

Abstract
Background
Titin is the largest mammalian (∼3000-4000 kDa) and myofilament protein which acts as a molecular spring in the cardiac sarcomere and determines systolic and diastolic function. Loss of titin in ischemic hearts has been reported, but the mechanism of titin degradation is not well understood. Matrix metalloproteinase-2 (MMP-2) is localized to the cardiac sarcomere and upon activation in ischemia/reperfusion injury proteolyzes specific myofilament proteins. Here we determine whether titin is an intracellular substrate for MMP-2 and if its degradation during ischemia/reperfusion contributes to cardiac contractile dysfunction.
Methods and Results
Immunohistochemistry and confocal microscopy in rat and human hearts showed discrete co-localization between MMP-2 and titin in the Z-disc region of titin and that MMP-2 is mainly localized to titin near the Z-disc of the cardiac sarcomere. Both purified titin or titin in skinned cardiomyocytes were proteolyzed when incubated with MMP-2 in a concentration-dependent manner and this was prevented by MMP inhibitors. Isolated rat hearts subjected to ischemia/reperfusion injury showed cleavage of titin in ventricular extracts by gel electrophoresis which was confirmed by reduced titin immunostaining in tissue sections. Inhibition of MMP activity with ONO-4817 prevented ischemia/reperfusion-induced titin degradation and improved the recovery of myocardial contractile function. Titin degradation was also reduced in hearts from MMP-2 knockout mice subjected to ischemia/reperfusion in vivo, compared to wild type controls.
Conclusions
MMP-2 localizes to titin at the Z-disc region of the cardiac sarcomere and contributes to titin degradation in myocardial ischemia/reperfusion injury.
Keywords: titin, MMP-2, sarcomere, contractile dysfunction
Introduction
Matrix metalloproteinase-2 (MMP-2) is a zinc-dependent protease that is best known for its ability to degrade the extracellular matrix in both physiological and pathological conditions. MMP-2 is synthesized as a zymogen by a variety of cells including cardiac myocytes and is activated either by proteases1 (such as by action of MMP-14) or by post-translational modifications to the full length enzyme stimulated by enhanced oxidative stress. For example, peroxynitrite, which is generated in early reperfusion following ischemia2, directly activates several MMPs3 including MMP-24 via a non-proteolytic mechanism involving the S-glutathiolation of a critical propeptide cysteine in its autoinhibitory domain.
MMPs are best recognized for their role in tissue remodeling by proteolyzing various components of the extracellular matrix in both health and disease, i.e. in angiogenesis, embryogenesis, wound healing5, atherosclerosis6, aortic aneurysm7 and myocardial infarction8. More recent studies, however, show that MMP-2 is involved in several acute biological processes independent of its actions on extracellular matrix proteins. This includes platelet activation9, regulation of vascular tone10 and myocardial stunning injury immediately after reperfusion of the ischemic heart11. Indeed, several reports indicate that MMP-2 does not exclusively degrade extracellular matrix components12,13.
In normal cardiac myocytes MMP-2 is found in discrete subcellular compartments including the thin and thick myofilaments of the cardiac sarcomere14,15, the cytoskeleton16,17, nuclei18, mitochondria14 and caveolae19 (for review see20). MMP-2 is activated in rat hearts subjected to myocardial oxidative stress injury and is responsible for the degradation of specific sarcomeric and cytoskeletal proteins including troponin I14,21, myosin light chain-115 and α-actinin17. Inhibition of MMPs activity reduced both the loss of contractile function as well as the degradation of these substrates, to which MMP-2 was co-localized. Furthermore, transgenic mice with myocardial specific expression of a mutant, constitutively active MMP-2, in the absence of additional injury, show significantly impaired cardiac contractile function, disrupted sarcomeres, profound myofilament lysis, breakdown of Z-band registration and reduced troponin I level22.
Titin, the largest known mammalian protein (3,000-4,000 kDa), forms an intrasarcomeric elastic filament that is thought to serve as a framework for the ordered assembly of other myofilament proteins23. In the sarcomere the titin molecule spans the distance from the Z-disc to the M-line region (half the length of the sarcomere). Moreover, the I-band region of titin is comprised of distinct elastic segments that allow titin to behave as a molecular spring, contributing to the passive tension of myofibrils and maintaining the structural and functional stability of the sarcomere. Titin is an important determinant of both systolic and diastolic function and the Frank-Starling mechanism of the heart24. Although loss and/or disorganization of titin in ischemic and failing human hearts has been reported25,26, the mechanism of titin degradation has not been extensively studied in hearts subjected to ischemia/reperfusion (I/R) injury. Since MMP-2 is localized to sarcomeric and cytoskeletal proteins and is activated in myocardial I/R injury, we address here whether MMP-2 targets titin to contribute to the pathogenesis of myocardial I/R injury.
Methods
Titin isolation and purification
Titin was prepared as described previously27,28. See Online Data Supplement.
Skinned cardiomyocyte isolation
Skinned cardiomyocytes were isolated as described in the Online Data Supplement.
Cleavage of native titin in skinned cardiomyocytes
Skinned cardiomyocytes were incubated with human recombinant MMP-2 catalytic domain (Enzo Life Sciences, 4-120 nmol/L) with or without MMP inhibitors (10 μmol/L o-phenanthroline or ONO-4817) at 37°C for 60 min. This concentration of o-phenanthroline inhibits MMP-2 activity under similar in vitro conditions29. The samples were denatured with 2× urea sample buffer (8 mol/L urea, 2 mol/L thiourea, 3% SDS, 75 mmol/L DTT, 0.03% bromophenol blue, and 0.05 mol/L Tris-HCl, pH 6.8) at 60°C for 10 min, and the proteins were electrophoresed by 1% SDS-agarose and stained with Coomassie brilliant blue.
