

Abstract
The molecular chaperone Hsp90 binds and hydrolyses ATP, but how this ATPase activity regulates the interaction of Hsp90 with a polypeptide substrate is not yet understood. Using the glucocorticoid receptor ligand binding domain as a substrate, we show that dissociation of Hsp90 from bound polypeptide depends on the Hsp90 ATPase and is blocked by geldanamycin, a specific ATPase inhibitor. The co-chaperone p23 greatly stimulates Hsp90 substrate release with ATP, but not with the non-hydrolysable nucleotides ATPγS or AMP-PNP. Point mutants of Hsp90 with progressively lower ATPase rates are progressively slower in ATP-dependent substrate release but are still regulated by p23. In contrast, ATPase-inactive Hsp90 mutants release substrate poorly and show no p23 effect. These results outline an ATP-driven cycle of substrate binding and release for Hsp90 which differs from that of other ATP-driven chaperones. Conversion of the ATP state of Hsp90 to the ADP state through hydrolysis is required for efficient release of substrate polypeptide. p23 couples the ATPase activity to polypeptide dissociation and thus can function as a substrate release factor for Hsp90.
Keywords: ATP/glucocorticoid receptor/Hsp90/molecular chaperones/protein folding
Introduction
The 90 kDa heat shock protein of the eukaryotic cytosol, Hsp90, is an abundant and highly conserved homodimeric chaperone protein required for the folding and regulation of various signal transduction molecules, including the steroid hormone receptors (Buchner, 1999; Caplan, 1999). Hsp90 acts as part of a multichaperone system together with Hsc70 and numerous other protein cofactors, but the biochemical mechanism of Hsp90 function is still poorly understood. Here, we focus on how the interaction of Hsp90 with substrate polypeptides is regulated by the Hsp90 ATPase activity.
Purified Hsp90 can bind denatured proteins (Jakob et al., 1995; Freeman and Morimoto, 1996; Young et al., 1997; Scheibel et al., 1998), and this chaperone activity is independent of nucleotide although Hsp90 is now known to be an ATPase (Obermann et al., 1998; Panaretou et al., 1998). Limited proteolysis of Hsp90 revealed three major domains, of which the N-terminal domain was shown to contain a binding pocket for ATP and for the competitive inhibitor geldanamycin (GA), an ansamycin drug (Prodromou et al., 1997; Stebbins et al., 1997). Both the N-terminal and the extreme C-terminal domains of Hsp90 display chaperone activity by preventing the aggregation of denatured proteins in vitro. The chaperone activity of the N-terminal domain is reduced in the presence of GA or ATP (Young et al., 1997; Scheibel et al., 1998), suggesting that substrate binding by Hsp90 is nucleotide regulated. ATP and GA also reduce the binding of Hsp90 to phenyl-Sepharose, suggestive of a conformational change (Csermely et al., 1993; Sullivan et al., 1997).
Physiologically, Hsp90 appears to recognize substrates in a near-native state rather than fully denatured polypeptides (Smith, 1993; Jakob et al., 1995). Hsp90 substrates can be co-immunoprecipitated from cell lysates as multichaperone complexes along with Hsp90, Hsc70 and various cofactor proteins (Bresnick et al., 1989; Smith et al., 1990; Schneider et al., 1996). Time course experiments with the progesterone receptor showed that Hsp90 binding is preceded by the interaction of Hsc70 and the adaptor protein Hop with the receptor. This step is followed by the recruitment of Hsp90 and the cofactor p23 into a complex from which Hsp90 can be released by the addition of ATP (Smith, 1993; Johnson and Toft, 1994). Similarly, ATP-dependent release of Hsp90 has been observed with multichaperone complexes containing non-native firefly luciferase (Schneider et al., 1996). GA blocked the dissociation of Hsp90 from these complexes (Schneider et al., 1996) by displacing ATP from Hsp90 (Obermann et al., 1998; Panaretou et al., 1998), consistent with substrate release being regulated directly via the Hsp90 ATPase domain. However, the interpretation of these experiments is complicated by the presence of Hop, which binds to both Hsp90 and Hsc70 via tetratricopeptide repeat (TPR) domains (Chen and Smith, 1998; Scheufler et al., 2000), and thus an indirect regulation of Hsp90 via the Hsc70 ATPase could not be ruled out.
The Hsp90 cofactor p23 forms complexes with the ATP-bound form of Hsp90 (Sullivan et al., 1997; Fang et al., 1998; Obermann et al., 1998), but the functional significance of this interaction is still unclear. While the exact binding site for p23 on Hsp90 is unknown, the N-terminal ATPase and the C-terminal dimerization sequences of Hsp90 are both required for binding (Chen et al., 1998). By itself, p23 can act as a general chaperone to prevent protein aggregation (Bose et al., 1996; Freeman et al., 1996), although a specific role of p23 in stabilizing steroid receptor complexes has been proposed (Hutchison et al., 1995). Null mutants in Saccharomyces cerevisiae of Sba1, the p23 homologue, show increased sensitivity to ansamycins and slower growth (Bohen, 1998; Fang et al., 1998).
In this study, we examined the direct interaction of Hsp90 with the ligand binding domain (LBD) of glucocorticoid receptor (GR) as a substrate. Our analysis shows that in addition to ATP binding, the conversion of Hsp90 from the ATP state to the ADP state through hydrolysis is necessary for efficient substrate release. Furthermore, our results indicate that p23 functions as a substrate release factor dependent on the Hsp90 ATPase activity.
Results
Generation of functional Hsp90–substrate complexes
Although isolated Hsp90 can recognize unfolded proteins in a nucleotide-independent manner, substrate loading onto Hsp90 in whole-cell lysates requires ATP and is probably mediated by the Hsc70 chaperone system (Smith et al., 1990; Schneider et al., 1996; Chen and Smith, 1998; Prodromou et al., 1999). To study the interaction of Hsp90 with substrate without interference from any substrate loading mechanism, we analysed chaperone complexes that had formed at steady state in a complete cytosolic extract (reticulocyte lysate, RL).
The model substrate used was a C-terminally myc-tagged version of the LBD of the rat GR, purified as a non-native polypeptide and partially refolded by slow removal of denaturant. The LBD produced by this procedure is monomeric (not shown), and contains the polypeptide sequence that is both necessary and sufficient for recognition of the full-length GR by Hsp90 (Howard et al., 1990). Upon addition of LBD to RL containing ATP, LBD–chaperone complexes formed that could be immunoprecipitated specifically with anti-myc antibody (Figure 1A). The predominant chaperone proteins in these complexes were identified as Hsp90, Hsc70 and Hop by immunoblotting (Figure 1A, right panel), and Hip by protein sequencing. Hsp90 and the great majority of Hsc70 remained bound to the LBD after a 10 min incubation of the isolated complex in buffer. However, upon incubation in buffer with ATP, a significant amount of the bound Hsp90 eluted from the complex (Figure 1A), reproducing the known behaviour of Hsp90 complexed with progesterone receptor or with unfolded luciferase (Smith, 1993; Schneider et al., 1996). The isolated LBD–chaperone complexes therefore provide a functional and partially pure Hsp90 system.
