

Abstract
Cyclophilin d (cypD)-deficient mice exhibit resistance to focal cerebral ischemia and to necrotic but not apoptotic stimuli. To address this disparity, we investigated isolated brain and in situ neuronal and astrocytic mitochondria from cypD-deficient and wild-type mice. Isolated mitochondria were challenged by high Ca2+, and the effects of substrates and respiratory chain inhibitors were evaluated on permeability transition pore opening by light scatter. In situ neuronal and astrocytic mitochondria were visualized by mito-DsRed2 targeting and challenged by calcimycin, and the effects of glucose, NaCN, and an uncoupler were evaluated by measuring mitochondrial volume. In isolated mitochondria, Ca2+ caused a large cypD-dependent change in light scatter in the absence of substrates that was insensitive to Ruthenium red or Ru360. Uniporter inhibitors only partially affected the entry of free Ca2+ in the matrix. Inhibition of complex III/IV negated the effect of substrates, but inhibition of complex I was protective. Mitochondria within neurons and astrocytes exhibited cypD-independent swelling that was dramatically hastened when NaCN and 2-deoxyglucose were present in a glucose-free medium during calcimycin treatment. In the presence of an uncoupler, cypD-deficient astrocytic mitochondria performed better than wild-type mitochondria, whereas the opposite was observed in neurons. Neuronal mitochondria were examined further during glutamate-induced delayed Ca2+ deregulation. CypD-knock-out mitochondria exhibited an absence or a delay in the onset of mitochondrial swelling after glutamate application. Apparently, some conditions involving deenergization render cypD an important modulator of PTP in the brain. These findings could explain why absence of cypD protects against necrotic (deenergized mitochondria), but not apoptotic (energized mitochondria) stimuli.
Keywords: Brain, Calcium, Microscopic Imaging, Mitochondria, Mitochondrial Apoptosis, Cyclophilin D, Permeability Transition Pore
Introduction
In 2005, four independent groups reported the beneficial effect of cyclophilin D (cypD)2 absence in transgenic mice, for an array of in vitro and in vivo pathologic stimuli (1–4). Ever since, the contribution of cypD in a variety of diseases has been strongly suggested or proven (for review, see Ref. 5), a momentum that was assisted by the wide availability of cypD knock-out (KO) mice. These studies converged to the conclusion that cypD-mediated mitochondrial permeability transition pore (PTP) regulates some forms of necrotic, but not apoptotic death. The notion in which PTP is involved in necrosis but not apoptosis has been originally suggested by the group of Crompton and colleagues (6).
A major difference among prerequisites for the manifestation of necrosis versus apoptosis is energy availability; a sufficient decline in energy reserves, primarily in ATP concentration, is a switch for a cell to die by necrosis rather than apoptosis (7, 8). Such an extensive decrease in ATP is invariably associated with loss of mitochondrial membrane potential, ΔΨm (9, 10). Mindful of the large increases in intracellular Ca2+ during cell injury (11) and the loss of ΔΨm preceding cell death (12), the conundrum appears that excessive Ca2+ induces PTP under conditions unfavorable for electrophoretic Ca2+ uptake by mitochondria (13). Some studies address this by proposing that in ischemia-reperfusion, Ca2+-induced PTP occurs during reperfusion of the affected tissue, but in several experimental models mimicking pathology, mitochondrial damage caused by excessive Ca2+ uptake did not involve restoration of bioenergetic functions. Partial resolution of this apparent contradiction came from an insightful work by the group of Bernardi demonstrating that the threshold for PTP induction by Ca2+ is modulated by the proton electrochemical gradient (14–18). Specifically, they have shown that the more depolarized mitochondria are, the higher the likelihood that they will exhibit PTP induced by Ca2+. Later on, the same group extended its findings by showing that pyridine nucleotides and dithiol oxidation of specific sites also modulate the pore (19) and that electron flow through complex I is a modulator of PTP opening upon Ca2+ uptake (20), concepts with inherent connection to the proton electrochemical gradient.
Brain mitochondria in relation to Ca2+-induced PTP deserve further attention, primarily because they reside within excitable cells exhibiting ample routes to Ca2+ and because unlike liver or heart mitochondria, there is still no universally accepted consensus here: claims of Ca2+ inducing PTP in brain mitochondria range from a partial (21) to a complete effect (22), and the disagreement extends to the degree of cyclosporin A (cys A) sensitivity (11, 22–24). Furthermore, because experimental conditions strongly shape the outcome and characteristics of brain mitochondrial PTP (11, 25), it becomes imperative to investigate PTP in mitochondria within neurons and astrocytes. In the present study we have identified bioenergetic conditions in isolated brain mitochondria that allow the demonstration of a cypD dependence upon Ca2+-induced PTP opening and applied them to in situ neuronal and astrocytic mitochondria.
EXPERIMENTAL PROCEDURES
Isolation of Brain Mitochondria from WT and CypD-KO Mice
C57BL/6J WT and KO for cypD littermate mice were a gift from Drs. Nika Danial and Anna Schinzel, from Howard Hughes Medical Institute and Dana-Farber Cancer Institute, Harvard Medical School. Mice were cross-bred for eight generations prior to harvesting brain tissues from WT and KO age-matched animals for the purpose of mitochondrial isolation and culturing of neurons and astrocytes. Nonsynaptic brain mitochondria from adult male WT and KO for cypD mice (aged 87–115 days) were isolated on a Percoll gradient as described previously (26) with minor modifications detailed in Ref. 24. All animal procedures were carried out according to the local animal care and use committee (Egyetemi Allatkiserleti Bizottsag) guidelines.
