

Abstract
Objective
Both platelet and neutrophil activation occur in sickle cell disease (SCD) but the interdependence of these events is unknown. The goal of this study was to determine the role of platelets in stimulating mouse and human neutrophil activation and pulmonary injury in SCD.
Methods and Results
Platelet activation and binding to leukocytes was measured in control and SCD mice and patients. Relative to controls, blood obtained from SCD mice or patients contained significantly elevated platelet-neutrophil aggregates (PNAs). Both platelets and neutrophils found in sickle PNAs were activated. Multi-spectral imaging (ImageStream) and conventional flow cytometry revealed a subpopulation of activated neutrophils with multiple adhered platelets that expressed significantly more CD11b and exhibited greater oxidative activity than single neutrophils. On average, wild type and sickle PNAs contained 1.1 and 2.6 platelets per neutrophil, respectively. Hypoxia/reoxygenation induced a further increase in platelet-neutrophil aggregates in SCD mice and additional activation of both platelets and neutrophils. Pretreatment of SCD mice with clopidogrel or P-selectin antibody reduced the formation of PNAs and neutrophil activation and decreased lung vascular permeability.
Conclusions
In sum, our findings suggest that platelet binding activates neutrophils and contributes to a chronic inflammatory state and pulmonary dysfunction in SCD. Inhibition of platelet activation may be useful to decrease tissue injury in SCD, particularly during the early stages of vaso-occlusive crises.
Keywords: platelet activation, sickle cell disease (SCD), inflammation, neutrophil activation, oxidative burst, clopidogrel, P-selectin
INTRODUCTION
Sickle cell disease (SCD) is the most common genetic hematological disorder in the United States. Vaso-occlusion characteristic of SCD has been viewed historically as resulting from deformed red blood cells (RBCs) that mechanically obstruct capillaries to produce tissue hypoxia.1 Present therapies for SCD are geared toward decreasing the concentration or polymerization rate of sickle hemoglobin.2 Recently, a modified paradigm has emerged suggesting that the wide spectrum of clinical manifestations of SCD results in part from recurrent episodes of disseminated microvascular ischemia/reperfusion (I/R) injury.3, 4 I/R triggers vascular inflammation characterized by increased adhesion of leukocytes5–8 and sickle RBCs9 to vascular endothelium as well as activation of coagulation,10–12 blood platelets,13–20 neutrophils,7 monocytes8, 21–23 and NKT cells.24 Since it has seen demonstrated that blockade of P-selectin-mediated platelet-leukocyte aggregation is beneficial in the animal models of vascular injury,25 we reasoned that platelet-leukocyte aggregation might contribute to the vascular inflammation and tissue injury that occurs in SCD. While increased formation of platelet-monocyte21 and platelet-RBC15 aggregates in SCD is well established, there are conflicting data regarding the occurrence of platelet-neutrophil aggregates (PNAs) in patients with SCD.18, 21 The question of neutrophil activation in SCD is an important one, since activated neutrophils play a major role in evoking vascular injury during I/R by adhering to blood vessels and releasing reactive oxygen species26. In vitro, binding of activated washed platelets to purified neutrophils results in their activation27–29 and in vivo studies demonstrate increased formation of neutrophil-platelet aggregates as a result of inflammation.30, 31
In this study, we investigated platelet-neutrophil aggregation in SCD using blood obtained from NY1DD sickle mice32, 33 and SCD patients. We found that both mice and people with SCD have markedly increased platelet-neutrophil aggregates (PNAs) compared to appropriate controls. Anti-platelet agents such as clopidogrel or anti-P-selectin antibodies as well as platelet depletion strongly suppressed formation of platelet-leukocyte aggregates and platelet-dependent neutrophil activation and pulmonary injury in sickle mice, indicating that anti-platelet therapy may be helpful for limiting vascular inflammation and injury in SCD.
MATERIALS AND METHODS
Human subjects
Peripheral venous blood samples were obtained from consenting adult SCD patients (hemoglobin S homozygotes) and age and race matched healthy control subjects (normal hemoglobin A) during a routine health examination at the Adult Hemoglobinopathy Clinic at the Washington University. All patients were at steady state, i.e. reported no more than typical pain at the time of phlebotomy. The human protocol was approved by the Institutional Review Boards of the Washington University and the University of Virginia.
Mice
Transgenic NY1DD mice on a C57BL6 genetic background were obtained from Robert P. Hebbel (University of Minnesota) and used as a model for SCD.33 NY1DD mice are deficient in mouse β-globin and express a fused human α-βS-globin transgene. These mice have a normal hematocrit at baseline, but exhibit multiple organ damage and leukocytosis.32 H/R evokes hemolysis associated with the development of increased inflammation. Congenic C57BL/6 female mice 8–12 weeks old were used as wild-type (WT) sex and age matched controls (Jackson Laboratory, Bar Harbor, ME). Experimental procedures were approved by the University of Virginia Animal Care and Use Committee.
Assessment of platelet activation in whole blood
Attempts to prepare platelets from sickle mice were complicated by the existence of platelet-platelet and platelet-leukocyte aggregates. Hence, platelet function was assessed in whole blood. Details about mouse blood collection and assessment of platelet activation in whole blood are described in the Supplemental Methods.
Platelet-leukocyte aggregates in whole blood
For mouse studies, heparin-anticoagulated blood was incubated with rat anti–mouse CD16/CD32 antibody to block the Fc III/II receptor. A 4-color flow cytometry assay was developed to analyze simultaneous platelet binding to neutrophils and monocytes in whole blood. Rat anti-mouse CD41-PE or rat anti-mouse GPIX-FITC antibodies were used to label platelets. The combinations of rat anti-mouse CD11b-APC, CD45-PerCP-Cy5.5 and GR-1-FITC antibodies and isotype controls (all from BD Biosciences PharMigen) were used for labeling leukocytes. Erythrocytes were lysed with High-Yield Lysing Solution (Caltag Labs). Neutrophils and monocytes were distinguished from other cells by their size and granularity as well as by anti-CD11b-APC and anti-GR-1-FITC antibody binding. Within the CD11b/CD45 double positive gate, platelet-neutrophil aggregate formation was calculated from CD41-PE/GR-1-FITC double-positive events, and platelet-monocyte aggregates from CD41-PE positive, GR-1-FITC negative events. The activation of platelets bound to leukocytes was assessed by counting CD62P-PE positive events in the CD45-PerCP-Cy5.5/GPIX-FITC double-positive gate.