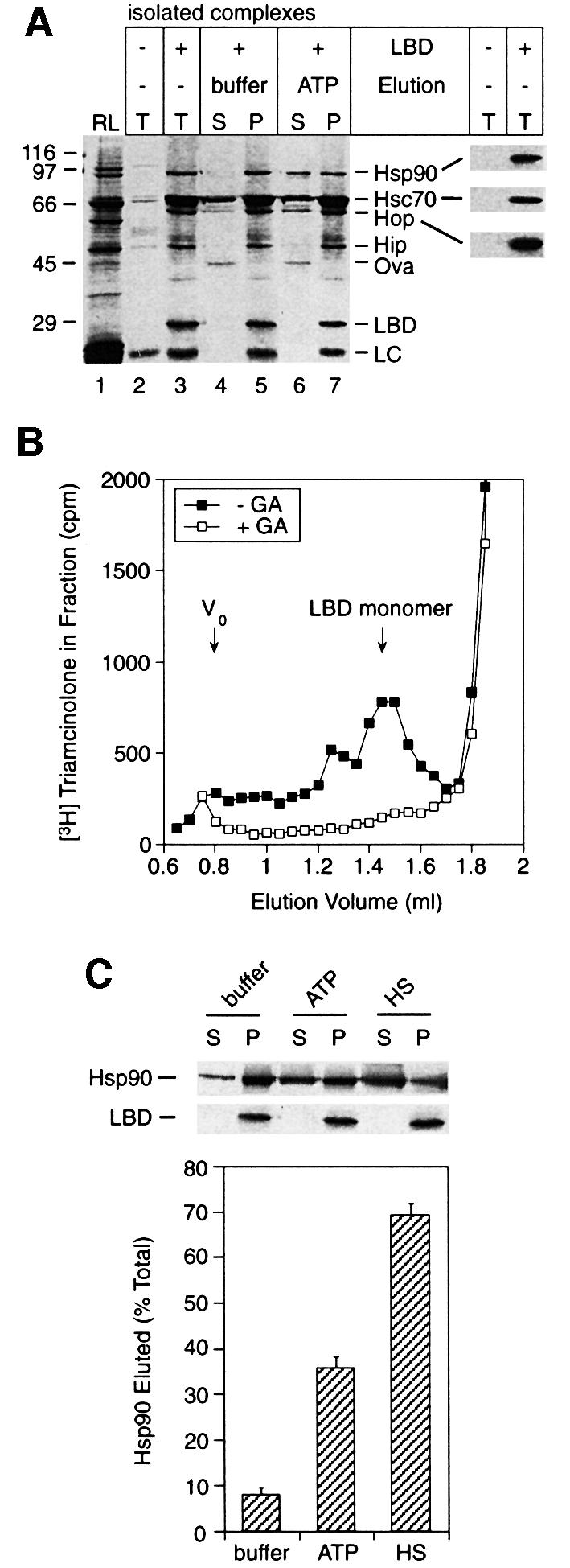
Fig. 1. ATP-dependent release of Hsp90 bound to substrate. (A) Coomassie stained SDS–PAGE of LBD–chaperone complexes. Lane 1, total RL; lanes 2–3, total immunoprecipitation (T) from RL without (lane 2) and with (lane 3) added LBD; lanes 4–7, isolated LBD complexes were eluted for 10 min with buffer alone (lanes 4–5) or containing 1 mM ATP (lanes 6–7) and supernatants (S) separated from the pellets (P). Ovalbumin (Ova) was added to the supernatant as a recovery control. LC, IgG light chain. Molecular weight standards are marked on the left. Right, lanes 2 and 3 immunoblotted for Hsp90, Hsc70 and Hop. (B) LBD was incubated with RL, ATP and [3H]triamcinolone acetonide with or without 90 µM GA, followed by gel filtration. Radioactivity in each fraction is plotted and the elution volumes of the void (V0) and LBD monomer are marked. (C) Radiolabelled Hsp90 was eluted from LBD complexes for 10 min with buffer alone or containing 1 mM ATP or 500 mM NaCl (HS). A representative phosphoimager scan (top) and Coomassie staining of LBD (middle) are shown. Bottom, the amount of Hsp90 released as a percentage of total Hsp90 in the LBD complex. Error bars in all figures show standard deviations from the mean of at least three experiments.
As a further test that LBD is handled by the chaperones in a physiological manner, the refolding of LBD in the lysate was assayed. Native LBD was detected by binding of the hormone analogue triamcinolone acetonide, which binds specifically and with high affinity to the LBD of full-length GR. Following the incubation of LBD in RL containing ATP and radiolabelled triamcinolone, the mixture was separated by gel filtration and the triamcinolone in each fraction was quantified. A clear peak of radioactivity coincided with the elution volume of monomeric LBD (Figure 1B), indicating that the LBD could be refolded. When LBD was incubated with triamcinolone in lysate pre-treated with GA, no native polypeptide was detected (Figure 1B). Folded LBD was also not observed when incubated in lysate without ATP, or in buffer alone (not shown). Thus, similar to the full-length receptor, LBD is dependent on the Hsp90 in the lysate to reach and maintain its native state. Moreover, the Hsp90 isolated with the LBD complexes is in the process of refolding LBD and should provide a ‘snap-shot’ of the functional ATP-driven folding cycle.
To improve the detection of Hsp90 in LBD–chaperone complexes, the complexes were formed in RL supplemented with a small amount of radiolabelled Hsp90. The results obtained with this labelled Hsp90 were quantifiable and highly reproducible. Again, incubation of immunoprecipitated LBD complexes with buffer alone produced little release of Hsp90 (8% of total), whereas buffer with ATP eluted ∼35% of total bound Hsp90 (Figure 1C). Maximal elution (60–70% of total) was obtained by incubation with high salt (500 mM NaCl), or with ATP and p23 (see below). The remaining 30% of Hsp90 was apparently trapped non-productively.
Hsp90 may be in direct contact with LBD in the isolated complexes, or indirectly bound via Hop and Hsc70. Because Hop is detected stoichiometrically with Hsp90 in the LBD–chaperone complexes (Figure 1A), it must provide the main connection to Hsc70 in these complexes. The only other protein stoichiometric to Hsp90 in the LBD–chaperone complexes is Hip (Figure 1A), which regulates Hsc70 and cannot itself anchor Hsp90 (Höhfeld et al., 1995). To determine the fraction of indirectly bound Hsp90, we used our recent analysis of the domains of Hop that bind to Hsc70 and Hsp90. The TPR1 domain of Hop binds specifically to Hsc70, while the TPR2A domain binds to Hsp90 (Scheufler et al., 2000). In experiments with purified proteins, full-length Hop bound fusion proteins of glutathione S-transferase (GST) with the C-terminal 12 kDa domain of Hsp90 (Young et al., 1998) or with the C-terminal 25 kDa of Hsc70 (Figure 2A). Both of these interactions were effectively disrupted by the subsequent addition of an excess of purified TPR2A and TPR1, respectively (Figure 2A).
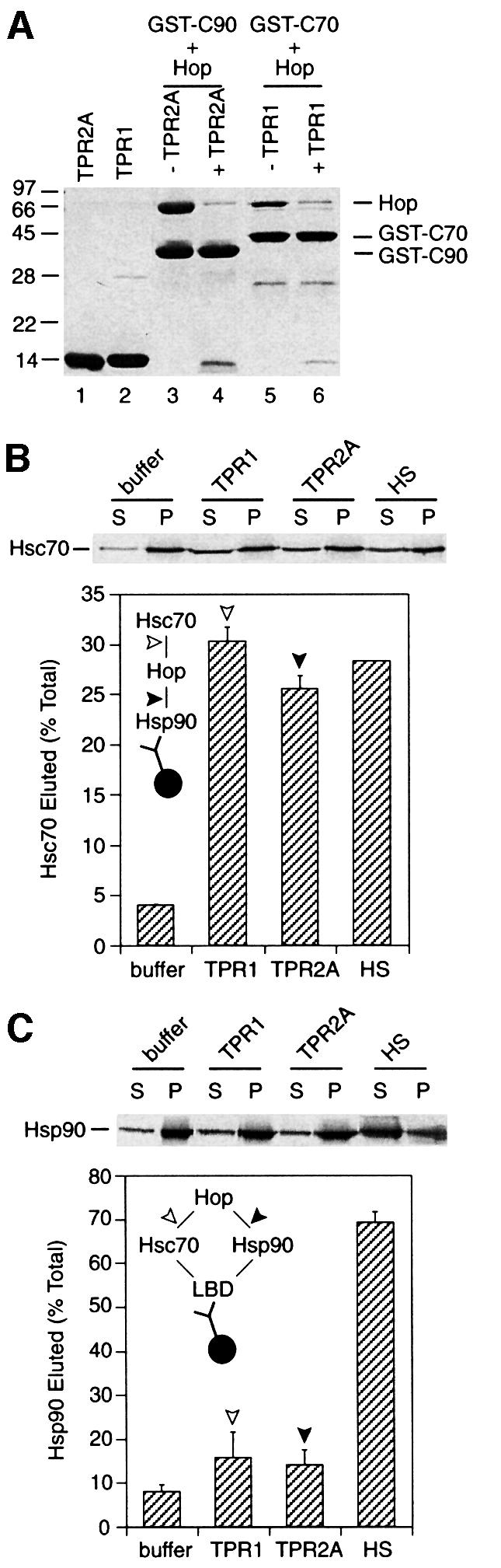
Fig. 2. Hsp90 remains bound to substrate after disruption of the Hop–Hsc70 linkage. (A) Lanes 1–2, Coomassie staining of TPR2A (lane 1) and TPR1 (lane 2). Lanes 3–6, binding reactions containing 2 µM Hop and 2 µM GST–C90 (lanes 3–4) or GST–C70 (lanes 5–6) were recovered with glutathione–agarose after incubation without (lanes 3 and 5) or with a 25-fold excess of TPR2A (lane 4) or TPR1 (lane 6) and detected by Coomassie staining. (B) Radiolabelled Hsc70 from RL was co-precipitated with Hsp90 and eluted from immune pellets with buffer alone or containing 20 µM TPR1, 20 µM TPR2A or 500 mM NaCl (HS). Top, phosphoimager scan. Inset, schematic diagram of the experiment showing the interactions disrupted by TPR1 (open arrowhead) and TPR2A (closed arrowhead). (C) Radiolabelled Hsp90 was eluted from LBD complexes with buffer alone or containing 20 µM TPR1, 20 µM TPR2A or 500 mM NaCl (HS). Top, phosphoimager scan. Inset, schematic diagram as in (B).