Ca2+ Uptake of Isolated Mitochondria
Mitochondria-dependent removal of medium Ca2+ was followed using the impermeant hexapotassium salt of the fluorescent dye Calcium Green 5N (Molecular Probes, Portland, OR). Calcium Green 5N (500 nm) was added to a 2-ml medium containing mitochondria (0.125 mg/ml) and 120 mm KCl, 10 mm Tris, 5 mm KH2PO4, 1 mm MgCl2, pH 7.6. Substrates were added where indicated. All experiments were performed at 37 °C. Fluorescence intensity was measured in a Hitachi F-4500 fluorescence spectrophotometer (Tokyo, Japan) using 517-nm excitation and 535-nm emission wavelengths.
Mitochondrial Swelling
Swelling of isolated mitochondria was assessed by measuring light scatter at 520 nm in a GBC UV/visible 920 spectrophotometer. Mitochondria were added at a final concentration of 0.125 mg/ml to 2 ml of medium containing 120 mm KCl, 10 mm Tris, 5 mm KH2PO4, 1 mm MgCl2, pH 7.6. Substrates were added where indicated. At the end of each experiment, the nonselective pore-forming peptide alamethicin (40 μg) was added as a calibration standard to cause maximal swelling. All experiments were performed at 37 °C.
ΔΨm Determination in Isolated Mitochondria
ΔΨm was estimated fluorometrically with safranine O (27). Mitochondria (0.25 mg) were added to 2 ml of incubation medium containing 120 mm KCl, 10 mm Tris, 5 mm KH2PO4, 1 mm MgCl2, pH 7.6, and 2.5 μm safranine O. Fluorescence was recorded in a Hitachi F-4500 spectrofluorometer at a 5-Hz acquisition rate, using 495- and 585-nm excitation and emission wavelengths, respectively. Experiments were performed at 37 °C. To convert safranine O fluorescence into millivolts, a voltage-fluorescence calibration curve was constructed. To this end, safranine O fluorescence was recorded in the presence of 2 nm valinomycin and stepwise increasing K+ (in the 0.2–120 mm range) which allowed calculation of ΔΨm by the Nernst equation assuming a matrix K+ = 120 mm (27).
Matrix Ca2+ Imaging of Isolated Mitochondria
Visualization of isolated mitochondria under epifluorescence imaging (Nikon Plan Fluor 100 × 1.3 NA) was achieved by loading mitochondria with fura-2/AM (8 μm for 20 min at 30 °C). Mitochondria were diluted to 1 mg/ml, and 5 μl was dropped on a coverslip, allowed to stand for 4 min prior to starting the perfusion. Image sequences (10 s/ratio frame, 50-ms exposure time, 2 × 2 binning) were acquired using an Micromax cooled digital CCD camera (Princeton Instruments) mounted on a Nikon Diaphot 200 inverted microscope (Nikon Corp., Tokyo, Japan). Image acquisition was controlled by Metafluor 3.5 (Universal Imaging Corp., West Chester, PA). The perfusate (120 mm KCl, 10 mm Tris, 5 mm KH2PO4, 1 mm MgCl2, pH 7.6, 50 ml/h flow rate) was temperature-controlled at 37 °C.
Cell Cultures and Transfections
Mixed primary cultures of cortical neurons and astrocytes were prepared from cypD-KO or wild-type (WT) mice pups (P 0–1). Cells were grown on poly-l-ornithine-coated 8-well LabTek II chambered coverglasses (Nunc, Rochester, NY) for 7–12 days, at a density of ∼105 cells/well in Neurobasal medium containing 2% B27 supplement and 2 mm glutamine (Sigma). Cultures were transfected at 7–9 days with mito-DsRed2 using Lipofectamine 2000 (Invitrogen) in Neurobasal medium at a 3:2 ratio of Lipofectamine (μl) to plasmid DNA (μg). Experiments were carried out at day 1–2 after transfection. Typical transfection rates were low, and therefore individual, nonoverlapping cells were visualized.
Imaging of Cultured Cells
Time lapse epifluorescence microscopy was carried out to image cells expressing mito-DsRed2 at 37 °C without superfusion in a medium containing 120 mm NaCl, 3.5 mm KCl, 1.3 mm CaCl2, 20 mm HEPES, 15 mm glucose at pH 7.4. For some experiments (detailed below) neurons were loaded with fura-FF/AM (2 μm) for 20 min before imaging. Experiments were performed on an Olympus IX81 inverted microscope equipped with a 60 × 1.4 NA oil immersion lens, a Bioprecision-2 xy-stage (Ludl Electronic Products Ltd., Hawthorne, NY), and a 75W xenon arc lamp (Lambda LS; Sutter Instruments, Novato, CA). For DsRed2 an 535/20 nm exciter, a 555LP dichroic mirror, and an 570LP emitter (Omega Optical, Brattleboro, VT) were used. Time lapses of z-series of 16 planes of 512 × 512-pixel frames (digitized at 14bit with no binning, 250-ms exposure time, yielding 0.1-μm pixel size and 0.8-μm z-spacing) were acquired using an ORCA-ER2 cooled digital CCD camera (Hamamatsu Photonics, Hamamatsu, Japan) under control of MetaMorph 6.0 software (Molecular Devices, Sunnyvale, CA). For fura-FF a 340/26-nm and a 387/11-nm exciter (Semrock, Rochester, NY), a 405LP dichroic mirror, and a 475LP emitter (Omega Optical) were used, and single plane images were recorded after each DsRed2 z-stack. Mitochondrial swelling was measured by the thinness ratio (TR) technique (28). Briefly, the TR technique measures changes of average diameters of thread-like or punctate structures in fluorescence images using a pair of (high and low frequency) bandpass spatial filters. A calibration image series of mito-DsRed2 fluorescence showing mitochondrial swelling by valinomycin (200 nm) was recorded and used to train a spatial bandpass filter set in Image Analyst MKII (Image Analyst Software, Novato, CA). To calculate TR, for each time point the z-stack was mean intensity-projected, and the projection image was duplicated. Then, both images were spatially filtered, and the absolute value of pixels was taken. The TR was calculated as the ratio of the average fluorescence intensity in the high frequency bandpass-filtered over the low frequency bandpass-filtered image. Mitochondrial swelling causes the loss of high spatial frequency image details, therefore a decrease in the TR value. Base-line normalized TR is given as δTR = (TR − TR0)/TR0.
