

Abstract
Crystal structure analysis of Flavivirus methyltransferases uncovered a flavivirus-conserved cavity located next to the binding site for its cofactor, S-adenosyl-methionine (SAM). Chemical derivatization of S-adenosyl-homocysteine (SAH), the product inhibitor of the methylation reaction, with substituents that extend into the identified cavity, generated inhibitors that showed improved and selective activity against dengue virus methyltransferase (MTase), but not related human enzymes. Crystal structure of dengue virus MTase with a bound SAH derivative revealed that its N6-substituent bound in this cavity and induced conformation changes in residues lining the pocket. These findings demonstrate that one of the major hurdles for the development of methyltransferase-based therapeutics, namely selectivity for disease-related methyltransferases, can be overcome.
Keywords: Enzyme Inhibitors, Enzyme Structure, S-adenosylmethionine (SAM), Viral Protein, Viral Replication, X-ray Structure, Flavivirus, Inhibitors, Methyltransferase, Rational Design
Introduction
Methyltransferases (MTases)3 play key roles in normal physiology and human diseases through methylating DNA, RNA, and proteins. Almost all MTases use S-adenosyl-l-methionine (SAM) as a methyl donor and generate S-adenosyl-l-homocysteine (SAH) as a by-product. Pharmacological modulation of MTases by small molecules represents a novel approach to therapeutic intervention in cancer and other diseases (1). However, because the core domains of various MTases are conserved, designing inhibitors that specifically block the disease-related MTase without affecting other MTases, has been challenging. The ability to rationally design and generate selective inhibitors would have profound implications for development of new medicines for many methyltransferase-mediated diseases.
Dengue virus (DENV), from genus Flavivirus in the family Flaviviridae, is the most prevalent mosquito-borne viral pathogen that infects humans. The four serotypes of DENV (DENV-1 to -4) pose a public health threat to 2.5 billion people worldwide, and cause 50–100 million human infections each year. Neither vaccine nor antiviral therapy is currently available for DENV. The flavivirus MTase methylates the guanine N7 and ribose 2′-O positions of the viral RNA cap in a sequential manner (i.e. GpppA-RNA → m7GpppA-RNA → m7GpppAm-RNA) (2, 3). Recent studies have shown that flavivirus MTase is critical for viral replication and, therefore, represents a valid target for antiviral therapeutics (4–6). We therefore examined the feasibility to design inhibitors that specifically modulate flavivirus MTase.
EXPERIMENTAL PROCEDURES
Preparation of DENV-3 MTases
The DNA fragment representing the MTase domain of DENV-3 was cloned into expression vector pGEX4T1 (Amersham Biosciences). Ala-substitution mutant MTases were prepared using a standard overlapping PCR procedure. Recombinant MTases, containing an N-terminal GST, were expressed in Escherichia coli. BL21 cells and purified through a GSTPrepTM FF 16/10 column (GE Healthcare). The GST tag was then cleaved by thrombin and removed from the MTases using the GST column. The MTases were further purified through gel filtration to ensure protein purity was >95%. The proteins were then concentrated using Amicon Ultra tube (MW cut-off of 10 kDa; Millipore).
Construction and Analysis of Mutant DENV-2
DENV-2 (strain TSV01) genome-length cDNA clones with specific mutations were constructed by using an infectious cDNA clone pACYC FLTSV. The methods for mutagenesis of the cDNA clone, in vitro transcription, RNA transfection, immunofluorescence assay, and plaque assay were reported previously (7).
Preparation of Human RNA MTase (hRNMT)
The 1st strand cDNA of hRNMT was synthesized using primer hRNMT-RT-REV (supplemental Table S1) and cellular RNA extracted from HuH7 cells. The cDNA encoding hRNMT amino acids 1–476 was PCR-amplified using primers hRNMT-Xho-FOR and hRNMT-Bam-REV (supplemental Table S1) from the RT reaction. The PCR product was cloned into expression vector pET15b (Novagen). The hRNMT protein, with an N-terminal His tag, was expressed in BL21 cells, and purified through a 5-ml HiTrap-chelating HP column (Amersham Biosciences).
DENV MTase Assays
Scintillation proximity-based N-7 and 2′-O methylation assays were used to determine the inhibitory activities of compounds, as previously reported (8, 9). The standard deviation is calculated by the non-biased n-1 method: Standard deviation (S.D.) = √((nΣ×2 − (Σ(×)2)/(n(n−1))). Nonlinear regression (curve fit) and the equation below for sigmoidal dose-response (variable slope) from GraphPad Prism version 3.02 (GraphPad Prism, Inc.) were used to interpolate values for IC50 values in Equation 1,
where X is the logarithm of concentration. Y is the response. Y starts at Bottom and goes to Top with a sigmoid shape. This is identical to the “four parameter logistic equation” (10). For determination of Ki values, the Cheng-Prusoff equation (11) shown as Equation 2 was used.
