

Abstract
FoxO1, a forkhead box O class transcription factor, is abundant in insulin-responsive tissues. Akt, downstream from phosphatidylinositol 3-kinase in insulin signaling, phosphorylates FoxO1 at Thr24, Ser256, and Ser319, negatively regulating its function. We previously reported that dehydroepiandrosterone-stimulated phosphorylation of FoxO1 in endothelial cells requires cAMP-dependent protein kinase α (PKA-α). Therefore, we hypothesized that FoxO1 is a novel direct substrate for PKA-α. Using an immune complex kinase assay with [γ-32P]ATP, purified PKA-α directly phosphorylated wild-type FoxO1 but not FoxO1-AAA (mutant with alanine substitutions at known Akt phosphorylation sites). Phosphorylation of wild-type FoxO1 (but not FoxO1-AAA) was detectable using phospho-specific antibodies. Similar results were obtained using purified GST-FoxO1 protein as the substrate. Thus, FoxO1 is a direct substrate for PKA-α in vitro. In bovine aortic endothelial cells, interaction between endogenous PKA-α and endogenous FoxO1 was detected by co-immunoprecipitation. In human aortic endothelial cells (HAEC), pretreatment with H89 (PKA inhibitor) or siRNA knockdown of PKA-α decreased forskolin- or prostaglandin E2-stimulated phosphorylation of FoxO1. In HAEC transfected with a FoxO-promoter luciferase reporter, co-expression of the catalytic domain of PKA-α, catalytically inactive mutant PKA-α, or siRNA against PKA-α caused corresponding increases or decreases in transactivation of the FoxO promoter. Expression of vascular cellular adhesion molecule-1 mRNA, up-regulated by FoxO1 in endothelial cells, was enhanced by siRNA knockdown of PKA-α or treatment of HAEC with the PKA inhibitor H89. Adhesion of monocytes to endothelial cells was enhanced by H89 treatment or overexpression of FoxO1-AAA, similar to effects of TNF-α treatment. We conclude that FoxO1 is a novel physiological substrate for PKA-α in vascular endothelial cells.
Keywords: Akt PKB, Endothelium, Protein Kinase A (PKA), Protein-Protein Interactions, Transcription Factors, FoxO1, Phosphorylation, VCAM-1
Introduction
Forkhead box O1 (FoxO1) is a member of the FoxO transcription factor family that plays important roles in regulation of glucose homeostasis, cellular proliferation, differentiation, and vascular homeostasis in response to insulin and other growth factors (1–3). Mice lacking FoxO1 die in utero from improper vascular development (4). Overexpression of FoxO1 in primary endothelial cells impairs cell migration and tube formation, whereas knockdown of FoxO1 using siRNA enhances angiogenic function (5). Moreover, siRNA knockdown of FoxO1 in human coronary artery endothelial cells reduces VEGF-induced vascular cellular adhesion molecule-1 (VCAM-1)2 expression and monocyte adhesion to endothelial cells (6). FoxO1 function is regulated, in part, by post-translational modifications including phosphorylation, acetylation, and ubiquitination (7–9). Phosphorylation of FoxO1 at a number of specific regulatory sites results in translocation of FoxO1 from the nucleus to the cytosol that impairs its transcriptional activity (2). Akt, a serine/threonine kinase downstream from PI3K in insulin signaling pathways, phosphorylate FoxO1 at Thr24, Ser256, and Ser319 to promote nuclear exclusion of FoxO1. Thus, insulin negatively regulates FoxO1 functions via phosphorylation by Akt (10, 11). In addition to Akt, other kinases including SGK phosphorylate FoxO1 to regulate its function in a similar manner. For example, SGK phosphorylates FoxO1 at Thr24 and Ser319 (11, 12). Similar to Akt, SGK is activated by a variety of growth and survival factors including insulin (13, 14).
cAMP-dependent protein kinase (PKA) is a key regulator of many processes involved with cell growth and development. PKA is activated when cAMP binds to the regulatory subunit of PKA, resulting in release of the catalytic subunit that then phosphorylates a variety of protein substrates including ion channels, key metabolic enzymes, and transcription factors (15).
In a previous study, we reported that dihydroepiandrosterone treatment of primary endothelial cells acutely increases phosphorylation of FoxO1 in a PKA-dependent manner to reduce expression of ET-1 by interfering with the binding of FoxO1 to the human ET-1 promoter (3). Therefore, in the present study, we tested the hypothesis that FoxO1 is a novel direct substrate for PKA-α that helps to regulate endothelial function in response to activation of PKA-α.
MATERIALS AND METHODS
Plasmid Constructs
pcDNA3 expression vectors containing cDNA for FLAG-tagged FoxO1 constructs were kindly provided by Dr. Eric Tang (University of Michigan Medical School, Ann Arbor, MI). These included constructs containing the full-length open reading frame of human wild-type FoxO1 (FoxO1-WT) and the constitutively nuclear mutant FoxO1-AAA (three Akt phosphorylation sites replaced by alanine, T24A/S256A/S319A). pcDNA3 expression vectors for HA-tagged PKA were kindly provided by Dr. Susan S. Taylor (University of California, San Diego, CA). These included constructs containing the full-length open reading frame of the wild-type PKA-catalytic domain (PKA-cat-WT) and a mutant with a kinase-dead PKA-catalytic domain.
In Vitro Kinase Assays
HEK293 cells cultured in 60-mm dishes were transiently transfected with empty vector pcDNA3, FLAG-tagged FoxO1-WT, or FoxO1-AAA using Lipofectamine Plus (Invitrogen) for 3 h according to the manufacturer's protocol. Two days after transfection, the cell lysates were prepared in cell lysis buffer (Cell Signaling Technology, Danvers, MA; buffer 9803) containing complete protease inhibitors (Roche Applied Science). Then recombinant FoxO1-WT and FoxO1-AAA were immunoprecipitated from cell lysates (1 mg of total protein in each sample) using anti-FLAG antibodies (1 μg) and protein A-agarose beads (Millipore; Temecula, CA) at 4 °C overnight in intraperitoneal reaction buffer (20 mm Tris-Cl, pH 7.4, 1 mm EDTA, 10% glycerol, 1 mm DTT, 150 mm NaCl). The immunocomplex samples were washed twice with cell washing buffer (20 mm Tris-Cl, pH 7.4, 1 mm EDTA, 10% glycerol, 1 mm DTT, 150 mm NaCl, 0.1% Triton X-100). The samples were then incubated in kinase assay buffer (Cell Signaling Technology; buffer 9802) containing 10 μCi of [γ-32P]ATP in the presence or absence of purified PKA-α protein (0.1 μg; Cell Signaling Technology) used as the enzyme for 30 min at 30 °C. The reaction was stopped by adding Laemmli sample buffer and boiling for 5 min. The samples were then subjected to 10% SDS-PAGE, transferred to nitrocellulose membranes, and exposed to x-ray film for autoradiography or phosphor screens for PhosphorImager analysis (Storm 860; GE/Amersham Biosciences) to detect phosphorylated FoxO1. In parallel, aliquots of samples from each group were immunoblotted for FoxO1 and PKA-α to demonstrate the appropriate presence or absence of substrate and enzyme in each experimental group. In some experiments, cold (nonradioactive) ATP was used, and the final samples were immunoblotted with phospho-specific antibodies that detect phosphorylated FoxO1 at Thr24, Ser256, or Ser319. The in vitro kinase assays described above were repeated using commercially acquired purified GST-FoxO1 protein (1 μg; Millipore, Temecula, CA) or GST alone (control) as the substrates to rule out the presence of contaminating kinases that may have been present in immune complex substrate preparations.
Co-immunoprecipitation Experiments
HEK293 cells were co-transfected with empty vector (pcDNA3), FLAG-tagged FoxO1-WT, and/or HA-tagged PKA-α. Cell lysates were prepared using ice-cold cell lysis buffer (Cell Signaling Technology) with complete protease inhibitors (Roche Applied Science). Cell lysates were precleared with protein A/G-agarose beads to minimize nonspecific binding. As an additional control for nonspecific binding, the samples were also immunoprecipitated with nonimmune rabbit IgG. Recombinant PKA-α was immunoprecipitated from cell lysates (1 mg of total protein for each sample) using anti-HA or anti-PKA-α antibodies. The samples were then subjected to SDS-PAGE and immunoblotted using antibodies against FoxO1 or HA. In parallel, aliquots of samples from each group were immunoblotted with antibodies against FLAG, FoxO1, HA, and β-actin to demonstrate the appropriate absence or presence of enzyme (PKA-α), substrate (FoxO1), and loading control.
