

Abstract
Cancer is a complex, multiscale process, in which genetic mutations occurring at a subcellular level manifest themselves as functional changes at the cellular and tissue scale. The multiscale nature of cancer requires mathematical modeling approaches that can handle multiple intra- and extracellular factors acting on different time and space scales. Hybrid models provide a way to integrate both discrete and continuous variables that are used to represent individual cells and concentration or density fields, respectively. Each discrete cell can also be equipped with sub-models that drive cell behavior in response to microenvironmental cues. Moreover, the individual cells can interact with one another to form and act as an integrated tissue. Hybrid models form part of a larger class of individual-based-models that can naturally connect with tumor cell biology and allow for the integration of multiple interacting variables both intrinsically and extrinsically and are therefore perfectly suited to a systems biology approach to tumor growth.
Keywords: hybrid models, individual cell based models, multiscale models, tumor growth, systems biology
Mathematical and computational modeling of tumor growth is not new – in fact it goes back over 50 years. However, to some extent it has largely been ignored by the biological and medical communities. There are multiple reasons for this but two of the most significant revolve around the reductionist focus of biology and the lack of directly testable hypotheses from the models. By necessity much of the models of cancer were general, phenomenological and not specific to a type of cancer and therefore were plagued by a lack of experimental data to both parameterize and validate. That is not to say they were not useful. At their heart most mathematical models are mechanistic – focusing on the core processes that drive tumor growth and integrating them leading to predictions that are holistic by definition. This further contributed to the lack of biological interest in combining laboratory experiments with computational simulations. Most of the experimental biologists working in this field were more focused on the reductionist route revolving around specific genetic mutations or signaling pathways that were found to be important in cancer development. This led to the data explosion that motivated the advent of early systems biology and the development of bioinformatics. Mathematical biology and the mechanistic cancer models it produced were somewhat left behind, but little by little they have matured moving from simple non-spatial growth laws (gompertz) all the way to hybrid multiscale models discussed in this review. See also a list of previously published reviews in the Future Reading section. In the last few years mathematical and computational models of cancer have become more accepted by the biological community both as means to motivate experimentation but also as a route to integrate multiple experimental measurements to generate testable predictions. This shift has been partly driven by the emergence of new modeling approaches (such as hybrid models) but also by the refocusing of the biological community on cancer as a system. Mathematical and computational models of cancer have almost always viewed cancer as a system of multiple interacting variables and processes and therefore should really be considered part of systems biology.
In this review we will focus on the recent development of hybrid models of tumor growth. Whilst not an exhaustive review we have tried to incorporate all of the most up to date models, constraining our search to key references within the last five years. Hybrid models integrate both continuous and discrete variables and are able to incorporate biological phenomena on various temporal and spatial scales. These models represent cells as individual discrete entities and often use continuous concentration or density fields to model cell intracellular and extracellular environments. By their very nature, hybrid models are ideal for examining direct interactions between individual cells and between the cells and their microenvironment, but they also allow us to analyze the emergent properties of complex multi-cellular systems (such as cancer). It is worth noting that since these interactions take place on the intra- and inter-cellular levels, but are manifested by changes on the tissue level, the emergent behavior of growing multi-clonal tumors are almost impossible to infer intuitively. Hybrid models can facilitate our understanding of the underlying bio-physical processes in tumor growth. For example, by using high-throughput simulation techniques, we can examine the impact that changes in specific cell interactions (or their microenvironment) have on tumor growth and treatment. Hybrid models are often multiscale by definition integrating processes on different temporal and spatial scales, such as gene expression, intracellular pathways, intercellular signaling, cell growth or migration. There are two general classes of hybrid models, those that are defined upon a lattice and those that are off lattice. The structure of this review will be to view these two broad classes in terms of increasing cellular complexity. We will then revisit these models in terms of the level of biological detail of the tumor growth process they recapitulate. Finally, we will discuss the critical role that integration needs to play if we want to make a direct impact on cancer research and treatment both from the perspective of integrating models with experiments but also from the perspective of integrating multiple modeling approaches.
Hybrid models complexity
Hybrid models can be divided into two classes that depend reciprocally on the number of cells these models can handle and the included details of each individual cell structure, i.e. models dealing with large cell populations but with simplified cell geometry, and those that model small colonies of fully deformable cells (Fig.1). Simplified geometry models are capable of handling large numbers of cells (thousands to millions) and still treat them as individual entities that can both act independently of other cells (individual cell cycle, cell mutations, cell phenotype) and interact with their immediate neighbors (cooperate or compete). With these kind of models one can simulate tumor growth up to clinically relevant sizes, thus allowing for incorporation of different kinds of tumor treatments, and enabling us to test in silico new and preexisting treatment protocols. Models with deformable cells allows us to investigate the intimate interactions between individual cells and between cells and their environment. Various cellular processes can be represented in these models in a more realistic way, by incorporating for example, the time- and space-dependent enlargement of growing cells, the orientation of cell division, the elongation during cell migration. Both classes however can be coupled with additional equations, such as ordinary differential (ODE), partial differential (PDE) and/or stochastic equations, to describe signaling or metabolic pathways, as well as mechanical or molecular details of cell life processes. Technically, hybrid models can also be divided into two classes, on- and off-lattice (Fig.1), however this term actually refers only to the imposed positions of the cells (a square, hexagonal or cubic lattice vs. unconstrained locations in the two or three dimensional space) but the underlying chemical or physical fields are typically defined on regular grids in both kinds of models (as the simplest way to solve standard reaction-diffusion equations). We elaborate on both classes of model below, discussing in briefly the different models that fit in each class and how they have been applied to tumor growth.
Figure 1. Reciprocal relation between the numbers of cells handled by the models and the level of included cellular details.