Western Blotting
Isolated brain and liver mitochondria were solubilized at a concentration of 10 mg/ml in a buffer containing 50 mm Tris-HCl, pH 8.0, 150 mm NaCl, 1.0% Igepal CA-630, 0.5% sodium deoxycholate, 0.1% SDS, plus a mixture of protease inhibitors (Protease Inhibitor Mixture Set I; Calbiochem). Solubilized mitochondria were rapidly frozen and stored at −70 °C until further manipulation. Upon thawing, the protein concentration of the samples was estimated by the Bradford assay. Subsequently, samples were mixed with 50 mm dithiothreitol and NuPage loading buffer (Invitrogen) and heated to 70° for 10 min. The samples were then loaded (25 μg/lane) on a 4–12% BisTris gel, and separated by SDS-PAGE in the presence of N,N-dimethylformamide and sodium bisulfite (10% w/v). Separated proteins were transferred to a methanol-activated polyvinylidene difluoride membrane. Immunoblotting was performed as recommended by the manufacturer. Mouse monoclonal anti-cypD (Mitosciences, Eugene, OR) primary antibody was used at 1 μg/ml. Immunoreactivity was detected using the appropriate peroxidase-linked secondary antibodies (1:10,000; Jackson Immunochemicals Europe, Cambridgeshire, UK) and enhanced chemiluminescence detection reagent (ECL system; Amersham Biosciences). Upon completion of Western blotting the blots were stained with Ponceau S (0.5% Ponceau S (w/v) in 5% acetic acid), and loading of individual lanes was assessed by densitometric analysis (Scion Image, version alpha 4.0.3.2; Scion Corp., Frederick, MD).
Reagents
Standard laboratory chemicals, cys A, calcimycin, and alamethicin were from Sigma. Ru360 was from Calbiochem. SF 6847 was from BIOMOL. Calcium Green 5N 6K+ salt was from Invitrogen. Mito-DsRed2 was purchased from Clontech. Mitochondrial substrate stock solutions were dissolved in bi-distilled water and titrated to pH 7.0 with KOH.
RESULTS
Effect of Substrate Availability, Cyclosporin A, or Genetic Deletion of Cyclophilin D on Ca2+-induced PTP in Isolated Brain Mitochondria
Electrophoretic Ca2+ uptake for induction of PTP is allowed either in the presence of respiratory substrates or in a substrate-free medium containing KSCN; diffusion of the lipophilic SCN− anion provides the driving force for electrophoretic Ca2+ accumulation (29, 30). However, in the original studies by Hunter and Haworth it was shown that PTP can be induced by Ca2+ in the absence of respiratory substrates (31), a phenomenon that has been subsequently reproduced (32–34), reviewed in (35). Furthermore, it was also shown that substrates delay Ca2+-induced PTP (33, 34), in accordance to the Bernardi scheme mentioned above. To reproduce these findings for our studies, isolated brain mitochondria were challenged by CaCl2, in the presence and absence of glutamate and malate, and light scattering was recorded spectrophotometrically at 520 nm. A three-pulse CaCl2 protocol was used for this and all subsequent similar experiments: 20 μm CaCl2 was given at 100 s, followed by 200 μm CaCl2 at 300 s and again at 500 s. Orange lines appearing in Figs. 1, 2, and 4 are control trace lines obtained from WT mitochondria not exposed to CaCl2. As shown in Fig. 1A, addition of 20 μm CaCl2 to substrate-supplemented or substrate-starved brain mitochondria of WT mice did not cause a decrease in light scatter; instead, a cessation in the initial base-line decrease was observed. However, the subsequent 200 μm CaCl2 pulse induced a large decrease in light scatter in substrate-starved but not substrate-supplemented mitochondria. The next 200 μm CaCl2 pulse given at 500 s did not induce any further changes in substrate-starved mitochondria, but caused a decrease in substrate-supplemented mitochondria. As shown in Fig. 1B, the effect of the first 200 μm CaCl2 pulse was cys A-sensitive; however, the second addition of 200 μm CaCl2 overrode the protective effect of cys A, consistent with the findings by Brustovetsky and Dubinsky (23). In Fig. 1B the effect of the pore-forming peptide alamethicin is also shown, so that the extent of changes in light scatter induced by CaCl2 can be better appreciated compared with maximum changes. Subsequent experiments benefitted from the availability of cypD-KO mice. We isolated mitochondria from the brains of WT and cypD-KO mice (see Fig. 1C). As shown in Fig. 1D, results obtained from substrate-starved mitochondria from cypD-KO mice were strikingly similar to those obtained from cys A-treated WT mice (Fig. 1B). The presence of substrates, however, did not provide additional protection in the cypD-KO mitochondria (Fig. 1E). Maximum swelling rates pooled from all experiments (expressed as percentage of swelling rate/minute and accounting for the condition producing the highest swelling rate as “maximum”) for each condition and after each Ca2+ pulse is shown in Fig. 1G. These results are in accord with earlier reports on various types of mitochondria and conditions, showing that high Ca2+ loads can induce PTP in the absence of substrates. In our hands, absence of substrates prevented isolated mitochondria from building a membrane potential of higher than −10 mV (data not shown). At this ΔΨm value, mitochondrial Ca2+ uptake is unfavorable (13). Indeed, recordings of extramitochondrial Ca2+ by Calcium Green 5N revealed that in the absence of substrates (Fig. 1F, traces c, d, and e) mitochondria were unable to perform Ca2+ sequestration, yet exhibited large changes in light scatter. Electron microscopy imaging of mitochondria that exhibited large changes in light scatter confirmed that this was due to swelling (data not shown). We therefore considered the possibilities that Ca2+ was either inducing cypD-sensitive swelling by acting on an extramitochondrial site, or because high amounts of CaCl2 were required, Ca2+ was entering mitochondria simply by a chemical gradient.