Cell Culture
Bovine aortic endothelial cells (BAEC) (Cell Applications, San Diego, CA) or human aortic endothelial cells (HAEC) (Lonza, Walkersville, MD) in primary culture were grown in endothelial growth medium EGM-MV (BAEC; Lonza) or EGM-2 (HAEC; Lonza) and used between passages 3 and 6 as described previously (3). Endothelial cells were grown in 60-mm dishes and serum-starved overnight (BAEC) or for 6 h (HAEC) in endothelial basal medium (Lonza). Some groups of cells were treated with forskolin (20 μm) or prostaglandin E2 (PGE2, 500 nm) for 30 min or PKA-α inhibitor H89 (20 μm) from Sigma) before treatment with forskolin or PGE2 as indicated in the figure legends.
Immunoblotting
Total cell lysates were made with cell lysis buffer (Cell Signaling Technology) containing complete protease inhibitors (Roche Applied Science). Samples (30 μg of total protein) were immunoblotted according to standard methods using antibodies against FoxO1, phospho-FoxO1-Thr24, phospho-FoxO1-Ser256, phospho-FoxO1-Ser319, PKA (PKA-α catalytic domain), α-tubulin, HA, FLAG, or β-actin. All of the antibodies were obtained from Cell Signaling Technology except anti-β-actin (Sigma). Immunoblotting results were quantified by scanning densitometry (GE Healthcare) and normalized to α-tubulin or β-actin expression.
siRNA Knockdown of PKA-α
HAEC were transfected with siRNA specifically targeting the catalytic subunit of human PKA with four sequences: 5′-GAA CAC ACC CUG AAU GAA A-3′; 5′-GAA CAC AGC CCA CUU GGA U-3′; 5′-CAA GGA CAA CUC AAA CUU A-3′; and 5′-GCU AAG GGC AAA UGA ACG A-3′ (catalogue number L-004649-00; human gene PRKACA; Dharmacon, Chicago, IL) or control scrambled siRNA (catalogue number D-001810-10–20; Dharmacon) using Lipofectamine Plus reagent for 3 h. One day after transfection, the cells were serum-starved overnight and then treated with vehicle or forskolin (20 μm, 30 min). The cell lysates were subjected to immunoblotting with antibodies against PKA-α, FoxO1, or α-tubulin.
FoxO-responsive Luciferase Reporter Assay
A FoxO-responsive luciferase reporter construct containing a constitutively expressing Renilla element as an internal control (SA Biosciences, Frederick, MD; catalogue number CCS-1022L) was used in this assay. The FOXO-responsive luciferase construct encodes the firefly luciferase reporter gene under control of a minimal CMV promoter and tandem repeats of a FOXO transcriptional response element. BAEC were cultured in 24-well plate to 80% confluence. Each well of BAEC was co-transfected with 800 ng of FOXO-responsive luciferase construct and 200 ng of empty vector, pcDNA3-wild-type PKA-catalytic domain, or kinase-dead PKA-catalytic domain constructs using Lipofectamine Plus (Invitrogen) for 3 h according to the manufacturer's protocol. Transfected cells were cultured in complete medium for 24 h followed by serum-starving overnight. The cells were next treated with vehicle or forskolin (20 μm) for 6 h. In some experiments, HAEC were transfected with the FOXO-responsive luciferase construct in the presence of 100 nm of siRNA specifically targeting PKA or nontargeting control siRNA using Effectene transfection reagent (Invitrogen) for 3 h according to the manufacturer's protocol. Two days after transfection, the cells were washed with PBS and lysed in 100 μl of passive lysis buffer (Promega, Madison, WI). Firefly luciferase activity and Renilla luciferase activity were measured using the dual-luciferase reporter assay system (Promega) according to the manufacturer's protocol and a Berthold Biolumat model LB 9501 luminometer (Berthold Technologies, Oak Ridge, TN). Firefly luciferase activity was normalized to Renilla luciferase activity.
Quantitative Real Time PCR
HAEC were transfected with either scrambled control siRNA or siRNA specifically targeting human PKA-α. In other experiments, the cells were transiently transfected with FoxO1-WT or FoxO1-AAA and then cultured in complete medium for 48 h followed by serum-starvation for 1 h. The cells were then treated with vehicle or H89 (10 μm) for 6 h. Subsequently, RNA was isolated using total RNA and Turbo-DNase kits (Ambion, Inc., Austin, TX). For quantitative real time PCR analysis, total RNA from HAEC was isolated using the RNeasy mini kit (Qiagen). One μg of total RNA was reverse transcribed into cDNA using a high capacity cDNA archive kit (Applied Biosystems, Foster City, CA). All of the primers for mRNA analysis were obtained from Integrated DNA Technologies (Coralville, IA). The primers used were: for human VCAM-1, 5′-TTC CTC AGA TTG GTG ACT CCG-3′ (forward) and 5′-AAA ACT CAC AGG GCT CAG GGT CAG-3′ (reverse); and for human β-actin, 5′-CTG GCA CCC AGC ACA ATG AAG-3′ (forward) and 5′-TAG AAG CAT TTG CGG TGG ACG-3′ (reverse). Quantitative real time PCR was performed using a QuantiTect SYBR Green PCR kit (Qiagen) and analyzed with the ABI Prism 7900HT sequence detection system (Applied Biosystems; Foster City, CA). Quantitative PCR was performed as follows: step 1: 50 °C for 2 min; step 2: 95 °C for 15 min; and step 3: 95 °C for 15 s, 59 °C for 30 s, and 72 °C for 30 s for 45 cycles. The data and calculation of cycle threshold were analyzed using SDS 2.2 software (Applied Biosystems). mRNA expression for VCAM-1 was normalized to β-actin mRNA expression.
Monocyte Adhesion Assay
HAEC were grown to 90% confluence in Lab-Tek chamber slides and serum-starved for 1 h in endothelial basal medium. The cells were then treated without or with TNF-α (10 ng/ml) in the presence of vehicle for 5 h. In some experiments, HAEC were transiently transfected with empty vector pcDNA3, FLAG-tagged FoxO1-WT, or FoxO1-AAA using Lipofectamine Plus (Invitrogen) for 3 h according to the manufacturer's protocol. Transfected cells were cultured in complete medium for 48 h. The cells were serum-starved for 1 h followed by treatment with H89 (10 μm) or TNF-α vehicle (10 ng/ml) in the presence of vehicle for 5 h. U937 monocytes (American Type Culture Collection, Manassas, VA) were grown in DMEM containing 10% FBS and then labeled with 5 μm calcein-AM (Invitrogen) for 15 min. Labeled U937 cells (6 × 105) were incubated for 30 min at 37 °C. Co-cultured cells were washed three times with PBS and fixed in 2% formaldehyde. Prior to visualization, the cells were washed three times with PBS and treated with ProLong Gold anti-fade reagent (Invitrogen). Images of monocytes adhering to HAEC were obtained with an Olympus IX81 inverted microscope with attached CCD camera.
Statistical Analysis
The data are expressed as the means ± S.E. from multiple independent experiments. Unpaired t test (two-tailed) was performed for statistical analyses of bar graphs where appropriate. Differences with p value < 0.05 were considered statistically significant.
RESULTS
FoxO1 Is a Novel Substrate for PKA-α in Vitro
Previously, we demonstrated that FoxO1 is downstream from PKA-α in mediating DHEA action in vascular endothelial cells (3). Therefore, we hypothesized that FoxO1 may be a direct substrate for PKA-α. To evaluate this possibility, we performed an in vitro immune complex kinase assay using purified PKA-α as the enzyme and recombinant wild-type FLAG-tagged FoxO1 (FoxO1-WT) or mutant FoxO1 (FoxO1-AAA, three Akt phosphorylation sites replaced by alanine, T24A/S256A/S319A) immunoprecipitated from transfected HEK293 cells as the substrate in the presence of [γ-32P]ATP (Fig. 1). Significant and substantial phosphorylation of FoxO1-WT was observed in the presence (Fig. 1A, lane 3), but not in the absence (Fig. 1A, lane 2), of purified PKA-α. By contrast, significant phosphorylation of FoxO1-AAA was not detectable (Fig. 1A, lane 4) in the presence of purified PKA-α and [γ-32P]ATP. Similarly, phosphorylation of FoxO1 was undetectable in control samples where PKA-α was incubated with anti-FLAG immunoprecipitates from HEK293 cells transfected with empty pcDNA vector alone (Fig. 1A, lane 1). These results suggest that FoxO1 is a novel direct substrate for PKA-α. Moreover, three previously identified FoxO1 phosphorylation sites for other kinases at Thr24, Ser256, and Ser319 (16) are among the major potential targets for direct phosphorylation by PKA-α because FoxO1-AAA did not undergo substantial detectable phosphorylation by PKA-α.