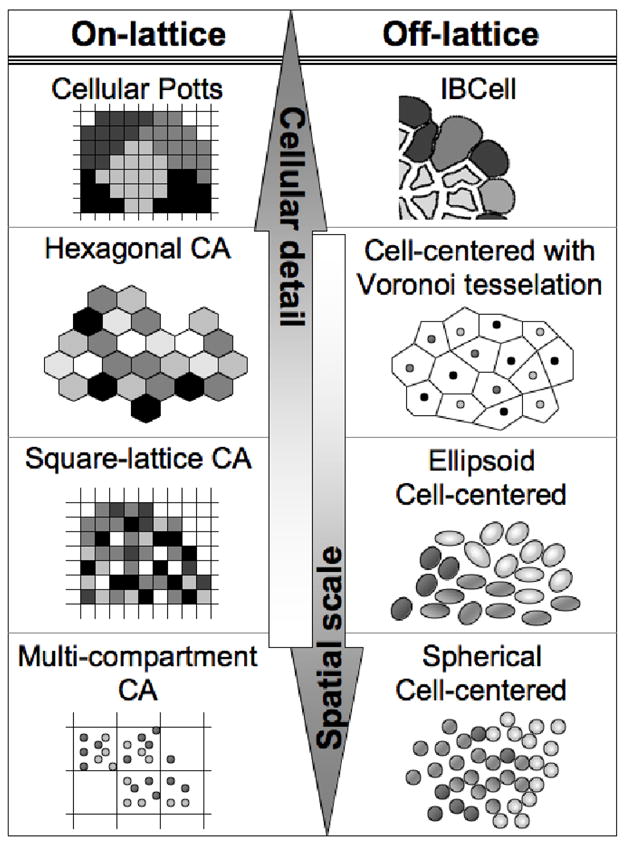
In each class (on-lattice and off-lattice) the models complexity rises from cells represented by single points to fully deformable bodies
On-lattice models
On-lattice models are constrained by a lattice structure (square, hexagonal, cubical) that defines the locations of cells and cell-cell interaction neighborhoods. Technically, they may seem to be more straightforward to program than the off-lattice models, since usually the underlying grid is common for modeling both cellular locations and the chemical/metabolic fields. Also, the algorithms for cellular neighborhood and cellular microenvironment are easier to handle in computational implementation since cell neighborhood relation is defined by the fixed number of surrounding grid sites, and therefore the search for cell neighbors (for example to determine cell-cell adhesion or communication) is simplified. However, the common underlying grid implies that changes in the chemical fields are modeled on the cell scale (unless a multi-grid approach is used), and therefore the “jumps” in these values may not reflect the smooth changes in chemical gradients. Another disadvantage of the lattice-based models is a limited number of directions in which the cells can move and communicate with their neighbors. Several examples of on-lattice models are shown in Fig.2a–h.
Figure 2. Snapshots from simulations of various hybrid models of tumor growth.
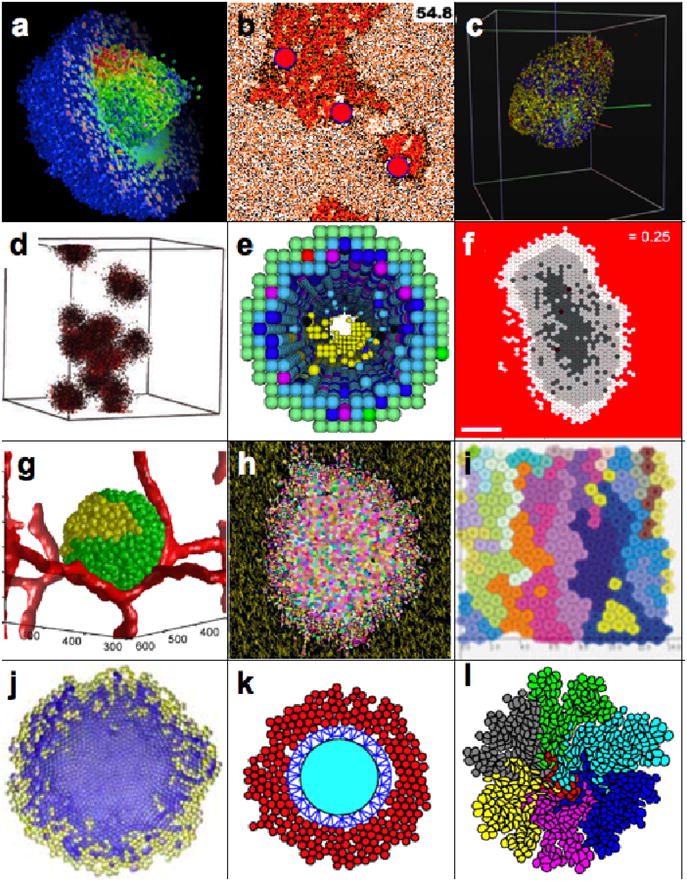
a) 3D tumor spheroid, simulated by a hybrid cellular automaton; reprinted from [8] with permission from Birkhauser-Verlag. b) Tumor invasion in prostate ducts simulated by a hybrid cellular automaton; Reprint permission requested from [16]. c) 3D tumor spheroid simulated by an agent-based on-lattice model; reprinted from [10] with permission from Springer. d) 3D tumor self-metastatic spheroids simulated by a hybrid cellular automaton; reprinted from [5] with permission from Nature Publishing Group. e) 3D model of ductal carcinoma in situ simulated by a square-grid cellular automaton; reprinted from [18] with permission from Elsevier. f) 2D tumor spheroids simulated by a hexagonal cellular automaton; reprinted from [34] with permission from BioMed Central, the Open Access Publisher. g) 3D vascularized tumor spheroid simulated by Potts model; reprinted from [46] with permission from Public Library of Science, open access article. h) 2D tumor spheroid in a heterogeneous environment composed on ECM fibers simulated by Potts model; reprinted from [42] with permission from Elsevier. i) 2D model of colorectal tumor simulated using the particle model with Voronoi triangulation; reprinted from [61] with permission from John Wiley and Sons. j) 2D tumor spheroid modeled using the cell-centered off-lattice model; reprinted from [50] with permission from Springer. k) 2D hybrid model of tumor growth simulated by particle center-based ellipsoid model; reprinted from [56] with permission from World Scientific. l) 2D multiclonal tumor growth simulated by a model of deformable fluid-based cells; reprinted from [69] with permission from Birkhauser-Verlag.
Multi-compartment cellular automata may contain more than one cell in each grid site, however they have a predefined grid capacity and upon reaching this limit some cells need to be replaced to another grid site. The efficient algorithms for cell migration between neighboring grid sites need to be defined. In the context of tumor development the multi-compartment cellular automata (CA) were used to model multicellular spheroids growth [1] and the emergence of invasive tumors [2].
Square-lattice cellular automata assume the mutually exclusive space management, i.e. each grid site can accommodate at most one cell. Typically, each cell has ether four or eight potential neighbors (von Neumann neighborhood or Moore neighborhood, respectively). Cells move to the one of the unoccupied lattice sites (either randomly or via the directional stimulus, such as chemo- or haptotaxis). This kind of models has been widely used to simulate avascular [3,4,5,22,24,29,30] and vascularized tumor growth [6, 7], tumor cell invasion [8,9,10,20,31] and tumor interactions with various environmental factors, including oxygen, glucose, growth factors [11,12,13,14,21,23,28], stromal composition [15,16,17,25,27] and tissue architecture [18,19,26,32].
