

Abstract
Genome-based studies of metazoan evolution are most informative when phylogenetically diverse species are incorporated in the analysis. As such, evolutionary trends within and outside the phylum Nematoda have been less revealing by focusing only on comparisons involving Caenorhabditis elegans. Herein, we present a draft of the 64 megabase nuclear genome of Trichinella spiralis, containing 15,808 protein coding genes. This parasitic nematode is an extant member of a clade that diverged early in the evolution of the phylum enabling identification of archetypical genes and molecular signatures exclusive to nematodes. Comparative analyses support intrachromosomal rearrangements across the phylum, disproportionate numbers of protein family deaths over births in parasitic vs. a non-parasitic nematode, and a preponderance of gene loss and gain events in nematodes relative to Drosophila melanogaster. This sequence and the panphylum characteristics identified herein will advance evolutionary studies and strategies to combat global parasites of humans, food animals and crops.
Currently no complete genome sequence information exists from lineages spanning the phylum Nematoda (Supplementary Fig. 1). Yet, such information is essential to understanding evolution of the Nematoda analogous to the way that a basal chordate informed vertebrate evolution1. To this end, the genome sequence of Trichinella spiralis a food-borne, zoonotic parasite was generated to reveal molecular characters and evolutionary trends among this organism, evolutionarily distant parasitic and non-parasitic nematodes, and a member of the next closest sequenced relatives, the arthropods. In so doing, commonalities that link nematodes to other Metazoa were identified, as well as distinctions that define the Nematoda and differentiate T. spiralis from other species investigated. The Trichinella assembly is 64 million base pairs in length and encodes at least 15,808 proteins which make this genome substantially smaller than that of the prototypical nematode, Caenorhabditis elegans.
Trichinellosis is worldwide zoonotic disease. The nematode, Trichinella spiralis, the most common cause of human trichinellosis, is a member of a clade that diverged early in the evolution of the Nematoda. It differs substantially in biological and molecular characters from other crown groups2–4. The lineage giving rise to the genus Trichinella last shared a common ancestor approximately 275 million years ago (Lower Permian Period) whereas the diversification of extant Trichinella species occurred as recent as 16–20 million years ago (Miocene Epoch)5.
The life-cycle of Trichinella spp. (Supplementary Fig. 2) begins when muscle tissue containing first stage larvae (ML) is ingested by the new host. The ML rapidly develop to adults in the intestines where they mate and produce newborn larvae (NBL). The NBL migrate from the intestines through the lymphatic system and eventually to the blood where they search for striated skeletal muscle cells to invade, complete the cycle and become infectious. Intense inflammation is a primary cause of disease and involves myositis, myocarditis and encephalitis, the intensity of which depends on the number of parasites ingested. Currently, the genus consists of 8 distinct species and/or genotypes that are further categorized as encapsulated or non-encapsulated predicated upon the formation of a collagen envelope around the infected muscle cell. This capsule is believed to be a host-derived structure induced only by species that infect placental mammals and is unique to this genus. In addition to the formation of a collagen capsule, and contrary to most other parasitic nematodes, T. spiralis exhibits little host specificity, completes its entire life cycle in a single host, does not have a free-living stage, and lives as an intracellular parasite within a single striated muscle cell. As such, this genus presents biological characteristics that markedly differ from what is common among most other nematodes.
Herein we compared molecular characteristics of nematodes and other metazoans using the entire T. spiralis genome. The comparative approach identified conserved protein and gene sequences with apparent archetypical standing for the phylum Nematoda. We found that intrachromosomal rearrangements were common throughout the phylum; however, this was in contrast to other characters such as protein family deaths and births which showed a clear demarcation between parasitic and a non-parasitic nematode. In addition, unlike Drosophila melanogaster the levels of gene loss and gain in each nematode species indicate that these events may have played a substantially larger role in the evolution of this phylum. The identification of these and other conserved characteristics, predicated in part upon this work, will advance more targeted research on pathogens from a phylum harboring thousands of pathogens that infect humans, animals and plants. The advances may one day provide holistic strategies to treat and control diseases caused by pathogens from across the Nematoda.
RESULTS
Sequencing, assembly and gene organization
Data were generated from whole-genome shotgun sequencing and hierarchal map-assisted sequencing 6. The assembly totaled 64 Mb (Supplementary Note and Supplementary Table 1), which is in line with recent genome size estimates made by flow cytometry (1C = 71 Mb)6–7. The data provided coverage of 35-fold, with 15% of the supercontigs encompassing 90% of the genome. The T. spiralis fingerprint clone map enabled construction of nine ultracontigs comprised of 69 supercontigs representing 49 Mb or 76% of the genome.
The repeat content of the T. spiralis genome is estimated at 18%. The repeats have a low GC content (27%) relative to the genome overall (34%) and to protein coding regions (43%). The 15,808 protein-coding sequences occupy 26.6% of the genome at an average density of 272 genes per Megabase (Mb). Although 15% of C. elegans genes are organized in operons8, spatial relationships of genes in T. spiralis do not readily indicate the existence of operons (Supplementary Note). This observation validated prior studies indicating similar findings4. As such, the existence of operons in this nematode remains an open question. Further, T. spiralis lacks both the canonical SL1 trans-spliced leader found in most nematodes and the SL2 trans-spliced leader that is spliced onto transcripts from downstream genes in C. elegans operons. To date, at least 15 distinct spliced leaders encoded by 19 SL RNA genes have been identified in T. spiralis4; however, these putative splice leaders, exhibit sequence variability at nearly all base positions, and were found to be present in only 1% of the cDNAs examined. It is likely, therefore, that the canonical SL1 and SL2 spliced leader sequences were not part of the genetic repertoire in nematodes that diverged early in the evolution of the Nematoda. This hypothesis is supported in part by our inability to identify canonical SL1 and SL2 sequences among Trichuris muris EST as well (data not shown). After comparison to an extensive collection of proteins from other species, 45% (7,251) of the predicted protein coding genes were T. spiralis specific, of which 12% had EST confirmation (Supplementary Fig. 3). The amino acid (AA) composition of predicted proteins in T. spiralis is similar to that observed in other nematodes9, organisms (Supplementary Table 2), and taxa10. In agreement with previous studies11, nematodes show a correlation between AA usage and the degree of codon degeneracy (R=0.74).
Genome evolution
The availability of a genome from a member of the Dorylaimia expanded our abilities to evaluate genome evolution among highly divergent crown clades and to potentially identify factors underlying lineage diversification. We evaluated changes associated with nematode evolution in relation to: i) genome organization; ii) births and deaths of gene families; iii) gene duplications and deletions that have occurred within gene families; and iv) linear organization of orthologous genes.
Organizational characteristics were evaluated by comparing the genomes of T. spiralis and C. elegans. The number of predicted genes in T. spiralis is notably lower than the 20,140 genes identified in C. elegans even though the two genomes exhibit similar repeat content and gene density. A comparison of approximately ~3,400 predicted orthologous genes (based on reciprocal best BLAST hits) showed that T. spiralis has a significantly shorter average intron size (191 bp vs. 391 bp, P=6.5e–69), amidst an average exon size that is relatively similar for the two species (179 bp for T. spiralis and 226 bp for C. elegans, P=7.0e–3). Focusing only on predicted orthologous genes with 20 or more exons, the mean total length for all exons was significantly higher in C. elegans (P=0.001). Comparisons of Pfam domains contained in orthologous pairs showed C. elegans had significantly more domains compared to the orthologous T. spiralis genes (876 vs. 755, P<0.01). These differences coincide with the smaller size of the T. spiralis genome; however, we cannot rule out the possibility for higher numbers of gene fragments in T. spiralis resulting from less refined genome annotation.