Isolated working rat heart- ex vivo model of ischemia/reperfusion
Male Sprague–Dawley rats (300–350 g) were anesthetized with sodium pentobarbital (60 mg/kg, ip). Hearts were isolated and paced at 300 beats per minute during perfusion at 37°C as working hearts30 with 100 ml recirculating Krebs–Henseleit solution containing 118 mmol/L NaCl, 4.7 mmol/L KCl, 1.2 mmol/L KH2PO4, 1.2 mmol/L MgSO4, 25 mmol/L NaHCO3, 11 mmol/L glucose, 100 μU/ml insulin, 2.5 mmol/L Ca2+, 0.5 mmol/L EDTA and 0.1% bovine serum albumin; continuously gassed with 95% O2/5% CO2 (pH 7.4). The perfusate enters the left atrium at a hydrostatic preload pressure of 9.5 mmHg and the left ventricle ejects it against a hydrostatic afterload of 70 mmHg. Cardiac work (cardiac output × peak systolic pressure) was used as an index of mechanical function. After 25 min of equilibration, hearts were either aerobically perfused for 85 min (Control, n=6) or subjected to 25 min global, no-flow ischemia followed by 60 min aerobic reperfusion without (I/R, n=7) or with 50 μmol/L ONO-4817 (I/R + ONO-4817, n=8). ONO-4817, a selective MMP inhibitor (Ki in the nanomolar range for MMP-2 and almost no inhibitory activity up to 100 μmol/L against several other proteases of different classes31), was added to the perfusion buffer 10 min before the induction of ischemia. All hearts were perfused for a total of 110 min. At the end of perfusion the ventricles were rapidly frozen in liquid nitrogen and processed for titin analysis in ventricular extracts as described below.
Additional series of hearts (Control n=5, I/R n=5, and I/R + ONO-4817 n=4) were perfused and processed for immunohistochemistry and confocal microscopy analysis for assessment of titin immunostaining.
Another six hearts were briefly perfused for 10 min at 37°C with Krebs–Henseleit buffer at a constant hydrostatic pressure of 70 mmHg to clear them of blood, followed by processing for immunohistochemistry as described below to investigate the co-localization of titin and MMP-2 in the left ventricle.
This investigation conforms to the Guide to the Care and Use of Experimental Animals published by the Canadian Council on Animal Care.
In vivo model of ischemia/reperfusion
Ischemia/reperfusion was induced in vivo by modifying a previously described protocol32. Briefly, MMP-2 knockout and age-matched wild type male C57BL/6 mice were anaesthetized with isoflurane, intubated and kept on a heating pad to maintain body temperature at 37°C. The heart was exposed and the left anterior descending coronary artery (LAD) was temporarily ligated with a 7-0 silk suture, with a piece of 4-0 silk placed between the LAD and the 7-0 silk. After 30 min of LAD occlusion, reperfusion was initiated by releasing the ligature and removing the 4-0 silk. The loosened suture was left in place to help identify the ischemic area of the left ventricle. After 30 min of reperfusion, the hearts were excised, the ischemic and non-ischemic regions of the left ventricle were dissected out under a magnifying glass and flash frozen in liquid nitrogen for titin analysis.
Analysis of titin by gel electrophoresis
Titin was analyzed in ventricular extracts using 1% vertical SDS-agarose gel electrophoresis as previously described33. See Online Data Supplement for details.
Immunohistochemistry and confocal microscopy
a- Co-localization of titin and MMP-2
Rat hearts perfused aerobically for 10 min to flush them of blood, or left ventricular tissue from the explanted heart of a heart transplant patient were fixed with 4% paraformaldehyde in 0.1 mol/L sodium phosphate buffer (pH 7.4) and cryoprotected with 30% sucrose in 0.1 mol/L sodium phosphate buffer. Details of the double immunolabeling are provided in the Online Data Supplement.
b- Titin 9D10 immunostaining
At the end of the 110 min working heart perfusion protocol some Control, I/R or I/R + ONO-4817 hearts were fixed and cryoprotected (as described above) for 9D10 immunostaining as detailed in the Online Data Supplement.
Overlay assay to determine MMP-2 binding to titin
Skinned muscle fibers were incubated with trypsin to increase titin degradation (intact T1 to T2 fragment). The proteins in the samples were separated by gel electrophoresis and transferred to a PVDF membrane. These membranes were used in an overlay assay whereby the binding of human recombinant MMP-2 to titin T1 and T2 on the membrane was assessed. For details see the Online Data Supplement.
Statistical analysis
Results are expressed as mean ± S.E.M. for n hearts. As appropriate, one-way ANOVA or repeated measures two-way ANOVA followed by Tukey's post hoc test were used. Differences were considered significant at p < 0.05.
Results
Co-localization of MMP-2 and titin near the Z disc region of the cardiac sarcomere
We first investigated whether MMP-2 is localized to titin in the cardiac sarcomere. In this regard, we used two different anti-titin antibodies which target specific epitopes (supplement Figure S1A). The T12 antibody labels titin near the Z disc region of titin and the M8 antibody recognizes an epitope at the M line region of titin. Images obtained by immunohistochemistry followed by confocal microscopy showed that T12-immunoreactivity distributes near Z-lines and M8-immunoreactivity is alternatively distributed at M-lines without overlapping (supplement Figure S1B). Images obtained using anti-MMP-2 and anti-titin T12 in left ventricle sections from 10 min aerobically perfused rat hearts showed clear co-localization of MMP-2 to the Z disc region of titin (Figure 1). When using anti-titin M8, we observed a weaker localization of MMP-2 to this region of titin (Figure 1). These data suggest that MMP-2 localizes mainly near the Z-disc region of the sarcomere, with a secondary and weaker localization near the M-line portion in the titin molecule.
Figure 1.

Co-localization of MMP-2 and titin at the Z disc region of the left ventricular cardiac sarcomere in 10 min aerobically perfused rat hearts (longitudinal sections). MMP-2 shows better co-localization with T12 than M8 epitope of titin in the sarcomere of left ventricular myocardium. MMP-2-immunoreactivity reveals at Z-lines with high density as well as M-lines with low density. T12 epitope reveals at only Z-lines and M8 epitope reveals at only M-lines. a-c shows that high density of MMP-2 (green) co-localizes (yellow) with T12 epitope (red) at the Z-lines. d-f shows that low density of MMP-2 (red) co-localizes (yellow) with M8 epitope (green) at M-lines. Scale bar is 5 μm for all images except for the enlarged portion of c illustrating the Z- and M-lines.