 |
Human DNA MTase 1A (hDNMT1A) Assay
The hDNMT1A reaction contained 2 units of hDNMT1A MTase (New England BioLabs), 1 pmol annealed double-stranded hemi-DNA oligonucleotide (supplemental Table S1), 320 nm [3H-methyl]SAM in the hDNMT buffer (50 mm Tris/HCl, pH 7.8, 1 mm EDTA, 1 mm DTT, 5% glycerol, 100 μg/ml BSA, and 0.05% CHAPS). The reaction was assembled in a 96-well ½ area, white opaque plates (Corning). After incubation at 37 °C for 40 min, the reaction was stopped with 2× stop solution (containing streptavidin-SPA beads), and measured for [3H]methyl incorporation. Dose-responsive experiments were performed to determine the Ki values of inhibitors. The double-stranded hemi-DNA oligonucleotide was prepared by annealing 40-μm hDNMT sense and antisense primers (supplemental Table S1) in 1× assay buffer through denaturing at 95 °C for 3 min and cooling to room temperature.
Human RNA guanine-7-MTase (hRNMT) Assay
The hRNMT reaction contained 40 nm hRNMT MTase, 1 pmole RNA substrate (5′-GpppAGAACCUG-biotin-TEG-3 (TriLink BioTechnologies), 640 nm [3H-methyl]SAM in the hRNMT buffer (50 mm Tris/HCl, pH 7.5, 5 mm DTT, and 0.05% CHAPS). The RNA substrate and enzyme were first mixed in buffer in a single well in a 96-well ½ area, white opaque plates (Corning), followed by initiation of the reaction with [3H-methyl]SAM. The reaction was carried out at 22 °C for 10 min, stopped with 2× stop solution (containing streptavidin-SPA beads), and quantified for [3H]methyl incorporation.
Crystallization and Structure Determination
DENV-3 MTase was crystallized using the hanging-drop vapor diffusion method. 1 μl of reservoir solution (containing 22.5% PEG 8000, 0.2 m NaCl, 0.1 m Tris, pH 8.5, and 20 mm tri-sodium citrate) was mixed with an equal volume of MTase solution at 10 mg/ml. The unit cell parameters are given in Table 1. The asymmetric unit contains two MTase molecules. For the structure with compound, 4 mm of compound was first added to the protein solution from a stock of 50 mm (dissolved in 90% DMSO) prior to mixing with the reservoir solution. Crystals appeared overnight and grew to maximum size within a week. Crystals were frozen by transferring stepwise into a mother liquor containing an additional 10%, then 20% (v/v) glycerol as cryoprotectant, and frozen in liquid nitrogen. Diffraction data were collected at the PXII (X10SA) beam line of the Swiss Light Source, Paul Scherrer Institut, Switzerland. Indexing and scaling were performed using MOSFLM (12) and SCALA, part of the CCP4i suite (13). The structures were determined by molecular replacement using MOLREP from the CCP4i package using DENV2 MTase (PDB code 1L9K) (2) as a search probe. For the compound 10* structure, only one of the two molecules (molecule A) is bound to the inhibitor, while the other molecule C binds to the co-purifying SAH cofactor (Table 2). The bond restraints for the compound were built using SKETCHER from the CCP4i package. Manual rebuilding of the model was performed using COOT (14) and the structures were refined using REFMAC5. The quality of the structures was analyzed using PROCHECK (15).
TABLE 1.
Data collection and phasing statistics
| SAM | Compound 10* | |
|---|---|---|
| Space group | P21212 | P21212 |
| Cell parameters (a, b, c; Å) | 51.82, 61.10, 186.04 | 51.81, 61.10, 184.68 |
| Wavelength (Å) | 1.0 | 1.0 |
| Resolution range (Å) | 51.85–1.7 | 51.78–1.7 |
| No. of observed reflections | 455628 | 349066 |
| No. of unique reflectionsa | 66086 (9512) | 64661 (9441) |
| Completeness (%) | 100 (100) | 98.9 (99.8) |
| Multiplicity | 6.9 (6.9) | 5.4 (5.3) |
| Rmergeb | 0.076 (0.536) | 0.071 (0.424) |
| I/σ(I) | 16.1 (3.4) | 14.6 (3.2) |
| Solvent content (%) | 42.8 | 41.5 |
a The numbers in parentheses refer to the last (highest) resolution shell.
b Rmerge = ΣhΣi|Ihi − <Ih>|/Σh,i Ihi, where Ihi is the ith observation of the reflection h, while <Ih> is its mean intensity.
TABLE 2.