Delineating gene family emergence and extinction within phylogenetically related organisms can identify molecular determinants that underlie species (and pathogen) adaptation and lineage or species evolution. Such an approach has been used in analyzing nematode EST12–14. Here we measured potential emergence and extinction events of protein families across the Nematoda. The analysis included species from four major lineages that collectively span the phylum (C. elegans, Meloidogyne incognita15, Brugia malayi16 and T. spiralis). These species represent nematodes that are non-parasitic, parasitic in plants, and parasitic in animals, respectively, thus representing diverse trophic ecologies. Arthropod (Drosophila melanogaster17) and yeast (Saccharomyces cerevisae18) species were used as outgroups. Markov clustering19 of the complete protein catalog (87,406 proteins) comprising all six species generated 12,163 protein families (Supplementary Table 3). Inter-specific protein families overlaid onto species phylogeny identified 702 protein families at the node between Nematoda and the outgroups (Fig. 1a and Supplementary Table 4). Of these nematode families, 274 families were common among all four members of the Nematoda. We screened the genes in the 274 core nematode group (1,990 genes) against all available nematode ESTs/cDNAs and found that 73% shared homology to nematode transcriptome data from 27 nematode genera, and only 5% shared sequence homology to arthropods using the same cutoff value. These numbers do not preclude gains that may have occurred before the appearance of the Nematoda or gains relative to Drosophila that may still be present in other arthropods. In contrast, 88 protein family deaths were identified as common among the four nematodes relative to D. melanogaster. Protein family deaths outnumbered births for all three parasitic species, whereas in the non-parasitic species C. elegans, births outnumbered deaths four to one. The methods utilized here will allow future assessment of this tendency with availability of additional genomes from other parasitic and non-parasitic nematodes. Emergence of new protein families was observed in all nematode lineages, albeit less so for B. malayi. Accordingly, it is now possible to explore the relevance of protein families identified in the evolution of lineages within the Nematoda and across phyla.
Fig. 1.
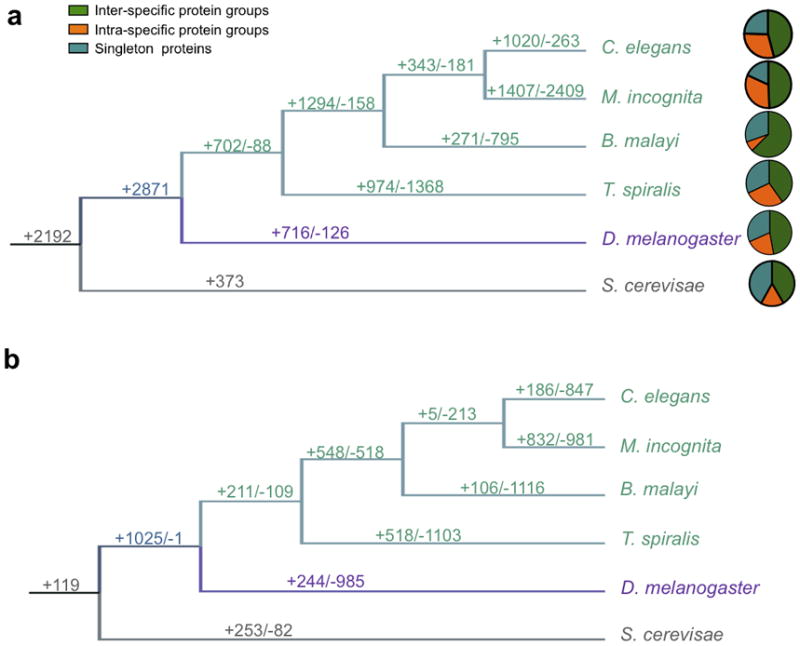
Protein and gene family changes associated with the origin and evolution of the Nematoda. (a) Protein family changes. At the branch of each lineage, the ‘+’ number indicates family birth events and the ‘-’ number indicates family death events represented by all members indicated for that lineage. For example, there are 702 protein family births ancestral to the phylum Nematoda and 88 protein family deaths in common among the four nematodes by comparison to arthropods (represented by D. melanogaster). These events were reconstructed from 12,206 inter-specific orthologous families (63,273 proteins). (b) Gene duplications and losses over the evolution of the common protein families. The gene duplication and loss events were reconstructed using 858 orthologous multi-member protein families (containing 8,260 proteins) conserved among all 6 species. At the branch of each lineage, the ‘+’ number indicates the number of gene duplication events and the ‘-’ number indicates the number of gene loss events for that lineage.
Similarly, quantitative changes in protein family members (duplications and deletions) can reflect evolutionary determinants of lineage and species diversity. We evaluated 858 families (8,260 genes) common to the four nematode species and two outgroup species defined above (Fig. 1b); 674 families had no obvious duplications or deletions, 70 had only deletions,105 had only duplications and nine had both. Nematode species had a higher number of events compared to D. melanogaster (Fig. 1b). Among the nematodes, M. incognita had the highest number of both duplications and deletions likely due to the 30% of the genome being duplicated resulting in more species-specific events15. An example for T. spiralis involves the secreted DNase II-like protein family, a member of which has been evaluated as a vaccine candidate20 and implicated in host-parasite interactions. The genome shows more extensive expansion of this family (estimated 125 genes) than previously realized (Supplementary Note and Supplementary Fig. 4).
To provide additional examples, we compared protein families in C. elegans with sequence homologues in T. spiralis. Ten families were relatively expanded and five families were contracted in T. spiralis (P<0.001) (Supplementary Table 5). These families can be grouped into i) those present prior to the separation of nematodes and arthropods (nine families) and ii) those putatively born coincident with this separation (six families), and possibly the origin of nematodes. The six protein families in this later group included four that are relatively expanded in T. spiralis; a retrotransposon (2:201 Ce:Ts), a translation initiation factor 2C, putatively related to lipid metabolism (2:140 Ce:Ts), a zinc finger C2H2 type protein (1:14, Ce:Ts), and a hypothetical protein (1:44, Ce:Ts) associated with defective egg laying in C. elegans. Two protein families are relatively contracted in T. spiralis; a major sperm protein (33:1, Ce:Ts ), and a protein of unknown function, DUF1647, (18:1, Ce:Ts).
Comparisons of orthologous protein families outlined in sections ii and iii facilitated assessment of a nematode genome (T. spiralis) from a basally positioned clade (clade 2), with those from highly divergent clades (clades 8, 9, 12)21 and an outgroup member (D. melanogaster). Results consistently demonstrated similar and extensive levels of disparity in orthologous family sizes between T. spiralis and either C.elegans or D. melanogaster, while members of clades 8, 9, and 12 showed higher levels of shared attributes with C. elegans only (Fig. 2). Information in the next section provide independent measures, based on genome organization, to support this data which previously was indicated by rRNA sequence comparisons21.
Fig. 2.