FIGURE 1.

Effect of substrate availability on Ca2+-induced PTP and modulation by cyclosporin A or genetic deletion of cyclophilin D. A, B, D, and E, traces of light scatter recorded spectrophotometrically in mitochondrial suspensions at 520 nm during CaCl2 additions at the concentrations indicated in the panels. Conditions are given in the panels. C, Western blot (upper panel) of brain and liver mitochondria from WT versus cypD-KO mice probed for cypD immunoreactivity. Lower panel, Ponceau S staining of the same blot shown in the upper panel. MWM, molecular weight marker. D and E are aligned on the y axis. A, D, E, and F are aligned on the x axis. F, mitochondrial Ca2+ uptake followed by Calcium Green 5N hexapotassium salt fluorescence (noncalibrated). The black striped region is expanded on the y axis, for the sake of clarity; a, WT plus glutamate plus malate; b, WT plus glutamate plus malate plus cys A; c, WT, no substrates; d, WT, no substrates plus cys A; e, WT, no substrates plus Ru360. Results are representative of at least four independent experiments. G, maximum swelling rates pooled from all individual experiments (expressed as percentage of swelling rate per minute and accounting for the condition producing the highest swelling rate as maximum) for each condition and after each Ca2+ pulse. Error bars represent S.E.; a is statistically significant from b, c, d, and e, p < 0.001; d is statistically significant from e, p < 0.05, one-way ANOVA on Ranks.
FIGURE 2.

Effect of SF 6847 and/or inhibitors of the Ca2+ uniporter on mitochondrial Ca2+ uptake; modulation by cyclosporin A or genetic deletion of cyclophilin D. Traces of light scatter recorded spectrophotometrically at 520 nm during CaCl2 additions at the concentrations are indicated in the panels, to mitochondrial suspensions. Conditions of the suspensions are given in the panels. All panels are aligned on the x axis. Results are representative of at least four independent experiments. E, maximum swelling rates pooled from all individual experiments (expressed as in Fig. 1) for each condition and after each Ca2+ pulse. Error bars represent S.E.; a is statistically significant from b, c, d, e, f, and g, p < 0.001; d is statistically significant from e, p < 0.05, one-way ANOVA on Ranks.
FIGURE 4.

Effect of respiratory chain inhibition on Ca2+-induced PTP. Traces of light scatter recorded spectrophotometrically at 520 nm during CaCl2 additions at the concentrations indicated in the figures, to mitochondrial suspensions are shown. All experiments were performed on WT mice A: a, no substrates; b, plus glutamate plus malate; c, plus glutamate plus malate plus 1 μm rotenone; d, no substrates + 1 μm rotenone; e, no substrates + 1 mm KCN; f, plus glutamate plus malate plus 1 mm KCN. B: a, no substrates; b, no substrates + 1 μm piericidin A; c, no substrates + 1 μm rotenone. C: a, no substrates; b, no substrates + 0.2 μm stigmatellin; c, no substrates + 2 μm stigmatellin; d, no substrates + 0.5 μm myxothiazol; e, no substrates + 10 μm myxothiazol; f, no substrates + 1 mm KCN. Results are representative of at least four independent experiments. D, maximum swelling rates pooled from all individual experiments (expressed as in Figs. 1 and 2) for each condition and after each Ca2+ pulse. Error bars represent S.E.; a is statistically significant from c, p < 0.05; b is statistically significant from d, p < 0.05, one-way ANOVA on Ranks.
Effect of an Uncoupler and/or an Inhibitor of the Ca2+ Uniporter on Mitochondrial Ca2+ Uptake and Light Scatter
To address the site of action of Ca2+ on the light scatter, we pretreated mitochondria with the Ca2+ uniporter inhibitor, Ru360 (36). As shown in Fig. 2A, WT mitochondria still exhibited high Ca2+-induced changes in light scatter in the presence of Ru360, at a concentration that was found to prevent the uptake of extramitochondrial Ca2+ (Fig. 1F, trace e). The lack of effect of Ru360 was also observed in the presence of cys A (Fig. 2A), or when the effect of Ca2+ was compared in WT versus cypD-KO mitochondria (Fig. 2B). To depolarize mitochondria completely, 1 μm SF 6847 was added to the medium, and the effects of Ca2+ and Ru360 were recorded. As shown in Fig. 2C, the presence of the uncoupler failed to provide extra protection against high Ca2+-induced swelling. Furthermore, the presence of the uncoupler negated the protective effects of substrates in WT mitochondria (Fig. 2D). Maximum swelling rates pooled from all experiments (expressed as in Fig. 1) for each condition and after each Ca2+ pulse are shown in Fig. 2E.
Effect of Ca2+ Uniporter Inhibitors on Mitochondrial Matrix Ca2+ Accumulation of Isolated Mitochondria Imaged under Wide Field Epifluorescence
The failure of Ru360 to protect against the Ca2+-induced large changes in light scatter shown in Fig. 2, A and B, could be explained by assuming that Ca2+ acted on the extramitochondrial side. To provide further evidence for this, we loaded isolated mitochondria with fura-2 and imaged them under wide field epifluorescence (Fig. 3A). This experimental setup (i) benefits from the spatial resolution in fura-2 imaging, avoiding a “contaminant” signal of leaked fura-2 in the extramitochondrial space and (ii) provides a valid quantitative signal of matrix [Ca2+] in the submicromolar range. Surprisingly, isolated mitochondria perfused with a buffer containing 0.1 mm CaCl2 showed robust increases in matrix-entrapped fura-2 fluorescence that exhibited only a partial sensitivity to Ru360 (10 μm) and Ruthenium red (10 μm) (Fig. 3B), arguing against the assumption that Ca2+ was acting exclusively on an extramitochondrial site when inducing changes in light scatter.