FIGURE 1.
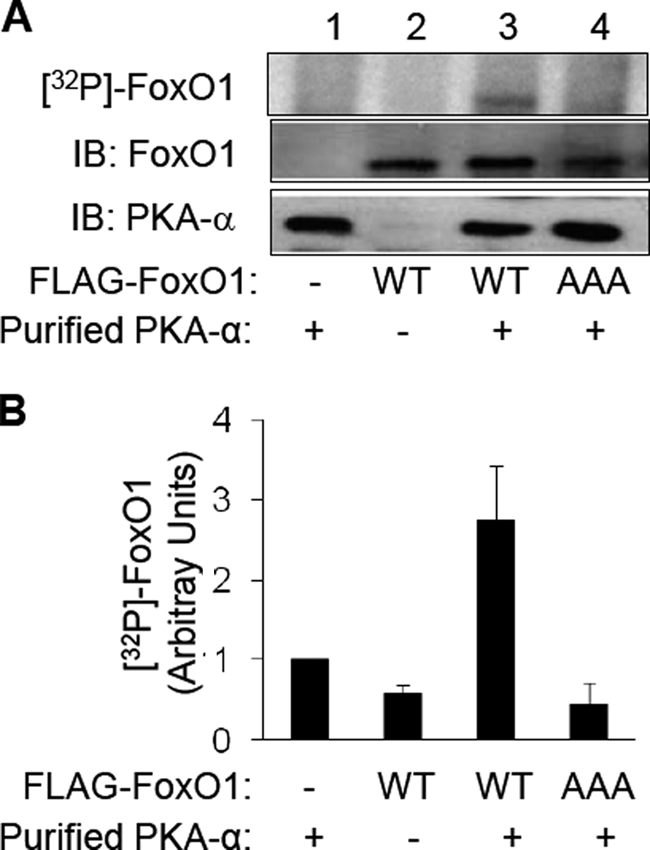
PKA-α directly phosphorylates FoxO1 in vitro. HEK293 cells were transfected with empty vector (pcDNA3) or expression vectors for FLAG-tagged FoxO1-WT or FoxO1-AAA. Two days after transfection, recombinant FoxO1 was immunoprecipitated from cell lysates (1 mg of total protein) using an anti-FLAG antibody. These recombinant FoxO1 proteins were used as the substrate along with purified PKA-α as the enzyme for an in vitro kinase assay as described under “Materials and Methods.” A, autoradiogram of a representative immune complex kinase assay is shown in the top panel. Samples from the kinase assay were immunoblotted (IB) using antibodies against FoxO1 (middle panel) or PKAα (bottom panel), demonstrating the appropriate absence or presence of the substrate (FoxO1) and the kinase (PKA-α). B, 32P-labeled FoxO1 from three independent experiments as shown in A was quantified using a Phospho-Imager and normalized to FoxO1 expression and is represented as the mean ± S.E. (32P-labeled FoxO1-WT in the presence of purified PKA-α is significantly greater that 32P-labeled FoxO1 in all other groups, p < 0.03; 32P-labeled FoxO1 in the absence of PKA-α is not statistically different from phosphorylated FoxO1-AAA in the presence of PKA-α, p > 0.7).
To determine which of the three FoxO1 phosphorylation sites at Thr24, Ser256, and Ser319 are direct targets for PKA-α, we repeated the in vitro immune complex kinase assay represented in Fig. 1 with unlabeled ATP and examined the phosphorylation status of FoxO1 by immunoblotting anti-FLAG immunoprecipitates of transfected HEK293 cells using phospho-specific antibodies that detect phosphorylation of FoxO1 at Thr24, Ser256, or Ser319 (Fig. 2). As expected, phospho-specific antibodies were unable to detect phosphorylation of FoxO1 in the anti-FLAG immunoprecipitates from HEK293 cells transfected with empty pcDNA vector (Fig. 2A, lane 1), or in the samples where the mutant FoxO1-AAA was incubated with PKA-α. Importantly, we detected significant phosphorylation of FoxO1 at Thr24, Ser256, or Ser319 in the presence (Fig. 2A, lane 3), but not in the absence (Fig. 2A, lane 2), of purified PKA-α. Therefore, all three known Akt phosphorylation sites are also direct phosphorylation sites for PKA-α.
FIGURE 2.
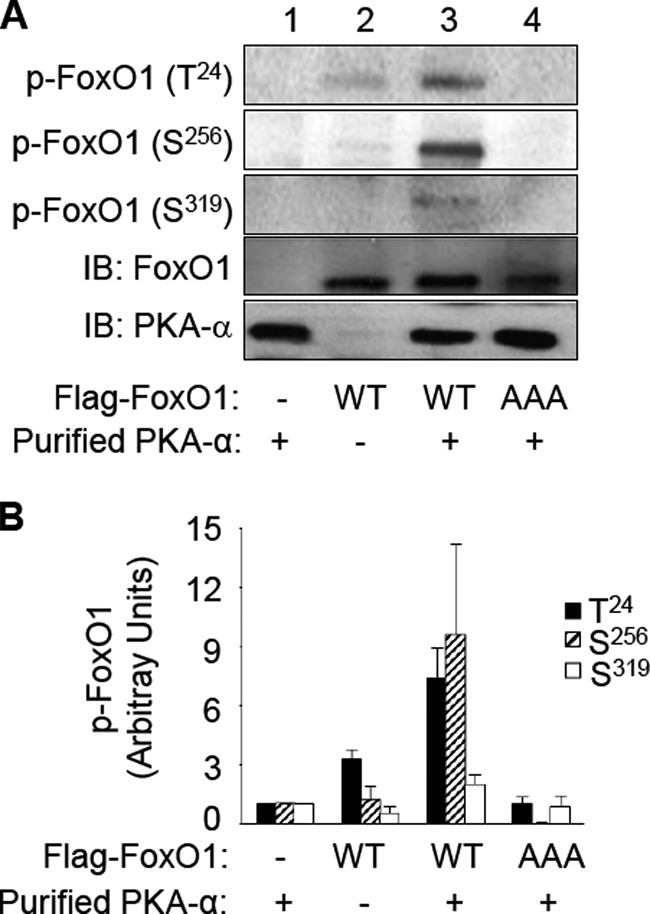
PKA-α specifically phosphorylates FoxO1 at Thr24, Ser256, and Ser319in vitro. Recombinant FoxO1-WT and FoxO1-AAA were obtained and subjected to an in vitro kinase assay with PKA-α as described in the legend to Fig. 1. A, samples were subjected to immunoblotting (IB) using antibodies that specifically detect phospho-FoxO1 at Thr24, Ser256, or Ser319. Samples from the kinase assay were also immunoblotted using antibodies against FoxO1 or PKAα to demonstrate the appropriate absence or presence of the substrate (FoxO1) and the kinase (PKA-α). Representative immunoblots are shown from a single experiment that was repeated independently four times. B, results from four independent experiments as shown in A were quantified using scanning densitometry and normalized to total FoxO1 expression and are represented as the means ± S.E. In the presence of PKA-α, wild-type FoxO1 (but not FoxO1-AAA) was significantly phosphorylated at Thr24 and Ser256 (when compared with all other corresponding groups, p < 0.01). Phosphorylation of wild-type FoxO1 at Ser319 in the presence of PKA-α was also significantly greater than that in the absence of PKA-α (compare the sixth and ninth bars; p < 0.05).