Refinement statistics
| SAM | Compound 10* | |
|---|---|---|
| Resolution range (Å) | 30–1.7 | 30–1.7 |
| Completeness (%) | 100 | 98.57 |
| No of reflections | ||
| Used for refinement | 62658 | 61298 |
| Used for Rfree calculation | 3345 | 3279 |
| No of non hydrogen atoms (Average B factors, Å2) | ||
| Protein chain A | 2051 (18.6) | 2074 (19.3) |
| Protein chain C | 2062 (18.0) | 2076 (19.9) |
| Compound | − | 34 (16.4) |
| SAM (chain B; D) | 27 (14.1); 27 (14.8) | − |
| SAH | − | 26 (20.7) |
| Water molecules | 689 (32.8) | 718 (33.3) |
| Rfactora (%) | 0.192 | 0.197 |
| Rfreeb(%) | 0.220 | 0.231 |
| RMS deviations from ideality | ||
| Bond lengths (Å) | 0.013 | 0.014 |
| Bond angles (°) | 1.64 | 1.57 |
| Ramanchandran plot | ||
| Residues in most favoured regions (%) | 92.9 | 93.8 |
| Residues in additional allowed regions (%) | 6.7 | 5.7 |
| Residues in generously allowed regions (%) | 0.0 | 0.0 |
| Residues in disallowed regions (%) | 0.5 | 0.5 |
| PDB code | 3P8Z | 3P97 |
a Rfactor = Σ||Fobs| − |Fcalc||/Σ|Fobs|.
b Rfree was calculated with 5% of reflections excluded from the refinement.
Binding of RNA to DENV MTase Measured by Competitive Fluorescence Polarization
To measure the displacement of GTP-bodipy binding to DENV MTase by RNA, we adapted the method used by (17). Briefly, 100 nm GTP-bodipy and 300 nm MTase in 50 mm Tris/HCl, pH 7.5 plus 20 mm NaCl were incubated with 2-fold serial dilutions of RNA ranging from 0–3 μm in black 96-well ½ area microplate (Corning). Fluorescence polarization was measured using Synergy 4 microplate reader (Biotek) with λex = 485 nm and λem = 528 nm. IC50 (concentration of ligand needed to displace 50% of GTP-bodipy) was determined by fitting the dose response curve with Equation 3,
 |
where FP0 is the fluorescence polarization units (mP) obtained when no GTP-bodipy in bound and FPT the fluorescence polarization units (mP) when all GTP-bodipy is bound. [L] is the ligand concentration and n represents the Hill slope obtained.
Ki for ligand was defined by the Cheng-Prusoff equation (11) shown in Equation 4,
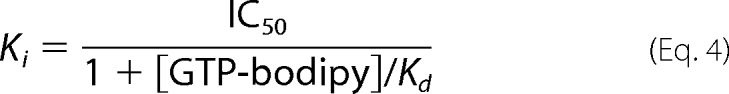 |
where Kd is the dissociation constant of GTP-bodipy for MTase (Kd = 0.8 ± 0.2 μm).
Thermo-denaturation Assay
Three μm of MTase were incubated with 0.01X SYPRO® Orange protein gel stain (Invitrogen) in the absence or presence of 50 μm sinefungin (SF) in 1× assay buffer (50 mm Bis/Tris-HCl, pH 7.5 and 20 mm NaCl) for 10 min in a 96-well PCR white plate (Bio-Rad). The plate was then sealed with Microseal®B Adhesive sealer (Bio-Rad) and heated from 25 to 85 °C with increments of 0.5 °C using iQTM5 Multicolor Real-Time PCR Detection System (Bio-Rad). Excitation and emission wavelengths were 485 nm and 625 nm, respectively. MTase melting temperature (Tm) in the presence or absence of SF was determined from the graph of -dRFU/dT versus temperature, where RFU is relative fluorescence unit, and T is the temperature in degree Celsius.
RESULTS
DENV MTase-SAM Co-crystal Complex
We determined the crystal structure of DENV-3 MTase (representing the N-terminal 272 amino acids of the viral NS5 protein) in complex with methyl donor SAM at a resolution of 1.7 Å (PDB code 3P8Z), with a final Rfactor of 19.2% and an Rfree of 22% (Fig. 1; Tables 1 and 2). The structure of the SAM-MTase complex is almost identical to that of the SAH-MTase complex (2), with an overall Cα root-mean-square deviation (RMSD) of 0.49 Å (Fig. 1A). The current structure also closely resembles MTase structures from other members of the Flavivirus family (reviewed in Ref. 18). All residues forming the SAM/SAH-binding pocket are well superimposed (data not shown), indicating that the extra methyl group in the SAM molecule does not affect the ligand-protein interaction. Because no SAM was present during the crystallization, its presence in the structure must have originated from E. coli during protein expression. Biochemical analysis of the purified MTase also suggested that the SAM molecule was co-purified with the protein (supplemental Fig. S1).
FIGURE 1.