MMP-2 binds and cleaves titin in a concentration-dependent manner
In silico mapping of MMP-2 cleavage sites in both human and mouse N2B titin revealed multiple putative sites in both I-band and A-band titin regions, including near the Z-line terminus of titin. These sites show more than 60% homology to the three MMP-2 cleavage motifs (supplemental Figure S2). Moreover, human recombinant MMP-2 was able to bind to titin prepared from skinned muscle fibers as shown by the overlay assay method (supplemental Figure S3). Next we tested the susceptibility of purified titin to proteolytic degradation by MMP-2 in vitro. Incubation of titin with MMP-2 (60 min, 37°C) at increasing MMP-2:titin molar ratios (1:500, 1:50 and 1:5) caused titin degradation in a concentration-dependent manner (supplement Figure S4A). Inhibition of MMP-2 activity with GM-6001 or ONO-4817 prevented titin cleavage by MMP-2 (supplement Figure S4B). To determine whether MMP-2 directly cleaves cardiac titin in situ, we incubated skinned mouse cardiomyocytes with increasing concentration of MMP-2 (60 min, 37°C). This resulted in concentration-dependent cleavage of cardiac titin (T1) as shown by the increased level of the degradation product of titin (T2) with increasing MMP-2 concentration (Figure 2A). Inhibition of MMP-2 activity with o-phenanthroline or ONO-4817 prevented titin cleavage by MMP-2 (Figure 2B).
Figure 2.

In vitro incubation (60 min, 37°C) of skinned cardiomyocytes with MMP-2. A, MMP-2 cleaved cardiac titin (T1) in a concentration-dependent manner (4-120 nmol/L) as shown by the appearance of the titin degradation product (T2). B, The cleavage of titin by MMP-2 was prevented by inhibiting the activity of MMP-2 with MMPs inhibitors o-phenanthroline (o-ph) or ONO-4817. T2/T1 band density ratio is indicated below each lane. MHC (myosin heavy chain) as loading control.
Effect of ONO-4817 on functional recovery of ischemic-reperfused hearts
Isolated working rat hearts were perfused for 110 min under one of three conditions: (1) aerobic perfusion (Control); (2) 25 min of global, no-flow ischemia and 60 min of aerobic reperfusion (I/R); or (3) I/R in the presence of a selective MMP inhibitor, ONO-4817 (I/R + ONO-4817, Figure 3A). Control hearts showed no significant loss of mechanical function over 110 min of aerobic perfusion (Figure 3). I/R hearts showed markedly reduced recovery of mechanical function during reperfusion in comparison with control hearts (Figure 3B). The recovery of cardiac work during reperfusion was significantly improved following MMP inhibition with ONO-4817 compared to the I/R group (Figure 3B).
Figure 3.

Mechanical recovery of isolated perfused working rat hearts subjected to 25 min global, no-flow ischemia followed by 60 min reperfusion without (I/R) or with 50 μmol/L ONO-4817 (I/R + ONO-4817) in comparison with aerobically perfused control hearts. A, Schematic representation of the perfusion protocols for Control (n=6), I/R (n=7) and I/R + ONO-4817 (n=8) groups. B, Time course of changes in cardiac work of isolated working rat hearts. **p<0.001, *p<0.05 versus corresponding values of I/R group, repeated measures two-way ANOVA.
Myocardial ischemia/reperfusion causes titin cleavage, an effect diminished by an MMP inhibitor
To investigate whether MMP-2 can cleave titin in the intact heart under pathophysiological conditions, titin content was assessed using 1% vertical SDS-agarose gels in ventricular extracts prepared from the Control, I/R or I/R + ONO-4817 hearts. Ventricular extracts from control hearts revealed a titin band at ∼3000 kDa (Figure 4A). I/R caused a significant increase in T2 band density, an effect abolished in the I/R + ONO-4817 hearts (Figure 4A). Quantification of total titin/MHC ratio showed that I/R did not significantly change this ratio as compared to control hearts (Figure 4B) whereas T2/MHC ratio significantly increased in I/R hearts. ONO-4817 abolished the I/R-induced increase in the T2/MHC ratio (Figure 4C). These observations were further confirmed by immunohistochemistry experiments using the anti-titin 9D10 antibody, raised against the PEVK domain in the spring region of titin. Titin immunostaining was significantly reduced by I/R whereas ONO-4817 treatment preserved titin immunostaining to a level comparable to control (Figure 4D).
Figure 4.

Titin degradation in ischemic-reperfused rat hearts. A, Representative SDS-agarose gel for analysis of titin in ventricular extracts. Titin (T1) and titin degradation product (T2) in ventricular homogenates from control, I/R and I/R + ONO-4817 hearts analyzed using a 1% vertical SDS-agarose gel. MHC (myosin heavy chain). BLV (bovine left ventricle) was used as a standard and shows N2BA and N2B isoforms of titin; note that the majority of rat heart titin is the N2B isoform. Each lane is an extract from individually perfused hearts. B, Ratio of total titin (T1 + T2) to MHC content (n=6 in each group). C, Ratio of T2 titin to MHC content (n=6 in each group). *p<0.05 versus control (One-way ANOVA, Tukey's post hoc test). D, Representative left ventricular cryosections immunostained against titin epitope 9D10. Titin immunostaining using the 9D10 antibody (raised against the proline-glutamate-valine-lysine, PEVK, domain) was decreased in I/R hearts compared to control, whereas staining intensity was comparable between I/R + ONO-4817 and control hearts. Scale bar is 10 μm for all images. Images are representative of at least four individual hearts investigated under each condition.
Titin degradation is reduced in hearts from MMP-2 knockout mice subjected to I/R injury in vivo
We next determined whether genetic ablation of MMP-2 could influence titin degradation in cardiac muscle. Mouse hearts subjected in vivo to LAD ligation for 30 min followed by 30 min reperfusion exhibited titin degradation which was significantly less in MMP-2 knockout hearts than in wild type control hearts (Figure 5).
Figure 5.