Human blood samples were collected by venipuncture into Vacutainer-ACD tubes and prepared for flow cytometry as described above. PNAs in these samples were identified as CD14-negative and CD41+/CD16+ events and platelet-monocyte aggregates as CD16-negative and CD41+/CD14+ events in the CD11b+/CD45+ leukocyte gate. The activation of platelets bound to neutrophils was determined by counting CD62P+ events on PNAs. Fluorescence intensity was measured with a CyAn™ ADP LX 9 Color Analyzer (DakoCytomation) and data analysis performed with FlowJo software. Details about antibodies used in human studies are given in the Supplemental Methods.
Multi-spectral Imaging Flow Cytometry (MIFC)
Platelet-neutrophil aggregates were imaged by MIFC using an ImageStream flow cytometer (Amnis Corporation, Seattle, WA). At least 5,000 cells were collected from each sample and data were analyzed using IDEAS image analysis software (Amnis Corp, Seattle, WA). Platelets were labeled using anti-CD41-PE antibody. Neutrophil images were identified as GR-1-FITC+/HI scatter and CD11b+ events. Neutrophil-bound platelets were identified using a GR-1 membrane mask and the Small Spot Intensity feature of the IDEAS software. The number of platelets bound per neutrophil was determined using the Spot Count and Peak Intensity algorithms of the IDEAS software and confirmed by examination of images positive for CD41and GR-1.
H2O2 production by neutrophils
We developed a new 5-color flow cytometry assay to assess oxidative burst simultaneously in platelet-neutrophil aggregates and single neutrophils in whole blood. Respiratory burst was monitored by dihydrorhodamine (DHR) 123 which produces fluorescent rhodamine123 after oxidation by hydrogen peroxide (H2O2). Heparinized whole blood (0.5 mL) from WT or sickle mice treated for 3 days with clopidogrel or vehicle and then subjected to H/R was incubated for 30 minutes with a cocktail of DHR 123 and appropriate fluorescent antibodies at 37°C and then transferred to ice. DHR 123 fluorescence was identified in both CD45+/CD11b+/GR1+/CD41+ and CD45+/CD11b+/GR1+/CD41− regions and analyzed with a CyAn™ADP LX9 Color Analyzer.
Hypoxia/reoxygenation and drug treatment of mice
Mice were placed in a hypoxia chamber (8% O2 -92% N2; Coy Laboratory Products, Inc) for 3 h and then reoxygenated for 3 h in ambient air. The number of platelet-leukocyte aggregates did not change during hypoxia (data not shown) but increased to a maximum 3 h after reoxygenation. Some mice received carotid artery injections of 100 μg of blocking anti-P-selectin antibody (RB40.34, BD Biosciences) or control IgG just before the reoxygenation period. In other experiments, vehicle or clopidogrel (30 mg/kg/day) were administrated to mice by oral gavage for 3 days before H/R.
Platelet depletion effects on pulmonary neutrophil accumulation and vascular permeability
Platelets were depleted by a single intravenous injection of anti-GPIbα antibody (2μg/g; Emfret Analytics).34 After 24 hours circulating platelets were reduced in sickle mice by 85 % (from 950 ± 40 × 103/μL to 142 ± 8 × 103/μL). Subsequently, mouse lungs were harvested and neutrophils/lung counted by flow cytometry as described.24 Pulmonary vascular permeability was assessed by Evans blue dye (EBD) extravasation as described in the Supplemental Methods.
Data analysis
Representative data shown in the figures are typical of 3 or more replicate experiments. Where applicable, results are presented as means ± SD. WT and NY1DD mice, or African-American healthy controls and SCD patients were compared using the Student’s t test.
RESULTS
Activation of αIIbβ3 integrin and increased membrane expression of CD40L and P-selectin on sickle platelets
There are several mouse models of SCD.35 One frequently used model is the Berkeley sickle mouse, which expresses exclusively human sickle hemoglobin and develops severe SCD.36, 37 However, these mice exhibit marked thrombocytopenia38 which does not parallel the increase in platelet number usually noted in human SCD at baseline.39 Hence, we focused our studies on the NY1DD mouse model of SCD that develops moderate SCD.32, 33 Like SCD patients, NY1DD mice exhibit a pro-inflammatory phenotype with leukocytosis and increased platelet number.24 Since platelet activation is well established in human SCD,19, 20 we first determined if platelet activation is replicated in NY1DD mice by employing the JON/A antibody that recognizes the activated conformation of the murine fibrinogen receptor, αIIbβ3.40 The percentage of JON/A-positive platelets was significantly greater in sickle (6.4%) compared to WT mouse blood (3%), (p < 0.01) and was increased significantly more in SCD than in WT mice after stimulation for 10 seconds with ADP (Figure 1A). Surface expression of CD40L was used as additional marker of platelet activation. Compared to WT, unstimulated sickle blood contained more CD40L+ platelets (P < 0.001, Figure 1B). Stimulation of heparinized blood for 15 seconds with the strong platelet agonist convulxin (50 nM), induced a rapid increase in CD40L in WT platelets, but a significantly greater activation in sickle platelets. Convulxin triggered a 2.3 ± 0.3 and a 3.5 ± 0.2 fold increase in surface expression of CD40L in WT and SCD mice, respectively, based on the geometric mean of fluorescence intensity (GMFI).
Figure 1. Increased activated αIIbβ3 integrin and CD40L on platelets in SCD mouse blood.

(A) Activation of the αIIbβ3 integrin in platelets at rest and following stimulation with 1 μM ADP were measured by binding of JON/A-PE antibody. Following stimulation, blood samples were diluted and immediately analyzed by flow cytometry. The percentages of the total platelet population that is activated (JON/A+) in resting and ADP-stimulated WT and SCD blood are plotted as means ± SD, n = 5 for all groups. (B) Expression of platelet CD40L in resting and convulxin-stimulated blood was determined from binding of anti-CD40L-PE antibody. Platelets were identified by FSC and SSC characteristics and by GPIX-FITC binding. The percentages of the total platelet population that is CD40L+ in blood from WT and SCD mice are plotted as means ± SD from 3–4 independent experiments.
