C., Hantke K., Braun V. (2008) Mol. Microbiol. 69, 926–937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Danese P. N., Silhavy T. J. (1997) Genes Dev. 11, 1183–1193 [DOI] [PubMed] [Google Scholar]
- 13. Arié J. P., Sassoon N., Betton J. M. (2001) Mol. Microbiol. 39, 199–210 [DOI] [PubMed] [Google Scholar]
- 14. Bothmann H., Pluckthun A. (2000) J. Biol. Chem. 275, 17100–17105 [DOI] [PubMed] [Google Scholar]
- 15. Ramm K., Plückthun A. (2001) J. Mol. Biol. 310, 485–498 [DOI] [PubMed] [Google Scholar]
- 16. Saul F. A., Arié J. P., Vulliez-le Normand B., Kahn R., Betton J. M., Bentley G. A. (2004) J. Mol. Biol. 335, 595–608 [DOI] [PubMed] [Google Scholar]
- 17. Betton J. M. (2007) in The Periplasm (Ehrmann M. ed) pp. 141–149, American Society for Microbiology, Washington, D. C [Google Scholar]
- 18. Kleinschmidt J. H. (2007) in The Periplasm (Ehrmann M. ed) pp. 30–66, American Society for Microbiology, Washington, D. C [Google Scholar]
- 19. Mogensen J. E., Otzen D. E. (2005) Mol. Microbiol. 57, 326–346 [DOI] [PubMed] [Google Scholar]
- 20. Schaller K., Dreher R., Braun V. (1981) J. Bacteriol. 146, 54–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kabsch W. (2010) Acta. Crystallogr. D Biol. Crystallogr. 66b, 125–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Potterton E., Briggs P., Turkenburg M., Dodson E. (2003) Acta. Crystallogr. D Biol. Crystallogr. 59, 1131–1137 [DOI] [PubMed] [Google Scholar]
- 23. Emsley P., Cowtan K. (2004) Acta. Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 24. Laskowski R. A., Moss D. S., Thornton J. M. (1993) J. Mol. Biol. 231, 1049–1067 [DOI] [PubMed] [Google Scholar]
- 25. Kabsch W., Sander C. (1983) Biopolymers 22, 2577–2637 [DOI] [PubMed] [Google Scholar]
- 26. Thumm G., Olschläger T., Braun V. (1988) Plasmid 20, 75–82 [DOI] [PubMed] [Google Scholar]
- 27. Nossal N. G., Heppel L. A. (1966) J. Biol. Chem. 241, 3055–3062 [PubMed] [Google Scholar]
- 28. Tilby M., Hindennach I., Henning U. (1978) J. Bacteriol. 136, 1189–1191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Pilsl H., Glaser C., Gross P., Killmann H., Olschläger T., Braun V. (1993) Mol. Gen. Genet. 240, 103–112 [DOI] [PubMed] [Google Scholar]
- 30. Kadner R. J., Heller K. J. (1995) J. Bacteriol. 177, 4829–4935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Harkness R. E., Braun V. (1990) Mol. Gen. Genet. 222, 37–40 [DOI] [PubMed] [Google Scholar]
- 32. Olschläger T., Turba A., Braun V. (1991) Mol. Microbiol. 5, 1105–1111 [DOI] [PubMed] [Google Scholar]
- 33. Gross P., Braun V. (1996) Mol. Gen. Genet. 251, 388–396 [DOI] [PubMed] [Google Scholar]
- 34. Olschläger T. (1991) Arch. Microbiol. 156, 449–454 [PubMed] [Google Scholar]
- 35. Guzman L. M., Belin D., Carson M. J., Beckwith J. (1995) J. Bacteriol. 177, 4121–4130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Benson S. A., Bremer E., Silhavy T. J. (1984) Proc. Natl. Acad. Sci. U.S.A. 81, 3830–3834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Schiene-Fischer C., Habazettl J., Schmid F. X., Fischer G. (2002) Nat. Struct. Biol. 9, 419–424 [DOI] [PubMed] [Google Scholar]
- 38. Fischer G., Aumüller T. (2003) Rev. Physiol. Biochem. Pharmacol. 148, 105–150 [DOI] [PubMed] [Google Scholar]
- 39. Weisshoff H., Frost K., Brandt W., Henklein P., Mügge C., Frömmel C. (1995) FEBS Lett. 372, 203–209 [DOI] [PubMed] [Google Scholar]
- 40. Harrison R. K., Stein R. L. (1990) Biochemistry 29, 3813–3816 [DOI] [PubMed] [Google Scholar]
- 41. Chen R., Henning U. (1996) Mol. Microbiol. 19, 1287–1294 [DOI] [PubMed] [Google Scholar]
- 42. Justice S. S., Hunstad D. A., Harper J. R., Duguay A. R., Pinkner J. S., Bann J., Frieden C., Silhavy T. J., Hultgren S. J. (2005) J. Bacteriol. 187, 7680–7686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Qu J., Mayer C., Behrens S., Holst O., Kleinschmidt J. H. (2007) J. Mol. Biol. 374, 91–105 [DOI] [PubMed] [Google Scholar]
- 44. Rizzitello A. E., Harper J. R., Silhavy T. J. (2001) J. Bacteriol. 183, 6794–6800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Rouvière P. E., Gross C. A. (1996) Genes Dev. 10, 3170–3182 [DOI] [PubMed] [Google Scholar]
- 46. Schäfer U., Beck K., Müller M. (1999) J. Biol. Chem. 274, 24567–24574 [DOI] [PubMed] [Google Scholar]
- 47. Sklar J. G., Wu T., Kahne D., Silhavy T. J. (2007) Genes Dev. 21, 2473–2484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Stymest K. H., Klappa P. (2008) FEBS 275, 3470–3479 [DOI] [PubMed] [Google Scholar]
- 49. Vertommen D., Ruiz N., Leverrier P., Silhavy T. J., Collet J. F. (2009) Proteomics 9, 2432–2443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Studier F. W., Moffatt B. A. (1986) J. Mol. Biol. 189, 113–130 [DOI] [PubMed] [Google Scholar]
- 51. Mende J., Braun V. (1990) Mol. Microbiol. 4, 1523–1533 [DOI] [PubMed] [Google Scholar]
- 52. Hanahan D. (1983) J. Mol. Biol. 166, 557–580 [DOI] [PubMed] [Google Scholar]
- 53. Tabor S., Richardson C. C. (1985) Proc. Natl. Acad. Sci. U.S.A. 82, 1074–1078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Braun M., Endriss F., Killmann H., Braun V. (2003) J. Bacteriol. 185, 5508–5518 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.