The effectiveness of the TPR domain competitors on immunoprecipitated complexes was tested using radiolabelled Hsc70 generated by in vitro translation in RL and then co-precipitated with antibodies against Hsp90 without the addition of any exogenous substrate polypeptide. Incubation of the immune pellets with either TPR1 or TPR2A was as efficient as high salt (500 mM NaCl) at releasing Hsc70 (Figure 2B). Complexes of Hop with Hsp90 or Hsc70 are completely disrupted with high salt (Young et al., 1998; A.Brinker, personal communication), indicating that the remaining salt-resistant Hsc70 was bound non-specifically to the antibody matrix or via some intermediate other than Hop. Thus, the purified TPR1 and TPR2A domains could be used to disrupt effectively Hsc70–Hop and Hsp90–Hop interactions in isolated complexes.
When isolated LBD–chaperone complexes were incubated with a large excess of TPR1 and TPR2A to disrupt the Hsc70–Hop and Hsp90–Hop interactions, only a small amount of additional Hsp90 elution was observed with TPR1 and TPR2A (16 and 14%, respectively) compared with the buffer control (8%) (Figure 2C). Also, no further elution of Hsp90 was observed with a mixture of both TPR1 and TPR2A (not shown). This demonstrates that only a small fraction of Hsp90 in the isolated complexes is connected indirectly to LBD by a Hsc70–Hop linkage (<10%), while the remainder is directly bound to the LBD polypeptide. Having clearly established this, the mechanism that regulates substrate binding and release of Hsp90 could be unambiguously analysed.
The nucleotide state of Hsp90 regulates substrate binding
The ability of different nucleotides to effect the release of Hsp90 from LBD–chaperone complexes was tested. The greatest level of release was produced by ATP (∼35% of bound Hsp90), followed by the non-hydrolysable analogue ATPγS (∼30%) (Figure 3A). In contrast, the non-hydrolysable nucleotide AMP-PNP was inefficient in producing a release of Hsp90 above the buffer control, similar to ADP (Figure 3A). However, like ATPγS, AMP-PNP was found to inhibit the Hsp90 ATPase activity competitively (not shown), suggesting that the analogue binds to Hsp90 efficiently. The ability of ATP and ATPγS to cause Hsp90 release was inhibited by GA, confirming that these nucleotides are acting on the ATP binding domain of Hsp90 (Figure 3A). Unlike AMP-PNP, ATPγS can be hydrolysed by many ATPases, albeit more slowly, which may explain the similarity of its effect on Hsp90 to that of ATP. This was addressed in experiments described below. Adding a large excess of the TPR2A domain of Hop, to disrupt the Hsp90–Hop interaction, did not alter the general behaviour of Hsp90 in response to nucleotides (Figure 3B). The overall level of Hsp90 release was, however, slightly higher in the presence of TPR2A, reflecting the small amount of Hsp90 bound via Hsc70.

Fig. 3. The nucleotide state of Hsp90 regulates substrate binding. (A) Radiolabelled Hsp90 was eluted from LBD complexes with buffer alone or buffer containing the indicated nucleotide (1 mM), either without or with 90 µM GA. Top, phosphoimager scan. (B) The experiment in (A) was repeated but with 20 µM TPR2A added to the elution buffers. (C) The elution of radiolabelled Hsc70 was tested similarly to (A). (D) The elution of Hsp90 from LBD complexes with 1 mM ATP, ATPγS, AMP-PNP or with buffer alone was monitored over time.
As an important control for the Hsp90 specificity of the observed nucleotide effects, the release of Hsc70 from LBD–chaperone complexes was monitored. LBD complexes containing radiolabelled Hsc70 were formed in RL and the elution with nucleotides was quantified. The pattern of Hsc70 elution was markedly different from that of Hsp90: efficient Hsc70 release from the immobilized substrate was only observed with the non-hydrolysable analogues ATPγS and AMP-PNP, while contrary to Hsp90, ATP produced little release (Figure 3C). Release of Hsc70 with ATPγS and AMP-PNP was expected based on the dissociation of Hsc70–polypeptide complexes upon ATP binding (reviewed in Hartl, 1996; Bukau and Horwich, 1998). Also, the weak release of Hsc70 by ATP is consistent with the effect of substrate on stimulating the hydrolysis of Hsc70-bound ATP, thereby generating the ADP state that binds substrate strongly (Figure 3C). This state may be further stabilized by the Hsc70 cofactor Hip (Höhfeld et al., 1995), which is also present in the LBD–chaperone complex (Figure 1A). The different behaviour of Hsc70 and Hsp90 again demonstrates that the interaction between Hsp90 and LBD is not regulated by the Hsc70 ATPase. Furthermore, these results establish that substrate binding by the two chaperones is regulated independently by distinct ATP-driven mechanisms.
To obtain further insight into the nucleotide regulation of Hsp90, the kinetics of Hsp90 elution from LBD complexes was examined. The release of Hsp90 was found to be surprisingly slow. With ATP, Hsp90 eluted with a half-time of ∼1.3 min (Figure 3D). In the presence of ATPγS, Hsp90–LBD complexes dissociated even more slowly with an estimated half-time of 3.5 min (Figure 3D). For comparison, Hsc70–peptide complexes can dissociate within seconds (Schmid et al., 1994). AMP-PNP produced only slightly more release of Hsp90 compared with buffer after longer incubation (Figure 3D).
The different results obtained with ATPγS and AMP-PNP may suggest that one or both of the analogues cannot properly substitute for ATP in stabilizing the ATP-bound conformation of Hsp90. Also, slow hydrolysis of ATPγS by Hsp90 may explain its intermediate effect between ATP and AMP-PNP. To resolve these issues and more precisely characterize the nucleotide regulation of substrate binding by Hsp90, we followed two experimental approaches. First, we tested the effect of the nucleotide-dependent Hsp90 co-chaperone p23 on Hsp90 release. Secondly, we examined several Hsp90 mutants that have reduced or inactivated ATPase activities.
p23 is an ATP-dependent substrate release factor for Hsp90
The interaction of p23 with Hsp90 is regulated by nucleotide, with optimal complex formation occurring in the presence of ATPγS or AMP-PNP (Sullivan et al., 1997; Fang et al., 1998; Obermann et al., 1998). We therefore tested the ATP-dependent release of Hsp90 from LBD complexes in the presence of purified p23. In the absence of nucleotide, p23 did not cause release of Hsp90 above the buffer control (Figure 4A); however, p23 together with ATP released ∼60% of total bound Hsp90 (Figure 4A), almost twice the amount of Hsp90 released with ATP alone (35%) and close to the maximum elution obtained with high salt (70%) (Figure 1C). The enhanced Hsp90 release in the presence of p23 and ATP was fully inhibited by GA, but not affected by the addition of the TPR2A domain of Hop (not shown). In contrast, p23 had no effect on the moderate release of Hsp90 with ATPγS, or the low level of release with AMP-PNP (Figure 4A). Because both non-hydrolysable analogues stabilize the p23–Hsp90 complex, we conclude that p23 does not sterically displace substrate from Hsp90, or prevent substrate binding onto Hsp90. ADP, either alone or with inorganic phosphate, did not release Hsp90 in the presence or absence of p23 (Figure 4A), suggesting that Hsp90 must cycle through the actual ATP-bound state to dissociate efficiently from substrate. Thus, substrate release is likely to be related to the conversion of Hsp90 from the ATP- to the ADP-bound state, and p23 must act on Hsp90 at this step.