FIGURE 3.
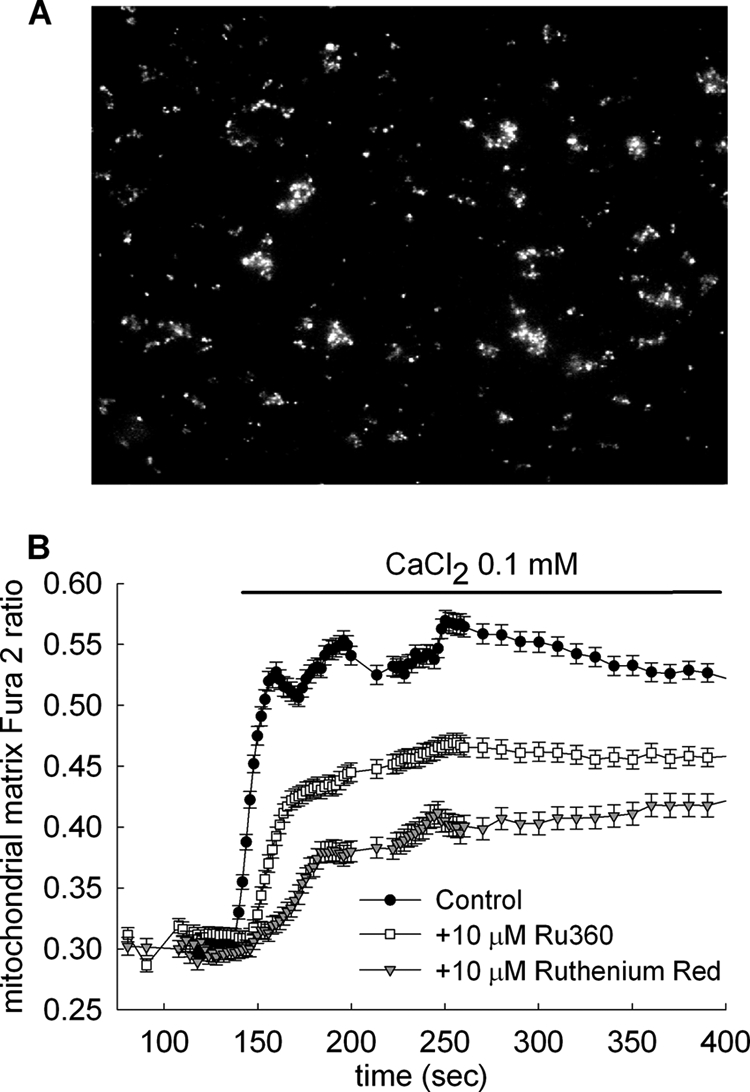
Effect of the Ca2+ uniporter inhibitors Ru360 and Ruthenium red on mitochondrial matrix Ca2+ accumulation of isolated WT brain mitochondria imaged under wide field epifluorescence. A, wide field epifluorescence image of isolated brain mitochondria from WT mice, loaded with fura-2/AM. B, time recordings of mitochondrial-trapped fura-2 fluorescence of immobilized mitochondria, perfused by 0.1 mm CaCl2 in the presence or absence of Ca2+ uniporter inhibitors as indicated in the panel, in the absence of exogenous substrates. Results are representative of at least four independent experiments.
Effect of Respiratory Chain Inhibition on Ca2+-induced PTP
To address the contribution of respiratory chain components to the protective effect of substrates against the Ca2+-induced changes in light scatter, we pretreated mitochondria with complex I (rotenone or piericidin A), complex III (myxothiazol or stigmatellin), and complex IV (KCN) inhibitors. The emerging picture depicted from Fig. 4 was that rotenone or piericidin (traces c and d of Fig. 4A and traces b and c of Fig. 4B) afforded protection against Ca2+-induced changes in light scatter, irrespective of the presence or absence of substrates, whereas myxothiazol, stigmatellin, and KCN not only failed to confer protection, but also negated the protective effect of substrates (traces e and f of Fig. 4A and traces b–f of Fig. 4C; see also Fig. 4D). High concentrations of myxothiazol and stigmatellin (10 μm and 2 μm, respectively) that also block complex I (37) failed to afford protection, as opposed to rotenone and piericidin A. However, rotenone and piericidin A (as well as rolliniastatin used in Ref. 19) bind to a different site than myxothiazol and stigmatellin (37). Maximum swelling rates pooled from all experiments (expressed as in Figs. 1 and 2) for each condition and after each Ca2+ pulse are shown in Fig. 4D. Inhibition of PTP by rotenone has been reported previously (19, 31, 33, 38). Because the protective effect of substrates was negated by cyanide, we decided to use this regimen for testing mitochondrial swelling within neurons and astrocytes because the in situ availability of substrates is much less amenable to manipulation.