Hexagonal-lattice cellular automata use a hexagonal grid to define cell positions and cell neighborhood consisting of six symmetrically located cells surrounding the host cell. This kind of model has been used to simulate multicellular tumor growth [33,34,38], to investigate different arrangements of growing ductal carcinoma in situ [35], and to the reproduce patterns of migrating glioma cells [36,37].
Potts models or GGH (Glazier-Granier-Hogeweg) models extend the square CA by allowing individual cells to occupy several 2D square or 3D cubical lattice sites that together define cell volume and cell surface area. Cell shape deformation and direct cell-cell interactions are based on the concept of Monte Carlo simulations and energy minimalization. This model has been used to model tumor growth and invasion [39,40,41,42,43], and angiogenic vascularization [44,45,46].
Off-lattice models
Off-lattice models, in comparison to on-lattice models, have a more realistic representation of cell spatial locations in the sense that they do not need to be uniformly spaced on a fixed grid. However, to determine cell placement of daughter cells during cell division some additional steps need to be undertaken to account for mutually exclusive cell areas or volumes (i.e. non-overlapping). Moreover, the main advantage of the lattice-free models, i.e., the freedom to move each cell in any direction, brings a risk that an inappropriately chosen discrete time step will result in cell collisions (especially in densely packed populations). The main disadvantage of this kind of model is that one needs to design special algorithms to efficiently handle cell-cell neighborhoods, and some interpolation techniques need to be applied to transfer values between the cellular off-lattice individuals and the chemical fields that are usually computed on regular grids. Several examples of on-lattice models are shown in Fg.2i–l.
Spherical cell-centered models represent the cells as single points with either the springs or energy potentials defined between the neighboring cells to keep them within a minimum predefined distance (circular or spherical geometry). These have been used to simulate tumor cell colonies growing in the form of monolayers and spheroids [47,48,53,54], various patterns of ductal carcinoma in situ [55], tumor invasion [49,50,51] and blood vessel intravasation [52].
Ellipsoidal cell-centered models are similar in concept to the spherical cell-centered models, however, each cell in this model is defined by two axes with different lengths to form an elliptical shape. Thus allow for a more intuitive definition of cell orientation and polarization [56,57].
Voronoi tesselation technique can be used for the cell-centered models to overlay polygonal shapes around cell nuclei, that in turn leads to a variable number of cell neighbors, but also defines cell-cell interactions based on variable contact between neighboring cells, e.g. based on the length of adherent cell sides. This model has been used to simulate growth of multicellular spheroids [58,59] and the development of colorectal tumors [60,61].
Fluid based elastic cell models allow for modeling cell plasticity, geometrical adaptability and cell deformation during migration, polarization and differentiation. Moreover, since cell elastic boundaries are discretized, all boundary points may be treated as cell membrane receptors/sensors and various cell-cell and cell-ECM (extracellular matrix) interactions can be defined based on cell membrane receptor dynamics. These kind of models has been used to simulate growth of multicellular spheroids [62,63], various cellular patterns in developing ductal carcinoma in situ [64,69], invasive tumors [65,68] as well as to model normal development of epithelial ductal monolayers and their various mutants [66,67].
Biological complexity
Cancer development is a complex multiscale process that depends on both the intrinsic factors (such as genetic mutation, gene expression, cell adaptability, robustness and phenotypic evolution) and on extrinsic cues sensed from the cell microenvironment (such as multiple metabolite and nutrient gradients, different densities and alignments of ECM fibers or diverse tissue architectures). Experimentally, cancer evolution and development are generally only considered at the gene or protein scale, however, recently there has been a great deal of interest in the impact of this evolution at the cellular scale. After all, selection occurs upon the cellular phenotype even if mutations take place in the genotype. This selection pressure is often driven by changes in the tumor microenvironment. Hybrid models seem particularly well suited to investigate the evolutionary aspects of cancer and various strategies have been developed to model evolution of both cell phenotypes and genotypes, as well as the complex interactions between cancer cells and their surrounding microenvironment. Evolution of cell phenotypes is often modeled using deterministic flow charts in which a decision to enter the specific cellular process (such as cell growth, division, death or movement) is determined sequentially by comparing cell status (e.g. cell age, nutrients level, the number of cell neighbors or the configuration of cell membrane receptors) to predetermined thresholds [5,16,18,20,66]. Another approach involves the introduction of random mutations that determine the evolution of a given cellular phenotype (e.g. doubling time, death rate or sensitivity to contact inhibition), or cell interactions with external factors (such as concentration of metabolites or ECM degradation) [19,20]. Such interactions can be also modeled using the neural networks [12,21] or systems of ODEs defining certain signaling pathways or protein networks [10,16, 40,61].
Evolution of cell phenotypes depends not only on cell genotype, but also on cues sensed by the cells from their neighborhood. Moreover, the evolving cells modify also their immediate vicinity, and these mutual interactions may lead to the emergence of certain microenvironments promoting tumor development. The establishment of a three-layered structure (consisting of a proliferating rim, a ring of quiescent cells and a necrotic core) that arises in tumor spheroids as a consequence of nutrient depletion has been reproduced by virtually every kind of modeling approach, and has become a test problem for every newly developed mathematical model of solid tumor growth [see 3,20,22,34,40,42,46,49,53,56,61,68,69,70]. Gradients of nutrients, such as oxygen or glucose, are not the only chemical species present in the stroma surrounding normal and tumor tissues. In fact tumor cells are exposed to various enzymes (more than 20 kinds of MMPs: matrix metalloproteinases, and TIMPs: tissue inhibitors of matrix metalloproteinases), a multitude of growth factors and a range of chemokines. Mathematical models were used extensively to investigate relations between gradients of various metabolites and the emerging morphologies of developing tumors [13,16,20,21,23,24,34,40,43,56,61,68]. In addition to responding to various chemical factors tumor cells can mechanically interact with other tumor cells as well as with various other stromal cells, such as fibroblasts, macrophages, immune cells. Tumor cell behavior depends also on the interactions with its physical environment, e.g. variable densities and alignment of different ECM fibers (such as collagen, laminin, elastin or fibronectin). The intimate adhesive relations between neighboring tumor cells, cells and the ECM, and interactions between tumor cells and other stromal cells have been addressed by multiple investigators [7,16,17,20,25,37,42,45,48,49,51]. The initiation and progression of most tumors depends strongly on the architecture of the host tissue. Various computational models have addressed the issues arising from confined microenvironments such as the structure of epithelial ducts [18,38,61,62,64] or brain geometry [19,71].