Titin degradation in MMP-2 knockout (KO) and wild type (WT) mouse hearts subjected to ischemia/reperfusion (I/R) in vivo. A, Representative 1% vertical SDS-agarose gel shows titin (T1) isoforms (N2BA and N2B) and titin degradation product T2 in left ventricle from Sham or in ischemic regions from I/R groups in either wild type or MMP-2 knockout mice. B, Quantification of T2 titin to total titin ratios (n=6 in each group). *p<0.01 versus Sham control (One-way ANOVA, Tukey's post hoc test).
MMP-2 localizes near the Z-disc region of titin in the human heart
Immunostaining of sections prepared from the left ventricle of an explanted heart from a patient undergoing heart transplantation showed co-localization of MMP-2 and titin mainly near the Z-disc, with a weaker co-localization at the M-line. In comparison to the rat heart, MMP-2 immunostaining in the human heart was more diffuse yet still showed a sarcomeric staining pattern (Figure 6).
Figure 6.
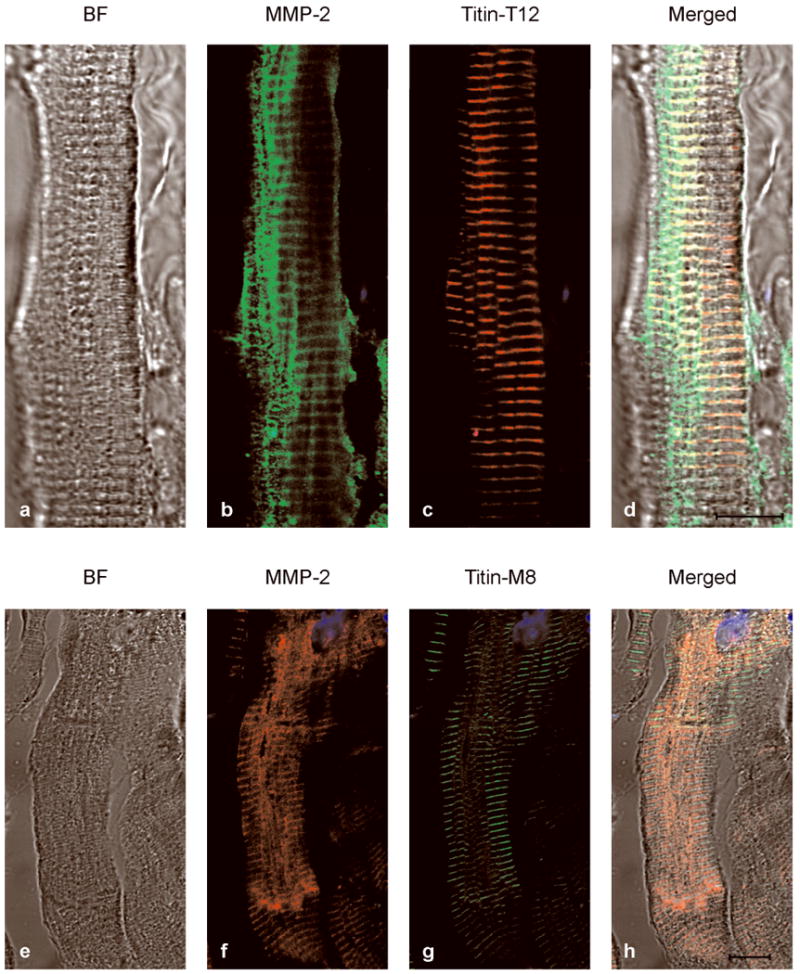
Co-localization of MMP-2 and titin near the Z-disc in diseased human heart. Left ventricle sections were used from the explanted failing heart from a patient receiving a heart transplant. a-d shows that high density of MMP-2 (green) co-localizes (yellow) with T12 epitope (red) at the Z-lines. e-h shows that low density of MMP-2 (red) co-localizes (yellow) with M8 epitope (green) at M-lines. BF (bright field images). Scale bar is 10 μm.
Discussion
In this study we demonstrated that the giant sarcomeric protein, titin, is a target of the proteolytic activity of MMP-2 in the setting of acute myocardial I/R injury. Immunohistochemical analysis shows that MMP-2 clearly co-localizes with titin near the Z-disc region of the sarcomere in both rat and human hearts. We established that under in vitro conditions MMP-2 is able to bind to and cleave titin in a concentration-dependent manner. The proteolytic action of MMP-2 is blocked by the selective MMP inhibitors, GM-6001 and ONO-4817, verifying that the cleavage is indeed due to MMP activity. ONO-4817 not only improves the functional recovery after I/R in isolated rat hearts but also prevents the significant increase in the titin degradation product T2 caused by I/R injury, indicating that titin degradation is reduced when MMP activity is inhibited. Furthermore, hearts from MMP-2 knockout mice subjected to in vivo I/R injury show less titin degradation in comparison to wild type controls. Titin proteolysis has been observed in various human heart diseases associated with increased myocardial oxidative stress including dilated cardiomyopathy, the terminally failing heart and Chagas' cardiomyopathy25,26,34, however the protease(s) responsible for this was not identified.
MMPs are best known as proteases responsible for the degradation and remodeling of extracellular matrix proteins in both physiological and pathological conditions, including various cardiac pathologies. However, the discovery of the intracellular localization14,16,18,22 and functions of MMP-2 to proteolyze troponin I14,22 myosin light chain-115 and α-actinin17 during myocardial oxidative stress injury challenged the canonical notion of extracellular-only actions of this enzyme. In previous studies we showed that peroxynitrite biosynthesis in ischemic-reperfused rat hearts peaks within the first minute of reperfusion2 and the peak in MMP-2 activity follows at 2 to 5 minutes of reperfusion11. Infusion of peroxynitrite into isolated perfused rat hearts35 or isolated cardiomyocytes36 caused a time- and concentration-dependent contractile dysfunction which was abrogated with MMP inhibitors. In vitro, peroxynitrite was shown to directly activate MMP-2 via a non-proteolytic mechanism involving S-glutathiolation of the propeptide cysteine sulphydryl group4. Indeed, this intracellular activity of MMP-2 upon I/R injury caused proteolytic degradation of specific sarcomeric (troponin I14 and myosin light chain-115) and cytoskeletal (α-actinin17) proteins which are susceptible to its proteolytic activity.