P-selectin on activated platelets is a major mediator of adhesion to leukocytes.41 In unstressed mice, the percentage of P-selectin positive platelets was very low (<1%) in both WT and sickle mice (Figure 2A). The percentage of P-selectin positive-platelets increased in response to convulxin to 34% in WT mice and to 50% in SCD mice (Supplemental Figure Ia). By contrast, platelets aggregated with leukocytes were partially P-selective positive, 5.8 ± 0.3% in WT mice, and 16.0 ± 1.8% in SCD mice (Figure 2B). Convulxin treatment increased the percentage of P-selectin positive platelets in platelet–leukocyte aggregates to 44% in WT mice and to 71% in sickle mice (p<0.005, Supplemental Figure Ib).
Figure 2. Increased P-selectin on platelet-leukocyte aggregates in sickle mice.

Platelets and leukocytes were recognized by their forward and side scatter characteristics. Platelet singlets were further identified with anti-GPIX-FITC. Platelet-leukocyte complexes were identified by anti-CD45-PerCP-Cy5.5 and anti-GPIX-FITC positive fluorescence. P-selectin expression was assessed using anti-CD62P-PE. Representative histograms showing P-selectin expression on (A) platelet singlets and (B) platelet-leukocyte heteroaggregates, n ≥ 5. Data are expressed as means ± SD.
Increased platelet-leukocyte aggregation in SCD
The percentage of platelets found in PNAs or platelet-monocyte aggregates in mouse peripheral blood was assessed by 4-color flow cytometry. At baseline, about 2.5-fold more sickle than WT neutrophils and 1.7-fold more sickle than WT monocytes had adhered CD41+-platelets (Figure 3A and 3B). Neutrophils aggregated with platelets were more activated in sickle than WT blood based on higher expression of CD11b (MFI 459 ± 39 vs 233 ± 50, p< 0.001, n = 10). We also detected eosinophil-platelet aggregates that were also significantly increased in sickle mice compared to WT (Supplemental Figure II). Once formed, platelet-leukocyte aggregates were stable and did not change in number for up to 4 hours following addition of heparin or anti-P-selectin blocking antibody, RB40.34 to blood (data not shown).
Figure 3. Increased platelet interactions with leukocytes in NY1DD sickle mice and SCD patients.

(A) Representative FACS analysis of whole blood platelet-neutrophil (N) and platelet-monocyte (M) aggregates in WT and SCD mice. CD45+ leukocytes: neutrophils (N) and monocytes (M) were gated by forward and side light scatter (not shown) and the differential expression of GR-1-FITC, and CD11b-APC. The percentage of neutrophils and monocytes that are found in heteroaggregates with platelets (CD41+) is greater in SCD than WT mice. (B) Quantitative analysis of CD41+ neutrophils, monocytes and eosinophils in WT and sickle mice blood. Results are presented as means ± SD, n=10 for both groups, ***p<0.001, *p<0.05. (C) Increased formation of platelet-neutrophil and platelet-monocyte aggregates in blood obtained from the adult SCD patients compared to healthy controls. Results are presented as means ± SD, n=7 for both groups, ** p<0.005, *p<0.05.
Since the number of PNAs in blood from children with SCD was previously reported to be similar to controls,18 we studied platelet-neutrophil interactions in adults with SCD using 5-color flow cytometry. The percentage of neutrophils and monocytes found aggregated with platelets in blood obtained from SCD patients vs. controls was, 2.5-fold and 2.1-fold higher (Figure 3C). Human neutrophils in PNAs were more activated in SCD patients than in control subjects based on the expression of CD11b (MFI 311 ± 80 vs 210 ± 57, p<0.05, n =7). The number of P-selectin+ platelets in blood of SCD patients was not different from controls. However, the surface expression of P-selectin given as MFI on PNAs was significantly greater in SCD patients than controls 63.2 ± 13.3 vs 24.8 ± 2, p<0.05, n = 7. In sum, as in mice, patients with SCD were found to have increased numbers of PNAs, and aggregation resulted in activation of both platelets and neutrophils.
ImageStream analysis of platelet-neutrophil aggregates
We used ImageStream fluorescence imaging to confirm the existence of platelet-neutrophil aggregates and determine the number of platelets attached to each neutrophil (Figure 4). The percentage of neutrophils with adherent platelets was significantly higher in SCD mice than in WT (15.3 ± 2.4 % vs. 5.8 ±0.9 %). These numbers varied with age, sex and handling of mice, so it was important to use age and sex-matched mice. In this experiment, the average number of adherent platelets per neutrophil-platelet aggregate was about 2.4-fold as high in SCD compared to WT mice (Figure 4C and 4D). This was confirmed by measuring the mean fluorescence intensity of anti-CD41 on adhered platelets, indicative of the number of platelets per neutrophil, which was 2.3-fold higher in sickle mice compared to WT. The expression of CD11b was 1.7-fold higher in neutrophils aggregated with platelets compared to single neutrophils in sickle mice. There was no significant difference in CD11b expression between platelet-free and platelet+-neutrophils in WT mice. In SCD, 40% of platelet-neutrophil aggregates included more than 2 (up to 6) platelets, while in WT mice, only 20% of aggregates had more than 1 adhered platelet.
Figure 4. Images of platelet-neutrophil aggregates (PNAs).

(A) Peripheral blood cells were labeled with CD11b-Cy5-PE, GR-1-FITC and CD41-PE. Platelet-neutrophil aggregates were identified as GR-1 HI/CD11b+/CD41+ events and analyzed using the IDEAS™ software (AMNIS). (B) Quantitative analysis of CD41+ neutrophils in WT and SCD mouse blood shown in panel (D). Results are presented as means ± SD, n =3, *** p<0.001. (C) The number of platelets bound per neutrophil was determined by the Spot Count and Peak Intensity features of IDEAS software and calculated as the number of total platelets attached to CD41+ neutrophils divided by the number of CD41+ neutrophils in WT or SCD blood. Data are presented as means ± SD from 3 separate experiments, ** p<0.005 (D) Images of platelet-neutrophil aggregates are shown as Bright-Field illumination images (gray) and as composite images for six representative CD41+ neutrophils (of 21 WT and of 59 sickle neutrophils imaged). The composite images represent an overlay of the images from the 3 fluorescent channels (FITC, PE, and Cy5-PE) and demonstrate platelets (red/yellow) bound to neutrophils (green).