The immune complex kinase assays described above using immunoprecipitated recombinant FoxO1-WT did not undergo detectable phosphorylation in the absence of PKA-α (Figs. 1 and 2). Therefore, it seems very unlikely that our immunoprecipitated recombinant FoxO1 substrates were contaminated with minute amounts of other kinases such as Akt or SGK that may provide an alternative explanation for the phosphorylation of FoxO1 we observed. Nevertheless, it remains possible that PKA-α may be required to activate any such contaminating kinases present and that these putative contaminating kinases may be responsible for the phosphorylation of FoxO1 rather than PKA-α. To definitively rule out this unlikely possibility, we repeated our in vitro kinase assays using commercially obtained purified GST-FoxO1 fusion protein as the substrate using a 1:20 molar ratio of kinase (PKA-α) to substrate (GST-FoxO1) (Fig. 3). Importantly, under these cleaner conditions, using a GST fusion purified protein substrate rather than an immune complex purified substrate, our in vitro kinase assay results were identical. That is, the autoradiogram from the kinase assay using [γ-32P]ATP as well as the kinase assay with cold ATP assessed with phospho-specific antibodies against FoxO1 demonstrated phosphorylation of FoxO1 only in the presence of PKA-α. Moreover, the control GST protein alone did not undergo phosphorylation in the presence of PKA-α. Taken together, these results confirm that FoxO1 is a novel direct substrate for PKA-α in vitro.
FIGURE 3.

PKA-α directly phosphorylates purified GST-FoxO1 protein at Thr24, Ser256, and Ser319in vitro. Commercially obtained purified GST-FoxO1 protein or GST control protein (1 μg, as described under “Materials and Methods”) was used as the substrate along with purified PKA-α (0.1 μg) as the enzyme and for in vitro kinase assays as in Fig. 1. A, autoradiogram from a representative kinase assay using [γ-32P]ATP (top panel). Aliquots from each kinase assay were also immunoblotted using antibodies against FoxO1 (middle panel) or PKA-α (bottom panel), demonstrating the appropriate absence or presence of the substrate (GST-FoxO1) and the kinase (PKA-α). B, 32P-labeled GST-FoxO1 from three independent experiments as shown in A was quantified using a Phospho-Imager and normalized to GST-FoxO1 expression and is represented as the mean ± S.E. 32P-Labeled GST-FoxO1 in the presence of purified PKA-α is significantly greater than that in all other groups; p < 0.03). C, samples from in vitro kinase assays conducted without [γ-32P]ATP were subjected to immunoblotting using antibodies that specifically detect phospho-FoxO1 at Thr24, Ser256, or Ser319. Aliquots from each kinase assay were also immunoblotted using antibodies against FoxO1 or PKA-α to demonstrate the appropriate absence or presence of the substrate (GST-FoxO1) and the kinase (PKA-α). Representative immunoblots are shown from a single experiment that was repeated independently three times. D, results from three independent experiments as shown in C were quantified using scanning densitometry and normalized to total GST-FoxO1 expression and are represented as the means ± S.E. In the presence of PKA-α, GST-FoxO1 was significantly phosphorylated at Thr24, Ser256, and Ser319 (when compared with all other corresponding groups; p < 0.004).
PKA-α Interacts with FoxO1 in Intact Cells
Because FoxO1 is a direct substrate for PKA-α in vitro, we next inquired whether interactions of FoxO1 and PKA-α were detectable in intact cells. We addressed this in two ways. First, we used co-immunoprecipitation experiments to detect interactions between recombinant HA-tagged PKA-α and recombinant FLAG-tagged FoxO1 in HEK293 cells co-transfected with FLAG-tagged FoxO1 or empty pcDNA vector in the presence of HA-tagged PKA-α or empty pcDNA vector (Fig. 4A). Immunoblotting of cell lysates from transfected cells with anti-FLAG, anti-HA, and anti-β-actin antibodies demonstrated the appropriate absence or presence of HA-tagged PKA-α and FLAG-tagged FoxO1 comparable protein content in each group of samples (Fig. 4A, bottom three panels). In the co-immunoprecipitation experiment, we demonstrated substantial interaction between recombinant HA-tagged PKA-α and recombinant FLAG-tagged FoxO1 by immunoblotting anti-HA immunoprecipitates with anti-Foxo1 antibodies (Fig. 4A, top panel, lane 4). In our negative control, no FoxO1 was detected in anti-HA immunoprecipitates from cells co-transfected with HA-PKA-α and empty pcDNA vector (Fig. 4A, top panel, lane 3).
FIGURE 4.

Recombinant and endogenous PKA-α and FoxO1 interact with each other in intact cells. A, HEK293 cells were co-transfected with empty vector or FLAG-tagged FoxO1-WT in the absence or presence of HA-tagged PKA-α. Cell lysates from each group (1 mg total protein) were subjected to immunoprecipitation (IP) using anti-HA antibodies followed by immunoblotting (IB) using antibodies against FoxO1 or HA. Samples of whole cell lysates from the same experiments were also immunoblotted using antibodies against FLAG, HA, or β-actin to demonstrate the appropriate absence or presence of enzyme and substrate in each sample and comparable loading of samples. Representative immunoblots are shown for experiments that were repeated independently three times. B, total cell lysates prepared from nontransfected BAEC were subjected to immunoprecipitation using antibodies against PKA-α or FoxO1. The samples were also subjected to immunoprecipitation using nonimmune IgG as a negative control. Samples from each group were then subjected to immunoblotting using anti-FoxO1 antibodies. Total cell lysates (lane 4) were also immunoblotted with anti-FoxO1 antibodies to confirm the presence of endogenous FoxO1 (lane 4). Representative immunoblots are shown for experiments that were repeated independently three times.
Second, more relevant to physiological conditions, we determined whether endogenous FoxO1 and endogenous PKA-α interact in intact cells by performing co-immunoprecipitation experiments in untransfected BAEC in primary culture (Fig. 4B). Importantly, we detected endogenous FoxO1 by immunoblotting PKA-α immunoprecipitates with anti-FoxO1 antibodies (Fig. 4B, lane 1). The presence of endogenous FoxO1 in BAEC and the specificity of our FoxO1 antibody for detecting FoxO1 were demonstrated by FoxO1 immunoblotting of BAEC lysates and lysates immunoprecipitated with anti-FoxO1 antibody (Fig. 4B, lanes 4 and 2). In our negative control group (Fig. 4B, lane 3), we were unable to detect FoxO1 by immunoblotting samples of BAEC lysates subjected to immunoprecipitation with nonimmune IgG (Fig. 4B, lane 3). The interactions of both recombinant and endogenous FoxO1 and PKA-α that we observed in intact cells are consistent with our findings that PKA-α directly phosphorylates FoxO1 in vitro (Figs. 1–3). Under similar conditions, when we stimulated cells with forskolin (an activator of PKA-α), we did not observe any change in the magnitude of interactions between endogenous FoxO1 and endogenous PKA-α in HAEC as detected by co-immunoprecipitation (data not shown). Nevertheless, our results raise the possibility that PKA-α may directly phosphorylate FoxO1 under physiological conditions in intact vascular endothelial cells.
PKA-α Stimulates Phosphorylation of FoxO1 in Intact Endothelial Cells to Regulate Its Transcriptional Activity
Because FoxO1 is a direct substrate of PKA-α in vitro (Figs. 1–3) and endogenous FoxO1 and PKA-α interact in intact vascular endothelial cells (Fig. 4B), we next evaluated whether activation of PKA results in increased phosphorylation of FoxO1 in intact vascular endothelial cells. HAEC were serum-starved overnight, pretreated with vehicle or H89 (PKA inhibitor; 20 μm, 30 min), and then treated with vehicle or forskolin (compound that increases cAMP to activate PKA; 20 μm, 30 min (17)) (Fig. 5, A and B). Cell lysates were then subjected to immunoblotting with antibodies against phospho-FoxO1 (Ser256). In the absence of H89 pretreatment, when compared with vehicle-treated control cells, forskolin treatment caused a significant increase in the amount of FoxO1 phosphorylated at Ser256 (Fig. 5, A and B, compare lanes 1 and 2). This effect of forskolin was blocked by pretreatment of cells with H89 (Fig. 5, A and B, lane 3).
FIGURE 5.