Fig. 4. p23 is an ATP-dependent substrate release factor for Hsp90. (A) Radiolabelled Hsp90 was eluted from LBD complexes with buffer alone or buffer containing the indicated nucleotide (1 mM) with 1 mM phosphate (Pi) where marked. Elutions were either without or with 5 µM p23. (B) The elution of Hsp90 with RL and 1 mM ATP, or with the indicated nucleotide (1 mM) with or without 5 µM p23 was monitored over time. Curves shown in dashed lines are taken from Figure 3. (C) Hsp90 was eluted with 1 mM ATP and varying concentrations of p23. (D) Hsp90 was eluted with or without 1 mM ATP in the presence of full-length (FL) p23 or the p23 mutants 1–128 and 1–120 having the C-terminal 32 and 40 amino acids, respectively, deleted.
The kinetics of p23-enhanced Hsp90 release was examined (Figure 4B). p23 addition clearly increased the release of Hsp90 from LBD even at early time points (Figure 4B), although the estimated half-time of release, 1.5 min, was similar to that observed with ATP alone (Figure 4B). In contrast, there was no effect of p23 on the slower kinetics of release observed in the presence of ATPγS with an identical estimated half-time of 3.5 min. The kinetics of Hsp90 release with AMP-PNP was also not altered by p23 (Figure 4B). Interestingly, the level and rate of Hsp90 release observed with ATP and p23 were close to those measured upon re-addition of total cell lysate to the isolated LBD–chaperone complexes with ATP (Figure 4B), indicating that p23 alone is sufficient to recover most of the substrate-releasing activity normally present in cell lysate. The effect of p23 on the level but not the half-time of Hsp90 elution with ATP is consistent with a mechanism in which the co-chaperone increases the efficiency of substrate release with each ATPase cycle by Hsp90.
We also tested the ATP-dependent release of Hsp90 at varying concentrations of p23. A significant effect was already observed at p23 concentrations as low as 100 nM (Figure 4C), less than the concentration of p23 in cell lysate (∼500 nM). These concentrations of p23 are substoichiometric to the concentrations of Hsp90 in cell lysates (2–5 µM) and in the substrate release reaction (estimated at 0.5 µM), which suggests that p23 acts only transiently on Hsp90. Interestingly, deletion mutants of p23 that had the C-terminus containing its chaperone activity removed (Weikl et al., 1999) were as active as full-length p23 in enhancing Hsp90 substrate release (Figure 4D). Thus, the transient substrate-releasing activity of p23 on Hsp90 is independent of its chaperone activity, where p23 must act stoichiometrically with substrate.
These results indicate that p23 functions as a substrate release factor for Hsp90, and that neither ATPγS nor AMP-PNP can substitute for ATP in this function. Because both nucleotide analogues support stable complexes of p23 and Hsp90, simply the binding of p23 onto Hsp90 is not sufficient to produce the complete release of substrate observed with ATP. This suggests that a further step, such as efficient ATP hydrolysis followed by conversion to the ADP-bound state, may be required. p23 may then act either by assisting the conversion between these states, or by more tightly coupling the conversion to binding and release of substrate polypeptide.
Effects of p23 and of point mutations in Hsp90 on the Hsp90 ATPase
Using recombinant yeast Hsp90 (Hsp82) and p23 (Sba1) we reported previously that p23 does not significantly change the overall ATPase activity of Hsp90 (Obermann et al., 1998). However, it is possible that p23-dependent changes in the rates of individual steps within the ATPase cycle may occur, but be masked by another rate-limiting step. To address this, a more extensive quantitative analysis of the Hsp90 ATPase activity in the presence of p23 was performed. Because recombinant human Hsp90 could not be purified from Escherichia coli in an enzymatically active form, we used the recombinant yeast Hsp90, into which point mutations could be introduced, together with yeast p23. The molecular structures of the ATPase domains from yeast and human Hsp90 are virtually identical (Prodromou et al., 1997; Stebbins et al., 1997), and the enzymatic mechanisms of the proteins are expected to be the same. Indeed, yeast p23 was as active as human p23 in the Hsp90 substrate release assay described above (not shown). To allow further comparison with the Hsp90 substrate release assay, the enzymatic analysis was carried out in the same physiological buffer conditions.
The Hsp90 ATPase activity at 30°C was monitored at early time points to determine the initial hydrolysis rate, with or without a 4-fold molar excess of p23 (Figure 5A). The ATPase rates determined at varying concentrations of ATP with or without p23 were fitted to the Michaelis–Menten model of enzyme kinetics (Figure 5B), and the kinetic constants are listed in Table I. The kcat for the reaction was similar to previously reported values (Obermann et al., 1998), and was not significantly different in the absence or presence of p23 (0.53 and 0.51 min–1, respectively). In addition, p23 did not greatly affect the KM values of the reaction. Because the KM constant represents the apparent affinity of Hsp90 for ATP, these data indicate that p23 does not act as an ATP loading factor for Hsp90. The KM constants were somewhat higher than previous estimates, as a result of the higher ionic strength of the physiological buffer system used in this study.

Fig. 5. Effect of p23 and point mutations on the Hsp90 ATPase. (A) Purified Hsp90 (5 µM) was incubated at 30°C in 2 mM ATP containing [α-32P]ATP either without or with 10 µM p23. ADP production over time was measured and plotted after subtraction of the low GA-inhibited background. (B) ATPase rates of Hsp90 were determined as in (A) at different concentrations of ATP, either without or with 10 µM p23. The rates are plotted against ATP concentration and fitted to the Michaelis–Menten equation v = Vmax/(1 + KM/[ATP]). (C) ATPase rates in 2 mM ATP of wild-type Hsp90 (WT) or the indicated point mutants were measured at 30 or 37°C.
Table I. Effects of p23 and of point mutations in Hsp90 on the Hsp90 ATPase.
| Hsp90 | kcat (min–1) | kcat (min–1) + p23 | KM (mM) | KM (mM) + p23 |
|---|---|---|---|---|
| WT | 0.53 ± 0.03 | 0.51 ± 0.03 | 0.83 ± 0.13 | 0.77 ± 0.13 |
| E33D | 0.42 ± 0.04 | 0.42 ± 0.04 | 0.48 ± 0.16 | 0.31 ± 0.12 |
| T101I | 0.35 ± 0.02 | 0.34 ± 0.03 | 2.09 ± 0.10 | 1.79 ± 0.25 |
| E33A | <0.01 | <0.01 | n.d. | n.d. |
| D79N | <0.01 | <0.01 | n.d. | n.d. |
n.d., not determined.
We next tested whether p23 could affect the ATP hydrolysis activity of Hsp90 mutants with ATPase defects. A mutation in the catalytic residue of Hsp90, E33D, has been shown to reduce the ATPase rate of Hsp90 without drastically changing the affinity for ATP (Obermann et al., 1998). Under the conditions used in this study, a similar reduction in kcat was observed, while the KM was in fact lower than for wild-type (WT) Hsp90 (Table I). In the presence of p23, the kinetic constants remained essentially unchanged (Table I). Because the catalytic step of ATP hydrolysis is negatively affected by the E33D mutation, it is unlikely that p23 acts to stimulate this step.
In addition to the engineered mutation E33D, several point mutations within the N-terminal ATP binding domain of Hsp90 produce a temperature-sensitive (ts) phenotype in yeast (Kimura et al., 1994; Nathan and Lindquist, 1995). Interestingly, two of these ts mutations (A97T and T101I) are located close together in a highly conserved sequence of Hsp90 that can fold either as an α-helix or as a loop, and that may regulate access to the nucleotide binding cavity (Stebbins et al., 1997). These mutations are also in the section of the ATPase domain proposed to form a ‘lid’ enclosing bound ATP (Prodromou et al., 1997). In the ATPase cycle, the alternate conformations of this segment may be related to loading of ATP or release of bound ADP and phosphate after hydrolysis.