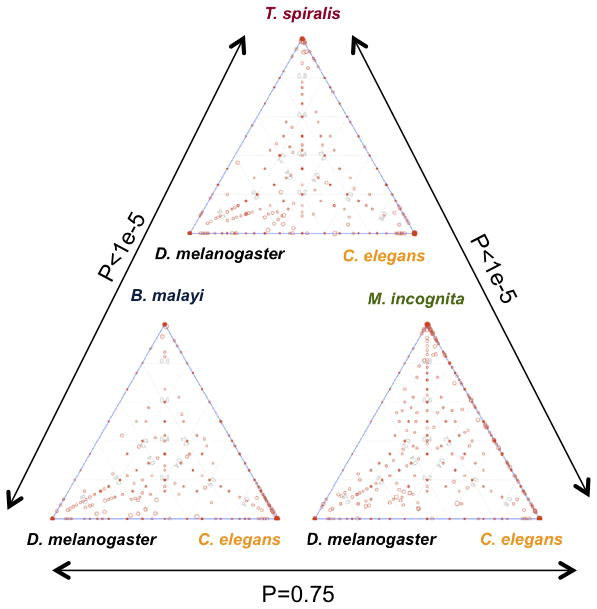
Comparison of orthologous protein families among nematodes that span the phylum. Orthologous families comprised of each of the three parasites and D. melanogaster and C. elegans are plotted separately. The size of the dot represents the size of the orthologous family; the position represents the composition of the family based on the three represented species. With the assumption that evolutionarily close species have similar orthologous family size (fewer duplications and deletions), these plots illustrate that T. spiralis is equally distinct from both C. elegans and D. melanogaster while the two other parasites share greater commonality with C. elegans. P-values (derived using Chi-square test in pair-wise plot comparison) indicate a greater number of families present in C. elegans compared to D. melanogaster, and show that significantly fewer families are biased to C. elegans when T. spiralis is present in the orthologous family
Next we evaluated the nematode genomes across the phylum regarding extent and limits to evolutionary changes and functional associations that may depend on gene arrangements. Comparisons between C. elegans and B. malayi (~350 million years of separation) indicated that intra- rather than inter-chromosomal rearrangements preferentially characterize genome evolution evident between these species16. We used the T. spiralis genes organized on the six longest ultracontigs to extend this analysis. As for B. malayi, T. spiralis genes showed macrosyntenic relationships with predicted orthologs from C. elegans (P<0.0001) albeit to a lesser extent (Fig. 3a). Because T. spiralis is diploid only in females of these species (female 2n=12 [XX], male 2n=11 [XO]), the correlation coefficient was calculated also when the X chromosome was excluded. This resulted in improved support for macrosynteny. This non-random distribution of orthologous genes is consistent with that observed in several nematode species22–24.
Fig. 3.
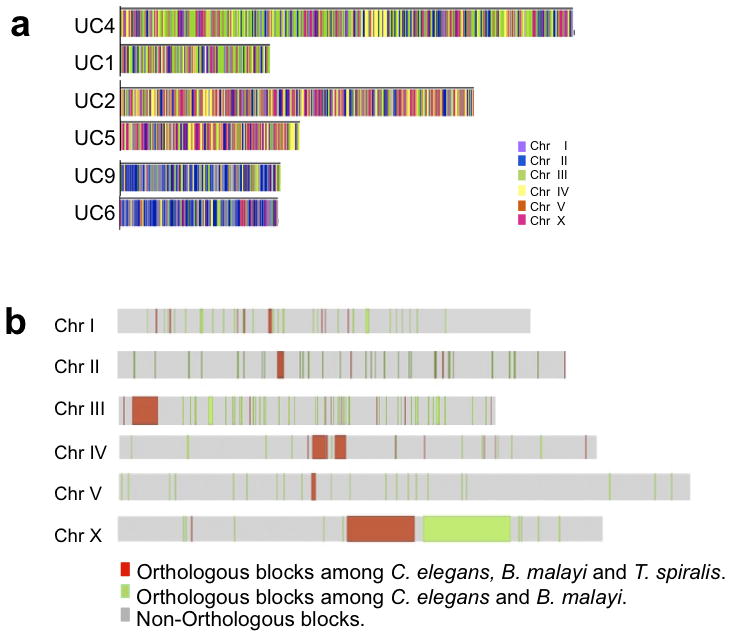
Genes from T. spiralis show macrosyntenic relationships with predicted orthologs from other nematodes. (a) T. spiralis genes on the six largest ultracontigs with orthologs in C. elegans, colored to indicate the C. elegans chromosome on which the ortholog is located. The correlation was strong (R=0.95, R=0.76 and R=0.99), and even stronger when the X chromosome was excluded (R=0.97, R=0.97 and R=0.99). As example, R=0.95 indicates that both T. spiralis Ultracontigs 1 and 4 are strongly associated with one predominant C. elegans chromosome, Chr III, and not a result of random gene distribution. (b) Orthologous segments shared among nematode species shown on the C. elegans chromosomes. Red segments are considered to be ancestral orthologous segments among nematodes. The size of segments corresponds to the C. elegans orthologous segment that might be different than the orthologous segment in the other two species (Supplementary Table 7).
Assuming a constant tendency towards randomness, genome re-assortment is expected to occur at a rate commensurate with evolutionary distance. Using syntenic blocks of C. elegans for standardization, we measured dynamics of nematode chromosome re-assortment among multiple nematode pairs25. The highest syntenic conservation score was observed between C. elegans and C. briggsae (0.752), less so between C. elegans and B. malayi (0.508), and the least between C. elegans and T. spiralis (0,28) (Supplementary Table 6). Because sequences for non-C. elegans genomes have varying levels of fragmentation, it was not possible to use entirely complementary gene sets in the pairwise comparisons (orthologous genes on different scaffolds were not considered). Nevertheless, the relative syntenic conservation values were consistent with the perceived evolutionary distance of the species investigated. The approximate 72% of the T. spiralis genome organization that lacked demonstrable congruence with the C. elegans genome provided a tentative estimate on the limits of evolutionary diversity of this kind across the Nematoda.
Despite an anticipated tendency toward randomization, existence of syntenic blocks suggests functional constraints to genome evolution. This possibility was investigated with a high-level orthology map created with coding exons as anchors26 from C. elegans, B. malayi and T. spiralis. We identified 196 orthologous segments (Supplementary Table 7); 155 were shared among C. elegans and B. malayi, five were shared among B. malayi and T. spiralis and 36 segments were shared among all three species, putatively defined as ancestral orthologous segments. No segments were shared exclusively between C. elegans and T. spiralis (Fig. 3b). The results are again consistent with the perceived evolutionary distance among these organisms based on all pairwise comparisons. The genes within the 36 ancestral segments accounted for ~50% of the genes in all segments for C. elegans and B. malayi, but 97% of the genes in T. spiralis. Over half of the ancestral segments are located on C. elegans chromosomes III and IV. These ancestral segments tended to localize more centrally in the chromosomes (P=0.001)27. This tendency was also suggested by the two-species orthologous segments, although less evident (different at P=0.1). The overall patterns highlighted likely reflect basic properties that influence the evolution of genome organization in nematodes.
Nematode species from the lineages evaluated span recent and early radiation events within the phylum Nematoda. Hence, the quantitative and qualitative measures of genomic diversity will help to define both the extent and limits of genome organizational diversity across the Nematoda and help clarify molecular determinants of nematode lineages and species. Nevertheless, the results based on Markov clustering of predicted orthologous protein families will exclude other forms of diversity such as nucleotide substitutions, insertions and deletions. As such, the documented differences reflect but a small component of the total genomic diversity within the Nematoda.
Molecular determinants archetypical of the phylum Nematoda
Molecular determinants for traits that characterize the archetypical nematode have been evaluated 12,14. To identify proteins and protein sequences that are broadly conserved among the four nematodes that span the phylum, we further compared worm derived proteins to those of arthropod and yeast outgroups. The 12,163 orthologous protein families were partitioned into: 1) orthologous protein sequences that are broadly conserved among all of the four nematode species and any of the two outgroups (2,517 families, 14,801 nematode proteins); 2) those conserved exclusively among the four nematodes (274 families, 1,990 nematode proteins); and 3) those that are conserved between any nematode and any outgroup (4,980 families, 30,729 proteins) (Supplementary Table 3). We evaluated 328 protein families represented by a single copy gene in all six species by querying the C. elegans database for RNAi phenotypes. The exclusion of multi-member protein families from this evaluation precluded cases where compensation by other family members might obscure RNAi phenotypes. Of the 328 C. elegans genes, 232 (71%) had associated RNAi phenotypes (significant enrichment at P<0.00001) consistent with a gene set essential to core cellular and biochemical functions of eukaryotes (Supplementary Table 8).