A cavity in DENV MTase. A, superposition of the crystal structures of the DENV-3 MTase-SAM complex (yellow) and the DENV-2 MTase-SAH complex (green; PDB code: 1L9K (2)). SAM and SAH are shown in stick representation. B, surface representation of the DENV-3 MTase. The additional cavity adjacent to the adenine of the SAM molecule is colored in yellow. The MTase protein was set to partial transparency to view the complete molecule of SAM. SAM is shown as stick representation. C, stereo view of amino acids involved in the formation of the additional cavity. Residues and the SAM molecule are labeled and shown in stick representation. The images were produced using PyMOL.
Mutation of Residues Lining the Hydrophobic Pocket Affect Virus Replication
Structure examination located the additional hydrophobic cavity (colored in yellow, Fig. 1B) above the adenine base that was previously identified in West Nile virus (WNV) MTase (19). As shown in Fig. 1C, the cavity is lined by the conserved amino acids Phe-133, Ile-147, Gly-148, Glu-149, Arg-160, Arg-163, Val-164, and Leu-182, among which Phe-133 and Ile-147 also contribute to the SAM-binding pocket. Functional analysis of the identified cavity was performed through Ala mutations of these residues in the context of genome-length RNA of DENV-2. After transfection into BHK-21 cells, wild-type (WT), and mutant RNAs generated varying amounts of envelope protein-expressing cells in the order of WT>F133A≈L182A >G148A>R163A>R160A (Fig. 2A). All RNAs produced infectious viruses with plaque sizes of WT≈F133A≈L182A>G148A≈R163A>R160A (Fig. 2B). Sequencing analysis showed that all mutant viruses retained the engineered changes with no other mutations (data not shown). The growth kinetics of F133A and L182A viruses was similar to that of WT virus, whereas the G148A, R160A, and R163A viruses replicated slower than the WT virus (Fig. 2C). At 24 h post-infection, the viral titers of these latter three mutant viruses were about 1,000-fold less than that of WT virus (Fig. 2C). Similar results were obtained from three independent transfection experiments. The negative effect of the R160A or R163A substitution on viral replication was further confirmed using a luciferase-reporting replicon (supplemental Fig. S2). In vitro enzymatic assays using recombinant MTases (supplemental Fig. S3) showed that most of the mutations reduced either N-7 (R160A, R163A) or 2′-O (F133A, L182A) methylation activities, while both activities were significantly decreased in mutant G148A (Table 3). Thermo-denaturation analysis suggests that the defects in cap methylations are not due to misfolding of the mutant MTases as their melting temperatures are not significantly reduced compared with the WT protein. In addition, their ability to bind RNA and sinefungin was not markedly different from WT protein (Table 3). Overall, the data support results obtained with WNV that the identified cavity is important for Flavivirus cap methylations as well as replication in cells (19).
FIGURE 2.

Functional analysis of the identified cavity in DENV-2 replication. A, immunofluorescence analysis (IFA). BHK-21 cells were electroporated with WT and mutant genome-length RNAs. The mutant genome-length RNAs contained Ala substitution at indicated positions. At various time points post-transfection (p.t.), the cells were subjected to IFA using mouse antibody 4G2 (against DENV E protein) and anti-mouse immunoglobulin G conjugated with FITC (green) as the primary and secondary antibodies, respectively. Nuclei were counterstained with DAPI in blue. B, plaque morphology of WT and mutant viruses from supernatants collected from Vero cells after 5 days post-infection. C, growth kinetics WT and mutant viruses. Vero cells were infected with the indicated viruses at an MOI of 0.1. Culture fluids of the infected cells were determined for viral titers using plaque assay. Note the scale of viral titer from 0 to 102 PFU/ml is different from the scale of 102 to 108 PFU/ml.
TABLE 3.
Effects of Ala substitutions of DENV MTase hydrophobic pocket residues on DENV-3 MTase activities
| DENV-3 MTase | N7 activitya | 2′-O activitya | MTase melting temperature, Tm (°C) [+sinefungin, °C]b | RNA bindingc, Ki |
|---|---|---|---|---|
| % | % | μm | ||
| F133A | 100 ± 1.2 | 65 ± 3.2 | 38 [+1.5] | 0.017 |
| G148A | 33 ± 4.5 | 40 ± 5.3 | 35 [+1] | 0.033 |
| R160A | 63 ± 4.47 | 102 ± 5.0 | 40 [+4] | 0.031 |
| R163A | 59 ± 4.8 | 100 ± 5.1 | 40 [+4] | 0.021 |
| L182A | 95 ± 5.9 | 18 ± 5.0 | 37.5 [+1] | 0.052 |
| WT | 100 | 100 | 40 [+4] | 0.048 |
a Recombinant WT and mutant MTases were assayed for N7 and 2′-O methyltransferase activities by quantifying the conversion of GpppA-RNA → m7GpppA-RNA and m7GpppA-RNA → m7GpppAm-RNA, respectively (4, 8). Methylation efficiencies of mutant MTases were compared with that of the WT MTase (set at 100%). An average of three experiments is shown.
b The stabilities of the mutant and WT proteins were assessed by determination of their melting temperature, and their abilities to bind the SAM mimetic, sinefungin, and RNA substrate. Protein melting temperatures were obtained in thermodenaturation assays in the presence or absence of 50 μm sinefungin containing SYPRO® Orange protein gel stain (10).