The mutant Hsp90-T101I was constructed, purified and characterized. At the permissive temperature (30°C), Hsp90-T101I was found to have a kcat (0.35 min–1) reduced by ∼40% compared with WT Hsp90, and a higher KM value (2.09 versus 0.83 mM; Table I). This apparent reduced affinity for ATP is consistent with the mutation being in a conformationally sensitive region of the N-terminal domain. When the mutant was tested in the presence of p23, the kcat was unaffected while the KM was slightly but not significantly reduced (Table I). Taken together, these results suggest that p23 is not a regulator of the Hsp90 ATPase but is itself regulated in its functions by the Hsp90 ATPase cycle.
Two additional mutations in the Hsp90 ATPase domain have previously been shown to eliminate the ATPase activity. The E33A mutation, which removes the catalytic carboxyl group, allows binding but not hydrolysis of ATP by Hsp90, while the D79N mutation abolishes both ATP binding and hydrolysis (Obermann et al., 1998; Panaretou et al., 1998). As expected, p23 could not restore ATPase activity to either of these mutant Hsp90 proteins (Table I).
The WT and mutant Hsp90 proteins were also assayed at 37°C, the non-permissive temperature for the T101I mutant in vivo (Kimura et al., 1994; Nathan and Lindquist, 1995). The ATPase activity of WT Hsp90 was almost doubled at the higher temperature (Figure 5C). The activity of the E33D mutant was also increased at 37°C, although it was still lower than that of WT Hsp90 (Figure 5C). However, the ATPase activity of the T101I mutant was not significantly stimulated at 37°C compared with 30°C and was less than half that of WT Hsp90 at 37°C (Figure 5C). This finding suggests that the ts phenotype seen with the T101I mutation in vivo is a consequence of inadequate ATPase activity. It then remained to establish how the ATPase activities of WT and mutant Hsp90 relate to substrate binding and release.
Substrate release of Hsp90 is linked to the ATP hydrolysis rate
The enzymatic analysis of the Hsp90 ATPase activity provided us with a series of mutants that reduced or abolished the ATPase rate. Using these mutations, we tested whether the release of Hsp90 from substrate is linked to the binding or the hydrolysis of ATP. To permit their analysis in the functional LBD release assay described above (Figure 4), mutations equivalent to those characterized in yeast Hsp90 were introduced into human Hsp90. The resulting series of mutant Hsp90 proteins comprised: Hsp90-E47D and Hsp90-T115I, which have progressively lower ATPase rates compared with WT; Hsp90-E47A, which can bind but not hydrolyse ATP; and Hsp90-D93N, which is unable to bind ATP.
All of the mutant forms of Hsp90, when generated by in vitro translation and added to a normal LBD binding reaction, were co-precipitated with the LBD in significant amounts similar to WT Hsp90. Binding was direct since there was little elution with the Hop competitor TPR2A, and WT levels of Hsp90 release were produced upon elution with 500 mM NaCl (Figure 6A and data not shown). In the presence of ATP, the E47D mutant, having a moderate defect in the ATPase, was released less efficiently compared with WT Hsp90 (∼27% of total, compared with 35%) (Figure 6A). The T115I mutant, which has a still lower ATPase rate than Hsp90-E47D, released more poorly, at a level close to the ATPase-inactive E47A mutant (∼20%) (Figure 6A). This effect was not due to the lower affinity of the T115I mutant for ATP because it was also seen at higher ATP concentrations (5 mM and above), where the ATPase of Hsp90-T115I is close to Vmax (see also Figure 6C). As expected, the D93N mutant, which could not bind ATP, was not released from LBD significantly above the buffer control (Figure 6A). Thus, a progressive reduction in the ATPase rate of Hsp90 leads to a corresponding reduction in ATP-dependent release from substrate. Because the apparent affinity (KM) of Hsp90-E47D for ATP is expected to be at least as good as that of WT (Table I), the lower substrate release activity of this mutant is unlikely to be due to reduced binding of ATP and is attributed to the defect in ATP hydrolysis.
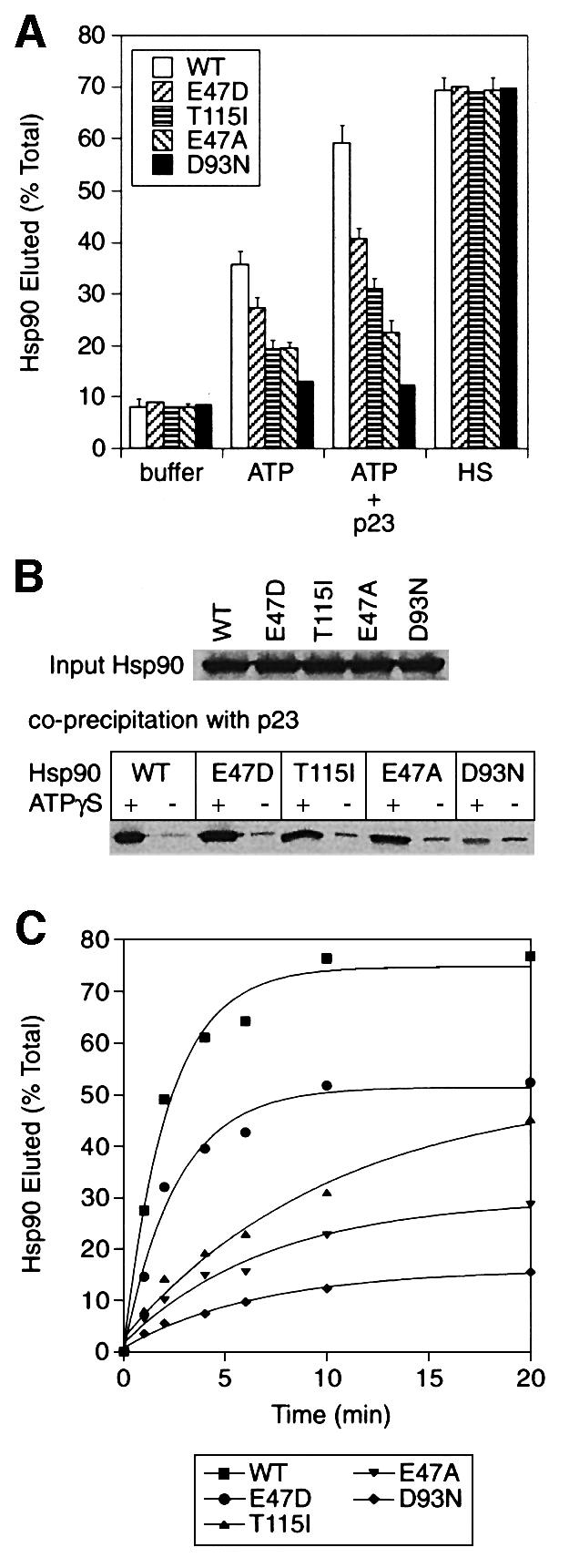
Fig. 6. Substrate release of Hsp90 is linked to the ATP hydrolysis rate. (A) LBD complexes containing radiolabelled WT or mutant Hsp90 were eluted with buffer alone or buffer containing 1 mM ATP, 1 mM ATP and 5 µM p23, or 500 mM NaCl (HS). (B) Radiolabelled WT or mutant Hsp90 translated in RL was co-precipitated with antibodies against p23 either with or without 5 mM ATPγS. Top, input Hsp90. (C) The elution from LBD complexes of WT Hsp90 and the indicated point mutants with 5 mM ATP and 5 µM p23 was monitored over time.
The correlation between the level of substrate release and ATPase activity was preserved in the presence of p23 (Figure 6A). Compared with WT Hsp90 (60% release), progressively less E47D and T115I were released upon addition of ATP and p23 (∼40 and 32%, respectively), but still above the level of release with ATP alone (Figure 6A). However, addition of p23 had little effect on the release of the non-hydrolysing mutant Hsp90-E47A (Figure 6A). As expected, the D93N mutant showed no response to p23 and ATP (Figure 6A). Based on these results, the level of substrate release of Hsp90 is correlated with the rate of ATP hydrolysis and not simply with ATP binding, and efficient ATP hydrolysis is also necessary for p23 stimulation of substrate release. It was noted that while ATP caused the release of some Hsp90-E47A from substrate complexes, the level of release achieved was lower than that observed with WT Hsp90 and ATPγS (see Figure 4A). Thus, hydrolysis of ATPγS probably increases the level of Hsp90 release from substrate somewhat above that expected for the true ATP-bound state of Hsp90, but this hydrolysis is too slow to allow a significant effect of p23.