Forskolin-stimulated phosphorylation of FoxO1 is prevented by pretreatment with PKA inhibitor H89 and by siRNA knockdown of PKA-α in HAEC. A, HAEC were serum-starved overnight and pretreated with vehicle or H89 (20 μm, 30 min) followed by treatment with vehicle or forskolin (20 μm, 30 min). Total cell lysates were subjected to immunoblotting using antibodies against phospho-FoxO1 (Ser256), FoxO1, or α-tubulin. Representative immunoblots are shown for experiments that were repeated independently four times. B, results from three independent experiments as shown in A were quantified using scanning densitometry and normalized to total FoxO1 expression and are represented as the means ± S.E. Forskolin treatment (second bar) significantly stimulated phosphorylation of FoxO1 (when compared with control (first bar), p < 0.01). This effect of forskolin was completely inhibited in cells pretreated with H89 (third bar versus first bar, p > 1.0). C, HAEC were transfected with nontargeting siRNA (control, lanes 1 and 3) or siRNA specifically targeting PKA-α (lanes 2 and 4) as described under “Materials and Methods.” 24 h after transfection, HAEC were serum-starved overnight and then treated with vehicle (Me2SO, lanes 1 and 2) or forskolin (20 μm, lanes 3 and 4) for 30 min. Total cell lysates from each group were subjected to immunoblotting using antibodies against phospho-FoxO1 (Ser256), PKA-α, FoxO1, or α-tubulin. Representative immunoblots are shown for experiments that were repeated independently four times. D, results from three independent experiments as shown in C were quantified by scanning densitometry. The amounts of phospho-FoxO1 were normalized to total FoxO1 (mean ± S.E.). For samples transfected with control siRNA, forskolin treatment significantly increased p-FoxO1 (Ser256) (p < 0.02). Phosphorylation of FoxO1 (Ser256) in cells transfected with siRNA targeting endogenous PKA-α in either the absence or presence of forskolin treatment was significantly decreased when compared with corresponding groups of cells transfected with control siRNA (p < 0.03).
To further investigate the role of PKA in phosphorylation of FoxO1 in intact cells, we evaluated FoxO1 phosphorylation in HAEC treated without or with forskolin in the absence or presence of targeted siRNA knockdown of PKA-α (Fig. 5, C and D). HAEC were transiently transfected with nontargeting control siRNA (Fig. 5, C and D, lanes 1 and 3) or siRNA specifically targeting PKA-α (Fig. 5, C and D, lanes 2 and 4). 24 h after transfection, HAEC were serum-starved overnight and then treated with vehicle (Me2SO, Fig. 5, C and D, lanes 1 and 2) or forskolin (20 μm, Fig. 5, C and D, lanes 3 and 4) for 30 min. Cell lysates from each group were subjected to immunoblotting using antibodies against phospho-FoxO1 (Ser256), PKA-α, FoxO1, or α-tubulin. As expected, when compared with cells transfected with nontargeting control siRNA (Fig. 5C, second panel, lanes 1 and 3), cells transfected with siRNA targeting PKA had substantially reduced expression of PKA (Fig. 5C, second panel, lanes 2 and 4). Expression of FoxO1 or α-tubulin was not significantly different among the four groups of cells (Fig. 5C, third and fourth panels). Similar to previous experiments (Fig. 5, A and B), in HAEC transfected with nontargeting control siRNA, forskolin treatment significantly increased phosphorylation of FoxO1 at Ser256 (Fig. 5, C and D, compare lanes 1 and 3). By contrast, in HAEC transfected with siRNA targeting PKA, the effect of forskolin treatment to increase phosphorylation of FoxO1 was substantially impaired (Fig. 5, C and D, compare lanes 2 and 4). When we repeated experiments shown in Fig. 5 using the more physiological agonist PGE2 (500 nm, 30 min) instead of forskolin, we obtained similar results (Fig. 6). That is, PGE2-stimulated phosphorylation of FoxO1 at Ser256 was substantially impaired by pretreatment of cells with PKA inhibitor H89 or siRNA knockdown of PKA. Taken together, our results suggest that forskolin or PGE2 treatment increases phosphorylation of FoxO1 in vascular endothelial cells through activation of PKA that directly phosphorylates FoxO1.
FIGURE 6.

PGE2-stimulated phosphorylation of FoxO1 is prevented by pretreatment with PKA inhibitor H89 and by siRNA knockdown of PKA-α in HAEC. A, HAEC were serum-starved overnight and pretreated with vehicle or H89 (20 μm, 30 min) followed by treatment with vehicle or PGE2 (500 nm, 30 min). Total cell lysates were subjected to immunoblotting using antibodies against phospho-FoxO1 (Ser256), FoxO1, or α-tubulin. Representative immunoblots are shown for experiments that were repeated independently three times. B, results from three independent experiments as shown in A were quantified using scanning densitometry and normalized to total FoxO1 expression and are represented as the means ± S.E. PGE2 treatment (second bar) significantly stimulated phosphorylation of FoxO1 (when compared with control (first bar), p < 0.03). This effect of PGE2 was completely inhibited in cells pretreated with H89 (third bar versus first bar, p > 0.9). C, HAEC were transfected with nontargeting siRNA (control, lane 1) or siRNA specifically targeting PKA-α (lanes 2 and 3) as described under “Materials and Methods.” 24 h after transfection, HAEC were serum-starved overnight and then treated with vehicle (lane 1) or PGE2 (500 nm, lanes 2 and 3) for 30 min. Total cell lysates from each group were subjected to immunoblotting using antibodies against phospho-FoxO1 (Ser256), PKA-α, FoxO1, or α-tubulin. Representative immunoblots are shown for experiments that were repeated independently three times. D, results from three independent experiments as shown in C were quantified by scanning densitometry. The amounts of phospho-FoxO1 were normalized to total FoxO1 (means ± S.E.). For samples transfected with control siRNA, PGE2 treatment significantly increased p-FoxO1 (Ser256) (p < 0.02). Phosphorylation of FoxO1 (Ser256) in cells transfected with siRNA targeting endogenous PKA-α in the presence of PGE2 treatment was significantly decreased when compared with PGE2-treated cells transfected with control siRNA (p < 0.03).
Because phosphorylation of FoxO1 in endothelial cells is mediated by PKA-dependent signaling, we next tested whether PKA-α regulates FoxO1 promoter activity. HAEC were transiently co-transfected with a FoxO-responsive promoter luciferase reporter construct along with pcDNA empty vector (control), the active catalytic subunit of PKAα (cat-WT), or a kinase-dead mutant of the catalytic subunit of PKAα (cat-KD) (Fig. 7A). Two days after transfection, the cell lysates were subjected to a dual-luciferase reporter assay. When compared with control cells co-transfected with empty vector, expression of cat-WT diminished transactivation of the FoxO response promoter (Fig. 7A, compare first and second bars). By contrast, expression of cat-KD significantly enhanced transactivation of the FoxO response promoter (Fig. 7A, compare first and third bars). Thus, increasing the amount of activated PKA reduced transcriptional activity of FoxO1 while impairing the catalytic activity of PKA enhanced transcriptional activity of FoxO1. To further evaluate the role of PKA to regulate FoxO1 transcriptional activity, we transiently co-transfected HAEC with the FoxO-responsive promoter luciferase reporter construct along with either nontargeting control siRNA or siRNA specifically targeting PKA-α (Fig. 7B). Consistent with results using cat-KD (Fig. 7A), siRNA knockdown of PKA-α enhanced transcriptional activity of FoxO1 relative to results from the control group (Fig. 7B). These data support the idea that direct phosphorylation of FoxO1 by PKA-α regulates transcriptional activity of FoxO1.
FIGURE 7.