Bridging scales and models
In principle, it is possible to build a model that will span multiple scales from the genotype and various biochemical reactions to the details on cell morphology, and the collective behavior of millions of individual cells forming the whole tumor tissue. However, such a model may acquire structural complexity that is comparable with biological cells and far less effective computationally than the real living organism. It is therefore more desirable to find ways to bridge independent models rather than build a single “mega-model” that encompasses all the complexity of tumor development. This bridging may be in terms of separate models that consider distinct parts of the cancer process or the same process but on different scales. Our group has undertaken such an approach to address genetic, mechanistic and evolutionary mechanisms of disruption of tissue homeostasis and initiation of tumor growth [26], as well as to investigate how the local tumor microenvironment can select for cells with an invasive advantage [9,65]. Similarly, the questions of VEGF (vascular endothelial growth factor) transport in healthy and cancerous vascular systems were investigated by Popel and collaborators using a multicompartment model [72,73]. The emergence of glycolytic phenotype in carcinogenesis was addressed by Gatenby and collegues using a combination of approaches including cellular automata, evolutionary dynamics, information or competition theories [74,75,76,77]. The advantage of applying several distinct models in answering the same scientific question is manifold. If these models produce similar (or comparable) outcomes, the common assumptions underlying the investigated phenomena can be identified, and used to infer underlying mechanisms which can then be further investigated experimentally. If these models result in different outcomes, further investigation can be carried to determine which features specific to each model have influenced the contrasting results and how this relates to the underlying biology. Again, this may lead to further experimentation to confirm or rule out the contrasting results.
Conclusion
As we hinted at in the opening section of this article, computational models developed and implemented without real experimental data to neither parameterize nor validate their predictions was one of the major limitations in them gaining biological acceptance. What has recently become clear is that there is not only a need for greater integration between models and experimentation but a requirement [27,28,78,79]. This dialogue must go both ways--experiments should drive models and models should drive experiments. Models can utilize experimental data and produce novel hypotheses but without the experimental testing to validate or negate such hypotheses, it becomes a very limited academic exercise. Although to be fair, it can be very difficult to find appropriate collaborators motivated to provide such experimental support.
Models need to drive experimentation and to some extent this requires an understanding of the experimental systems that are currently being used by the cancer research community. The schematic presented in Fig.3 highlights the multiple scales that are experimentally studied in cancer research by means of the experimental systems that are utilized. If we truly want to build integrated models then we need to think of what sorts of experiments will be needed to drive our models and validate them. From our personal experience this leads to a significant shift in thinking in relation to which components are incorporated into a model and which are not. It also dictates what type of model should be utilized and this review wouldn’t be about hybrid models if we didn’t believe that hybrid approaches are perfectly suited to facilitate such integration. Due to their cell centric nature hybrid models naturally connect with cell biology and readily incorporate microenviromental components. The interface between tumor cells and their microenvironment being one of the critical drivers of cancer progression, the other being the intracellular changes that result from mutations, altered intra- and intercellular signaling or protein trafficking – which can also be captured using hybrid models [10,12,40,51,66].
Figure 3. A schematic of modeling scales and techniques.
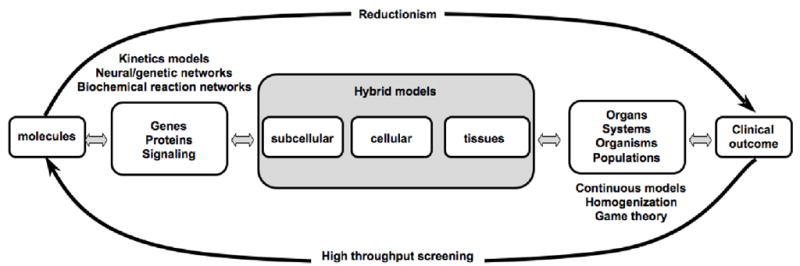
Multiple biological scales can be bridged by various types of mathematical models.
It is worth restating that cancer is a mutliscale process whereby mutations at the molecular scale effect protein formation which effects signaling pathways which modulate cell behavior that transforms the tissue leading to damaged organs and potentially death. This complex multiscale process can be broken down into smaller units that are more amenable to both experimental and theoretical approaches. This again brings into focus the bridging nature of mathematical models that are critical for understanding how the different biological scales of cancer impact upon one another. The models we have focused on this review bridge several scales both above and below the fundamental unit of the cell (Fig. 3), however, they cannot bridge all – this most certainly will require different modeling approaches such as continuous (e.g. [80]) or statistical models (e.g. [81]). In addition, there is an unspoken void between in vitro and in vivo models and between in vivo and the clinic. In silico models have the power to link these approaches and in doing so can give some insight into the processes that translate well between them and those that don’t. This is a severely understudied area for modeling in cancer research and should be a ripe focus for future work.
Acknowledgments
Both authors were partially supported by the National Cancer Institute Integrative Cancer Biology Program (U54 CA143970) and by NCI Physical Sciences-Oncology Centers Program (U54 CA143970).