The binding of the Hsp90 mutants to p23 was analysed to exclude the possibility that the defects in p23-induced substrate release could arise from an inability of the Hsp90 point mutants to bind p23. Indeed, in the presence of ATPγS, which stabilizes the Hsp90–p23 complex (Obermann et al., 1998), all of the radiolabelled Hsp90 point mutants were co-precipitated from RL with antibodies against p23 at levels comparable to WT Hsp90, except for the D93N mutant, which is unable to bind nucleotide (Figure 6B). As expected, only background levels of p23 binding were observed in the absence of nucleotide (Figure 6B). This confirms the conclusion above (see Figure 4): that binding of p23 to Hsp90 is by itself not sufficient to enhance substrate release. Rather, stimulation of substrate release by p23 requires the ATPase activity of Hsp90.
If indeed the efficiency of Hsp90 release from substrate is limited by the rate of ATP hydrolysis and not just ATP or p23 binding, this should be reflected in the actual rate of the release reaction. Therefore, the elution of WT and mutant Hsp90 proteins from LBD complexes was monitored over time under conditions expected to produce the maximum rate of elution (p23 and ATP). To ensure that ATP binding is not limiting, the proteins were eluted in a high concentration of ATP (5 mM). Compared with WT Hsp90 having a half-time of elution of ∼1.2 min, the E47D mutant had a slower rate of release (half-time of elution ∼1.8 min), corresponding to its reduced ATPase activity (Figure 6C). The T115I mutant, with a more severe ATPase defect, had a further reduced rate of release (half-time of elution >4 min) (Figure 6C). The elution of the non-hydrolysing Hsp90-E47A mutant from substrate was even slower, while the D93N mutant, unable to bind ATP, showed very little elution (Figure 6C). Thus, the kinetics of Hsp90 release from substrate is clearly linked to the rate of ATP hydrolysis rather than to the efficiency of ATP binding. This supports the mechanism suggested above, in which the conversion of Hsp90 from the ATP to the ADP state through hydrolysis is required for efficient release of substrate polypeptide.
Hydrolysis of ATP by Hsp90 is required not only to induce its fast release from substrate, but also for the stimulation of release by the co-chaperone p23 (Figure 6A). Because p23 has no effect on the ATP hydrolysis rate of Hsp90 (Figure 5B; Table I), it is unlikely to change the speed of the conversion between the ATP and ADP states of Hsp90, which leads to release of bound substrate. The mechanism of action of p23 is more probably that of a coupling factor, transmitting the conformational changes in the Hsp90 ATPase domains upon hydrolysis to other parts of the Hsp90 homodimer involved in handling the substrate polypeptide. The inability of the mutant Hsp90 proteins to release substrate properly by this regulated mechanism will greatly hinder or even abolish the folding of Hsp90-dependent polypeptides, and is likely to explain the ts or lethal phenotypes of these mutant Hsp90 forms in vivo.
Discussion
It is now firmly established that Hsp90 has an ATPase activity that is absolutely required for its function in vivo (Obermann et al., 1998; Panaretou et al., 1998). However, exact information on how this activity relates mechanistically to substrate binding and release has not been available. Our results now provide an outline of the ATPase-driven substrate binding cycle of Hsp90 (Figure 7): (1) the nucleotide-free state of the Hsp90 dimer is bound to substrate; (2) binding of ATP to Hsp90 causes only a slow release of substrate, but allows interaction of the Hsp90 complex with p23; (3) conversion of Hsp90 to the ADP state through hydrolysis of the bound ATP leads to fast release of substrate, and the efficiency of this release is enhanced by p23; and (4) after dissociation of ADP, Hsp90 can return to the nucleotide-free state. Transient stabilization of the nucleotide-free state of Hsp90 by the co-chaperone Hop has been proposed to allow loading of a fresh substrate protein by a mechanism involving Hop and Hsc70 (Prodromou et al., 1999).

Fig. 7. The ATP-driven substrate binding cycle of Hsp90. (1) Substrate protein (S) is stably bound by the nucleotide-free state of Hsp90. (2) Binding of ATP onto Hsp90 results in slow release of substrate. (3) Conversion of Hsp90 to the ADP state through ATP hydrolysis produces fast and complete release of substrate, enhanced by p23. (4) Substrate may be loaded onto Hsp90 at the nucleotide-free state via Hsc70 and Hop (Prodromou et al., 1999).
A key feature of this substrate binding cycle is that efficient release of substrate is linked to ATP hydrolysis. Notably, Hsp90 has recently been classified as a member of a superfamily of ATPases that includes the DNA gyrase subunit B (GyrB), the MutL DNA mismatch repair protein, and the histidine kinases EnvZ and CheA (Prodromou et al., 1997; Tanaka et al., 1998; Ban et al., 1999; Bilwes et al., 1999). Our model of the Hsp90 ATPase cycle is consistent with the biochemical mechanisms of these related proteins. For GyrB, binding of ATP causes homodimerization of the ATPase domain, forming a ‘clamp’ on a DNA strand. Hydrolysis of the bound ATP releases the DNA and resets the enzyme (Berger et al., 1996; Kampranis et al., 1999). MutL has an analogous mechanism in which ATP binding supports homodimer formation and hydrolysis opens up the protein by dissociating the dimer (Ban et al., 1999). Thus, we expect Hsp90 to follow a similar cycle whereby the ATP-bound state ‘closes’ the chaperone on a substrate polypeptide, and hydrolysis releases the substrate into solution. Intriguingly, electron microscopy and cross-linking data suggest that ATP causes the otherwise separate N-terminal ATPase domains of the Hsp90 homodimer to come together (Maruya et al., 1999; Prodromou et al., 2000).
Simply maintaining Hsp90 in the ADP-bound state is not sufficient to dissociate the complex with substrate (Figure 3). Rather, conversion from the ATP state to the ADP state is necessary for this step, whereby the rate of substrate release correlates with the rate of the Hsp90 ATPase (Figure 6). Conformational changes in GyrB upon binding and hydrolysis of ATP are tightly linked to the topoisomerase reaction, which drives DNA supercoiling (Kampranis et al., 1999). For the signal transducer CheA, large molecular motions coupled to ATP hydrolysis transmit signals between the regulatory and catalytic domains (Bilwes et al., 1999). By analogy, the hydrolysis of ATP by Hsp90 and the subsequent conversion from the ATP state to the ADP state may lead to a specific conformational motion that drives the release of substrate from the chaperone.
The ATPase cycle of Hsp90 thus differs significantly from that of Hsp70. Nucleotide binding and hydrolysis by Hsp70 switch the chaperone back and forth between the ATP state with fast on- and off-rates for peptide substrate, and the ADP state with high affinity for peptide (reviewed in Hartl, 1996; Bukau and Horwich, 1998). In contrast, the ATP state of Hsp90 still interacts with substrate polypeptide relatively tightly, as the rate of substrate release achieved by ATP binding without hydrolysis is too slow to be physiologically relevant (half-time >4 min; Figure 6C).
Our results suggest that p23 acts as a coupling factor for the ATP-dependent substrate release of Hsp90. In this role, p23 would increase the efficiency with which ATP binding and hydrolysis by Hsp90 are converted into the conformational changes that govern the interaction with substrate. For example, p23 may regulate the dimerization of the ATPase domains of Hsp90. The release factor activity of p23 would not be absolutely required for Hsp90 function, as some dissociation from substrate is observed without the co-chaperone (Figure 4). This agrees with the slow growth but non-lethal phenotype observed in an S.cerevisiae p23 null strain (Bohen, 1998; Fang et al., 1998). However, by enhancing the nucleotide regulation of Hsp90, p23 would in general improve the efficacy of Hsp90 function. Indeed, studies on the GR in vitro suggested that p23 increases hormone binding activity in cell lysates in an ATP-dependent manner (Hutchison et al., 1995). The substrate release function of p23 also explains the timing of its appearance in the Hsp90–receptor complexes at the specific step of complex dissociation (Johnson and Toft, 1994).