MMP-2 is localized within the cardiac sarcomere including near to the Z-discs14-16. These previous observations are supplemented by the current data showing clear co-localization of MMP-2 near the Z disc region of titin using the T12 clone in rat (Figure 1) and human (Figure 6) hearts. Several studies show that titin interacts with α-actinin at the Z-disc of the sarcomere and this interaction plays a crucial role in Z-disc assembly and sarcomeric integrity37-39. Interestingly, MMP-2 was found to not only co-localize with α-actinin in the Z-disc of cardiac sarcomeres16,17 but also degrades it following peroxynitrite infusion into isolated rat hearts17. The M8 titin antibody (raised against the M-line region of titin) shows a weaker localization of MMP-2 to this region of titin. Although our data do not rule out the possible localization of MMP-2 also to other region(s) of titin, they do suggest that a main MMP-2 anchoring site is at/near the Z-disc of the sarcomere.
Titin is the third myofilament (in addition to thick and thin myofilaments) of the sarcomere that plays an important role in sarcomere integrity and cardiac muscle contraction23. Any alterations in its structure could severely affect the contractile performance of the heart. The increase in T2 titin and the decrease in titin immunostaining after I/R injury observed here (Figure 4) was associated with poor cardiac mechanical recovery during reperfusion (Figure 3). These effects are likely at least in part due to titin degradation by MMP-2, given the co-localization of MMP-2 with titin near the Z-disc of cardiac sarcomeres, the susceptibility of titin to degradation by MMP-2 and the reduction in I/R-induced titin degradation in hearts from MMP-2 knockout mice or in rat hearts where MMP activity was selectively blocked with ONO-4817. A significant increase in MMP-2 activity was seen in the heart after experimental Trypanosoma cruzi infection (the parasite responsible for Chagas' disease) and mortality was markedly reduced upon treatment with an MMP inhibitor, suggesting a role of MMP-2 in mediating acute Chagas' cardiomyopathy40. Putative titin degradation products were detected in the plasma of patients with Chagas' disease34, further supporting a role of MMP-2 in titin degradation. Moreover, myocardial infarction is associated with a significant right shift in the left ventricle pressure-volume relation (an observation which is consistent with titin degradation in the heart) and the broad spectrum MMP inhibitor PD-166793 was shown to protect against this shift41. Although cardiac mechanical function at the end of perfusion is inversely related with T2/MHC ratios in hearts (Figures 3B and 4C), caution is needed in relating this effect exclusively to titin degradation. As mentioned above, other sarcomeric/cytoskeletal proteins including troponin I, myosin light chain-1 and α-actinin are also susceptible to degradation by MMP-2 under conditions of myocardial oxidative stress injury. However, our work clearly suggests that titin proteolysis is an important factor that negatively impacts myocardial contractility upon I/R injury.
Titin content in rat ventricles was investigated here by SDS-agarose gel electrophoresis or immunofluorescence staining against titin epitopes at the PEVK domain. Our electrophoresis results showed the approximate 60% elevation in the T2/MHC ratio in the I/R group in comparison with aerobic control hearts. Immunofluorescence data also showed a reduction of titin immunostaining in I/R group using the 9D10 antibody. In addition to degradation, post-translational modifications of titin may have occurred upon I/R that led to diminished binding of titin antibodies to the specific epitopes. Post-translational modifications of many cardiac myofilament/cytoskeletal proteins during I/R, including actin42 and myosin light chain-143, have been reported in previous studies.
Our study does not rule out the possible action of other proteases in titin degradation. Calpains are most likely involved in sarcomeric protein degradation after ischemic episodes more severe than that observed in our model44. Indeed, calpain was shown to be able to cleave titin only after 24 h doxorubicin treatment of rat cardiomyocytes45. The ubiquitin-proteasome system is another proteolytic pathway that may be involved in titin degradation. Increased proteasome activities have been reported in various models of I/R injury46-48. Moreover, the E3 ubiquitin-ligase MURF1 is known to be associated with the M-line region of titin49 and ubiquitinates titin in yeast two-hybrid screens50. In a rat heart failure model, both a loss of titin51 and an increase in MMP-2 gene expression52 were observed in diaphragm muscle. However, in our short-term experiments we did not observe a significant loss in intact titin upon I/R injury. We speculate that MMP-2 activation results not only in titin cleavage but it may also trigger a cascade of proteolytic events leading to titin loss several hours after reperfusion.
In conclusion, the present results indicate that MMP-2 cleaves titin during either ex vivo or in vivo I/R injury. Furthermore, MMP-2 inactivation by pharmacologic or genetic approaches protects against titin degradation. Our previous findings of troponin I14, myosin light chain-115 and α-actinin17 cleavage by MMP-2, in addition to our current results with titin, suggest that MMP-2 plays a crucial role in the pathogenesis of acute I/R injury at the level of the sarcomere and cytoskeleton. Whether MMP-2 causes contractile protein alterations in other cardiac pathologies needs further investigation. Pharmacological inhibition of MMP activity could represent a useful strategy for the prevention and/or treatment of myocardial ischemia/reperfusion injury.
Clinical perspective.
In addition to the well known extracellular effects of matrix metalloproteinases (MMP), we provide evidence that MMP-2 is also localized inside the cardiac myocyte, near the Z-disk region of the sarcomere. We also show that upon acute ischemia and reperfusion injury, MMP-2 is activated and proteolyses titin, the largest known protein that plays a crucial role in both diastolic and systolic function of the heart. Titin contains several cleavage motifs for MMP-2, and its proteolysis is reduced in hearts protected by pharmacological inhibition of MMP activity and in MMP-2 deficient hearts. This study provides new insights into the pathophysiological mechanism of ischemia and reperfusion injury and suggests that MMP inhibitors might be a useful strategy for reducing reperfusion injury.
Supplementary Material
Acknowledgments
We acknowledge Tiffany Pecor, William Rogers, Sike Pan and Chanrasekhar Saripalli for technical assistance. We thank Dr. Xiuhua Wang for technical assistance with mouse heart experiments and Dr. Costas Schulze for help in procuring human heart tissue, and Dr. Eliana Lucchinetti for help with the in silico analysis. We thank Dawne Colwell for help with the illustrations. We thank Dr. Elisabeth Ehler (King's College, London UK) for anti-titin antibodies (T12 and M8 clones).