Effect of CypD Ablation on Ca2+-induced Mitochondrial Swelling within Neurons and Astrocytes
To address the role of cypD in the opening of brain PTP in situ, swelling of cypD-deficient and WT mitochondria within neurons and astrocytes in mixed cortical cultures was compared during Ca2+ overload, induced by the addition of calcimycin (1 μm 4Br-A23187). Mitochondria were visualized by wide field epifluorescence imaging of mitochondrially targeted DsRed2. Astrocytes grew as a monolayer on the bottom of the chamber while neurons grew as a monolayer above them. Cultures were prepared at the same age from the WT and KO mouse pups (P0 or 1), and measurements were performed in the same range of days in vitro; accordingly, the neurons/astrocytes ratio deviated minimally from one culture to another (2–2.6), deduced from counting cells from images of fura-loaded cultures. Neurons and astrocytes were distinguished by their different mitochondrial morphology (28). Neuronal mitochondria are typically more densely packed in the soma, therefore dendritic mitochondria were chosen for analysis. Astrocytes are flatter than neurons in culture, and they exhibited elongated, branched mitochondria of even thickness. Mitochondrial swelling was monitored by evaluating DsRed2-visualized mitochondrial morphology by calculation of changes in mean mitochondrial diameters using the TR technique. In these assays the onset of swelling was defined by the sudden decrease in the thinness ratio. As shown in Fig. 5A, both WT and cypD-KO mitochondria within neurons and astrocytes exhibited significant morphological alterations due to swelling in response to Ca2+ overload induced by the addition of calcimycin within 600–800 s. When NaCN was co-applied with calcimycin in a glucose-free medium in the presence of 2 mm 2-deoxyglucose, swelling of mitochondria was almost immediate in both WT and cypD-KO neurons. However, Ca2+ overload of uncoupled mitochondria (by co-application of 1 μm SF 6847) triggered swelling at a significantly earlier time in cypD-KO than in WT neurons. In contrast to the neurons, Ca2+ overload of uncoupled mitochondria triggered swelling at a significantly earlier time in WT than in cypD-KO astrocytes (Fig. 5B). In the absence of glucose and concomitant presence of 2-deoxyglucose and NaCN, in situ mitochondria are almost certainly completely depolarized. We used calcimycin at 1 μm concentration that likely affects only the Ca2+ permeability of the plasma membrane. Experiments on cultured neurons and astrocytes loaded with fura-2 (Kd = 225 nm) (39) versus fura-6F (Kd = 2.47 μm) (24) revealed that the increase in cytosolic Ca2+ by 1 μm calcimycin was in the 1–2 μm range in contrast to the 1.3 mm in the medium (data not shown); thus the amount of calcimycin distributed in the plasma membrane was very small and unlikely for it to distribute in the inner mitochondrial membrane.
FIGURE 5.

Effect of cypD ablation on Ca2+-induced mitochondrial swelling within neurons and astrocytes. Time elapsed between calcimycin application and mitochondrial swelling was detected by wide field imaging of mito-DsRed2 expressing neurons (A) and astrocytes (B) in mixed cortical cultures from WT (black bars) or cypD-KO (gray bars) mice. The onset of swelling was determined by detection of an increase of mean diameter of mitochondria in the microscopic view field by calculation of the TR. Co-application of 5 mm NaCN and calcimycin was performed in a medium without glucose supplemented with 2 mm 2-deoxyglucose. Bars indicate means ± S.E. (error bars) of 4–12 cells (*, p < 0.05 significance by Kruskal-Wallis ANOVA on Ranks).
Effect of CypD Ablation on in Situ Mitochondrial Swelling of Neurons Challenged by Glutamate
To expose neuronal in situ mitochondria to high Ca2+ challenge by an alternative mechanism, neurons were exposed to excitotoxic levels of glutamate and glycine, in the absence of Mg2+. Mitochondrial swelling was monitored by TR values calculated from wide field fluorescence images of mitochondrially targeted DsRed2. Glutamate exposure triggered a biphasic mitochondrial swelling response (shown for a WT neuron) indicated by the decrease of the TR (Fig. 6A, black circles). Swelling comprised an initial, first and a well separated, delayed second drop. The first drop in the TR invariably coincided with the initial [Ca2+]i response to glutamate and the second drop to the secondary, irreversible rise of [Ca2+]i, termed delayed calcium deregulation (40–42) (Fig. 6, A and B, black bars). In cultures prepared from cypD-KO mice (Fig. 6B, gray bars) the first phase of mitochondrial swelling was detected only in 60% of the neurons. Furthermore, only 40% of cypD-KO neurons exhibited the secondary swelling of mitochondria during delayed Ca2+ deregulation (Fig. 6B). Finally, the initial swelling of mitochondria was significantly delayed in cypD-KO neurons compared with wild type, whereas the time of onset of the secondary mitochondrial swelling was not statistically different (Fig. 6C).
FIGURE 6.

Effect of cypD ablation on glutamate-evoked biphasic mitochondrial swelling in cortical neurons. A, wide field fluorescence time lapse recording in a fura-FF/AM-loaded WT cortical neuron expressing mito-DsRed. Glutamate (300 μm) plus glycine (10 μm) in the absence of [Mg2+]e evoked a rise of [Ca2+]i indicated by the increasing ratio of fura-FF 340/380-nm fluorescence intensities (triangles). Mitochondrial swelling was measured by calculating TR of mito-DsRed fluorescence images (circles), where swelling is marked by the decreasing TR (arrows). The first and second drops of the TR always coincided with the initial response to glutamate and to the delayed Ca2+ deregulation, respectively. Representative traces of 10 recordings are shown. B, quantification of the observation of first and second drops in neurons from WT (black bars) and cypD-KO mice (gray bars) using Fisher's exact test. Total (n) corresponds to the number of cells observed and first and second drops to the observation of the drops in the TR in recordings similar to A. C, onset of mitochondrial swelling defined as the time elapsed between the application of glutamate and the sudden decrease of the TR; bars show mean ± S.E. (error bars) of 10 cells (*, p < 0.05 significance by Kruskal-Wallis ANOVA on Ranks).
DISCUSSION
There are hundreds of publications addressing permeability transition with relation to various aspects of bioenergetics; here we attempted to shed light on the phenomenon where cypD-dependent PTP precedes necrotic events during which there is energy crisis, but not apoptosis, a mechanism that is dependent on energy provision. We have narrowed our investigations to brain mitochondria in isolation and within their natural environment (both neurons and astrocytes).