Design and Synthesis of Selective Inhibitors of Flavivirus MTase
The structural and functional results prompted us to design inhibitors that can specifically bind to the identified cavity of the flavivirus MTase. Because this cavity connects to the SAM-binding pocket at the adenine-holding site, we postulated that substituents extending from the adenine base of the SAH molecule would interact with the identified cavity. Such SAH derivatives would have the potential to selectively inhibit DENV MTase without suppressing host MTases. To test this hypothesis, we synthesized a series of SAH analogs, each with a substitution at the N-6 position of the adenine base (Fig. 3). This N6 position was selected for two reasons: (i) It is in the most optimal geometrical position for making extensions to occupy the hydrophobic cavity, and (ii) the N6 is the most chemically tractable group in the adenine ring for modifications. Compound synthesis was detailed in supplemental materials. The compounds were assayed as inhibitors of N7 and 2′-O methyltransferase activities using DENV-3 MTase. For ease of synthesis, initial structure-activity relationship (SAR) was assessed with compounds synthesized as epimeric mixtures, except for compounds 1* and 3* (Symbol * indicates enantiopure; Table 4). The SAR showed that, compared with SAH, a substitution with small alkyl groups (compounds 1* and 2) or a phenyl ring (compound 3*) reduced the anti-MTase activity. Further extension of the substituent showed that a benzyl group was optimal (compound 4), retaining the activity of SAH. Lengthening the spacer by one carbon between the phenyl ring and the adenine N6 reduced the activity (compound 5). Addition of a methyl group on the aryl ring at different positions indicated that the meta position is optimal, leading to an increase in potency of almost 4-fold over the starting point (compare compound 7 with 6 and 8). Next, halogen substituents were introduced at the meta position (compounds 9–12); these compounds exhibited potencies similar to the methyl substituent analog (compound 7). Compared with the SAH starting point, compounds 7 and 9–12 consistently showed better potencies; this conclusion was further supported by the results from enantiopure compounds 10* and SAH* (Table 5). For each compound, the Ki value for N-7 methylation was higher than that for 2′-O methylation; this difference could result from the distinct mechanisms of the two cap methylations (20, 21). The parent compound, SAH*, also displayed similar differences in N7 and 2′-O inhibition. Nonetheless, the general SAR trend was the same in N-7 and 2′-O methyltransferase activities. In stark contrast to SAH and compounds 1–3, the compounds with extended phenyl rings (compounds 4 and 5), and N-6 benzylic substituents (compounds 6–12) had either partially or completely lost the activity against human RNA guanine-7-MTase (hRNMT), DNA MTase 1A (hDNMT1A; Tables 4 and 5), and SET7/9 histone methyltransferases (hHMT; data not shown). These results demonstrate that the N6 modified SAH derivatives can selectively inhibit DENV MTase.
FIGURE 3.

Structures of SAH derivatives that inhibit DENV-3 MTase.
TABLE 4.
Ki values of compound
Each compound was tested for its inhibition against DENV-3 MTase (for both N7 and 2′-O MTase activities), human RNA guanine-7-MTase (hRNMT), and human DNA MTase1A (hDNMT). Symbol * indicates that the compounds are enantiopure. Average results from three independent experiments are presented.
| Compound | DENV-3 MTase Ki |
hRNMT Ki | hDNMT Ki | |
|---|---|---|---|---|
| N7 | 2′-O | |||
| μm | μm | μm | μm | |
| SAH | 3.19 ± 0.04 | 0.57 ± 0.04 | 4.54 ± 0.02 | 6.18 ± 0.02 |
| 1* | 5.91 ± 0.01 | 1.51 ± 0.04 | 1.09 ± 0.05 | 2.75 ± 0.04 |
| 2 | 21.94 ± 0.05 | 2.07 ± 0.05 | 14.73 ± 0.01 | 2.06 ± 0.02 |
| 3* | 10.86 ± 0.07 | 3.45 ± 0.06 | 16.09 ± 0.04 | 1.90 ± 0.03 |
| 4 | 2.1 ± 0.05 | 0.52 ± 0.05 | 20.85 ± 0.06 | >50 |
| 5 | 8.26 ± 0.06 | 2.1 ± 0.03 | 26.18 ± 0.05 | 11.85 ± 0.02 |
| 6 | 6.03 ± 0.06 | 4.23 ± 0.07 | >50 | >50 |
| 7 | 0.85 ± 0.04 | 0.28 ± 0.03 | >50 | >50 |
| 8 | 1.76 ± 0.05 | 0.75 ± 0.05 | >50 | >50 |
| 9 | 2.15 ± 0.03 | 0.15 ± 0.04 | >50 | >50 |
| 10 | 0.82 ± 0.06 | 0.17 ± 0.02 | >50 | >50 |
| 11 | 0.77 ± 0.04 | 0.19 ± 0.03 | >50 | 25.07 ± 0.10 |
| 12 | 1.30 ± 0.04 | 0.20 ± 0.06 | >50 | 17.60 ± 0.04 |
TABLE 5.