Hypoxia/reoxygenation enhances P-selectin-dependent formation of platelet-neutrophil aggregates
H/R has been shown to exacerbate the inflammatory response in sickle mice.4, 42 To investigate whether H/R has an effect on the formation of platelet-neutrophil aggregates, WT and SCD mice were placed in a hypoxia chamber and exposed to 8% O2 and 92% N2 for 3 h followed by 3 h of reoxygenation in ambient air. Flow cytometric analysis revealed that H/R elicited a significant increase in platelet-neutrophil aggregates in blood from sickle mice (Figure 5A). The injection of anti-P selectin antibody just before reoxygenation led to a significant reduction in platelet-neutrophil aggregates following H/R. Treatment of SCD mice with clopidogrel or platelet depletion also resulted in significant suppression of CD41+-neutrophils both at baseline and following subsequent H/R in sickle mice (Figure 5A). Platelet binding to neutrophils has been reported to increase neutrophil adhesion to endothelium and may be responsible for endothelial activation.43, 44 To test the hypothesis that activated platelets contribute to endothelial dysfunction in SCD, platelets were depleted and pulmonary microvascular permeability was measured with Evans blue dye (EBD). Platelet depletion was associated with a decreased number of pulmonary neutrophils, from 1.71 ± 0.18×105/lung to 0.32 ± 0.11×105/lung (n=10, p<0.05), and with decreased pulmonary vascular leakage (Figure 5B) in sickle mice. These findings implicate platelet activation and exposure of platelet P-selectin as functionally important events mediating platelet-neutrophil aggregate formation that may contribute to vaso-occlusive crisis in SCD.
Figure 5. Anti-platelet treatments suppress H/R-induced formation of platelet-neutrophil aggregates, neutrophil activation and lung vascular permeability in SCD mice.
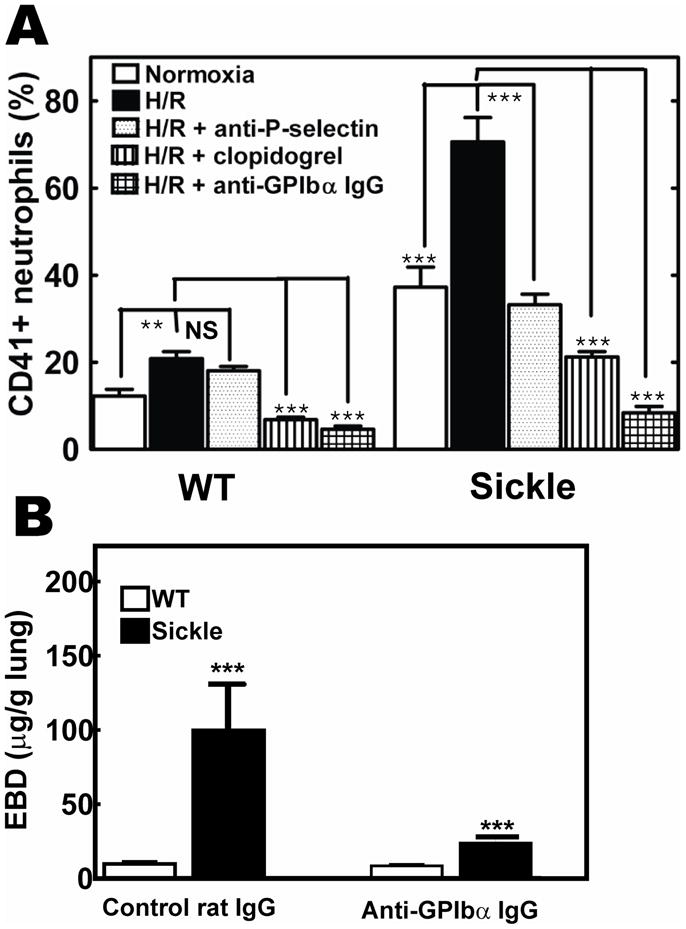
(A) WT and SCD mice were subjected to 3 h hypoxia (8% O2 -92% N2) followed by 3 h reoxygenation (H/R). Prior oral treatment with clopidogrel (3 days), injection of 100 μg of anti-P-selectin antibody or platelet depletion with anti GPIbα antibody prevented formation of platelet-neutrophil aggregates in SCD mice. (B), Platelet depletion with anti-GPIbα-antibody (24 hrs) reduced pulmonary vascular leak measured with Evans blue dye (EBD). Data are expressed as means ± SD (n=3). **p<0.005, ***p<0.001.
Adherent platelets promote neutrophil oxidative activity
We investigated whether platelet binding to neutrophils influences neutrophil activation in SCD mice by measuring oxidative burst and expression of CD11b adhesive receptors using 5-color flow cytometry. The oxidative burst that results in production of hydrogen peroxide (H2O2) was detected from the intracellular oxidation of the fluorescent dye DHR 123 (Figure 6). In SCD mice, PNAs (Figure 6A, CD41+) produced substantially more H2O2 (more oxidative burst; Figure 6B) and had significantly higher expression of CD11b (Figure 6C) than single neutrophils (Figure 6A, CD41-free), suggesting that platelet binding activates neutrophils in vivo.
Figure 6. Neutrophils with attached platelets (CD41+) have increased oxidative burst and CD11b expression in sickle mice.

(A) Gating strategy for evaluation of oxidative-burst in platelet+ or platelet-free neutrophils. Quantitative analysis in neutrophils of: (B) oxidative burst; and (C) CD11b expression. Results are expressed as means ± SD, n=5, *p<0.05, ***p<0.001.