The most important finding of this study is the dramatic hastening of the swelling of in situ neuronal and astrocytic mitochondria by glucose deprivation and NaCN co-application, upon calcimycin exposure. This extends the findings on isolated mitochondria (14–18) and this study, showing that a diminished electrochemical gradient primes mitochondria within their natural environment to undergo Ca2+-induced PTP. We created depolarizing conditions by three different means: (i) substrate deprivation (no electron flow), (ii) electron transport chain inhibitors (no electron flow), and (iii) presence of an uncoupler (high electron flow). Results were not entirely congruent between isolated versus in situ mitochondria (i.e. compare the presence versus absence of substrates or uncoupler), but in both experimental models the presence of cyanide prompted pore opening. Our findings support the notion that the lack of an electrochemical gradient not favoring electrophoretic Ca2+ uptake not only fails to protect in situ mitochondria from PTP, but it also subserves the purpose of decreasing a threshold to the point that either an increase in matrix Ca2+ concentration due to a mere diffusion and/or an extramitochondrial site with low affinity for Ca2+, induces pore opening. However, our findings are partially at odds with those obtained in Ref. 20, in which the effect of cyanide was found to disfavor PTP opening. The difference may stem from the fact that in that study isolated skeletal muscle mitochondria were investigated.
The swelling induced by high Ca2+ in energized in situ neuronal and astrocytic mitochondria was not cypD-dependent. To this we must stress that our results on isolated mitochondria point to the possibility of an Ru360/Ruthenium red-insensitive route for Ca2+ entry. Furthermore, our results also show that there are cell-specific differences of the same tissue regarding the bioenergetic contribution to Ca2+-induced PTP. For example, it is striking that the elapsed time upon calcimycin exposure until the onset of swelling of uncoupler-treated neuronal WT mitochondria was smaller than that recorded for cypD-KO mitochondria, and the exact opposite was observed in astrocytes.
There are ∼1500 proteins in mitochondria, and <3% of those are mitochondrially encoded (43), whereas the rest are nuclear-encoded. It is at least prudent to consider that the composition and/or regulation of the mitochondrial PTP exhibits tissue- or even cell-specific differences, as it has been suggested elsewhere (44, 45).
Acknowledgments
We thank Drs. Nika Danial and Anna Schinzel for providing the cyclophilin D KO mice and WT littermates and Katalin Zölde for excellent technical assistance.
This work was supported by grants from the Országos Tudományos Kutatási Alapprogram (OTKA), Magyar Tudományos Akadémia (MTA), Nemzeti Kutatási és Technológiai Hivatal (NKTH), and Egészségügyi Tudományos Tanács (ETT) (to V. A.-V.) and Semmelweis University Research Grant 63320, OTKA-NKTH Grant NF68294, OTKA Grant NNF78905, and ETT Grant 55160 (to C. C.).
- cypD
- cyclophilin D
- BisTris
- bis(2-hydroxyethyl)iminotris(hydroxymethyl)methane
- cys A
- cyclosporin A
- ΔΨm
- mitochondrial membrane potential
- PTP
- permeability transition pore
- TR
- thinness ratio.
REFERENCES
- 1. Baines C. P., Kaiser R. A., Purcell N. H., Blair N. S., Osinska H., Hambleton M. A., Brunskill E. W., Sayen M. R., Gottlieb R. A., Dorn G. W., Robbins J., Molkentin J. D. (2005) Nature 434, 658–662 [DOI] [PubMed] [Google Scholar]
- 2. Nakagawa T., Shimizu S., Watanabe T., Yamaguchi O., Otsu K., Yamagata H., Inohara H., Kubo T., Tsujimoto Y. (2005) Nature 434, 652–658 [DOI] [PubMed] [Google Scholar]
- 3. Schinzel A. C., Takeuchi O., Huang Z., Fisher J. K., Zhou Z., Rubens J., Hetz C., Danial N. N., Moskowitz M. A., Korsmeyer S. J. (2005) Proc. Natl. Acad. Sci. U.S.A. 102, 12005–12010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Basso E., Fante L., Fowlkes J., Petronilli V., Forte M. A., Bernardi P. (2005) J. Biol. Chem. 280, 18558–18561 [DOI] [PubMed] [Google Scholar]
- 5. Giorgio V., Soriano M. E., Basso E., Bisetto E., Lippe G., Forte M. A., Bernardi P. (2010) Biochim. Biophys. Acta 1797, 1113–1118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Li Y., Johnson N., Capano M., Edwards M., Crompton M. (2004) Biochem. J. 383, 101–109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Leist M., Single B., Castoldi A. F., Kühnle S., Nicotera P. (1997) J. Exp. Med. 185, 1481–1486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Volbracht C., Leist M., Nicotera P. (1999) Mol. Med. 5, 477–489 [PMC free article] [PubMed] [Google Scholar]