Given the wide range of polypeptides recognized by Hsp90, the requirement for p23 function may vary considerably among substrates. However, the ATP-driven substrate binding cycle of Hsp90 is the common core of its diverse cellular activities, which are still to be understood.
Materials and methods
Purification of proteins
DNA encoding the LBD (amino acids 518–795) of rat GR (Dr A.J.Caplan) was inserted into pMS-mycHis (Schneider et al., 1996). The LBD expressed in E.coli inclusion bodies was redissolved in 6 M guanidine hydrochloride and bound to a Ni-NTA–agarose column (Qiagen) and then refolded by a linear gradient into 300 mM KCl, 50 mM KH2PO4 pH 7.5 and 10% glycerol. LBD was eluted with 500 mM imidazole, 50 mM KH2PO4 pH 7.5 and 10% glycerol. Alternatively, LBD was selectively solubilized from inclusion bodies with 20 mM Tris–HCl pH 7.5 and 1% NP-40, bound to Ni-NTA agarose and eluted with 300 mM imidazole, 20 mM Tris–HCl pH 7.5 and 1% NP-40. The LBD was adjusted to 1% SDS and exchanged into buffer containing 100 mM KOAc, 20 mM HEPES–KOH pH 7.5, 1% NP-40, 1% sodium deoxycholate and 0.1% SDS. Both LBD preparations behaved identically in the assays used here.
The C-terminal domain of human Hsc70 (amino acid residues 153–413) was fused to GST (GST–C70) by inserting the corresponding DNA coding sequence into pGEX-2T (Pharmacia), and the expressed protein was purified by glutathione affinity chromatography. Recombinant human p23 in pET28 (Dr W.B.Pratt) was expressed and purified by chromatography on Q-Sepharose and Superdex 75 (Pharmacia). The TPR1 and TPR2A domains of human Hop (amino acid residues 1–119 and 223–353, respectively), full-length Hop, GST–C90 and S.cerevisiae Hsp90 and p23 were purified as described (Obermann et al., 1998; Young et al., 1998; Scheufler et al., 2000). The point mutation T101I was introduced into the plasmid encoding Hsp82 using the QuickChange mutagenesis system (Stratagene).
LBD binding and release
Rabbit RL (Green Hectares, WI) was desalted into 100 mM KOAc, 20 mM HEPES–KOH pH 7.5 and 5% glycerol (buffer B). For in vitro translation of Hsp90, DNA encoding human Hsp90α was inserted into pET15b (Novagen). All point mutations in Hsp90 were constructed from this plasmid using the QuickChange mutagenesis system (Stratagene), and Hsp90 was translated using TNT coupled RL (Promega).
Anti-myc monoclonal antibodies were covalently coupled to protein G–Sepharose (Pharmacia) (Schneider et al., 1996). Binding reactions contained 200 µg of LBD and anti-myc beads in 1 ml of buffer containing 500 mM NaCl, 20 mM Tris–HCl pH 7.5 and 2 mM EDTA (buffer H). After incubation, the beads were washed with buffer H and added to reactions containing 500 µl of RL, 500 µl of buffer B, 50 µl of labelled Hsp90, 5 mM MgOAc2 and 4 mM ATP. Steady-state binding of Hsp90 was attained after 10 min and reactions were stopped after 20 min by adding 10 U/ml apyrase (grade VII; Sigma), then split into 12 equal aliquots. The beads were washed with buffer B and resuspended in 100 µl of buffer B containing 5 mM MgOAc2 and other additions as indicated. After 10 min or otherwise indicated times, beads and supernatants were separated. The supernatants were precipitated with trichloroacetic acid for SDS–PAGE analysis, except for elutions in RL where 10 µl of the supernatants were analysed directly. The beads were eluted with SDS loading buffer. Quantitation was performed on a Fuji Film FLA-2000 phosphoimager.
For refolding of LBD, 0.1 µM LBD was incubated in RL with 5 mM MgOAc2, 1 mM ATP and 10 µCi of [3H]triamcinolone acetonide (Amersham) with or without 90 µM GA for 10 min at 30°C. The reaction was separated on a Superdex 200-3230 column (Pharmacia) equilibrated in buffer B, and the fractions analysed in a Packard Tri-Carb 1500 scintillation counter. The fractions were also immunoblotted to locate the LBD.
ATPase activity assays
The ATPase activity of Hsp90 (Hsp82) was determined as described (Obermann et al., 1998), except that reactions were in buffer B containing 10 mM MgOAc2, and between 0.2 and 10 mM ATP. Negative controls contained 90 µM GA. GA-inhibitable ADP production was fitted to a linear function to determine initial ATPase rates, and kinetic constants determined by direct fitting to the Michaelis–Menten equation using Kaleidagraph (Synergy Software).
Immunological methods
Immunoblots of isolated LBD–chaperone complexes were probed with antibodies against Hsp90, Hsc70 and Hop (Stressgen) and developed using the ECL detection system (Amersham Pharmacia). To co-precipitate Hsc70 with Hsp90, radiolabelled Hsc70 was generated by in vitro translation. The completed reactions were diluted with buffer B and immunoprecipitated with antibodies against Hsp90 (Stressgen) and protein A–Sepharose (Pharmacia). Immune pellets were washed with buffer B and eluted in 200 µl of buffer B with additions as indicated. Eluted supernatants and pelleted material were analysed as above. Radiolabelled Hsp90 was similarly co-precipitated with antibodies against p23 (Affinity BioReagents), except that immunoprecipitations contained either 10 mM MgOAc2 and 5 mM ATPγS, or apyrase.
Acknowledgments
Acknowledgements
The authors wish to thank Achim Brinker and Nick J.Hoogenraad for helpful discussion.
References
- Ban C., Junop,M. and Yang,W. (1999) Transformation of MutL by ATP binding and hydrolysis: a switch in DNA mismatch repair. Cell, 97, 85–97. [DOI] [PubMed] [Google Scholar]
- Berger J.M., Gamblin,S.J., Harrison,S.C. and Wang,J.C. (1996) Structure and mechanism of DNA topoisomerase II. Nature, 379, 225–232. [DOI] [PubMed] [Google Scholar]
- Bilwes A.M., Alex,L.A., Crane,B.R. and Simon,M.I. (1999) Structure of CheA, a signal-transducing histidine kinase. Cell, 96, 131–141. [DOI] [PubMed] [Google Scholar]
- Bohen S.P. (1998) Genetic and biochemical analysis of p23 and ansamycin antibiotics in the function of Hsp90-dependent signaling proteins. Mol. Cell. Biol., 18, 3330–3339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bose S., Weikl,T., Bügl,H. and Buchner,J. (1996) Chaperone functions of hsp90-associated proteins. Science, 274, 1715–1717. [DOI] [PubMed] [Google Scholar]
- Bresnick E.H., Dalman,F.C., Sanchez,E.R. and Pratt,W.B. (1989) Evidence that the 90-kDa heat shock protein is necessary for the steroid binding conformation of the L cell glucocorticoid receptor. J. Biol. Chem., 267, 14047–14053. [PubMed] [Google Scholar]
- Buchner J. (1999) Hsp90 & Co.—a holding for folding. Trends Biochem. Sci., 24, 136–141. [DOI] [PubMed] [Google Scholar]
- Bukau B. and Horwich,A. (1998) The Hsp70 and Hsp60 chaperone machines. Cell, 92, 351–366. [DOI] [PubMed] [Google Scholar]
- Caplan A.J. (1999) Hsp90’s secrets unfold: new insights from structural and functional studies. Trends Cell Biol., 9, 262–268. [DOI] [PubMed] [Google Scholar]
- Chen S. and Smith,D.F. (1998) Hop as an adaptor in the heat shock protein 70 (Hsp70) and Hsp90 chaperone machinery. J. Biol. Chem., 273, 35194–35200. [DOI] [PubMed] [Google Scholar]