Multicompartment CA
- 1.Piotrowska MJ, Angus SD. A quantitative cellular automaton model of in vitro multicellular spheroid tumour growth. Journal of Theoretical Biology. 2009;258:165–178. doi: 10.1016/j.jtbi.2009.02.008. [DOI] [PubMed] [Google Scholar]
- 2.Basanta D, Hatzikirou H, Deutsch A. Studying the emergence of invasiveness in tumours using the game theory. Eur Phys J B. 2008;63:393–397. [Google Scholar]
Square-Lattice CA
- 3.Dormann S, Deutsch A. Modeling of Self-Organized Avascular Tumor Growth with a Hybrid Cellular Automaton. In Silico Biology. 2002;2:393–406. [PubMed] [Google Scholar]
- 4.Spencer SL, Gerety RA, Pienta KJ, Forrest S. Modeling Somatic Evolution in Tumorigenesis. PLoS Comput Biol. 2006;2(8):e108. doi: 10.1371/journal.pcbi.0020108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Enderling H, Hlatky L, Hahnfeldt P. Migration rules: tumours are conglomerates of self-metastases. British Journal of Cancer. 2009;100:1917–1925. doi: 10.1038/sj.bjc.6605071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stephanou A, McDougall SR, Anderson ARA, Chaplain MAJ. Mathematical Modelling of flow in 2D and 3D vascular networks: applications to anti-cancer and chemotherapeutic drug strategies. Math Comp Model. 2005;41:1137–1156. [Google Scholar]
- 7.Owen MR, Alarcon T, Maini PK, Byrne HM. Angiognesis and vascular remodelling in normal and cancerous tissues. J Math Biol. 2009;58:689–721. doi: 10.1007/s00285-008-0213-z. [DOI] [PubMed] [Google Scholar]
- 8.Anderson ARA. A hybrid multiscale model of tumour invasion: evolution and the microenvironment. In: Anderson A, Chaplain M, Rejniak KA, editors. Single-Cell-Based Models in Biology and Medicine. Chapter I.1 Birkhauser-Verlag; Basel, Switzerland: 2007. [Google Scholar]
- 9.Anderson ARA, Rejniak KA, Gerlee P, Quaranta V. Microenvironment driven invasion: a multiscale model investigation. J Math Biol. 2009a;58:579–624. doi: 10.1007/s00285-008-0210-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang L, Wang Z, Sagotsky JA, Deisboeck TS. Multiscale agent-based cancer modeling. J Math Biol. 2009a;58:545–559. doi: 10.1007/s00285-008-0211-1. [DOI] [PubMed] [Google Scholar]
- 11.Gatenby RA, Smallbone K, Maini PK, Rose F, Averill J, Nagle RB, Worrall L, Gillies RJ. Cellular adaptations to hypoxia and acidosis during somatic evolution of breast cancer. British Journal of Cancer. 2007;97:646–653. doi: 10.1038/sj.bjc.6603922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gerlee P, Anderson ARA. Evolution of cell motility in an individual-based model of tumour growth. Journal of Theoretical Biology. 2009;259:67–83. doi: 10.1016/j.jtbi.2009.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Silva AS, Yunes JA, Gilles RJ, Gatenby RA. The potential of systemic buffers in reducing intratumoral extracellular pH and acid-mediated invasion. Cancer Research. 2009;69:2677–2684. doi: 10.1158/0008-5472.CAN-08-2394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang L, Chen LL, Deisboeck TS. Multi-scale, multi-resolution brain cancer modeling. Mathematics and Computers in Simulation. 2009;79:2021–2035. doi: 10.1016/j.matcom.2008.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Anderson ARA, Weaver AM, Cummings PT, Quaranta V. Tumor morphology and phenotypic evolution driven by selective pressure from the microenvironment. Cell. 2006;127:905–915. doi: 10.1016/j.cell.2006.09.042. [DOI] [PubMed] [Google Scholar]
- 16.Basanta D, Strand DW, Lukner RB, Franco OE, Cliffel DE, Ayala GE, Hayward SW, Anderson ARA. The Role of Transforming Growth Factor-B–Mediated Tumor-Stroma Interactions in Prostate Cancer Progression: An Integrative Approach. Cancer Res. 2009;69:7111–7120. doi: 10.1158/0008-5472.CAN-08-3957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.De Pillis LG, Mallet DG, Radunskaya AE. Spatial tumor-immune modeling. Computational and Mathematical Methods in Medicine. 2006;7:159–176. [Google Scholar]
- 18.Bankhead A, Magnuson N, Heckendorn R. Cellular automaton simulation examining progenitor hierarchy structure effects on mammary ductal carcinoma in situ. J Theor Biol. 2007;246:491–498. doi: 10.1016/j.jtbi.2007.01.011. [DOI] [PubMed] [Google Scholar]
- 19.Gevertz J, Torquato S. Growing heterogeneous tumors in silico. Phys Rev E. 2009;80:051919. doi: 10.1103/PhysRevE.80.051910. [DOI] [PubMed] [Google Scholar]
- 20.Anderson ARA. A hybrid mathematical model of solid tumour invasion: the importance of cell adhesion. Math Med Biol. 2005;22:163–186. doi: 10.1093/imammb/dqi005. [DOI] [PubMed] [Google Scholar]
- 21.Gerlee P, Anderson ARA. A hybrid cellular automaton model of clonal evolution in cancer: the emergence of the glycolytic phenotype. 2008;250:705–722. doi: 10.1016/j.jtbi.2007.10.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kansal AR, Torquato S, Chiocca EA, Deisboeck TS. Emergence of a subpopulation in a computational model of tumor growth. J Theor Biol. 2000;207:431–441. doi: 10.1006/jtbi.2000.2186. [DOI] [PubMed] [Google Scholar]
- 23.Smallbone K, Gatenby RA, Gillies RJ, Maini PK, Gavaghan DJ. Metabolic changes during carcinogenesis: potential impact on invasiveness. J Thoer Boil. 2007;244:703–713. doi: 10.1016/j.jtbi.2006.09.010. [DOI] [PubMed] [Google Scholar]
- 24.Zhang L, Strouthos CG, Wang Z, Deisboeck TS. Simulating brain tumor heterogeneity with multiscale agent-based model: linking molecular signatures, phenotypes and expansion rate. Math Comp Model. 2009c;49:307–319. doi: 10.1016/j.mcm.2008.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Enderling H, Alexander NR, Clark ES, Branch KM, Estrada L, Crooke C, Jourquin J, Lobdell N, Zaman MH, Guelcher SA, Anderson ARA, Weaver AM. Dependence of Invadopodia Function on Collagen Fiber Spacing and Cross-Linking: Computational Modeling and Experimental Evidence. Biophys J. 2008;95:2203–2218. doi: 10.1529/biophysj.108.133199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Anderson ARA, Basanta D, Gerlee P, Rejniak KA. Evolution, regulation and disruption of homeostasis and its role in carcinogenesis. In: Deisboeck TS, Stamatakos G, editors. Multiscale Cancer Modeling. Chapman & Hall; 2010. [Google Scholar]