- 9. Chinopoulos C., Adam-Vizi V. (2010) Biochim. Biophys. Acta 1802, 221–227 [DOI] [PubMed] [Google Scholar]
- 10. Chinopoulos C., Gerencser A. A., Mandi M., Mathe K., Töröcsik B., Doczi J., Turiak L., Kiss G., Konràd C., Vajda S., Vereczki V., Oh R. J., Adam-Vizi V. (2010) FASEB J. 24, 2405–2416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chinopoulos C., Adam-Vizi V. (2006) FEBS J. 273, 433–450 [DOI] [PubMed] [Google Scholar]
- 12. Fiskum G., Starkov A., Polster B. M., Chinopoulos C. (2003) Ann. N.Y. Acad. Sci. 991, 111–119 [DOI] [PubMed] [Google Scholar]
- 13. Vajda S., Mándi M., Konràd C., Kiss G., Ambrus A., Adam-Vizi V., Chinopoulos C. (2009) FEBS J. 276, 2713–2724 [DOI] [PubMed] [Google Scholar]
- 14. Bernardi P. (1992) J. Biol. Chem. 267, 8834–8839 [PubMed] [Google Scholar]
- 15. Petronilli V., Cola C., Bernardi P. (1993) J. Biol. Chem. 268, 1011–1016 [PubMed] [Google Scholar]
- 16. Petronilli V., Cola C., Massari S., Colonna R., Bernardi P. (1993) J. Biol. Chem. 268, 21939–21945 [PubMed] [Google Scholar]
- 17. Petronilli V., Costantini P., Scorrano L., Colonna R., Passamonti S., Bernardi P. (1994) J. Biol. Chem. 269, 16638–16642 [PubMed] [Google Scholar]
- 18. Scorrano L., Petronilli V., Bernardi P. (1997) J. Biol. Chem. 272, 12295–12299 [DOI] [PubMed] [Google Scholar]
- 19. Costantini P., Chernyak B. V., Petronilli V., Bernardi P. (1996) J. Biol. Chem. 271, 6746–6751 [DOI] [PubMed] [Google Scholar]
- 20. Fontaine E., Eriksson O., Ichas F., Bernardi P. (1998) J. Biol. Chem. 273, 12662–12668 [DOI] [PubMed] [Google Scholar]
- 21. Kristián T., Weatherby T. M., Bates T. E., Fiskum G. (2002) J. Neurochem. 83, 1297–1308 [DOI] [PubMed] [Google Scholar]
- 22. Hansson M. J., Månsson R., Mattiasson G., Ohlsson J., Karlsson J., Keep M. F., Elmér E. (2004) J. Neurochem. 89, 715–729 [DOI] [PubMed] [Google Scholar]
- 23. Brustovetsky N., Dubinsky J. M. (2000) J. Neurosci. 20, 8229–8237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chinopoulos C., Starkov A. A., Fiskum G. (2003) J. Biol. Chem. 278, 27382–27389 [DOI] [PubMed] [Google Scholar]
- 25. Chinopoulos C., Adam-Vizi V. (2010) FEBS J. 277, 3637–3651 [DOI] [PubMed] [Google Scholar]
- 26. Sims N. R. (1990) J. Neurochem. 55, 698–707 [DOI] [PubMed] [Google Scholar]
- 27. Akerman K. E., Wikström M. K. (1976) FEBS Lett. 68, 191–197 [DOI] [PubMed] [Google Scholar]
- 28. Gerencser A. A., Doczi J., Töröcsik B., Bossy-Wetzel E., Adam-Vizi V. (2008) Biophys. J. 95, 2583–2598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Selwyn M. J., Dawson A. P., Dunnett S. J. (1970) FEBS Lett. 10, 1–5 [DOI] [PubMed] [Google Scholar]
- 30. Nicolli A., Petronilli V., Bernardi P. (1993) Biochemistry 32, 4461–4465 [DOI] [PubMed] [Google Scholar]
- 31. Hunter D. R., Haworth R. A. (1979) Arch. Biochem. Biophys. 195, 453–459 [DOI] [PubMed] [Google Scholar]
- 32. Yamamoto T., Yoshimura Y., Yamada A., Gouda S., Yamashita K., Yamazaki N., Kataoka M., Nagata T., Terada H., Shinohara Y. (2008) J. Bioenerg. Biomembr. 40, 619–623 [DOI] [PubMed] [Google Scholar]
- 33. Rigobello M. P., Turcato F., Bindoli A. (1995) Arch. Biochem. Biophys. 319, 225–230 [DOI] [PubMed] [Google Scholar]
- 34. Moore G. A., Jewell S. A., Bellomo G., Orrenius S. (1983) FEBS Lett. 153, 289–292 [DOI] [PubMed] [Google Scholar]
- 35. Leverve X. M., Fontaine E. (2001) IUBMB Life 52, 221–229 [DOI] [PubMed] [Google Scholar]
- 36. Matlib M. A., Zhou Z., Knight S., Ahmed S., Choi K. M., Krause-Bauer J., Phillips R., Altschuld R., Katsube Y., Sperelakis N., Bers D. M. (1998) J. Biol. Chem. 273, 10223–10231 [DOI] [PubMed] [Google Scholar]
- 37. Degli Esposti M. (1998) Biochim. Biophys. Acta 1364, 222–235 [DOI] [PubMed] [Google Scholar]
- 38. Chauvin C., De Oliveira F., Ronot X., Mousseau M., Leverve X., Fontaine E. (2001) J. Biol. Chem. 276, 41394–41398 [DOI] [PubMed] [Google Scholar]
- 39. Grynkiewicz G., Poenie M., Tsien R. Y. (1985) J. Biol. Chem. 260, 3440–3450 [PubMed] [Google Scholar]
- 40. Sattler R., Tymianski M. (2001) Mol. Neurobiol. 24, 107–129 [DOI] [PubMed] [Google Scholar]
- 41. Chinopoulos C., Gerencser A. A., Doczi J., Fiskum G., Adam-Vizi V. (2004) J. Neurochem. 91, 471–483 [DOI] [PubMed] [Google Scholar]
- 42. Nicholls D. G., Ward M. W. (2000) Trends Neurosci. 23, 166–174 [DOI] [PubMed] [Google Scholar]
- 43. Ryan M. T., Hoogenraad N. J. (2007) Annu. Rev. Biochem. 76, 701–722 [DOI] [PubMed] [Google Scholar]
- 44. Panov A., Dikalov S., Shalbuyeva N., Hemendinger R., Greenamyre J. T., Rosenfeld J. (2007) Am. J. Physiol. Cell Physiol. 292, C708–C718 [DOI] [PubMed] [Google Scholar]
- 45. Zorov D. B., Juhaszova M., Yaniv Y., Nuss H. B., Wang S., Sollott S. J. (2009) Cardiovasc. Res. 83, 213–225 [DOI] [PMC free article] [PubMed] [Google Scholar]