- Chen S., Sullivan,W.P., Toft,D.O. and Smith,D.F. (1998) Differential interactions of p23 and the TPR-containing protein Hop, Cyp40, FKBP52 and FKBP51 with Hsp90 mutants. Cell Stress Chaperones, 3, 118–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csermely P. et al. (1993) ATP induces a conformational change of the 90-kDa heat shock protein (hsp90). J. Biol. Chem., 268, 1901–1907. [PubMed] [Google Scholar]
- Fang Y., Fliss,A.E., Rao,J. and Caplan,A.J. (1998) SBA1 encodes a yeast Hsp90 cochaperone that is homologous to vertebrate p23 proteins. Mol. Cell. Biol., 18, 3727–3734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeman B.C. and Morimoto,R.I. (1996) The human cytosolic molecular chaperones hsp90, hsp70 (hsc70) and hdj-1 have distinct roles in recognition of a non-native protein and protein refolding. EMBO J., 15, 2969–2979. [PMC free article] [PubMed] [Google Scholar]
- Freeman B.C., Toft,D.O. and Morimoto,R.I. (1996) Molecular chaperone machines: chaperone activities of the cyclophilin Cyp-40 and the steroid aporeceptor-associated protein p23. Science, 274, 1718–1720. [DOI] [PubMed] [Google Scholar]
- Hartl F.U. (1996) Molecular chaperones in cellular protein folding. Nature, 381, 571–580. [DOI] [PubMed] [Google Scholar]
- Höhfeld J., Minami,Y. and Hartl,F.U. (1995) Hip, a novel cochaperone involved in the eukaryotic Hsc70/Hsp40 reaction cycle. Cell, 83, 589–598. [DOI] [PubMed] [Google Scholar]
- Howard K.J., Holley,S.J., Yamamoto,K.R. and Distelhorst,C.W. (1990) Mapping the HSP90 binding region of the glucocorticoid receptor. J. Biol. Chem., 265, 11928–11935. [PubMed] [Google Scholar]
- Hutchison K.A., Stancato,L.F., Owens-Grillo,J.K., Johnson,J.L., Krishna,P., Toft,D.O. and Pratt,W.B. (1995) The 23-kDa acidic protein in reticulocyte lysate is the weakly bound component of the hsp foldosome that is required for assembly of the glucocorticoid receptor into a functional heterocomplex with hsp90. J. Biol. Chem., 270, 18841–18847. [DOI] [PubMed] [Google Scholar]
- Jakob U., Lilie,H., Meyer,I. and Buchner,J. (1995) Transient interaction of Hsp90 with early unfolding intermediates of citrate synthase. J. Biol. Chem., 270, 7288–7294. [DOI] [PubMed] [Google Scholar]
- Johnson J.L. and Toft,D.O. (1994) A novel chaperone complex for steroid-receptors involving heat-shock proteins, immunophilins and p23. J. Biol. Chem., 269, 24989–24993. [PubMed] [Google Scholar]
- Kampranis S.C., Bates,A.D. and Maxwell,A.M. (1999) A model for the mechanism of strand passage by DNA gyrase. Proc. Natl Acad. Sci. USA, 96, 8414–8419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura Y., Matsumoto,S. and Yahara,I. (1994) Temperature-sensitive mutants of hsp82 of the budding yeast Saccharomyces cerevisiae. Mol. Gen. Genet., 242, 517–527. [DOI] [PubMed] [Google Scholar]
- Maruya M., Sameshima,M., Nemoto,T. and Yahara,I. (1999) Monomer arrangement in Hsp90 dimer as determined by decoration with N and C-terminal region specific antibodies. J. Mol. Biol., 285, 903–907. [DOI] [PubMed] [Google Scholar]
- Nathan D.F. and Lindquist,S. (1995) Mutational analysis of Hsp90 function: interactions with a steroid receptor and a protein kinase. Mol. Cell. Biol., 15, 3917–3925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obermann W.M.J., Sondermann,H., Russo,A.A., Pavletich,N.P. and Hartl,F.U. (1998) In vivo function of hsp90 is dependent on ATP binding and ATP hydrolysis. J. Cell Biol., 143, 901–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panaretou B., Prodromou,C., Roe,S.M., O’Brien,R., Ladbury,J.E., Piper,P.W. and Pearl,L.H. (1998) ATP binding and hydrolysis are essential to the function of the hsp90 molecular chaperone in vivo. EMBO J., 17, 4829–4836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prodromou C., Roe,S.M., O’Brien,R., Ladbury,J.E., Piper,P.W. and Pearl,L.H. (1997) Identification and structural characterization of the ATP/ADP binding site in the hsp90 molecular chaperone. Cell, 90, 65–75. [DOI] [PubMed] [Google Scholar]
- Prodromou C., Siligardi,G., O’Brien,R., Woolfson,D.N., Regan,L., Panaretou,B., Ladbury,J.E., Piper,P.W. and Pearl,L.H. (1999) Regulation of hsp90 ATPase activity by tetratricopeptide repeat (TPR)-domain co-chaperones. EMBO J., 18, 754–762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prodromou C., Panaretou,B., Chohan,S., Siligardi,G., O’Brien,R., Ladbury,J.E., Roe,S.M., Piper,P.W. and Pearl,L.H. (2000) The ATPase cycle of Hsp90 drives a molecular ‘clamp’ via transient dimerization of the N-terminal domains. EMBO J., 19, 4383–4392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheibel T., Weikl,T. and Buchner,J. (1998) Two chaperone sites in Hsp90 differing in substrate specificity and ATP dependence. Proc. Natl Acad. Sci. USA, 95, 1495–1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheufler C., Brinker,A., Bourenkov,G., Pegoraro,S., Moroder,L., Bartunik,H., Hartl,F.U. and Moarefi,I. (2000) Structure of TPR domain–peptide complexes: critical elements in the assembly of the Hsp70–Hsp90 multichaperone machine. Cell, 101, 199–210. [DOI] [PubMed] [Google Scholar]
- Schmid D., Baici,A., Gehring,H. and Christen,P. (1994) Kinetics of molecular chaperone action. Science, 263, 971–973. [DOI] [PubMed] [Google Scholar]
- Schneider C., Sepp-Lorenzino,L., Nimmesgern,E., Ouerfelli,O., Danishevsky,S., Rosen,N. and Hartl,F.U. (1996) Pharmacologic shifting of a balance between protein refolding and degradation mediated by hsp90. Proc. Natl Acad. Sci. USA, 93, 14536–14541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith D.F. (1993) Dynamics of heat shock protein 90–progesterone receptor binding and the disactivation loop model for steroid receptor complexes. Mol. Endocrinol., 7, 1418–1429. [DOI] [PubMed] [Google Scholar]
- Smith D.F., Schowalter,D.B., Kost,S.L. and Toft,D.O. (1990) Reconstitution of progesterone receptor with heat shock proteins. Mol. Endocrinol., 4, 1704–1711. [DOI] [PubMed] [Google Scholar]
- Stebbins C.E., Russo,A.A., Schneider,C., Rosen,N., Hartl,F.U. and Pavletich,N.P. (1997) Crystal structure of an hsp90–geldanamycin complex: targeting of a protein chaperone by an antitumor agent. Cell, 89, 239–250. [DOI] [PubMed] [Google Scholar]
- Sullivan W., Stensgard,B., Caucutt,G., Bartha,B., McMahon,N., Alnemri,E.S., Litwack,G. and Toft,D. (1997) Nucleotides and two functional states of hsp90. J. Biol. Chem., 272, 8007–8012. [DOI] [PubMed] [Google Scholar]
- Tanaka T. et al. (1998) NMR structure of the histidine kinase domain of the E.coli osmosensor EnvZ. Nature, 396, 88–92. [DOI] [PubMed] [Google Scholar]
- Weikl T., Abelmann,K. and Buchner,J. (1999) An unstructured C-terminal region of the Hsp90 co-chaperone p23 is important for its chaperone function. J. Mol. Biol., 293, 685–691. [DOI] [PubMed] [Google Scholar]
- Young J.C., Schneider,C. and Hartl,F.U. (1997) In vitro evidence that hsp90 contains two independent chaperone sites. FEBS Lett., 418, 139–143. [DOI] [PubMed] [Google Scholar]
- Young J.C., Obermann,W.M.J. and Hartl,F.U. (1998) Specific binding of tretratricopeptide-repeat proteins to the C-terminal 12 kDa domain of Hsp90. J. Biol. Chem., 273, 18007–18010. [DOI] [PubMed] [Google Scholar]