- 27.Anderson ARA, Quaranta V. Integrative mathematical oncology. Nat Rev Cancer. 2008;8:227–234. doi: 10.1038/nrc2329. [DOI] [PubMed] [Google Scholar]
- 28.Anderson ARA, Hassanein M, Branch KM, Lu J, Lobdell NA, Maier J, Basanta D, Weidow B, Narasanna A, Arteaga CL, Reynolds AB, Quaranta V, Estrada L, Weaver AM. Microenvironmental independence associated with tumor progression. Cancer Research. 2009b;69:8797–8806. doi: 10.1158/0008-5472.CAN-09-0437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wcislo R, Dzwinel W, Yuen DA, Dudek AZ. A 3-D model of tumor progression based on complex automata driven by particle dynamics. J Mol Model. 2009;15:1517–1539. doi: 10.1007/s00894-009-0511-4. [DOI] [PubMed] [Google Scholar]
- 30.Enderling H, Anderson ARA, Chaplain MAJ, Beheshti A, Hlatky LR, Hahnfeldt PJ. Paradoxical dependencies of tumor dormancy and progression on basic cell kinetics. Cancer Res. 2009;69:8814–8821. doi: 10.1158/0008-5472.CAN-09-2115. [DOI] [PubMed] [Google Scholar]
- 31.Sottoriva A, Verhoeff JJC, Borovski T, McWeeney SK, Naumov L, Medema JP, Sloot PMA, Vermeulen L. Cancer stem cell tumor model reveals invasive morphology and increased phenotypical heterogeneity. Cancer Res. 2010;70:46–56. doi: 10.1158/0008-5472.CAN-09-3663. [DOI] [PubMed] [Google Scholar]
- 32.Silva AS, Gatenby RA, Gillies RJ, Tunes JA. A quantitative theoretical model for development of malignancy in ductal carcinoma in situ. Journal of Theoretical Biology. 2010;262:601–613. doi: 10.1016/j.jtbi.2009.10.031. [DOI] [PubMed] [Google Scholar]
Hexagonal-Lattice CA
- 33.Deutsch A. Lattice-gas cellular autimata modeling of developing cell systems. In: Anderson A, Chaplain M, Rejniak KA, editors. Single-Cell-Based Models in Biology and Medicine. Chapter I.2 Birkhauser-Verlag; Basel, Switzerland: 2007. [Google Scholar]
- 34.Engelberg JA, Ropella GEP, Hunt CA. Essential operating principles for tumor spheroid growth. BMC Systems Biology BMC Systems Biology. 2008;2:110. doi: 10.1186/1752-0509-2-110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shumate SD, El-Shenawee M. Computational model of ductal carcinoma in-situ: the effects of contact inhibition on pattern formation. IEEE Transactions on biomedical engineering. 2009;5:1341–1347. doi: 10.1109/TBME.2008.2005638. [DOI] [PubMed] [Google Scholar]
- 36.Aubert M, Badoual M, Christov C, Grammaticos B. A model for glioma cell migration on collagen and astrocytes. J R Soc Interface. 2008a;5:75–83. doi: 10.1098/rsif.2007.1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Aubert M, Badoual M, Grammaticos B. A Model for Short- and Long-range Interactions of Migrating Tumour Cell Acta Biotheor. 2008b;56:297–314. doi: 10.1007/s10441-008-9061-x. [DOI] [PubMed] [Google Scholar]
- 38.Kim SHJ, Debnath J, Mostov K, Park S, Hunt CA. A computational approach to resolve cell level contributions to early glandular epithelial cancer progression. BMC Systems Biology. 2009;3:122. doi: 10.1186/1752-0509-3-122. [DOI] [PMC free article] [PubMed] [Google Scholar]
Cellular Potts models
- 39.Turner S, Sherratt JA, Cameron D. Tamoxifen treatment failure in cancer and the nonlinear dynamics of TGF-beta. Journal of Theoretical Biology. 2004;229:101–111. doi: 10.1016/j.jtbi.2004.03.008. [DOI] [PubMed] [Google Scholar]
- 40.Jiang Y, Pjesivac-Grbovic J, Cantrell C, Freyer JP. A Multiscale Model for Avascular Tumor Growth. Biophysical Journal. 2005;89:3884–3894. doi: 10.1529/biophysj.105.060640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Maree AFM, Grieneisen VA, Hogweg P. The cellular Potts model and biophysical properties of cells, tissues and morphogenesis. In: Anderson ARA, Chaplain MAJ, Rejniak KAR, editors. Single Cell Based Models in Biology and Medicine. Birkhauser; 2007. [Google Scholar]
- 42.Rubenstein BM, Kaufman LJ. The Role of Extracellular Matrix in Glioma Invasion: A Cellular Potts Model Approach. Biophys J. 2008;95:5661–5680. doi: 10.1529/biophysj.108.140624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Poplawski NJ, Agero U, Gens JS, Swat M, Glazier JA, Anderson ARA. Front Instabilities and Invasiveness of Simulated Avascular Tumors. Bulletin of Mathematical Biology. 2009;71:1189–1227. doi: 10.1007/s11538-009-9399-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Merks RMH, Perryn ED, Shirinifard A, Glazier JA. Contact-inhibited chemotaxis in de novo and sprouting blood-vessel growth. PLoS Comput Biol. 2008;4(9):e1000163. doi: 10.1371/journal.pcbi.1000163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bauer AL, Jackson TL, Jiang Y. Topography of extracellular matrix mediates vascular morphogenesis and migration speed in angiogenesis. PLOS Comput boil. 2009;5(7):e1000445. doi: 10.1371/journal.pcbi.1000445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shirinifard A, Gens JS, Zaitlen BL, Popławski NJ, Swat M, Glazier JA. 3D Multi-Cell Simulation of Tumor Growth and Angiogenesis. PLoS ONE. 2009;4(10):e7190. doi: 10.1371/journal.pone.0007190. [DOI] [PMC free article] [PubMed] [Google Scholar]
Particle center based models (spherical)
- 47.Drasdo D, Hoehme S. A single-cell-based model of tumor growth in vitro: monolayers and spheroids Phys. Biol. 2005;2:133–147. doi: 10.1088/1478-3975/2/3/001. [DOI] [PubMed] [Google Scholar]
- 48.Galle J, Sittig D, Hanisch I, Wobus M, Wandel E, Loeffler M, Aust G. Individual Cell-Based Models of Tumor-Environment Interactions. Multiple Effects of CD97 on Tumor Invasion. The American Journal of Pathology. 2006;169:1802–1811. doi: 10.2353/ajpath.2006.060006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Drasdo D, Hoehme S, Block M. On the role of physics in the growth and pattern formation of multi-cellular systems: what can we learn from individual – cell based models? J Stat Physics. 2007;128:287–345. [Google Scholar]