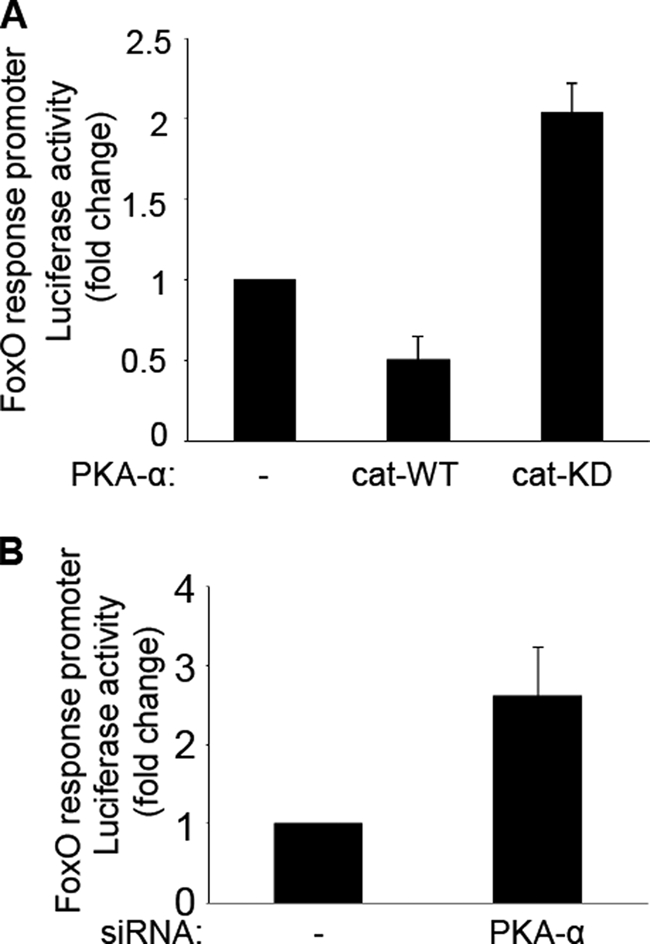
PKA-α regulates transcriptional activity of FoxO. A, HAEC were co-transfected with a FoxO promoter luciferase reporter and expression vectors for pcDNA (control), PKAα-cat-WT, or PKAα-cat-KD. Two days after transfection, the cell lysates were then subjected to dual-luciferase reporter assay as described under “Materials and Methods.” The data shown are the means ± S.E. of three independent experiments. Transcriptional activity of FoxO was substantially inhibited in cells transfected with PKA-α-cat-WT (when compared with the pcDNA control group, p < 0.03). By contrast, transcriptional activity of FoxO was substantially enhanced in cells transfected with PKA-α-cat-KD (p < 0.005). B, HAEC were co-transfected with nontargeting control siRNA or siRNA specifically targeting PKA-α along with the FoxO promoter luciferase reporter construct. Two days later, the lysates were subjected to a dual-luciferase reporter assay. The data shown are the means ± S.E. of three independent experiments. Transcriptional activity of FoxO was significantly increased by siRNA knockdown of endogenous PKA-α (when compared with control cells transfected with nontargeting siRNA; p < 0.05).
Modulation of VCAM-1 Expression through PKA-α/FoxO1 Regulates Endothelial Function
Because a previous study implicated FoxO1 in regulation of VCAM-1 expression in coronary artery endothelial cells (6), we used quantitative RT-PCR analysis to measure VCAM-1 mRNA expression in HAEC transfected with scrambled siRNA or siRNA targeting PKA-α (Fig. 8A). siRNA knockdown of PKA-α in HAEC increased expression of VCAM-1 mRNA 2-fold. Similarly, transfection of HAEC with FoxO1-AAA increased expression of VCAM-1 mRNA over that of cells transfected with FoxO1-WT in either the presence or absence of treatment with H89 (Fig. 8B, compare closed bars with open bars). Moreover, when compared with the corresponding control cells untreated with H89, treatment of cells with the PKA inhibitor H89 significantly increased expression of VCAM-1 mRNA in cells transfected with either FoxO1-WT or FoxO1-AAA (Fig. 8B, compare second group of bars with first group of bars). Taken together, our data suggest that FoxO1 modulation of VCAM-1 expression is regulated by direct phosphorylation of FoxO1 by PKA-α.
FIGURE 8.
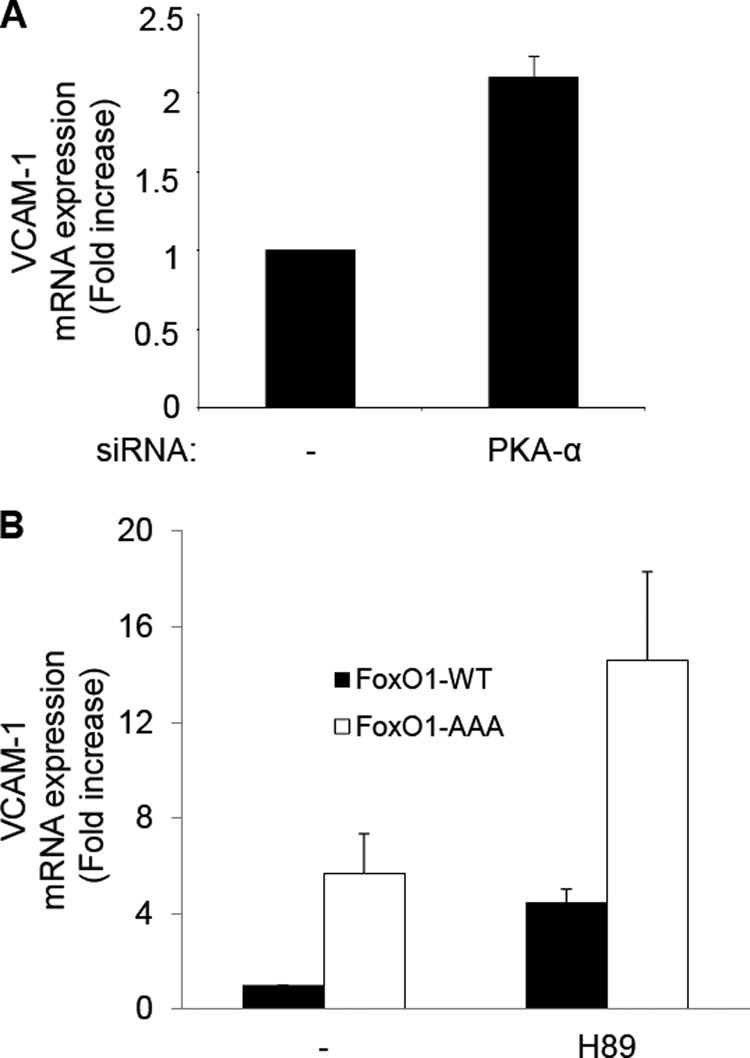
PKA-α regulates transcriptional activity of VCAM-1 through FoxO1. A, HAEC were transfected with nontargeting control siRNA (first bar) or siRNA specifically targeting PKA-α (second bar) as described under “Materials and Methods.” The data shown are the means ± S.E. of three independent experiments. When compared with the control group, the cells transfected with siRNA specifically targeting PKA-α caused a significant increase in expression of VCAM-1 mRNA (p < 0.02). B, HAEC were co-transfected with expression vectors for FoxO1-WT or FoxO1-AAA. 24 h after transfection, HAEC were serum-starved overnight and then treated with vehicle (Me2SO, first and second bars) or H89 (10 μm, third and fourth bars) for 6 h. 1 μg of total RNA was reverse-transcribed and subjected to quantitative real time PCR for VCAM-1 and β-actin using QuantiTect SYBR Green PCR with appropriate primer sets as described under “Materials and Methods.” VCAM-1 mRNA expression was normalized to β-actin mRNA expression. The data shown are the means ± S.E. of four independent experiments. In both the absence and presence of H89, cells transfected with FoxO1-AAA significantly increased expression of VCAM-1 mRNA (compare first and second bars or third and fourth bars; p < 0.03). Moreover, in cells co-transfected with FoxO1-WT, treatment with H89 significantly enhanced expression of VCAM-1 mRNA (compare first and third bars; p < 0.001). Finally, in cells transfected with FoxO1-AAA, H89 treatment caused a significant increase in VCAM-1 mRNA (compare second and fourth bars; p < 0.02).
To further investigate the functional consequences of PKA/FoxO1 regulation of VCAM-1 expression, we used a monocyte adhesion assay to evaluate pro-atherogenic actions of PKA and FoxO-1 in HAEC. TNF-α is known to increase expression of VCAM-1 and other adhesion molecules to increase monocyte adhesion to endothelial cells (18). As expected, when compared with vehicle treatment, TNF-α increased the number of labeled monocytes adhering to endothelial cells (Fig. 9A). In cells transfected with FoxO1-WT there was enhanced binding of monocytes to endothelial cells in the absence of TNF-α treatment (Fig. 9B), whereas transfection with the constitutively active FoxO1-AAA mutant enhanced monocyte binding even further (Fig. 9C). Adhesion of monocytes to endothelial cells in the absence of TNF-α was also augmented by treatment with H89. The effects of TNF-α and H89 were augmented further by transfection with FoxO1-WT or FoxO1-AAA. It is important to note that these experiments are semiquantitative at best and thus difficult to quantify using statistical analyses. Nevertheless, the representative data shown taken from multiple independent experiments demonstrate trends suggesting that phosphorylation of FoxO1 by PKA in vascular endothelial cells down-regulate expression of VCAM-1 to modulate decreased adhesion of monocytes to vascular endothelial cells.