Of the 2,517 nematode protein families (Fig. 4), 274 were detected in all four nematodes only (see Genome evolution section ii) and were collectively referred to as Nematode Orthologous Groups (NOGs)(Supplementary Table 9 and Supplementary Fig. 5). These NOGs were significantly enriched (P<0.00001) for genes with RNAi phenotypes in C. elegans and likely represent a gene set essential to core cellular and biochemical functions of nematodes.
Fig. 4.
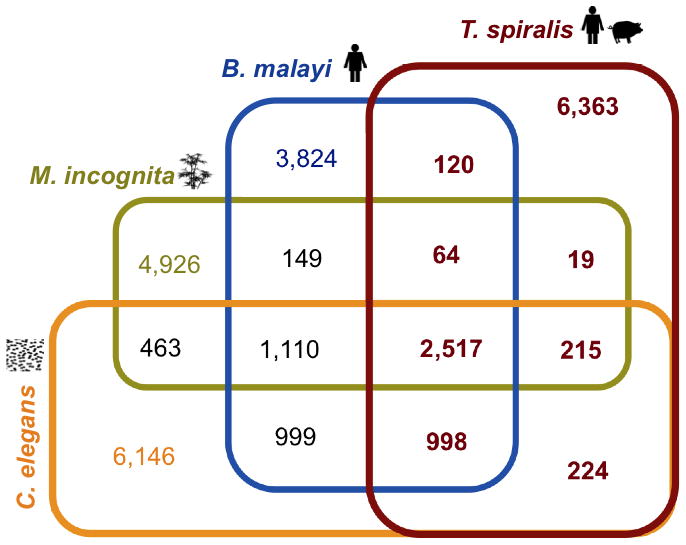
Distribution of orthologous families among the four nematode representatives spanning the phylum Nematoda. The lineages represented in the Nematoda are: Rhabditida (C. elegans), Tylenchina (M. incognita), Spirurina (B. malayi) and Dorylaimia (T. spiralis). The trophic ecology of each of the 4 nematode species used in this study for pan-phylum analysis is indicated next to the species name. The 2,517 orthologous groups are conserved in all four nematodes. Sixty-four orthologous groups are conserved among the parasitic species, but not the free-living C. elegans. Enrichment of functional categories related to certain orthologous groups compared to the complete functional repertoire for the 4 nematode species is presented in Supplementary Table 8 and Supplementary Table 9.
The 274 NOGs encoded 189 multi-copy gene families and 85 single copy gene families (scNOGs). Sixty-eight of the scNOGs had RNAi information and 21 had observable RNAi phenotypes (Table 1 and Supplementary Table 9). There was no enrichment of RNAi phenotypes in the C elegans genes in scNOGs compared to all C. elegans genes (p<0.05). Nevertheless, among the 21 genes with phenotypes, eight had known tissue localization and only one was neuronal. Of the remaining 64 genes, 17 had known expression patterns of which 10 were neuronal. Therefore, the biological significance of the scNOGs may be underestimated by RNAi information because nervous tissue is relatively insensitive to RNAi (e.g.28).
Table 1.
Pan-phylum single-copy genes withC. elegan s ortholog having severe RNAi phenotype
| T. spiralis gene | Ortholog in |
Descriptora | C. elegnas RNAib | Structural annotation |
|||
|---|---|---|---|---|---|---|---|
| B. malayi | C. elegans | M. incognita | TMc | SPd | |||
| Tsp_14949 | 14972.m07791 | F39H12.2 | Minc14650 | Hypothetical, WD40 repeat-like | Emb | - | - |
| Tsp_03879 | 14330.m00196 | F28F8.6 | Minc16561 | Machedo-Joseph protein | Emb | - | - |
| Tsp_09591 | 14058.m00575 | M05B5.2 | Minc04214 | Hypothetical protein | Lon Unc thin Gro | Y | Y |
| Tsp_02563 | 14972.m07706 | F53B6.1 | Minc01712a | Tetraspanin family protein NADH dehydrogenase (ubiquinone) 1 | Lva Dpy Bmd Bli | Y | - |
| Tsp_07476 | 14379.m00149 | W01A8.4 | Minc03402 | beta subcomplex 4 | Lva Emb Bmd | Y | - |
| Tsp_05829 | 14961.m05209 | ZK899.2 | Minc06660 | Hypothetical protein NADH dehydrogenase (ubiquinone) 1 | Lva Emb Lvl Gro | Y | - |
| Tsp_10274 | 13068.m00024 | F44G4.2 | Minc14463 | beta subcomplex 2 | Lva Emb RBS | - | - |
| Tsp_05872 | 14972.m06963 | ZK682.5 | Minc05446a | Leucine Rich Repeat family protein | Lva Gro | Y | Y |
| Tsp_05373 | 13756.m00013 | C45B2.7 | Minc06522 | Patched related family protein 4 | Prl Unc Lva Dpy Emb Lvl | Y | - |
| Tsp_09505 | 14968.m01485 | W08F4.6 | Minc18112 | Hypothetical protein | Prl Unc Lva Lvl Bmd Ela | - | - |
| Tsp_10877 | 14992.m10900 | T19B10.2 | Minc10356 | Hypothetical protein | Prl Unc Tsla Rup Gro | - | Y |
| Tsp_11032 | 13644.m00292 | C09H10.7 | Minc15358 | Hypothetical protein | Pvl Da, Emb Stp | - | - |
| Tsp_10369 | 13847.m00044 | F10E7.6 | Minc16059 | Hypothetical protein | Sck Clr Ela Gro | Y | - |
| Tsp_01966 | 14972.m07319 | W04G3.2 | Minc11161 | Lipocalin protein | Unc Lva Lvl Bmd | - | Y |
| Tsp_10030 | 14961.m05181 | Y8G1A.2 | Minc07816 | Innexin membrane protein | Unc Rup Stp Gro | Y | - |
Descriptor, annotation based on KEGG Orthology and Interpro.
RNAi phenotype description (www.wormbase.org).
TM, transmembrane.
SP, signal peptide for secretion.
Nematode-specific amino acid sequences in scNOG proteins may have practical significance for functional investigations. As such, we evaluated the scNOGs sequences for molecular features by forced alignment with non-nematode homologs i.e. human, chicken, frog and zebrafish, associated with the same Pfam entries. The scNOGs were categorized into two groups; i) those involving nematode-specific insertions and deletions (InDels)(e.g.29) relative to non-nematode homologues (15 proteins) (Supplementary Fig. 6a) and ii) those involving unique patterns of conservation independent of InDels (70 proteins) (Supplementary Fig 6b and Supplementary Fig. 7)(e.g.14). Sequence variation exclusive of conserved motifs was generally higher among the nematode proteins than among the vertebrate proteins, even though evolutionarily, each comparison spanned similar predicted lengths of time, consistent with a previous report30 (Supplementary Fig. 8). Therefore, pan-Nematoda specific conservation has persisted despite the high evolutionary rate in adjacent sequences of these NOGs.