DISCUSSION
Circulating blood leukocytes in SCD, particularly neutrophils,7, 45, 46 display an activated phenotype that may predispose them to endothelial adhesion and amplification of vascular inflammation and vaso-occlusion. In this study, we sought to determine if platelet activation that occurs in SCD15, 16, 47 actively contributes to SCD-associated neutrophil activation. Our data reveal for the first time an increased formation of PNAs in blood obtained from SCD-patients and from NY1DD sickle mice. In both sickle patients and NY1DD sickle mice the most activated neutrophils were those with adhered platelets as assessed by oxidative burst and increased expression of CD11b adhesive receptors (Figure 3). In sickle mice the formation of PNA was exacerbated following hypoxia/reoxygenation, suggesting that platelet activation and adhesion to neutrophils might occur as a result of H/R during vaso-occlusive crisis. Since, the most activated neutrophils were those with adhered platelets we hypothesized that it may be possible to reduce neutrophil activation in SCD by inhibiting platelet activation. Indeed, treatment of SCD mice with the anti-platelet drug clopidogrel, which blocks P2Y12 ADP receptors,48, 49 or with anti-P-selectin antibody significantly reduced the number of platelet-neutrophil aggregates and lowered overall neutrophil activation. Depletion of platelets with anti-GPIbα antibody also reduced the number of platelet-neutrophil aggregates and decreased pulmonary neutrophil infiltration and vascular permeability in sickle lung. This novel link between platelet and neutrophil activation in SCD provides a new perspective into SCD pathology and treatment.
In vitro evidence indicates that platelet binding to leukocytes likely changes both platelet and leukocyte activation.28, 50 Thrombotic processes associated with enhanced platelet activation and formation of platelet-leukocyte aggregates have been observed in several chronic inflammatory diseases.51, 52 Despite the fact that activation of platelets,15, 16, 17, 19 monocytes,21 neutrophils7, 46 and eosinophils53 have been observed in human SCD, the only previously reported increases in heteroaggregates that contain platelets are those formed between platelets and monocytes or platelets and sickle RBCs.21, 15 The analysis of platelet function in SCD is complicated by the fact that severe chronic inflammation may result in platelet depletion, margination and desensitization.54 Moreover, previous studies have not attempted to measure in vivo the ratio of platelets bound per neutrophil or the effect of platelet binding on the oxidative activity of individual neutrophils.16, 18, 21, 46 In a study of severe SCD in children with nocturnal hypoxia, the fraction of platelet-neutrophil aggregates was not significantly changed compared to controls.18 The discrepancy with our findings could be due partly to difference in the patient age and duration of the disease/or different techniques of blood sample preparation and analysis. We used 5-color flow cytometry of cell markers with confirmation by ImageStream analysis to define platelet-neutrophil aggregates in ACD-anticoagulated blood while Inwald et al18 used specific light scatter characteristic of neutrophils and CD61+-platelets in citrate-anticoagulated blood. In NY1DD mice with mild SCD we were able to consistently demonstrate that platelet binding to neutrophils has a significant impact on the overall activation of neutrophils which likely promotes adhesion to endothelial cells and induction of pulmonary vascular injury in SCD.5, 46, 50 This is in agreement with previous studies showing that ligation of the P-selectin glycoprotein ligand 1 (PSGL-1) on neutrophils by P-selectin Ig chimera leads to production of reactive oxygen intermediates and increased neutrophil expression of cell-surface CD11b.55 The anlaysis of platelet-leukocyte aggregates revealed higher numbers of platelets bound to neutrophils in SCD: on average, about 2.6 platelets/neutrophil in platelet-neutrophil aggregates in SCD vs. about 1.1 platelet/neutrophil in WT mice. Thus, the binding of multiple platelets associated with increase neutrophil adhesive and oxidative activities may represent a newly recognized mechanism contributing to neutrophil activation in peripheral blood in vivo.
Several investigators have reported that only activated platelets expressing surface P-selectin bind to leukocytes.41 To test the hypothesis that P-selectin is required for neutrophil activation in SCD we studied the effects of the anti-platelet drug clopidogrel and anti-P-selectin antibody on platelet-neutrophil aggregate formation in sickle mice at baseline and following H/R. H/R triggers vascular inflammation in transgenic sickle mice.4 We found that in NY1DD mice H/R enhances platelet activation and formation of platelet-neutrophil aggregates. Clopidogrel decreased the number of circulating platelet-neutrophil aggregates and suppressed oxidative burst in neutrophil- platelet aggregates in sickle mice at baseline and following H/R. Injection of P-selectin blocking antibody to H/R-treated sickle mice prior to reoxygenation almost completely prevented formation of new platelet-neutrophil aggregates, suggesting that H/R leads to P-selectin-dependent platelet-neutrophil binding. The inability of anti-P-selectin to dissociate already preexisting platelet-neutrophil aggregates in SCD is consistent with the idea that P-selectin bound to PSGL-1 is not accessible for anti-P-selectin antibody or that the initial platelet adhesion to neutrophils occurs via surface P-selectin, but stabilization of aggregates requires additional adhesive interactions such as binding of neutrophil CD11b to platelet GPIb.56 These observations provide strong evidence that platelets play a role to enhance neutrophil activation in vivo in SCD. The physiological significance of increased formation of platelet-leukocyte aggregates in peripheral blood in inflammatory diseases is unknown. Previous studies of P-selectin+-platelet-leukocyte aggregates indicate that leukocytes with attached platelets tether and roll on endothelial cells with higher avidity than single leukocytes, thereby exacerbating endothelial inflammation.57–59
Recently, we showed that NY1DD mice display baseline pulmonary injury.24 In the current study, we found that platelet depletion reduced the number of circulating platelet-neutrophil aggregates, significantly suppressed neutrophil migration to the lung and reduced lung vascular permeability, suggesting that platelets contribute to acute lung injury in SCD.43 Activated platelets with surface expression of P-selectin have been shown to be essential for neutrophil recruitment into kidney in acute post-ischemic renal failure60 and for leukocyte extravasation into airways after allergen challenge.61 Thus targeting platelet activation and membrane expression of P-selectin may be a useful strategy to reduce lung injury in SCD, particularly at the onset of vaso-occlusive crisis. In previous studies, chronic anti-platelet drug therapy (ticlopidin, low dose aspirin) and anticoagulant treatment (low dose heparin, warfarin) have not been effective in the prophylaxis of acute vaso-occlusive events in human SCD.62–64 However, these studies have not evaluated dose ranging, nor measured the levels of platelet activation or the extent of vascular damage before and after treatment. Hence it is not known if these treatments were sufficiently aggressive to reduce platelet activation and platelet-leukocyte aggregation.