Funding sources: This work was supported by the Canadian Institutes of Health Research (MOP-77526 to R.S., MOP-84279 to Z.K.) and the NIH (HL062881 to H.G.). M.A.A. is supported by Alberta Heritage Foundation for Medical Research (AHFMR) studentship award. R.S. was an AHFMR Scientist.
Footnotes
Disclosures: None.
References
- 1.Birkedal-Hansen H. Proteolytic remodeling of extracellular matrix. Curr Opin Cell Biol. 1995;7:728–735. doi: 10.1016/0955-0674(95)80116-2. [DOI] [PubMed] [Google Scholar]
- 2.Yasmin W, Strynadka KD, Schulz R. Generation of peroxynitrite contributes to ischemia-reperfusion injury in isolated rat hearts. Cardiovasc Res. 1997;33:422–432. doi: 10.1016/s0008-6363(96)00254-4. [DOI] [PubMed] [Google Scholar]
- 3.Okamoto T, Akaike T, Sawa T, Miyamoto Y, van der Vliet A, Maeda H. Activation of matrix metalloproteinases by peroxynitrite-induced protein S-glutathiolation via disulfide S-oxide formation. J Biol Chem. 2001;276:29596–29602. doi: 10.1074/jbc.M102417200. [DOI] [PubMed] [Google Scholar]
- 4.Viappiani S, Nicolescu AC, Holt A, Sawicki G, Crawford BD, Leon H, van Mulligen T, Schulz R. Activation and modulation of 72kDa matrix metalloproteinase-2 by peroxynitrite and glutathione. Biochem Pharmacol. 2009;77:826–834. doi: 10.1016/j.bcp.2008.11.004. [DOI] [PubMed] [Google Scholar]
- 5.Woessner JF., Jr Matrix metalloproteinases and their inhibitors in connective tissue remodeling. FASEB J. 1991;5:2145–2154. [PubMed] [Google Scholar]
- 6.Newby AC, Southgate KM, Davies M. Extracellular matrix degrading metalloproteinases in the pathogenesis of arteriosclerosis. Basic Res Cardiol. 1994;89 1:59–70. doi: 10.1007/978-3-642-85660-0_6. [DOI] [PubMed] [Google Scholar]
- 7.Thompson RW, Parks WC. Role of matrix metalloproteinases in abdominal aortic aneurysms. Ann N Y Acad Sci. 1996;800:157–174. doi: 10.1111/j.1749-6632.1996.tb33307.x. [DOI] [PubMed] [Google Scholar]
- 8.Creemers EE, Cleutjens JP, Smits JF, Daemen MJ. Matrix metalloproteinase inhibition after myocardial infarction: a new approach to prevent heart failure? Circ Res. 2001;89:201–210. doi: 10.1161/hh1501.094396. [DOI] [PubMed] [Google Scholar]
- 9.Sawicki G, Salas E, Murat J, Miszta-Lane H, Radomski MW. Release of gelatinase A during platelet activation mediates aggregation. Nature. 1997;386:616–619. doi: 10.1038/386616a0. [DOI] [PubMed] [Google Scholar]
- 10.Fernandez-Patron C, Radomski MW, Davidge ST. Vascular matrix metalloproteinase-2 cleaves big endothelin-1 yielding a novel vasoconstrictor. Circ Res. 1999;85:906–911. doi: 10.1161/01.res.85.10.906. [DOI] [PubMed] [Google Scholar]
- 11.Cheung PY, Sawicki G, Wozniak M, Wang W, Radomski MW, Schulz R. Matrix metalloproteinase-2 contributes to ischemia-reperfusion injury in the heart. Circulation. 2000;101:1833–1839. doi: 10.1161/01.cir.101.15.1833. [DOI] [PubMed] [Google Scholar]
- 12.McCawley LJ, Matrisian LM. Matrix metalloproteinases: they're not just for matrix anymore! Curr Opin Cell Biol. 2001;13:534–540. doi: 10.1016/s0955-0674(00)00248-9. [DOI] [PubMed] [Google Scholar]
- 13.Doucet A, Overall CM. Protease proteomics: revealing protease in vivo functions using systems biology approaches. Mol Aspects Med. 2008;29:339–358. doi: 10.1016/j.mam.2008.04.003. [DOI] [PubMed] [Google Scholar]
- 14.Wang W, Schulze CJ, Suarez-Pinzon WL, Dyck JR, Sawicki G, Schulz R. Intracellular action of matrix metalloproteinase-2 accounts for acute myocardial ischemia and reperfusion injury. Circulation. 2002;106:1543–1549. doi: 10.1161/01.cir.0000028818.33488.7b. [DOI] [PubMed] [Google Scholar]
- 15.Sawicki G, Leon H, Sawicka J, Sariahmetoglu M, Schulze CJ, Scott PG, Szczesna-Cordary D, Schulz R. Degradation of myosin light chain in isolated rat hearts subjected to ischemia-reperfusion injury: a new intracellular target for matrix metalloproteinase-2. Circulation. 2005;112:544–552. doi: 10.1161/CIRCULATIONAHA.104.531616. [DOI] [PubMed] [Google Scholar]
- 16.Coker ML, Doscher MA, Thomas CV, Galis ZS, Spinale FG. Matrix metalloproteinase synthesis and expression in isolated LV myocyte preparations. Am J Physiol. 1999;277:H777–H787. doi: 10.1152/ajpheart.1999.277.2.H777. [DOI] [PubMed] [Google Scholar]
- 17.Sung MM, Schulz CG, Wang W, Sawicki G, Bautista-Lopez NL, Schulz R. Matrix metalloproteinase-2 degrades the cytoskeletal protein alpha-actinin in peroxynitrite mediated myocardial injury. J Mol Cell Cardiol. 2007;43:429–436. doi: 10.1016/j.yjmcc.2007.07.055. [DOI] [PubMed] [Google Scholar]
- 18.Kwan JA, Schulze CJ, Wang W, Leon H, Sariahmetoglu M, Sung M, Sawicka J, Sims DE, Sawicki G, Schulz R. Matrix metalloproteinase-2 (MMP-2) is present in the nucleus of cardiac myocytes and is capable of cleaving poly (ADP-ribose) polymerase (PARP) in vitro. FASEB J. 2004;18:690–692. doi: 10.1096/fj.02-1202fje. [DOI] [PubMed] [Google Scholar]