The nematode specific amino acid sequences in NOGs may have fundamental importance across the Nematoda. For instance, the predicted subunit of an electron transfer complex (Supplementary Fig. 6a) has well defined insertions, and a severe RNAi phenotype is associated with the C. elegans member of this NOG. As such, comparative information from the vertebrate homolog may guide experiments to dissect the functional roles of the NOG insertions. Furthermore, a sequence containing amino acid insertions in one protein interaction partner may be compensated by deletions in the other protein interaction partner. We indeed identified that the interaction partner of the complex to which that protein belongs (long chain Acyl-CoA dehydrogenase, interaction that has been confirmed experimentally31) has deletions in the non-nematode protein (Supplementary Note, Supplementary Fig. 9 and Supplementary Fig 10).
This series of analyses identified genes and proteins that may have fundamental importance to all nematode species. Two categories of nematode-specific sequences are responsible for delineation as scNOGs. Therefore, scNOGs, and most likely other NOGs, contain pan-phylum nematode-specific sequences incorporated either into universally conserved protein structures or into protein structures that are unique to the Nematoda. Evidence reflecting biological significance highlights the potential for NOGs to serve as targets for control of parasitic nematodes that infect humans, animals and plants, while potentially limiting risk to the host.
Nematode core- and phylogenetically-restricted functional categories
A question of central importance is whether or not parasitic nematodes (and potentially other parasites) have independently evolved, or preferentially retained common solutions to challenges of parasitism despite their exploitation of widely divergent trophic ecologies (e.g.32). Much interest in this context has focused on: i) secretory proteins, ii) molecular functions, and iii) biochemical pathways that are conserved or taxonomically restricted.
Although not all secretory proteins from parasitic nematodes are involved in interactions with the host, constituents of this protein category are prime candidates for examining the host-pathogen interface. Here, we sought proteins that are broadly conserved among nematodes, or among parasitic nematodes. These proteins were sorted into orthologous protein groups shared among species representing diverse parasite lineages and then sub-grouped into those with secretory peptides (Supplementary Fig. 11). Predicted secretory protein orthologs were interrogated with previously identified secreted proteins using an orthogonal approach, based on excretory-secretory products in T. spiralis and B. malayi identified by tandem mass spectrographic analysis33–34. Only two proteins were identified as secretory and common to each parasite member (including vertebrate and plant parasites), but absent from the non-parasitic C. elegans: i) a serine peptidase member of the prolyl oligopeptidase family that can be critical for invasion of the mammalian host cells by protozoan parasites35; and ii) a cyanate hydratase that in other organisms hydrolyzes and detoxifies environmental cyanate36. Our results suggest that the number of conserved secretory proteins broadly involved in nematode interactions with hosts may be relatively few. Nevertheless, this number is likely to increase when reducing our analysis to sub-groupings of parasitic nematodes, as we found when proteomes for any two of the three parasitic species were interrogated here.
Among the T. spiralis genes analyzed, 35% (5,456/15,808) could be assigned one or more GO terms. Putative molecular functions were assigned to 90% of this 35%; biological processes to 68% and cellular components to 45%. The remaining two-thirds of genes in T. spiralis represent uncharacterized and possibly novel functions in the parasite. A set of 25 molecular functions were significantly enriched (at P<0.01) or depleted when intra- or inter-specific orthologous groups were compared to the complete repertoire of GO terms for T. spiralis (Supplementary Table 10 and Supplementary Fig. 12). Among the orthologous families confined only to T. spiralis and C. elegans, rhodopsin-like receptor activity was enriched, a possible consequence of the number of genes involved in G-protein coupled receptor protein signaling pathways. In orthologous groups with members only from T. spiralis and B. malayi, the enriched category involved steroid binding proteins.
Among a total of 71 molecular GO categories identified, 42 were enriched and 29 were depleted in the 2,517 nematode orthologous families (including C. elegans) by comparison to the complete proteomes of the four nematode species (Supplementary Table 11). When considering the 64 orthologous groups conserved among the three parasitic nematodes, nine GO categories were statistically enriched or depleted; ATP-binding was the only depleted category, whereas DNA-, and RNA-binding, aspartic-type endopeptidase and prolyl oligopeptidase activities were among those enriched (Supplementary Table 12). Therefore, commonalities in molecular functions may exist even among parasites from widely diverse ecological niches. Further light will be shed on genetic associations among parasitic and non-parasitic nematodes as more robust comparisons among species from each category begin to surface.
Guided by the possibility that parasitic nematodes undergo reductive genome evolution due to reliance on the metabolic capacity and homeostatic buffering of their host, we compared T. spiralis genes encoding enzymes to similar genes from the other parasites and the non-parasitic C. elegans (37–38, Supplementary Fig. 13) and the NemaCyc viewer (Supplementary Fig. 14). We found that the parasitic species had fewer KOs (KEGG orthology) associated with their genes (~522–548), compared to C. elegans (704) (Table 2 and Supplementary Table 13). The number of genes correlated with the number of associated KOs. Therefore, we examined the KOs in relation to nematode lineages used in this study. Among the 785 KOs associated with the nematode species evaluated herein, 337 were shared among all 4 species, i.e. Core Nematode KOs (CNKs). The pathway that had most of the KOs as CNKs was the energy metabolism (53% of all KOs were conserved across all 4 species); the least was the metabolism of cofactors and vitamins (34% of the KOs were in all 4 species). Among the energy metabolism pathways, there were 96 KOs related to oxidative phosphorylation, 52 of which were conserved among all 4 nematodes. This result supports previous observations in which parasite enzymes involved in oxidative phosphorylation exhibited significant sequence divergence from similar host proteins. These differences were largely associated with nematode-specific insertions14,29. Despite the high level of conservation, the number of CNKs among all 4 nematodes was very low (34%) suggesting that different adaptations distinguish nematodes with distinct modes of existence.
Table 2.
Genes and KEGG Orthologies (KOs) represented in metabolic patways in four nematodes
| Pathway | KOs in KEGG Reference pathway | Represented KOs in nematodes | Conserved KO in nematodes | C. elegans | M. incognita | B. malayi | T. spiralis | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Genes | KOs | Genes | KOs | Genes | KOs | Genes | KOs | ||||
| 1. Metabolism | 2258 | 785 | 337 | 2480 | 704 | 1822 | 525 | 1132 | 548 | 1069 | 515 |
| 1.1 Carbohydrate Metabolism | 550 | 192 | 92 | 626 | 167 | 499 | 130 | 294 | 133 | 252 | 145 |
| 1.2 Energy Metabolism | 408 | 131 | 71 | 235 | 123 | 210 | 97 | 144 | 107 | 123 | 87 |
| 1.3 Lipid Metabolism | 325 | 144 | 52 | 710 | 122 | 380 | 98 | 218 | 101 | 199 | 87 |
| 1.4 Nucleotide Metabolism | 174 | 78 | 35 | 306 | 74 | 294 | 52 | 182 | 51 | 182 | 53 |
| 1.5 Amino Acid Metabolism | 484 | 188 | 75 | 607 | 174 | 430 | 129 | 250 | 114 | 266 | 124 |
| 1.6 Metabolism of Other Amino Acids | 126 | 55 | 26 | 222 | 50 | 119 | 39 | 73 | 41 | 76 | 39 |
| 1.7 Glycan Biosynthesis and Metabolism | 160 | 83 | 30 | 163 | 74 | 153 | 54 | 95 | 63 | 89 | 55 |
| 1.8 Biosynth. of Polyketides and Nonrib. Peptides | 4 | 2 | 1 | 5 | 2 | 6 | 1 | 4 | 2 | 1 | 1 |
| 1.9 Metabolism of Cofactors and Vitamins | 301 | 91 | 31 | 392 | 80 | 298 | 55 | 174 | 57 | 185 | 56 |
| 1.10 Biosynthesis of Secondary Metabolites | 55 | 25 | 13 | 234 | 20 | 115 | 18 | 59 | 19 | 47 | 18 |
| 1.11 Xenobiotics Biodegradation and Metabolism | 178 | 61 | 27 | 548 | 55 | 249 | 40 | 125 | 37 | 119 | 38 |
DISCUSSION
Here we present the genome sequence of T. spiralis, a member of the Dorylaimia and a lineage that diverged early in the evolution of the phylum Nematoda. The draft sequence of T. spiralis covered over 90% of the estimated genome and expected genes. Coupled with genomes from nematode lineages depicting more recent episodes of divergence, the T. spiralis data provide new perspectives on genomic evolution that more broadly spans the Nematoda.