Our findings suggest that in addition to their procoagulant role, platelets may contribute directly to ongoing vascular inflammation in SCD by activating neutrophils. Consequently, therapies targeting platelet function and platelet interactions with other blood cells may help to control inflammation. The contribution of platelet activation to SCD pathophysiology should be further investigated using newly available anti-platelet agents with the evaluation of biomarkers, including P-selectin expression and platelet-leukocyte aggregate formation. The fact that H/R induces an increase in platelet-leukocyte interaction, which decreases to baseline levels after administration of clopidogrel or anti-P-selectin antibody, suggests that platelet-leukocyte cross-talkcontributes to vaso-occlusive events in SCD. Further studies will be required to revisit anti-platelet therapy for SCD with emphasis on inhibiting P-selectin-mediated platelet-leukocyte interactions.
Supplementary Material
Acknowledgments
We thank Dr. Robert Strieter (University of Virginia) for helpful comments, Dr. Joanne Lannigan and Mr. Mike Solga (University of Virginia Flow Cytometry Core Facility) for their valuable assistance.
Source of Funding
Supported by grant P01 HL073361 from the NationalInstitutes of Health (J. L).
Footnotes
Disclosures
The authors report no conflicts of interest related to this study.
References
- 1.Hebbel RP, Eaton JW, Steinberg MH, White JG. Erythrocyte/endothelial interactions and the vasocclusive severity of sickle cell disease. Prog Clin Biol Res. 1981;55:145–162. [PubMed] [Google Scholar]
- 2.Raghupathy R, Billett HH. Promising therapies in sickle cell disease. Cardiovasc Hematol Disord Drug Targets. 2009;9:1–8. doi: 10.2174/187152909787581354. [DOI] [PubMed] [Google Scholar]
- 3.Osarogiagbon UR, Choong S, Belcher JD, Vercellotti GM, Paller MS, Hebbel RP. Reperfusion injury pathophysiology in sickle transgenic mice. Blood. 2000;96:314–320. [PubMed] [Google Scholar]
- 4.Kaul DK, Hebbel RP. Hypoxia/reoxygenation causes inflammatory response in transgenic sickle mice but not in normal mice. J Clin Invest. 2000;106:411–420. doi: 10.1172/JCI9225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fadlon E, Vordermeier S, Pearson TC, Mire-Sluis AR, Dumonde DC, Phillips J, Fishlock K, Brown KA. Blood polymorphonuclear leukocytes from the majority of sickle cell patients in the crisis phase of the disease show enhanced adhesion to vascular endothelium and increased expression of CD64. Blood. 1998;91:266–274. [PubMed] [Google Scholar]
- 6.Turhan A, Weiss LA, Mohandas N, Coller BS, Frenette PS. Primary role for adherent leukocytes in sickle cell vascular occlusion: a new paradigm. Proc Natl Acad Sci U S A. 2002;99:3047–3051. doi: 10.1073/pnas.052522799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lard LR, Mul FP, de Haas M, Roos D, Duits AJ. Neutrophil activation in sickle cell disease. J Leukoc Biol. 1999;66:411–415. doi: 10.1002/jlb.66.3.411. [DOI] [PubMed] [Google Scholar]
- 8.Belcher JD, Marker PH, Weber JP, Hebbel RP, Vercellotti GM. Activated monocytes in sickle cell disease: potential role in the activation of vascular endothelium and vaso-occlusion. Blood. 2000;96:2451–2459. [PubMed] [Google Scholar]
- 9.Hebbel RP. Adhesive interactions of sickle erythrocytes with endothelium. J Clin Invest. 1997;100:S83–86. [PubMed] [Google Scholar]
- 10.Francis RB., Jr Platelets, coagulation, and fibrinolysis in sickle cell disease: their possible role in vascular occlusion. Blood Coagul Fibrinolysis. 1991;2:341–353. doi: 10.1097/00001721-199104000-00018. [DOI] [PubMed] [Google Scholar]
- 11.Key NS, Slungaard A, Dandelet L, Nelson SC, Moertel C, Styles LA, Kuypers FA, Bach RR. Whole blood tissue factor procoagulant activity is elevated in patients with sickle cell disease. Blood. 1998;91:4216–4223. [PubMed] [Google Scholar]
- 12.Ataga KI, Orringer EP. Hypercoagulability in sickle cell disease: a curious paradox. Am J Med. 2003;115:721–728. doi: 10.1016/j.amjmed.2003.07.011. [DOI] [PubMed] [Google Scholar]
- 13.Westwick J, Watson-Williams EJ, Krishnamurthi S, Marks G, Ellis V, Scully MF, White JM, Kakkar VV. Platelet activation during steady state sickle cell disease. J Med. 1983;14:17–36. [PubMed] [Google Scholar]
- 14.Beurling-Harbury C, Schade SG. Platelet activation during pain crisis in sickle cell anemia patients. Am J Hematol. 1989;31:237–241. doi: 10.1002/ajh.2830310404. [DOI] [PubMed] [Google Scholar]
- 15.Wun T, Paglieroni T, Tablin F, Welborn J, Nelson K, Cheung A. Platelet activation and platelet-erythrocyte aggregates in patients with sickle cell anemia. J Lab Clin Med. 1997;129:507–516. doi: 10.1016/s0022-2143(97)90005-6. [DOI] [PubMed] [Google Scholar]
- 16.Wun T, Paglieroni T, Rangaswami A, Franklin PH, Welborn J, Cheung A, Tablin F. Platelet activation in patients with sickle cell disease. Br J Haematol. 1998;100:741–749. doi: 10.1046/j.1365-2141.1998.00627.x. [DOI] [PubMed] [Google Scholar]
- 17.Tomer A, Harker LA, Kasey S, Eckman JR. Thrombogenesis in sickle cell disease. J Lab Clin Med. 2001;137:398–407. doi: 10.1067/mlc.2001.115450. [DOI] [PubMed] [Google Scholar]