- 50.Galle J, Hoffmann M, Aust G. From single cells to tissue architecture – a bottom-up approach to modelling the spatio-temporal organisation of complex multi-celluar systems. J Math Biol. 2009;58:261–283. doi: 10.1007/s00285-008-0172-4. [DOI] [PubMed] [Google Scholar]
- 51.Ramis-Conde I, Drasdo D, Anderson ARA, Chaplain MAJ. Modeling the influence of the cadherin-b-catenin pathway in cancer cell invasion: a multiscale approach. Biophys J. 2008;95:155–165. doi: 10.1529/biophysj.107.114678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ramis-Conde I, Chaplain MAJ, Anderson ARA, Drasdo D. Multi-scale modelling of cancer cell intravasation: the role of cadherins in metastasis. Phys Biol. 2009;6:016008. doi: 10.1088/1478-3975/6/1/016008. [DOI] [PubMed] [Google Scholar]
- 53.Galle J, Loeffler M, Drasdo D. Modeling the effects of deregulated proliferation and apoptosis on the growth dynamics of epithelial cell populations in vitro. Biophys J. 2005;88:62–75. doi: 10.1529/biophysj.104.041459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jeon J, Quaranta V, Cumming PT. An off-lattice hybrid discrete-continuum model of tumor growth and invasion. Biophysical Journal. 2010;98:37–47. doi: 10.1016/j.bpj.2009.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Norton K-A, Wininger M, Bhanot G, Ganesan S, Barnard N, Shinbrot T. A 2D mechanistic model of breast ductal cancinoma in situ (DCIS) morphology and progression. Journal of Theoretical Biology. 2010;263:393–406. doi: 10.1016/j.jtbi.2009.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
Particle center based models (ellipsoidal)
- 56.Kim Y, Stolarska MA, Othmer HG. A hybrid model for tumor spheroid growth in vitro, I: theoretical development and early results. Mathematical Models and Methods in Applied Sciences. 2007;17:1773–1798. [Google Scholar]
- 57.Stolarska MA, Kim Y, Othmer HG. Multi-scale models of cell and tissue dynamics. Phil Trans R Soc A. 2009;367:3525–3553. doi: 10.1098/rsta.2009.0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
Particle center based models with Voronoi tessellation
- 58.Schaller G, Meyer-Hermann M. Multicellular tumor spheroid in an off-lattice Voronoi-Delaunay cell model. Physical Review. 2005;71:051910. doi: 10.1103/PhysRevE.71.051910. [DOI] [PubMed] [Google Scholar]
- 59.Beyer T, Meyer-Hermann M. Multiscale modeling of cell mechanics and tissue organization. IEEE Eng Med Biol. 2009:38–45. doi: 10.1109/MEMB.2009.931790. [DOI] [PubMed] [Google Scholar]
- 60.Meineke FA, Potten CS, Loeffler M. Cell migration and organization in the intestinal crypt using a lattice-free model. Cell Prolif. 2001;34:253. doi: 10.1046/j.0960-7722.2001.00216.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.van Leeuwen IMM, Mirams GR, Walter A, Fletcher A, Murray P, Osborne J, Varma S, Young SJ, Cooper J, Doyle B, Pitt-Francis J, Momtahan L, Pathmanathan P, Whiteley JP, Chapman SJ, Gavaghan DJ, Jensen OE, King JR, Maini PK, Waters SL, Byrne HM. An integrative computational model for intestinal tissue renewal. Cell Prolif. 2009;42(5):617–636. doi: 10.1111/j.1365-2184.2009.00627.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Deformable cells
- 62.Rejniak KA. An immersed boundary framework for modelling the growth of individual cells: an application to the early tumour development. J Theor Biol. 2007;247:186–204. doi: 10.1016/j.jtbi.2007.02.019. [DOI] [PubMed] [Google Scholar]
- 63.Dillon RH, Owen M, Painter K. A single-cell-based model of multicellular growth using the immersed boundary method. AMS Contemporary Mathematics. 2008;466:1–15. [Google Scholar]
- 64.Rejniak KA, Dillon RH. A single cell based model of the ductal tumour microarchitecture. Computational and Mathematical Methods in Medicine. 2007;8:51–69. [Google Scholar]
- 65.Quaranta V, Rejniak KA, Gerlee P, Anderson ARA. Invasion emerges from cancer cell adaptation to competitive microenvironments: Quantitative predictions from multiscale mathematical models. Seminars in Cancer Biology. 2008;18:338–348. doi: 10.1016/j.semcancer.2008.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Rejniak KA, Anderson ARA. A computational study of the development of epithelial acini. I. Sufficient conditions for the formation of a hollow structure. Bull Math Biol. 2008a;70(3):677–712. doi: 10.1007/s11538-007-9274-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Rejniak KA, Anderson ARA. A computational study of the development of epithelial acini. II. Necessary conditions for structure and lumen stability. Bull Math Biol. 2008b;70(5):1450–1479. doi: 10.1007/s11538-008-9308-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Rejniak KA. A single-cell approach in modeling the dynamics of tumor microregions. Mathematical Biosciences and Engineering. 2005;2(3):643–655. doi: 10.3934/mbe.2005.2.643. [DOI] [PubMed] [Google Scholar]
- 69.Rejniak KA. Modelling the development of complex tissues using individual viscoelastic cells. In: Anderson A, Chaplain M, Rejniak KA, editors. Single-Cell-Based Models in Biology and Medicine. Birkhauser-Verlag; 2007b. [Google Scholar]
Other
- 70.Macklin P, McDougall S, Anderson ARA, Chaplain MAJ, Cristini V, Lowengrub J. Multiscale modelling and nonlinear simulation of vascular tumour growth. J Math Biol. 2009;58:765–798. doi: 10.1007/s00285-008-0216-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Szeto MD, Chakraborty G, Hadley J, Rockne R, Muzi M, Alvord EC, Krohn KA, Spence AM, Swanson KR. Quantitative Metrics of Net Proliferation and Invasion Link Biological Aggressiveness Assessed by MRI with Hypoxia Assessed by FMISO-PET in Newly Diagnosed Glioblastomas. Cancer Res 2009. 2009;69:4502–4509. doi: 10.1158/0008-5472.CAN-08-3884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Stefanini MO, Wu FTH, Mac Gabhann F, Popel AS. A compartment model of VEGF distribution in blood, healthy and diseased tissues. BMC Systems Biology. 2008;2(1):77. doi: 10.1186/1752-0509-2-77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wu FTH, Stefanini MO, Gabhann FM, Popel AS. A Compartment Model of VEGF Distribution in Humans in the Presence of Soluble VEGF Receptor-1 Acting as a Ligand Trap. PLOS ONE. 2009;4(4):e5108. doi: 10.1371/journal.pone.0005108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Fang JS, Gillies RD, Gatenby RA. Adaptation to hypoxia and acidosis in carcinogenesis and tumor progression. Seminars in Cancer Biology. 2008;18:330–337. doi: 10.1016/j.semcancer.2008.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Vincent TL, Gatenby RA. An evolutionary model for initiation, promotion, and progression in carcinogenesis. International Journal of Oncology. 2008;32:729–737. [PubMed] [Google Scholar]