Ki values of SAH* and compound 10* in various MTases
Symbol * indicates that the compounds are enantiopure. Average results from three independent experiments are presented.
| MTase | Activity | SAH* Ki | 10* Ki |
|---|---|---|---|
| μm | μm | ||
| DENV-3 WT | N7 | 0.65 ± 0.03 | 0.24 ± 0.07 |
| 2′-O | 0.28 ± 0.04 | 0.08 ± 0.01 | |
| DENV-3 F133A | N7 | 1.16 ± 0.02 | 0.0020 ± 0.0002 |
| 2′-O | 0.095 ± 0.010 | 0.007 ± 0.002 | |
| WNV WT | N7 | 2.44 ± 0.06 | 5.68 ± 0.05 |
| 2′-O | 0.020 ± 0.003 | 0.044 ± 0.010 | |
| hRNMT | N7 | 0.38 ± 0.05 | >50 |
| hDNMT | C5 | 1.38 ± 0.02 | >50 |
Because the identified cavity is conserved among flavivirus MTases, we examined whether the SAH analogs would inhibit flavivirus MTase other than DENV. As shown in Table 5, compound 10* also inhibited WNV MTase but was about 2.2-fold less potent than SAH*. Nevertheless, compound 10* did not inhibit host hRNMT or hDNMT (Table 5), indicating its selectivity for flavivirus MTase.
Crystal Structure of DENV-3 MTase Compound Complex
To provide structural evidence for mode-of-action, we solved the crystal structure of DENV-3 MTase in complex with compound 10* at a resolution of 1.7 Å (PDB code 3P97; Tables 1 and 2). The overall structure of the compound 10*-MTase complex is almost identical to that of the SAM-MTase complex, with a Cα RMSD of 0.16 Å (Fig. 4A). As shown in Fig. 4, A and B, the N6 benzyl ring of compound 10* fits snugly into the identified cavity without changing the binding of the SAH moiety. Remarkably, the benzyl ring of the compound induces a local conformational change within the cavity (Fig. 4, C and D): the side chains of Phe-133 flips away from the cavity by 100 degrees (measured at the Cα and Cβ torsion angle) to create space for the N6 benzylic ring, and the side chains of Arg-163 shifted by 3.5 Å (measured at the NH1 atom) to stack onto the benzylic ring, resulting in the formation of a cation-π interaction. Fig. 4E summarizes the interactions established between compound 10* and the DENV-3 MTase.
FIGURE 4.

Co-crystal structures of DENV-3 MTase in complex with compound 10*. A, superposition of the crystal structures of compound 10*-DENV-3 MTase (green) complex and the MTase-SAM complex (yellow). SAM is shown in stick representation. Compound 10* is colored in magenta. B, surface representation of the DENV-3 MTase, showing the binding of compound 10* (in stick presentation) in the identified cavity. C, representative omit (Fo − Fc) electron density map (green) at a level of 2.5 σ showing the bound compound 10* molecule. Amino acids Phe-133 and Arg-163 are labeled. D, compound-induced conformational change of amino acids. Compound 10* is shown in stick presentation (magenta). Residues Phe-133 and Arg-163 in the SAM-MTase complex are colored in yellow; the same residues in the compound 10*-MTase complex are shown in green; two arrows (red) indicate the compound-induced conformational change. E, schematic view of the interactions between compound 10* (brown) and the DENV-3 MTase (black).
Effect of Compound-induced Conformational Change on Efficacy
The observed flipping of Phe-133 in the co-crystal structure of DENV-3 MTase-compound complex suggests that this residue at its native conformation sterically blocks the binding of the N6 benzylic ring. If this is the case, substitution of Phe-133 to a residue with a smaller side chain would facilitate the binding of the benzylic ring of compound 10*; consequently, the mutant MTase would be more sensitive than the WT MTase to compound inhibition. To test this hypothesis, we prepared a mutant MTase containing a Phe → Ala substitution at amino acid position 133. Indeed, the Ki values of the F133A MTase were lower (120-fold for N7 activity and 11-fold for 2′-O activity) than that of the WT MTase (Table 5). These results demonstrate that the selective inhibition of compound 10* against DENV MTase is obtained through binding of the N6 benzylic ring to the identified pocket.