- 19.Chow AK, Cena J, El-Yazbi AF, Crawford BD, Holt A, Cho WJ, Daniel EE, Schulz R. Caveolin-1 inhibits matrix metalloproteinase-2 activity in the heart. J Mol Cell Cardiol. 2007;42:896–901. doi: 10.1016/j.yjmcc.2007.01.008. [DOI] [PubMed] [Google Scholar]
- 20.Schulz R. Intracellular targets of matrix metalloproteinase-2 in cardiac disease: rationale and therapeutic approaches. Annu Rev Pharmacol Toxicol. 2007;47:211–242. doi: 10.1146/annurev.pharmtox.47.120505.105230. [DOI] [PubMed] [Google Scholar]
- 21.Gao CQ, Sawicki G, Suarez-Pinzon WL, Csont T, Wozniak M, Ferdinandy P, Schulz R. Matrix metalloproteinase-2 mediates cytokine-induced myocardial contractile dysfunction. Cardiovasc Res. 2003;57:426–433. doi: 10.1016/s0008-6363(02)00719-8. [DOI] [PubMed] [Google Scholar]
- 22.Bergman MR, Teerlink JR, Mahimkar R, Li L, Zhu BQ, Nguyen A, Dahi S, Karliner JS, Lovett DH. Cardiac matrix metalloproteinase-2 expression independently induces marked ventricular remodeling and systolic dysfunction. Am J Physiol Heart Circ Physiol. 2007;292:H1847–H1860. doi: 10.1152/ajpheart.00434.2006. [DOI] [PubMed] [Google Scholar]
- 23.Granzier HL, Labeit S. The giant protein titin: a major player in myocardial mechanics, signaling, and disease. Circ Res. 2004;94:284–295. doi: 10.1161/01.RES.0000117769.88862.F8. [DOI] [PubMed] [Google Scholar]
- 24.Fukuda N, Granzier HL, Ishiwata S, Kurihara S. Physiological functions of the giant elastic protein titin in mammalian striated muscle. J Physiol Sci. 2008;58:151–159. doi: 10.2170/physiolsci.RV005408. [DOI] [PubMed] [Google Scholar]
- 25.Hein S, Scholz D, Fujitani N, Rennollet H, Brand T, Friedl A, Schaper J. Altered expression of titin and contractile proteins in failing human myocardium. J Mol Cell Cardiol. 1994;26:1291–1306. doi: 10.1006/jmcc.1994.1148. [DOI] [PubMed] [Google Scholar]
- 26.Morano I, Hadicke K, Grom S, Koch A, Schwinger RH, Bohm M, Bartel S, Erdmann E, Krause EG. Titin, myosin light chains and C-protein in the developing and failing human heart. J Mol Cell Cardiol. 1994;26:361–368. doi: 10.1006/jmcc.1994.1045. [DOI] [PubMed] [Google Scholar]
- 27.Kellermayer MS, Granzier HL. Calcium-dependent inhibition of in vitro thin-filament motility by native titin. FEBS Lett. 1996;380:281–286. doi: 10.1016/0014-5793(96)00055-5. [DOI] [PubMed] [Google Scholar]
- 28.Soteriou A, Gamage M, Trinick J. A survey of interactions made by the giant protein titin. J Cell Sci. 1993;104(Pt 1):119–123. doi: 10.1242/jcs.104.1.119. [DOI] [PubMed] [Google Scholar]
- 29.Nicolescu AC, Holt A, Kandasamy AD, Pacher P, Schulz R. Inhibition of matrix metalloproteinase-2 by PARP inhibitors. Biochem Biophys Res Commun. 2009;387:646–650. doi: 10.1016/j.bbrc.2009.07.080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schulz R, Panas DL, Catena R, Moncada S, Olley PM, Lopaschuk GD. The role of nitric oxide in cardiac depression induced by interleukin-1 beta and tumour necrosis factor-alpha. Br J Pharmacol. 1995;114:27–34. doi: 10.1111/j.1476-5381.1995.tb14901.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yamada A, Uegaki A, Nakamura T, Ogawa K. ONO-4817, an orally active matrix metalloproteinase inhibitor, prevents lipopolysaccharide-induced proteoglycan release from the joint cartilage in guinea pigs. Inflamm Res. 2000;49:144–146. doi: 10.1007/s000110050573. [DOI] [PubMed] [Google Scholar]
- 32.Kandalam V, Basu R, Abraham T, Wang X, Soloway PD, Jaworski DM, Oudit GY, Kassiri Z. TIMP2 deficiency accelerates adverse post-myocardial infarction remodeling because of enhanced MT1-MMP activity despite lack of MMP2 activation. Circ Res. 2010;106:796–808. doi: 10.1161/CIRCRESAHA.109.209189. [DOI] [PubMed] [Google Scholar]
- 33.Warren CM, Krzesinski PR, Greaser ML. Vertical agarose gel electrophoresis and electroblotting of high-molecular-weight proteins. Electrophoresis. 2003;24:1695–1702. doi: 10.1002/elps.200305392. [DOI] [PubMed] [Google Scholar]
- 34.Dhiman M, Nakayasu ES, Madaiah YH, Reynolds BK, Wen JJ, Almeida IC, Garg NJ. Enhanced nitrosative stress during Trypanosoma cruzi infection causes nitrotyrosine modification of host proteins: implications in Chagas' disease. Am J Pathol. 2008;173:728–740. doi: 10.2353/ajpath.2008.080047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang W, Sawicki G, Schulz R. Peroxynitrite-induced myocardial injury is mediated through matrix metalloproteinase-2. Cardiovasc Res. 2002;53:165–174. doi: 10.1016/s0008-6363(01)00445-x. [DOI] [PubMed] [Google Scholar]