- 76.Gatenby RA, Frieden BR. Application of Information Theory and Extreme Physical Information to Carcinogenesis. Cancer Research. 2002;62:3675–3684. [PubMed] [Google Scholar]
- 77.Gatenby RA. Application of Competition Theory to Tumour Growth: Implications for Tumour Biology and Treatment. European Journal of Cancer. 1996;32A:722–726. doi: 10.1016/0959-8049(95)00658-3. [DOI] [PubMed] [Google Scholar]
- 78.Wolkenhauer O, Auffray Ch, Baltrusch S, Blüthgen N, Byrne H, Cascante M, Ciliberto A, Dale T, Drasdo D, Fell D, Ferrell JE, Gallahan D, Gatenby R, Günther U, Harms BD, Herzel H, Junghanss Ch, Kunz M, van Leeuwen I, Lenormand P, Levi F, Linnebacher M, Lowengrub J, Maini PK, Malik A, Rateitschak K, Sansom O, Schäfer R, Schürrle K, Sers Ch, Schnell S, Shibata D, Tyson J, Vera J, White M, Zhivotovsky B, Jaster R. Systems biologists seek fuller integration of systems biology approaches in new cancer research programs. Cencer Research. 2010;70:12–13. doi: 10.1158/0008-5472.CAN-09-2676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wolkenhauer O, Fell D, De Meyts P, Bluthgen N, Herzel H, Le Novere N, Hofer T, Schurrle K, van Leeuwen I. SysBioMed report: Advancing systems biology for medical applications. IET Systems Biology. 2009;3(3):131–136. doi: 10.1049/iet-syb.2009.0005. [DOI] [PubMed] [Google Scholar]
- 80.Hinow P, Gerlee P, McCawley LJ, Quaranta V, Ciobanu M, Wang SZ, Graham JM, Ayati BP, Claridge J, Swanson KR, Loveless M, Anderson ARA. A spatial model of tumor-host interaction: application of chemotherapy. Mathematical Bioscience and Engineering. 2009;6(3):521–546. doi: 10.3934/mbe.2009.6.521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Komarova NL, Sadovsky AV, Wan FYM. Selective pressures for and against genetic instability in cancer: a time-dependent problem. J R Soc Interface. 2008;5:105–121. doi: 10.1098/rsif.2007.1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
Further Reading
Reviews: cancer growth
- 1.Preziosi L, editor. Cancer modeling and simulation. Champan & Hall; 2003. [Google Scholar]
- 2.Araujo RP, McElwain DLS. A History of the study of solid tumour growth: the contribution of mathematical modeling. Bulletin of Mathematical Biology. 2004;66:1039–1091. doi: 10.1016/j.bulm.2003.11.002. [DOI] [PubMed] [Google Scholar]
- 3.Byrne HM, Alarcon T, Owen MR, Webb SD, Maini PK. Modelling aspects of cancer dynamics: a review. Phil Trans R Soc A. 2006;364:1563–1578. doi: 10.1098/rsta.2006.1786. [DOI] [PubMed] [Google Scholar]
- 4.Anderson ARA, Chaplain MAJ, Rejniak KA, editors. Single-cell-based models in biology and medicine. Birkhauser-Verlag; 2007. [Google Scholar]
- 5.Sanga S, Frieboes HB, Zheng X, Gatenby R, Bearer EL, Cristini V. Predictive oncology: a review of multidisciplinary, multiscale in silico modeling linking phenotype, morphology and growth. Neuroimage. 2007;27:S120–S134. doi: 10.1016/j.neuroimage.2007.05.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bellomo N, Chaplain M, de Angelis E, editors. Selected topics in cancer modeling: genesis, evolution, immune competition and therapy. Birkhauser; 2008. [Google Scholar]
- 7.Wang Z, Deisboeck TS. Computational modeling of brain tumors: discrete, continuum or hybrid? Sci Model Simul. 2008;15:381–393. [Google Scholar]
- 8.Byrne H, Drasdo D. Individual-based and continuum models of growing cell populations: a comparison. J Math Biol. 2009;58:657–687. doi: 10.1007/s00285-008-0212-0. [DOI] [PubMed] [Google Scholar]
- 9.Tracqui P. Biophysical models of tumour growth. Rep Prog Phys. 2009;72:056701. [Google Scholar]
- 10.Lowengrub JS, Frieboes HB, Jin F, Chuang Y-L, Li X, Macklin P, Wise SM, Cristini V. Nonlinear modelling of cancer: bridging the gap between cells and tumours. Nonlinearity. 2010;23:R1–R91. doi: 10.1088/0951-7715/23/1/r01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Martins ML, Ferreira SC, Vilela MJ. Multiscale models fro the growth of avascular tumors. Physics of Life Reviews. 2007;4:128–156. [Google Scholar]
- 12.Edelman LB, Eddy JA, Price ND. In silico models of cancer. WIREs Syst Biol Med. 2009 doi: 10.1002/wsbm.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Byrne HM. Dissecting cancer through mathematics: from the cell to the animal model. Nature Reviews Cancer. 2010;10:221–230. doi: 10.1038/nrc2808. [DOI] [PubMed] [Google Scholar]
- 14.Rejniak KA, McCawley LJ. Current trends in mathematical modeling of tumor-microenvironment interactions: a survey of tools and applications. Experimental Biology and Medicine. 2010;235:411–423. doi: 10.1258/ebm.2009.009230. [DOI] [PubMed] [Google Scholar]
Reviews: integration of biological and mathematical modeling
- 1.Hunt CA, Ropella GEP, Lam TN, Tang J, Kim SHJ, Engelberg JA, Sheikh-Bahaei S. At the biological modeling and simulation frontier. Pharmaceutical Research. 2009;26(11):2369–2400. doi: 10.1007/s11095-009-9958-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Strand DW, Franco OE, Basanta D, Anderson ARA, Hayward SW. Perspectives on tissue interactions in development and disease. Curr Mol Med. 2010;10:95–112. doi: 10.2174/156652410791065363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Meier-Schellersheim M, Fraser IDC, Klauschen F. Multiscale modeling for biologists. WIREs Syst Biol Med. 2009;1:4–14. doi: 10.1002/wsbm.33. [DOI] [PMC free article] [PubMed] [Google Scholar]