The T. spiralis genome sequence and the accompanying genome-mining analysis address four key issues. First, details of genomic diversity that were deduced among species have outlined molecular determinants, where the magnitude of change likely reflects molecular elements that have figured decisively in both lineage and species evolution of the Nematoda (e.g.39–41). It has been argued that such drastic differences can be related to functional diversification, speciation and species adaptation. Given the modest number of nematode species with available genomes, we fully expect that as additional nematode genome sequences become available, much greater resolution of differences will occur. Nonetheless, results presented here helped resolve many specific genomic characteristics that can be further investigated in this context. Second, host characteristics may select for common parasite characteristics of otherwise widely disparate nematode species. The similarities in the steroid binding protein family common to the parasites of humans and mammals, T. spiralis and B. malayi, were distinct from a large family of related nuclear hormone receptors in C. elegans, many of which are homologous to steroid-binding receptors in other organisms42. This distinction provides support for convergent enrichment of common steroid binding receptors in the two parasites of humans and other mammals, possibly dictated by characteristics of the host environment, as previously suggested43. Third, the new databases guided discovery of genes and proteins that appear to have fundamental importance to all nematode species (archetypical characteristics). Accordingly, the NOGs were significantly enriched for genes with RNAi phenotypes in C. elegans. Success in circumscribing archetypical nematode characteristics from pan-phylum databases will serve to refocus research on characteristics that have the broadest application for controlling pathogens of humans, animals and plants. Fourth, these results provide a valuable resource to investigate the biology of the intracellular pathogen, T. spiralis. One example involves a DNase II gene family of T. spiralis, which includes secreted proteins previously implicated in host-parasite interactions and immune control20. The curious expansion and diversification of this family by comparison to other nematodes can now be related to unique characteristics of T. spiralis, and possibly the lineages it represents. A second example centers around why species within this genus have separated into those that generate protective capsules from those which do not; a character which is not host related. There are innumerable anticipated applications of the genome data towards elucidating the biology, methods for immune control and treatments of this parasite. The comparative value of this genome sequence will extend these applications well beyond this species and phylum.
METHODS
Sequencing, assembly and annotation
Rats were infected orally with ML of T. spiralis strain ISS 195. Infections were allowed to precede a minimum of 30 days, then the muscle tissue was digested and parasite collected. Genomic DNA was extracted from muscle larvae of T. spiralis using standard protocols. Whole genome shotgun, BAC and EST libraries were generated3,6. The assembly was performed using the PCAP package44. The physical map for T. spiralis was constructed using 26,784 clones (Supplementary Note).
The repeats were masked using RECON45 and RepeatMasker (see URLs1). Then the Ribosomal RNA genes were identified using RNAmmer (see URLs2). Transfer RNA genes were identified with tRNAscan-SE46. Non-coding RNAs were identified by sequence homology searches of the Rfam database (see URLs3). Protein-coding genes were predicted using a combination of ab initio programs47 and FgenesH (Softberry, Corp) and the evidence based program EAnnot48. A consensus gene set from the above prediction algorithms will be generated, using a logical, hierarchical approach. Gene product naming was determined by BER (see URLs4). Signal peptide for secretion and trans-membrane domain containing proteins were identified using PHOBIUS49.
Protein families and genome evolution
OrthoMCL 19 was used to predict orthologous groups of proteins. Phylogenetic trees were built for protein families with one member from each of the 6 species using PHYLIP (version 3.69; see URLs5) after aligning the family members with MUSCLE (version 3.7; 50). The consensus tree of the trees was used as the phylogeny of the species. Death and birth of each protein family overlaid over species phylogeny was constructed using PHYLIP-DOLLOP by treating each protein family as a character. Gene duplication and deletion events of the families having member from each of the 6 species were reconstructed using URec51 and a neighbor joining tree of each family was generated using PHYLIP-NEIGHBOR.
The dynamics of nematode chromosome re-assortment among multiple nematode pairs was measured using OrthoCluster25 and using syntenic blocks of C. elegans for standardization. For the identification of the ancestrial orthologous regions we used exons that are orthologous among species as map "anchors”52 (Supplementary Note).
Nematode-specific molecular features
A profile was built for each of the 85 scNOGs using HMMBUILD53. The profiles were calibrated using hmmcalibrate and each profile was used to search the Pfam (release 23.0). Hits better than 0.1 were considered. The selected non-nematode species were of evolutionary distances similar to C. elegans and T. spiralis: human, chicken, zebrafish and frog. After identification of the non-nematode families that were associated with same Pfam as the scNOGs the multi-fasta files were aligned using MUSCLE. These alignments were used to build distance matrix using PHYLIP-PROTDIST. RNAi source data was from Wormmart from Wormbase release 180. The core nematode groups were screened against nematode (~1.1 M ESTs and/or Roche/454 cDNAs) and arthropod (5.3 M ESTs) transcript data and sequence homology at 35 bits and 55% identity cut-off was accepted as significant.
Structural annotation and comparison of interaction partners
The three dimensional structure was modeled using the Rosetta3.0 software suite54–56. A total of 40,000 decoys were generated using the full-atom scoring method57 for each sequence. Several of the decoys with a small radius of gyration and low all-atom energy (i.e. the bottom of the energy well) were compared using TM-align58 and MAMMOTH59. The position of the insertions was mapped onto the models generated. The secondary structure predictions calculated for the Rosetta ab initio program were added to the sequence alignment generated by MUSCLE50. The functional significance of the insertions in the electron transfer complex was further dissected by comparing interacting proteins. Two protein-protein interaction databases, IntAct60 and MINT61, were used to see if this protein or its orthologs were involved in a protein-protein interaction.
Functional associations and taxonomic restrictions
Default parameters for InterProScan (v16.1) were used to search against the InterPro database62 and Gene Ontology (GO, 63) annotations were obtained with no additional curation (IEA associations only). These annotations have been displayed graphically by AmiGO and can be accessed at Nematode.net37. Significant enrichment of GO terms was computed based on the hypergeometric distribution using FUNC 64 (including false discovery rate, FDR). A probability refinement was done to remove the GO terms identified as significant due to their children terms. We used the false discovery rate (FDR) computed by FUNC to reduce false discovery. Therefore, unless specified otherwise, the GO term enrichment was selected based on both p-value <0.05 (after refinement) and FDR <0.1.
The gene products were associated with a specific biochemical pathway using the KEGG pathway mappings65. WU-BLAST matches of the genes against KEGG database version 46.0 was used for pathway mapping with a filter of 1e-10. Graphical presentation of the pathway associations was done using NemaPath38. The C. elegans NemaCyc viewer is based on mapping a BLASTP alignment of the KEGG’s genesDB against the predicted T. spiralis genes. Scores stronger than 1e-10 were considered.
Supplementary Material
Acknowledgments
We thank Asher Cutter and members from the Genome Center for discussion and helpful comments on the manuscript. This work was supported by a National Human Genome Research Institute grant to RKW (HG003079) and a National Institute of Allergy and Infectious Diseases grant to MM (81803) and to JA (14490).