DISCUSSION
We used DENV MTase as a model to prove the concept that selective inhibitors of disease-related MTase could be obtained. The selectivity was accomplished through designing SAH derivatives with substituents in the adenine moiety that extend into a cavity located above the SAM-binding pocket. The newly identified pocket is unique to the flavivirus MTases (18, 19) and was not found in the crystal structures of several human MTases available in the Protein Data Bank (data not shown). The starting compound, SAH, is a well-known product inhibitor, which is non-selective and inhibits eukaryotic, bacterial, and viral MTases (22–24). In our hands, it showed similar inhibitory properties against DENV, WNV, human RNA, DNA, and histone MTases (data not shown). Attachment of small alky or phenyl groups to the N6-position in its adenine base failed to improve its potency against DENV MTase or hRNMT, but moderately enhanced its activity against hDNMT by 2–3-fold. Our findings contradicts a previous report in which compound 3 was found to be inactive against hDNMT (25). Introduction of a benzyl ring retained its activity against DENV MTase and strongly reduced its activity against hRNMT. No activity was observed against hDNMT. Thus, some gain in selectivity for DENV MTase was possible through this modification. This result is in agreement with the hDNMT data obtained by Saavedra et al. (25). Extension of the benzyl group with a second carbon chain significantly reduced inhibition against all enzymes tested. It is possible that the hydrophobic pocket does not accommodate this longer extension; alternatively, the loss of activity may be due to greater flexibility of the carbon linker and resultant poorer binding into the cavity. Further probing with substituents in the aryl ring revealed that additions in the meta-position provided moderate improved potency for DENV MTase but gained selectivity against hRNMT, DNMT and histone MTases (data not shown). Substitution with either a methyl-, chloro- or fluoro- group in the meta-position resulted in about 4-fold better inhibitory properties compared with SAH. Replacement of a bromo- or iodo-group in this position exhibited similar inhibitory properties on DENV MTase, as the un-substituted aryl ring, indicating that bulky groups on the aryl ring were not preferred. On the other hand, substitution in the ortho position was not advantageous, while the para position conferred some improvement against DENV N7 activity only. Remarkably, additions in all three positions rendered the compounds either inactive or poorly active against hRNMT, hDNMT, and hHMT (data not shown).
Residue Phe-133 is conserved in the several mosquito- and tick-borne Flaviviruses, including DENV, WNV, and JEV (19). Mutation of this amino acid to Ala did not affect DENV N7 and 2′-O activity in vitro, nor impaired DENV growth in cells. Thus, this residue is not critical for virus replication. Furthermore, Phe-133 is rather flexible, as binding of compound 10* in DENV MTase induced its side chain to rotate away from the cavity, to accommodate its N6-substituted aryl ring. It is possible that other residues lining the pocket may have similar mobilities. Indeed, the improved potency of compound 10* over SAH* can be partially explained by the movement of residue Arg-163, to form a strong cation-π interaction with the N6-aryl ring of compound 10*.
Although the benzyl-SAH derivatives were selective for both DENV and WNV MTases, confirming the importance of the hydrophobic pocket (19), the most active compound, 10*, was not more potent in WNV compared with SAH*. Detailed examination of the hydrophobic pockets in both MTases did not reveal any obvious difference between the two sites, and the reason for the discrepancy in the potency of the compounds for the two virus enzymes is unknown. Nevertheless, both SAH* and compound 10* are significantly more potent on WNV 2′-O activity compared with the same activity in DENV (14 and 18-fold better, respectively, in WNV). In contrast, the opposite is true for their N7 activities. Both compounds respectively, showed 4- and 24-fold less potency in WNV than DENV. Thus, the observed difference in the two viral MTases for this class of compounds may be related to variations in their N7 and 2′-O mechanisms (20, 21). In summary, using a structure-guided approach, we have designed DENV selective and potent inhibitors by taking advantage of a specific pocket found in Flavivirus MTases, and provided a paradigm that may facilitate pharmacological therapeutics targeting other disease-related MTases.
Supplementary Material
Acknowledgments
We thank Jan Jiricek, Cheah Chen Seh, Daying Wen, and colleagues at Novartis Institutes for BioMedical Research in Shanghai for help. We thank the beamline scientists at the Swiss Light Source for assistance.
This work was supported in part by BMRC Grant 06/1/22/19/447 (to J. L.).
The atomic coordinates and structure factors (codes 3P8Z and 3P97) have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ (http://www.rcsb.org/).

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1–S3 and Table S1.
- MTase
- methyltransferase
- SAM
- S-adenosyl-methionine
- SAH
- S-adenosyl-homocysteine
- DENV
- Dengue virus
- hRNMT
- human RNA MTase
- RMSD
- root-mean-square deviation
- SAR
- structure-activity relationship.