FIGURE 9.

Overexpression of FoxO1 in human endothelial cells regulates TNF-α-stimulated expression of VCAM-1 and monocyte adhesion. HAEC were co-transfected with expression vectors for pcDNA (A), FoxO1-WT (B), or FoxO1-AAA (C). 24 h after transfection, HAEC were serum-starved for 1 h and then treated with vehicle, TNF-α (10 ng/ml), or H89 (20 μm) for 5 h. Calcein-AM-labeled U937 cells (6 × 105) were incubated for 30 min at 37 °C with confluent HAEC. Chambered wells were then washed three times with PBS, and the cells were fixed in 2% formaldehyde. Labeled monocytes adhering to HAEC were visualized using an epifluorescent microscope as described under “Materials and Methods.” B, overexpression of FoxO1-WT and treatment with H89 (PKA-α inhibitor) tended to increase adhesion of monocytes to endothelium when compared with the same treatment in cells transfected with control pcDNA. C, moreover, overexpression of FoxO1-AAA and treatment of PKA-α inhibitor H89 tended to increase adhesion of monocytes to endothelium when compared with control pcDNA. In the absence of TNF-α treatment, when compared with vehicle-treated control cells, TNF-α treatment tended to cause increased adhesion of monocytes to endothelium.
DISCUSSION
FoxO1 is an important transcription factor that regulates glucose homeostasis, cellular proliferation, differentiation, and vascular homeostasis (1–3). Molecular mechanisms regulating FoxO1 activity include phosphorylation of FoxO1 at Thr24, Ser256, and Ser319 by Akt that negatively regulates its function in insulin signaling by causing translocation of FoxO1 from the nucleus to the cytoplasm (10, 11). We recently reported that DHEA treatment of primary endothelial cells acutely increases phosphorylation of FoxO1 in a PKA-dependent manner that reduces expression of ET-1 by interfering with the binding of FoxO1 to the human ET-1 promoter (3). This raises the possibility that FoxO1 may be a direct substrate for PKA-α in endothelial cells, a finding that may be generalizable to other contexts with wide-reaching implications.
FoxO1 Is a Novel Direct Substrate for PKA-α in Vitro and in Intact Cells
Using an in vitro immune complex kinase assay, we found that purified PKA-α directly phosphorylated recombinant FoxO1-WT, whereas phosphorylation of recombinant mutant FoxO1-AAA (missing Akt phosphorylation sites Thr24, Ser256, and Ser319) was not detectable in our assay. We confirmed that Thr24, Ser256, and Ser319 are direct phosphorylation sites for PKA-α by using phospho-specific antibodies in similar experiments. Thus, in vitro, FoxO1 is a direct substrate for PKA-α that shares phosphorylation sites with Akt. The consensus phosphorylation motif for Akt substrates (R/K)XRXX(S*/T*) (19) is similar to the consensus phosphorylation motif for PKA substrates (R/K)XX(S*/T*) (20). The similarities between consensus phosphorylation sites for Akt and PKA are consistent with our data that directly demonstrate phosphorylation of Thr24, Ser256, and Ser319 in FoxO1 by PKA.
Experiments using purified GST-FoxO1 fusion proteins as substrates in additional in vitro kinase assays strongly rule out the remote possibility that minute amounts of other kinases such as Akt or SGK may be contaminating our immune complex kinase assays. Thus, we conclude that FoxO1 is a bona fide novel substrate for PKA-α in vitro. Our in vitro kinase assay data also support the idea that three phosphorylation sites (Thr24, Ser256, and Ser319) are direct substrates for PKA. However, in intact cells, it remains possible that PKA may be activating other kinases that then also directly phosphorylate some or all of these three phosphorylation sites on FoxO1. In experiments with purified kinase and substrate, we used a molar ratio of 1:20, providing limited information about the required stoichiometry in vitro. However, this does not provide much insight into the physiological stoichiometry of the reaction in vivo, an interesting topic that is beyond the scope of the present study.
To further support the role for FoxO1 as a bona fide physiological substrate for PKA in intact cells, we used co-immunoprecipitation assays to demonstrate that both recombinant and endogenous FoxO1 and PKA-α interact in intact primary endothelia cells. Moreover, we demonstrated that treatment of endothelial cells with forskolin, a known activator of PKA, increased phosphorylation of FoxO1, whereas pretreatment with the PKA inhibitor H89 blocked this effect. These results were more definitively substantiated using siRNA knockdown of PKA. In addition to the pharmacological agonist forskolin, more physiological agonists including PGE2 also generated similar results. Taken together with our in vitro kinase data, our results strongly suggest that endogenous PKA is required to directly phosphorylate endogenous FoxO1 in intact vascular endothelial cells in response to forskolin treatment. Thus, direct phosphorylation of FoxO1 by PKA is the likely mechanism used by DHEA to regulate ET-1 transcription that we described recently (3). Similarly, previous studies that demonstrate ligands including vasoactive intestinal peptide and follicle-stimulating hormone require activation of both PKA and phosphorylation of FoxO1 or other FoxO isoforms to mediate physiological functions may involve the novel mechanism described here of direct phosphorylation of FoxO1 by PKA (21, 22). Direct phosphorylation of FoxO1 by PKA may also contribute to interactions between PKA signaling and insulin/IGF-1-dependent signaling in regulation of adipogenesis by Rho-dependent pathways (23). Thus, direct phosphorylation of FoxO1 by PKA may have important and varied physiological consequences in a wide range of cellular contexts.
SGK is another kinase known to directly phosphorylate FoxO1 at Akt phosphorylation sites Thr24 and Ser319 (11, 12). Our present study indicates that PKA is an additional kinase that converges along with Akt and SGK to regulate FoxO1 through phosphorylation of similar sites. This contribution of PKA adds additional complexity in cellular signaling networks that may contribute further to ligand-specific biological actions. The physiological meaning of this additional complexity is not clear. However, this is not unprecedented because multiple kinases directly phosphorylating the same sites in a single substrate is not unique to FoxO1. For example, endothelial NOS is directly phosphorylated at Ser1179 by several kinases including Akt, AMPK, PKA, and PKC (24–27). One possible function for this redundancy with multiple kinases phosphorylating the same site on the same substrate may be to facilitate signal specificity from upstream agonists. Investigation of functional consequences and the relative importance of cross-talk between PKA, Akt, and SGK in phosphorylating FoxO1 is beyond the scope of the present study.
Expression of VCAM-1 in Vascular Endothelial Cells Is Regulated by Phosphorylation of FoxO1 by PKA-α
Phosphorylation of FoxO1 at Thr24, Ser256, and Ser319 results in translocation of FoxO1 from the nucleus to the cytoplasm to inhibit transcriptional function of FoxO1 (2). Consistent with PKA phosphorylating FoxO1 at Thr24, Ser256, and Ser319, we found that co-expression of PKA-WT inhibited transactivation of a FoxO1-responsive promoter reporter, whereas expression of kinase-dead mutant PKA-KD or siRNA knockdown of endogenous PKA-α enhanced transactivation of the FoxO1 reporter. Moreover, we observed similar effects with siRNA knockdown of PKA-α.
A previous study reported that siRNA knockdown of FoxO1 in human coronary artery endothelial cells reduces VEGF-induced VCAM-1 expression (6). In our study, expression of VCAM-1 mRNA in HAEC was significantly enhanced by siRNA knockdown of PKA-α or treatment of HAEC with PKA inhibitor H89. This effect on expression of VCAM-1 mRNA was further enhanced by co-transfection with a constitutively nuclear FoxO1 mutant (FoxO1-AAA). One likely explanation for our data is that direct phosphorylation of FoxO1 by PKA in endothelial cells results in translocation of FoxO1 to the cytoplasm where it can no longer promote transcriptional activity of the VCAM-1 promoter. VCAM-1 is an adhesion molecule induced by inflammatory cytokines including IL-1, TNF, and lipopolysaccharide (28, 29). VCAM-1 plays a critical role in increased adhesion of lymphocytes, monocytes, eosinophils, and basophils to vascular endothelium to promote atherogenesis and endothelial dysfunction (30). Our data establish links between PKA, FoxO1, VCAM-1 expression, and monocyte adhesion to vascular endothelium. Inhibition of PKA with H89 increased adhesion of monocytes to endothelial cells, similar to treatment of cells with TNF-α. This effect is further enhanced by transfecting cells with a constitutively nuclear mutant of FoxO1 (FoxO1-AAA) consistent with the increased expression of VCAM-1. Our findings are in agreement with and complementary to observations that elevated cAMP or overexpression of the catalytic subunit of PKA inhibits NF-κB-mediated gene expression, including VCAM-1, E-selectin, and tissue factor in THP-1 monocytes and human umbilical vein endothelial cells (31). Moreover, a recent study reports that the PI3K activity networks with cAMP and PKA signaling to regulate estrogen action signals (32). Because PKA-α phosphorylation sites on FoxO1 overlap with Akt phosphorylation sites, our findings add an additional layer of complexity in the signaling network comprised by PI3K and PKA interactions.