Footnotes
AUTHORS CONTRIBUTIONS
MM, DPJ, JPM, DSZ, ERM, and RKW initiated the project; JA and DSZ provided all the worms for the shotgun and DPJ for the cDNA sequencing; LF and RSF directed sequencing and sequence improvement, SY, PM and WCW assembled the genome and evaluated the assembly, VB, XZ and KP directed annotation, MM, ZW, SA, JM, YY and CMT contributed to most of the specific analysis presented in this manuscript; MM, DPJ, DSZ, SWC and MM directed the project and assembled the manuscript.
COMPETING FINANCIAL INTEREST
The authors have no competing financial interests.
Accession numbers
The Trichinella spiralis Whole Genome Shotgun project (project id 12603) has been deposited at DDBJ/EMBL/GenBank under the accession ABIR00000000. The version described in this paper is the second version, ABIR02000000 (contigs, ABIR02000001- ABIR02009267; scoffolds, GL622784-GL629646; proteins, EFV46182-EFV62561).
References
- 1.Putnam NH, et al. The amphioxus genome and the evolution of the chordate karyotype. Nature. 2008;453:1064–1071. doi: 10.1038/nature06967. [DOI] [PubMed] [Google Scholar]
- 2.Lavrov DV, Brown WM. Trichinella spiralis mtDNA: A nematode mitochondrial genome that encodes a putative ATP8 and normally structured tRNAs and has a gene arrangement relatable to those of coelomate metazoans. Genetics. 2001;157:621–637. doi: 10.1093/genetics/157.2.621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mitreva M, et al. Gene discovery in the adenophorean nematode Trichinella spiralis: an analysis of transcription from three life cycle stages. Mol Biochem Parasitol. 2004;137:277 – 291. doi: 10.1016/j.molbiopara.2004.05.015. [DOI] [PubMed] [Google Scholar]
- 4.Pettitt J, Müller B, Stansfield I, Connolly B. Spliced leader trans-splicing in the nematode Trichinella spiralis uses highly polymorphic, noncanonical spliced leaders. RNA. 2008;14:760–770. doi: 10.1261/rna.948008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zarlenga DS, Rosenthal BM, La Rosa G, Pozio E, Hoberg EP. Post-Miocene expansion, colonization, and host switching drove speciation among extant nematodes of the archaic genus Trichinella. Proc Natl Acad Sci U S A. 2006;103:7354–7359. doi: 10.1073/pnas.0602466103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mitreva M, Jasmer DP. Advances on sequencing the genome of the Clade I nematode Trichinella spiralis. Parasitology. 2008;135:869–880. doi: 10.1017/S0031182008004472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zarlenga DS, Rosenthal B, Hoberg E, Mitreva M. Integrating genomics and phylogenetics in understanding the history of Trichinella species. Vet Parasitol. 2009;159:210–213. doi: 10.1016/j.vetpar.2008.10.061. [DOI] [PubMed] [Google Scholar]
- 8.Blumenthal T, Gleason KS. Caenorhabditis elegans operons: form and function. Nat Rev Genet. 2003;4:112–120. doi: 10.1038/nrg995. [DOI] [PubMed] [Google Scholar]
- 9.Cutter AD, Wasmuth JD, Blaxter ML. The evolution of biased codon and amino acid usage in nematode genomes. Mol Biol Evol. 2006;23:2303–2315. doi: 10.1093/molbev/msl097. [DOI] [PubMed] [Google Scholar]
- 10.King JL, Jukes TH. Non-Darwinian evolution. Science. 1969;164:788–798. doi: 10.1126/science.164.3881.788. [DOI] [PubMed] [Google Scholar]
- 11.Mitreva M, et al. Codon usage patterns in Nematoda: analysis based on over 25 million codons in thirty-two species. Genome Biol. 2006;7:R75. doi: 10.1186/gb-2006-7-8-r75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wasmuth J, Schmid R, Hedley A, Blaxter M. On the extent and origins of genic novelty in the phylum Nematoda. PLoS Negl Trop Dis. 2008;2:e258. doi: 10.1371/journal.pntd.0000258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Parkinson J, et al. A transcriptomic analysis of the phylum Nematoda. Nat Genet. 2004;36:1259–1267. doi: 10.1038/ng1472. [DOI] [PubMed] [Google Scholar]
- 14.Yin Y, et al. Molecular determinants archetypical to the phylum Nematoda. BMC Genomics. 2009;10:114. doi: 10.1186/1471-2164-10-114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Abad P, et al. Genome sequence of the metazoan plant-parasitic nematode Meloidogyne incognita. Nat Biotech. 2008;26:909–915. doi: 10.1038/nbt.1482. [DOI] [PubMed] [Google Scholar]
- 16.Ghedin E, et al. Draft genome of the filarial nematode parasite brugia malayi. Science. 2007;317:1756 – 1760. doi: 10.1126/science.1145406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Adams MD, et al. The genome sequence of Drosophila melanogaster. Science. 2000;287:2185–2195. doi: 10.1126/science.287.5461.2185. [DOI] [PubMed] [Google Scholar]
- 18.Goffeau A, et al. Life with 6000 Genes. Science. 1996;274:546–567. doi: 10.1126/science.274.5287.546. [DOI] [PubMed] [Google Scholar]
- 19.Li L, Stoeckert CJ, Jr, Roos DS. OrthoMCL: Identification of ortholog groups for eukaryotic genomes. Genome Res. 2003;13:2178–2189. doi: 10.1101/gr.1224503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vassilatis DM, et al. Analysis of a 43-kDa glycoprotein from the intracellular parasitic nematode Trichinella spiralis. J Biol Chem. 1992;267:18459–18465. [PubMed] [Google Scholar]
- 21.Holterman M, et al. Phylum-wide analysis of SSU rDNA reveals deep phylogenetic relationships among nematodes and accelerated evolution toward crown Clades. Mol Biol Evol. 2006;23 :1792–1800. doi: 10.1093/molbev/msl044. [DOI] [PubMed] [Google Scholar]
- 22.Stein LD, et al. The genome sequence of Caenorhabditis briggsae: a platform for comparative genomics. PLoS Biol. 2003;1:E45. doi: 10.1371/journal.pbio.0000045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee KZ, Eizinger A, Nandakumar R, Schuster SC, Sommer RJ. Limited microsynteny between the genomes of Pristionchus pacificus and Caenorhabditis elegans. Nucl Acids Res. 2003;31:2553–2560. doi: 10.1093/nar/gkg359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Guiliano DB, et al. Conservation of long-range synteny and microsynteny between the genomes of two distantly related nematodes. Gen Biol. 2002;3:RESEARCH0057. doi: 10.1186/gb-2002-3-10-research0057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vergara IA, Chen N. Using OrthoCluster for the detection of synteny blocks among multiple genomes. Curr Protoc Bioinformatics. 2009;Chapter 6(Unit 6 10 16 10):11–18. doi: 10.1002/0471250953.bi0610s27. [DOI] [PubMed] [Google Scholar]
- 26.Dewey CN. Aligning multiple whole genomes with Mercator and MAVID. Methods Mol Biol. 2007;395:221–236. doi: 10.1007/978-1-59745-514-5_14. [DOI] [PubMed] [Google Scholar]
- 27.The C. elegans Sequencing Consortium. Genome sequence of the nematode C. elegans: A platform for investigating biology. Science. 282:2012–2018. 1998. doi: 10.1126/science.282.5396.2012. [DOI] [PubMed] [Google Scholar]
- 28.Fraser AG, et al. Functional genomic analysis of C. elegans chromosome I by systematic RNA interference. Nature. 2000;408:325–330. doi: 10.1038/35042517. [DOI] [PubMed] [Google Scholar]
- 29.Wang Z, et al. Systematic analysis of insertions and deletions specific to nematode proteins and their proposed functional and evolutionary relevance. BMC Evolut Biol. 2009;9:23. doi: 10.1186/1471-2148-9-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mushegian AR, Garey JR, Martin J, Liu LX. Large-Scale taxonomic profiling of eukaryotic model organisms: A comparison of orthologous proteins encoded by the human, fly, nematode, and yeast genomes. Gen Research. 1998;8:590–598. doi: 10.1101/gr.8.6.590. [DOI] [PubMed] [Google Scholar]