- 36.Leon H, Baczko I, Sawicki G, Light PE, Schulz R. Inhibition of matrix metalloproteinases prevents peroxynitrite-induced contractile dysfunction in the isolated cardiac myocyte. Br J Pharmacol. 2008;153:676–683. doi: 10.1038/sj.bjp.0707621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Young P, Ferguson C, Banuelos S, Gautel M. Molecular structure of the sarcomeric Z-disk: two types of titin interactions lead to an asymmetrical sorting of alpha-actinin. EMBO J. 1998;17:1614–1624. doi: 10.1093/emboj/17.6.1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Atkinson RA, Joseph C, Dal Piaz F, Birolo L, Stier G, Pucci P, Pastore A. Binding of alpha-actinin to titin: implications for Z-disk assembly. Biochemistry. 2000;39:5255–5264. doi: 10.1021/bi991891u. [DOI] [PubMed] [Google Scholar]
- 39.Gregorio CC, Trombitas K, Centner T, Kolmerer B, Stier G, Kunke K, Suzuki K, Obermayr F, Herrmann B, Granzier H, Sorimachi H, Labeit S. The NH2 terminus of titin spans the Z-disc: its interaction with a novel 19-kD ligand (T-cap) is required for sarcomeric integrity. J Cell Biol. 1998;143:1013–1027. doi: 10.1083/jcb.143.4.1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gutierrez FR, Lalu MM, Mariano FS, Milanezi CM, Cena J, Gerlach RF, Santos JE, Torres-Duenas D, Cunha FQ, Schulz R, Silva JS. Increased activities of cardiac matrix metalloproteinases matrix metalloproteinase (MMP)-2 and MMP-9 are associated with mortality during the acute phase of experimental Trypanosoma cruzi infection. J Infect Dis. 2008;197:1468–1476. doi: 10.1086/587487. [DOI] [PubMed] [Google Scholar]
- 41.Ikonomidis JS, Hendrick JW, Parkhurst AM, Herron AR, Escobar PG, Dowdy KB, Stroud RE, Hapke E, Zile MR, Spinale FG. Accelerated LV remodeling after myocardial infarction in TIMP-1-deficient mice: effects of exogenous MMP inhibition. Am J Physiol Heart Circ Physiol. 2005;288:H149–H158. doi: 10.1152/ajpheart.00370.2004. [DOI] [PubMed] [Google Scholar]
- 42.Eaton P, Byers HL, Leeds N, Ward MA, Shattock MJ. Detection, quantitation, purification, and identification of cardiac proteins S-thiolated during ischemia and reperfusion. J Biol Chem. 2002;277:9806–9811. doi: 10.1074/jbc.M111454200. [DOI] [PubMed] [Google Scholar]
- 43.Doroszko A, Polewicz D, Sawicka J, Richardson JS, Cheung PY, Sawicki G. Cardiac dysfunction in an animal model of neonatal asphyxia is associated with increased degradation of MLC1 by MMP-2. Basic Res Cardiol. 2009;104:669–679. doi: 10.1007/s00395-009-0035-1. [DOI] [PubMed] [Google Scholar]
- 44.Bolli R, Marban E. Molecular and cellular mechanisms of myocardial stunning. Physiol Rev. 1999;79:609–634. doi: 10.1152/physrev.1999.79.2.609. [DOI] [PubMed] [Google Scholar]
- 45.Lim CC, Zuppinger C, Guo X, Kuster GM, Helmes M, Eppenberger HM, Suter TM, Liao R, Sawyer DB. Anthracyclines induce calpain-dependent titin proteolysis and necrosis in cardiomyocytes. J Biol Chem. 2004;279:8290–8299. doi: 10.1074/jbc.M308033200. [DOI] [PubMed] [Google Scholar]
- 46.Kukan M. Emerging roles of proteasomes in ischemia-reperfusion injury of organs. J Physiol Pharmacol. 2004;55:3–15. [PubMed] [Google Scholar]
- 47.Stansfield WE, Moss NC, Willis MS, Tang R, Selzman CH. Proteasome inhibition attenuates infarct size and preserves cardiac function in a murine model of myocardial ischemia-reperfusion injury. Ann Thorac Surg. 2007;84:120–125. doi: 10.1016/j.athoracsur.2007.02.049. [DOI] [PubMed] [Google Scholar]
- 48.Zolk O, Schenke C, Sarikas A. The ubiquitin-proteasome system: focus on the heart. Cardiovasc Res. 2006;70:410–421. doi: 10.1016/j.cardiores.2005.12.021. [DOI] [PubMed] [Google Scholar]
- 49.Centner T, Yano J, Kimura E, McElhinny AS, Pelin K, Witt CC, Bang ML, Trombitas K, Granzier H, Gregorio CC, Sorimachi H, Labeit S. Identification of muscle specific ring finger proteins as potential regulators of the titin kinase domain. J Mol Biol. 2001;306:717–726. doi: 10.1006/jmbi.2001.4448. [DOI] [PubMed] [Google Scholar]
- 50.Witt SH, Granzier H, Witt CC, Labeit S. MURF-1 and MURF-2 target a specific subset of myofibrillar proteins redundantly: towards understanding MURF-dependent muscle ubiquitination. J Mol Biol. 2005;350:713–722. doi: 10.1016/j.jmb.2005.05.021. [DOI] [PubMed] [Google Scholar]
- 51.van Hees HW, Ottenheijm CA, Granzier HL, Dekhuijzen PN, Heunks LM. Heart failure decreases passive tension generation of rat diaphragm fibers. Int J Cardiol. 2010;141:275–283. doi: 10.1016/j.ijcard.2008.12.042. [DOI] [PubMed] [Google Scholar]
- 52.Carvalho RF, Dariolli R, Justulin LA, Junior, Sugizaki MM, Politi Okoshi M, Cicogna AC, Felisbino SL, Dal Pai-Silva M. Heart failure alters matrix metalloproteinase gene expression and activity in rat skeletal muscle. Int J Exp Pathol. 2006;87:437–443. doi: 10.1111/j.1365-2613.2006.00497.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.