In summary, we found that FoxO1 is a novel direct substrate for PKA-α in vascular endothelial cells with phosphorylation targets on FoxO1 that overlap with Akt, SGK, and other important kinases. This had functional consequences to regulate expression of VCAM-1 and adhesion of monocytes to endothelium, suggesting that activation of PKA-α in vascular endothelium may have anti-atherogenic actions. Our results are relevant to regulation of vascular homeostasis by PKA and may also be important for understanding mechanisms of action in other cellular contexts where PKA and FoxO1 are implicated.
Acknowledgments
We thank Dr. Eric Tang (University of Michigan Medical School, Ann Arbor, MI) for the gift of FLAG-tagged FoxO1 constructs and Dr. Susan S. Taylor (University of California, San Diego, CA) for the HA-tagged PKA constructs.
This work was supported in part by funds from the Intramural Research Program, National Center for Complementary and Alternative Medicine, National Institutes of Health (to M. J. Q.).
- VCAM
- vascular cellular adhesion molecule
- PKA
- cAMP-dependent protein kinase
- HAEC
- human aortic endothelial cell(s)
- PGE2
- prostaglandin E2
- BAEC
- bovine aortic endothelial cell(s)
- SGK
- serum/glucocorticoid regulated kinase
- DHEA
- dihydroepiandrosterone.
REFERENCES
- 1. Nakae J., Biggs W. H., 3rd, Kitamura T., Cavenee W. K., Wright C. V., Arden K. C., Accili D. (2002) Nat. Genet. 32, 245–253 [DOI] [PubMed] [Google Scholar]
- 2. Accili D., Arden K. C. (2004) Cell 117, 421–426 [DOI] [PubMed] [Google Scholar]
- 3. Chen H., Lin A. S., Li Y., Reiter C. E., Ver M. R., Quon M. J. (2008) J. Biol. Chem. 283, 29228–29238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Furuyama T., Kitayama K., Shimoda Y., Ogawa M., Sone K., Yoshida-Araki K., Hisatsune H., Nishikawa S., Nakayama K., Nakayama K., Ikeda K., Motoyama N., Mori N. (2004) J. Biol. Chem. 279, 34741–34749 [DOI] [PubMed] [Google Scholar]
- 5. Potente M., Urbich C., Sasaki K., Hofmann W. K., Heeschen C., Aicher A., Kollipara R., DePinho R. A., Zeiher A. M., Dimmeler S. (2005) J. Clin. Invest. 115, 2382–2392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Abid M. R., Shih S. C., Otu H. H., Spokes K. C., Okada Y., Curiel D. T., Minami T., Aird W. C. (2006) J. Biol. Chem. 281, 35544–35553 [DOI] [PubMed] [Google Scholar]
- 7. Kitamura Y. I., Kitamura T., Kruse J. P., Raum J. C., Stein R., Gu W., Accili D. (2005) Cell Metab. 2, 153–163 [DOI] [PubMed] [Google Scholar]
- 8. Myatt S. S., Lam E. W. (2007) Nat. Rev. Cancer 7, 847–859 [DOI] [PubMed] [Google Scholar]
- 9. van der Horst A., Burgering B. M. (2007) Nat. Rev. Mol. Cell Biol. 8, 440–450 [DOI] [PubMed] [Google Scholar]
- 10. Kops G. J., Burgering B. M. (1999) J. Mol. Med. 77, 656–665 [DOI] [PubMed] [Google Scholar]
- 11. Van Der Heide L. P., Hoekman M. F., Smidt M. P. (2004) Biochem. J. 380, 297–309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Burgering B. M., Medema R. H. (2003) J. Leukocyte Biol. 73, 689–701 [DOI] [PubMed] [Google Scholar]
- 13. Kobayashi T., Cohen P. (1999) Biochem. J. 339, 319–328 [PMC free article] [PubMed] [Google Scholar]
- 14. Park J., Leong M. L., Buse P., Maiyar A. C., Firestone G. L., Hemmings B. A. (1999) EMBO J. 18, 3024–3033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Taylor S. S., Kim C., Cheng C. Y., Brown S. H., Wu J., Kannan N. (2008) Biochim. Biophys. Acta 1784, 16–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Qu S., Altomonte J., Perdomo G., He J., Fan Y., Kamagate A., Meseck M., Dong H. H. (2006) Endocrinology 147, 5641–5652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Niisato N., Ito Y., Marunaka Y. (1999) Am. J. Physiol. 277, L727–L736 [DOI] [PubMed] [Google Scholar]
- 18. Bevilacqua M. P. (1993) Annu. Rev. Immunol. 11, 767–804 [DOI] [PubMed] [Google Scholar]
- 19. Kane S., Sano H., Liu S. C., Asara J. M., Lane W. S., Garner C. C., Lienhard G. E. (2002) J. Biol. Chem. 277, 22115–22118 [DOI] [PubMed] [Google Scholar]
- 20. Huang S. Y., Tsai M. L., Chen G. Y., Wu C. J., Chen S. H. (2007) J. Proteome Res. 6, 2674–2684 [DOI] [PubMed] [Google Scholar]
- 21. Anderson P., Gonzalez-Rey E. (2010) Mol. Cell. Biol. 30, 2537–2551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Richards J. S., Sharma S. C., Falender A. E., Lo Y. H. (2002) Mol. Endocrinol. 16, 580–599 [DOI] [PubMed] [Google Scholar]
- 23. Madsen L., Kristiansen K. (2010) Ann. N.Y. Acad. Sci. 1190, 1–14 [DOI] [PubMed] [Google Scholar]
- 24. Dimmeler S., Fleming I., Fisslthaler B., Hermann C., Busse R., Zeiher A. M. (1999) Nature 399, 601–605 [DOI] [PubMed] [Google Scholar]
- 25. Chen Z. P., Mitchelhill K. I., Michell B. J., Stapleton D., Rodriguez-Crespo I., Witters L. A., Power D. A., Ortiz de Montellano P. R., Kemp B. E. (1999) FEBS Lett. 443, 285–289 [DOI] [PubMed] [Google Scholar]
- 26. Boo Y. C., Sorescu G., Boyd N., Shiojima I., Walsh K., Du J., Jo H. (2002) J. Biol. Chem. 277, 3388–3396 [DOI] [PubMed] [Google Scholar]
- 27. Michell B. J., Chen Z., Tiganis T., Stapleton D., Katsis F., Power D. A., Sim A. T., Kemp B. E. (2001) J. Biol. Chem. 276, 17625–17628 [DOI] [PubMed] [Google Scholar]
- 28. Carlos T. M., Schwartz B. R., Kovach N. L., Yee E., Rosa M., Osborn L., Chi-Rosso G., Newman B., Lobb R., Rosso M. (1990) Blood 76, 965–970 [PubMed] [Google Scholar]
- 29. Iademarco M. F., Barks J. L., Dean D. C. (1995) J. Clin. Invest. 95, 264–271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Goldberg R. B. (2009) J. Clin. Endocrinol. Metab. 94, 3171–3182 [DOI] [PubMed] [Google Scholar]
- 31. Ollivier V., Parry G. C., Cobb R. R., de Prost D., Mackman N. (1996) J. Biol. Chem. 271, 20828–20835 [DOI] [PubMed] [Google Scholar]
- 32. Cosentino C., Di Domenico M., Porcellini A., Cuozzo C., De Gregorio G., Santillo M. R., Agnese S., Di Stasio R., Feliciello A., Migliaccio A., Avvedimento E. V. (2007) Oncogene 26, 2095–2103 [DOI] [PubMed] [Google Scholar]