- 18.Inwald DP, Kirkham FJ, Peters MJ, Lane R, Wade A, Evans JP, Klein NJ. Platelet and leucocyte activation in childhood sickle cell disease: association with nocturnal hypoxaemia. Br J Haematol. 2000;111:474–481. doi: 10.1046/j.1365-2141.2000.02353.x. [DOI] [PubMed] [Google Scholar]
- 19.Villagra J, Shiva S, Hunter LA, Machado RF, Gladwin MT, Kato GJ. Platelet activation in patients with sickle disease, hemolysis-associated pulmonary hypertension and nitric oxide scavenging by cell-free hemoglobin. Blood. 2007 doi: 10.1182/blood-2006-12-061697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lee SP, Ataga KI, Orringer EP, Phillips DR, Parise LV. Biologically active CD40 ligand is elevated in sickle cell anemia: potential role for platelet-mediated inflammation. Arterioscler Thromb Vasc Biol. 2006;26:1626–1631. doi: 10.1161/01.ATV.0000220374.00602.a2. [DOI] [PubMed] [Google Scholar]
- 21.Wun T, Cordoba M, Rangaswami A, Cheung AW, Paglieroni T. Activated monocytes and platelet-monocyte aggregates in patients with sickle cell disease. Clin Lab Haematol. 2002;24:81–88. doi: 10.1046/j.1365-2257.2002.00433.x. [DOI] [PubMed] [Google Scholar]
- 22.Shet AS, Aras O, Gupta K, Hass MJ, Rausch DJ, Saba N, Koopmeiners L, Key NS, Hebbel RP. Sickle blood contains tissue factor-positive microparticles derived from endothelial cells and monocytes. Blood. 2003;102:2678–2683. doi: 10.1182/blood-2003-03-0693. [DOI] [PubMed] [Google Scholar]
- 23.Brittain JE, Knoll CM, Ataga KI, Orringer EP, Parise LV. Fibronectin bridges monocytes and reticulocytes via integrin alpha4beta1. Br J Haematol. 2008;141:872–881. doi: 10.1111/j.1365-2141.2008.07056.x. [DOI] [PubMed] [Google Scholar]
- 24.Wallace KL, Marshall MA, Ramos SI, Lannigan JA, Field JJ, Strieter RM, Linden J. NKT cells mediate pulmonary inflammation and dysfunction in murine sickle cell disease through production of IFN-gamma and CXCR3 chemokines. Blood. 2009;114:667–676. doi: 10.1182/blood-2009-02-205492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Merhi Y, Provost P, Chauvet P, Theoret JF, Phillips ML, Latour JG. Selectin blockade reduces neutrophil interaction with platelets at the site of deep arterial injury by angioplasty in pigs. Arterioscler Thromb Vasc Biol. 1999;19:372–377. doi: 10.1161/01.atv.19.2.372. [DOI] [PubMed] [Google Scholar]
- 26.Thiagarajan RR, Winn RK, Harlan JM. The role of leukocyte and endothelial adhesion molecules in ischemia-reperfusion injury. Thromb Haemost. 1997;78:310–314. [PubMed] [Google Scholar]
- 27.Ruf A, Schlenk RF, Maras A, Morgenstern E, Patscheke H. Contact-induced neutrophil activation by platelets in human cell suspensions and whole blood. Blood. 1992;80:1238–1246. [PubMed] [Google Scholar]
- 28.Ott I, Neumann FJ, Gawaz M, Schmitt M, Schomig A. Increased neutrophil-platelet adhesion in patients with unstable angina. Circulation. 1996;94:1239–1246. doi: 10.1161/01.cir.94.6.1239. [DOI] [PubMed] [Google Scholar]
- 29.Kogaki S, Sawa Y, Sano T, Matsushita T, Ohata T, Kurotobi S, Tojo SJ, Matsuda H, Okada S. Selectin on activated platelets enhances neutrophil endothelial adherence in myocardial reperfusion injury. Cardiovasc Res. 1999;43:968–973. doi: 10.1016/s0008-6363(99)00140-6. [DOI] [PubMed] [Google Scholar]
- 30.Gawaz M, Fateh-Moghadam S, Pilz G, Gurland HJ, Werdan K. Platelet activation and interaction with leucocytes in patients with sepsis or multiple organ failure. Eur J Clin Invest. 1995;25:843–851. doi: 10.1111/j.1365-2362.1995.tb01694.x. [DOI] [PubMed] [Google Scholar]
- 31.Kirschenbaum LA, Adler D, Astiz ME, Barua RS, Saha D, Rackow EC. Mechanisms of platelet-neutrophil interactions and effects on cell filtration in septic shock. Shock. 2002;17:508–512. doi: 10.1097/00024382-200206000-00012. [DOI] [PubMed] [Google Scholar]
- 32.Fabry ME, Costantini F, Pachnis A, Suzuka SM, Bank N, Aynedjian HS, Factor SM, Nagel RL. High expression of human beta S- and alpha-globins in transgenic mice: erythrocyte abnormalities, organ damage, and the effect of hypoxia. Proc Natl Acad Sci U S A. 1992;89:12155–12159. doi: 10.1073/pnas.89.24.12155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fabry ME, Nagel RL, Pachnis A, Suzuka SM, Costantini F. High expression of human beta S- and alpha-globins in transgenic mice: hemoglobin composition and hematological consequences. Proc Natl Acad Sci U S A. 1992;89:12150–12154. doi: 10.1073/pnas.89.24.12150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nieswandt B, Bergmeier W, Rackebrandt K, Gessner JE, Zirngibl H. Identification of critical antigen-specific mechanisms in the development of immune thrombocytopenic purpura in mice. Blood. 2000;96:2520–2527. [PubMed] [Google Scholar]
- 35.Nagel RL, Fabry ME. The panoply of animal models for sickle cell anaemia. Br J Haematol. 2001;112:19–25. doi: 10.1046/j.1365-2141.2001.02286.x. [DOI] [PubMed] [Google Scholar]
- 36.Paszty C, Brion CM, Manci E, Witkowska HE, Stevens ME, Mohandas N, Rubin EM. Transgenic knockout mice with exclusively human sickle hemoglobin and sickle cell disease. Science. 1997;278:876–878. doi: 10.1126/science.278.5339.876. [DOI] [PubMed] [Google Scholar]