- 31.Schilling B, et al. Proteomic analysis of succinate dehydrogenase and ubiquinol-cytochrome c reductase (Complex II and III) isolated by immunoprecipitation from bovine and mouse heart mitochondria. Biochim Biophys Acta. 2006;1762:213–222. doi: 10.1016/j.bbadis.2005.07.003. [DOI] [PubMed] [Google Scholar]
- 32.Bird DM, Opperman CH. The secret(ion) life of worms. Gen Biology. 2009;10:205. doi: 10.1186/gb-2009-10-1-205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Robinson MW, Connolly B. Proteomic analysis of the excretory-secretory proteins of the Trichinella spiralis L1 larva, a nematode parasite of skeletal muscle. Proteomics. 2005;5:4525–4532. doi: 10.1002/pmic.200402057. [DOI] [PubMed] [Google Scholar]
- 34.Moreno Y, Geary TG. Stage- and Gender-Specific Proteomic Analysis of Brugia malayi excretory-secretory products. PLoS Negl Trop Dis. 2008;2:e326. doi: 10.1371/journal.pntd.0000326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Santana JM, Grellier P, Schrevel J, Teixeira ARL. A Trypanosoma cruzi-secreted 80 kDa proteinase with specificity for human collagen types I and IV. Biochem J. 1997;325:129–137. doi: 10.1042/bj3250129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sung YC, Fuchs JA. Characterization of the cyn operon in Escherichia coli K12. J Biol Chem. 1988;263:14769–14775. [PubMed] [Google Scholar]
- 37.Martin J, et al. Nematode. net update 2008: improvements enabling more efficient data mining and comparative nematode genomics. Nucleic Acids Res. 2009;37:D571–578. doi: 10.1093/nar/gkn744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wylie T, et al. NemaPath: online exploration of KEGG-based metabolic pathways for nematodes. BMC Genomics. 2008;9:525. doi: 10.1186/1471-2164-9-525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Panhuis TM, Clark NL, Swanson WJ. Rapid evolution of reproductive proteins in abalone and Drosophila. Philos Trans R Soc Lond: Biol Sci. 2006;361:261–268. doi: 10.1098/rstb.2005.1793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kocher TD. Adaptive evolution and explosive speciation: the cichlid fish model. Nat Rev Genet. 2004;5:288–298. doi: 10.1038/nrg1316. [DOI] [PubMed] [Google Scholar]
- 41.Givnish TJ, et al. Molecular evolution, adaptive radiation, and geographic diversification in the amphiatlantic family Rapateaceae: evidence from ndhF sequences and morphology. Evol Int J Org Evol. 2000;54:1915–1937. doi: 10.1111/j.0014-3820.2000.tb01237.x. [DOI] [PubMed] [Google Scholar]
- 42.Sluder AE, Mathews SW, Hough D, Yin VP, Maina CV. The nuclear receptor superfamily has undergone extensive proliferation and diversification in nematodes. Genome Res. 1999;9 :103–120. [PubMed] [Google Scholar]
- 43.Sluder AE, Maina CV. Nuclear receptors in nematodes: themes and variations. Trends Genet. 2001;17:206–213. doi: 10.1016/s0168-9525(01)02242-9. [DOI] [PubMed] [Google Scholar]
- 44.Huang X, Wang J, Aluru S, Yang SP, Hillier L. PCAP: A Whole-Genome Assembly Program. Genome Res. 2003;13:2164–2170. doi: 10.1101/gr.1390403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bao Z, Eddy SR. Automated de novo identification of repeat sequence families in sequenced genomes. Genome Res. 2002;12:1152–1155. doi: 10.1101/gr.88502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lowe TM, Eddy SR. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997;25:955–964. doi: 10.1093/nar/25.5.955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Korf I. Gene finding in novel genomes. BMC Bioinformatics. 2004;5:59. doi: 10.1186/1471-2105-5-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ding L, et al. EAnnot: a genome annotation tool using experimental evidence. Genome Res. 2004;14:2503–2509. doi: 10.1101/gr.3152604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kall L, Krogh A, Sonnhammer ELL. A combined transmembrane topology and signal peptide prediction method. J Mol Biol. 2004;338:1027–1036. doi: 10.1016/j.jmb.2004.03.016. [DOI] [PubMed] [Google Scholar]
- 50.Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gorecki P, Tiuryn J. URec: a system for unrooted reconciliation. Bioinformatics. 2007;23:511–512. doi: 10.1093/bioinformatics/btl634. [DOI] [PubMed] [Google Scholar]
- 52.Dewey CN. Aligning multiple whole genomes with Mercator and MAVID. Methods Mol Biol. 2007;395:221–236. doi: 10.1007/978-1-59745-514-5_14. [DOI] [PubMed] [Google Scholar]
- 53.Eddy SR. A new generation of homology search tools based on probabilistic inference. Genome Inform. 2009;23:205–211. [PubMed] [Google Scholar]
- 54.Andre I, Bradley P, Wang C, Baker D. Prediction of the structure of symmetrical protein assemblies. Proc Natl Acad Sci U S A. 2007;104:17656–17661. doi: 10.1073/pnas.0702626104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rohl CA, Strauss CE, Misura KM, Baker D. Protein structure prediction using Rosetta. Methods Enzymol. 2004;383:66–93. doi: 10.1016/S0076-6879(04)83004-0. [DOI] [PubMed] [Google Scholar]
- 56.Qian B, et al. High-resolution structure prediction and the crystallographic phase problem. Nature. 2007;450:259–264. doi: 10.1038/nature06249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Misura KM, Chivian D, Rohl CA, Kim DE, Baker D. Physically realistic homology models built with ROSETTA can be more accurate than their templates. Proc Natl Acad Sci U S A. 2006;103:5361–5366. doi: 10.1073/pnas.0509355103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhang Y, Skolnick J. TM-align: a protein structure alignment algorithm based on the TM-score. Nucleic Acids Res. 2005;33:2302–2309. doi: 10.1093/nar/gki524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ortiz AR, Strauss CE, Olmea O. MAMMOTH (matching molecular models obtained from theory): an automated method for model comparison. Protein Sci. 2002;11:2606–2621. doi: 10.1110/ps.0215902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kerrien S, et al. IntAct--open source resource for molecular interaction data. Nucleic Acids Res. 2007;35 :D561–565. doi: 10.1093/nar/gkl958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chatr-aryamontri A, et al. MINT: the Molecular INTeraction database. Nucleic Acids Res. 2007;35:D572–574. doi: 10.1093/nar/gkl950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mulder NJ, et al. InterPro, progress and status in 2005. Nucleic Acids Res. 2005:D201–205. doi: 10.1093/nar/gki106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.The Gene Ontology C. The gene ontology project in 2008. Nucl Acids Res. 2008;36:D440–444. doi: 10.1093/nar/gkm883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Prufer K, et al. FUNC: a package for detecting significant associations between gene sets and ontological annotations. BMC Bioinformatics. 2007;8:41. doi: 10.1186/1471-2105-8-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kanehisa M, et al. KEGG for linking genomes to life and the environment. Nucleic Acids Res. 2008;36:D480 – D484. doi: 10.1093/nar/gkm882. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.