REFERENCES
- 1. Copeland R. A., Solomon M. E., Richon V. M. (2009) Nat. Rev. Drug Discov. 8, 724–732 [DOI] [PubMed] [Google Scholar]
- 2. Egloff M. P., Benarroch D., Selisko B., Romette J. L., Canard B. (2002) EMBO J. 21, 2757–2768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ray D., Shah A., Tilgner M., Guo Y., Zhao Y., Dong H., Deas T., Zhou Y., Li H., Shi P. Y. (2006) J. Virol. 80, 8362–8370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Dong H., Chang D. C., Xie X., Toh Y. X., Chung K. Y., Zou G., Lescar J., Lim S. P., Shi P. Y. (2010) Virology 405, 568–578 [DOI] [PubMed] [Google Scholar]
- 5. Kroschewski H., Lim S. P., Butcher R. E., Yap T. L., Lescar J., Wright P. J., Vasudevan S. G., Davidson A. D. (2008) J. Biol. Chem. 283, 19410–19421 [DOI] [PubMed] [Google Scholar]
- 6. Zhou Y., Ray D., Zhao Y., Dong H., Ren S., Li Z., Guo Y., Bernard K. A., Shi P. Y., Li H. (2007) J. Virol. 81, 3891–3903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Qing M., Zou G., Wang Q. Y., Xu H. Y., Dong H., Yuan Z., Shi P. Y. (2010) Antimicrob. Agents Chemother. 54, 3686–3695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chung K. Y., Dong H., Chao A. T., Shi P. Y., Lescar J., Lim S. P. (2010) Virology 402, 52–60 [DOI] [PubMed] [Google Scholar]
- 9. Lim S. P., Wen D., Yap T. L., Yan C. K., Lescar J., Vasudevan S. G. (2008) Antiviral Res. 80, 360–369 [DOI] [PubMed] [Google Scholar]
- 10. Finney D. J. (1976) Biometrics 32, 721–740 [PubMed] [Google Scholar]
- 11. Cheng Y., Prusoff W. H. (1973) Biochem. Pharmacol. 22, 3099–3108 [DOI] [PubMed] [Google Scholar]
- 12. Leslie A. G. (2006) Acta Crystallogr. D Biol Crystallogr. 62, 48–57 [DOI] [PubMed] [Google Scholar]
- 13. Collaborative Computational Project, N. (1994) Acta Crystallogr. D Biol. Crystallogr. 50, 760–763 15299374 [Google Scholar]
- 14. Emsley P., Cowtan K. (2004) Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 15. Vaguine A. A., Richelle J., Wodak S. J. (1999) Acta Crystallogr. D Biol. Crystallogr. 55, 191–205 [DOI] [PubMed] [Google Scholar]
- 16. Deleted in proof.
- 17. Geiss B. J., Thompson A. A., Andrews A. J., Sons R. L., Gari H. H., Keenan S. M., Peersen O. B. (2009) J. Mol. Biol. 385, 1643–1654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bollati M., Alvarez K., Assenberg R., Baronti C., Canard B., Cook S., Coutard B., Decroly E., de Lamballerie X., Gould E. A., Grard G., Grimes J. M., Hilgenfeld R., Jansson A. M., Malet H., Mancini E. J., Mastrangelo E., Mattevi A., Milani M., Moureau G., Neyts J., Owens R. J., Ren J., Selisko B., Speroni S., Steuber H., Stuart D. I., Unge T., Bolognesi M. (2010) Antiviral Res. 87, 125–148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Dong H., Liu L., Zou G., Zhao Y., Li Z., Lim S. P., Shi P. Y., Li H. (2010) J. Biol. Chem. 285, 32586–32595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Fabrega C., Hausmann S., Shen V., Shuman S., Lima C. D. (2004) Mol. Cell 13, 77–89 [DOI] [PubMed] [Google Scholar]
- 21. Hodel A. E., Gershon P. D., Shi X., Quiocho F. A. (1996) Cell 85, 247–256 [DOI] [PubMed] [Google Scholar]
- 22. Borchardt R. T. (1980) J. Med. Chem. 23, 347–357 [DOI] [PubMed] [Google Scholar]
- 23. Hasobe M., McKee J. G., Borchardt R. T. (1989) Antimicrob Agents Chemother. 33, 828–834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Patnik D., Chin H. G., Estève P. O., Benner J., Jacobsen S. E., Pradhan S. (2004) J. Biol. Chem. 279, 53248–53258 [DOI] [PubMed] [Google Scholar]
- 25. Saavedra O. M., Isakovic L., Llewellyn D. B., Zhan L., Bernstein N., Claridge S., Raeppel F., Vaisburg A., Elowe N., Petschner A. J., Rahil J., Beaulieu N., MacLeod A. R., Delorme D., Besterman J. M., Wahhab A. (2009) Bioorg. Med. Chem. Lett. 19, 2747–2751 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.