- 37.Manci EA, Hillery CA, Bodian CA, Zhang ZG, Lutty GA, Coller BS. Pathology of Berkeley sickle cell mice: similarities and differences with human sickle cell disease. Blood. 2006;107:1651–1658. doi: 10.1182/blood-2005-07-2839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shet AS, Hoffmann TJ, Jirouskova M, Janczak CA, Stevens JR, Adamson A, Mohandas N, Manci EA, Cynober T, Coller BS. Morphological and functional platelet abnormalities in Berkeley sickle cell mice. Blood Cells Mol Dis. 2008;41:109–118. doi: 10.1016/j.bcmd.2008.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mohan JS, Lip GY, Bareford D, Blann AD. Platelet P-selectin and platelet mass, volume and component in sickle cell disease: relationship to genotype. Thromb Res. 2006;117:623–629. doi: 10.1016/j.thromres.2005.05.010. [DOI] [PubMed] [Google Scholar]
- 40.Bergmeier W, Schulte V, Brockhoff G, Bier U, Zirngibl H, Nieswandt B. Flow cytometric detection of activated mouse integrin alphaIIbbeta3 with a novel monoclonal antibody. Cytometry. 2002;48:80–86. doi: 10.1002/cyto.10114. [DOI] [PubMed] [Google Scholar]
- 41.de Bruijne-Admiraal LG, Modderman PW, Von dem Borne AE, Sonnenberg A. P-selectin mediates Ca(2+)-dependent adhesion of activated platelets to many different types of leukocytes: detection by flow cytometry. Blood. 1992;80:134–142. [PubMed] [Google Scholar]
- 42.Solovey A, Kollander R, Shet A, Milbauer LC, Choong S, Panoskaltsis-Mortari A, Blazar BR, Kelm RJ, Jr, Hebbel RP. Endothelial cell expression of tissue factor in sickle mice is augmented by hypoxia/reoxygenation and inhibited by lovastatin. Blood. 2004;104:840–846. doi: 10.1182/blood-2003-10-3719. [DOI] [PubMed] [Google Scholar]
- 43.Zarbock A, Singbartl K, Ley K. Complete reversal of acid-induced acute lung injury by blocking of platelet-neutrophil aggregation. J Clin Invest. 2006;116:3211–3219. doi: 10.1172/JCI29499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zennadi R, Chien A, Xu K, Batchvarova M, Telen MJ. Sickle red cells induce adhesion of lymphocytes and monocytes to endothelium. Blood. 2008;112:3474–3483. doi: 10.1182/blood-2008-01-134346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chang J, Shi PA, Chiang EY, Frenette PS. Intravenous immunoglobulins reverse acute vaso-occlusive crises in sickle cell mice through rapid inhibition of neutrophil adhesion. Blood. 2008;111:915–923. doi: 10.1182/blood-2007-04-084061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lum AF, Wun T, Staunton D, Simon SI. Inflammatory potential of neutrophils detected in sickle cell disease. Am J Hematol. 2004;76:126–133. doi: 10.1002/ajh.20059. [DOI] [PubMed] [Google Scholar]
- 47.Wun T, Paglieroni T, Field CL, Welborn J, Cheung A, Walker NJ, Tablin F. Platelet-erythrocyte adhesion in sickle cell disease. J Investig Med. 1999;47:121–127. [PubMed] [Google Scholar]
- 48.Evangelista V, Manarini S, Dell’Elba G, Martelli N, Napoleone E, Di Santo A, Lorenzet PS. Clopidogrel inhibits platelet-leukocyte adhesion and platelet-dependent leukocyte activation. Thromb Haemost. 2005;94:568–577. [PubMed] [Google Scholar]
- 49.Savi P, Nurden P, Nurden AT, Levy-Toledano S, Herbert JM. Clopidogrel: a review of its mechanism of action. Platelets. 1998;9:251–255. doi: 10.1080/09537109876799. [DOI] [PubMed] [Google Scholar]
- 50.Peters MJ, Dixon G, Kotowicz KT, Hatch DJ, Heyderman RS, Klein NJ. Circulating platelet-neutrophil complexes represent a subpopulation of activated neutrophils primed for adhesion, phagocytosis and intracellular killing. Br J Haematol. 1999;106:391–399. doi: 10.1046/j.1365-2141.1999.01553.x. [DOI] [PubMed] [Google Scholar]
- 51.May AE, Langer H, Seizer P, Bigalke B, Lindemann S, Gawaz M. Platelet-leukocyte interactions in inflammation and atherothrombosis. Semin Thromb Hemost. 2007;33:123–127. doi: 10.1055/s-2007-969023. [DOI] [PubMed] [Google Scholar]
- 52.Zarbock A, Polanowska-Grabowska RK, Ley K. Platelet-neutrophil-interactions: linking hemostasis and inflammation. Blood Rev. 2007;21:99–111. doi: 10.1016/j.blre.2006.06.001. [DOI] [PubMed] [Google Scholar]
- 53.Canalli AA, Conran N, Fattori A, Saad ST, Costa FF. Increased adhesive properties of eosinophils in sickle cell disease. Exp Hematol. 2004;32:728–734. doi: 10.1016/j.exphem.2004.04.010. [DOI] [PubMed] [Google Scholar]
- 54.von Hundelshausen P, Weber C. Platelets as immune cells: bridging inflammation and cardiovascular disease. Circ Res. 2007;100:27–40. doi: 10.1161/01.RES.0000252802.25497.b7. [DOI] [PubMed] [Google Scholar]
- 55.Blanks JE, Moll T, Eytner R, Vestweber D. Stimulation of P-selectin glycoprotein ligand-1 on mouse neutrophils activates beta 2-integrin mediated cell attachment to ICAM-1. Eur J Immunol. 1998;28:433–443. doi: 10.1002/(SICI)1521-4141(199802)28:02<433::AID-IMMU433>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 56.Pluskota E, Woody NM, Szpak D, Ballantyne CM, Soloviev DA, Simon DI, Plow EF. Expression, activation, and function of integrin alphaMbeta2 (Mac-1) on neutrophil-derived microparticles. Blood. 2008;112:2327–2335. doi: 10.1182/blood-2007-12-127183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Theilmeier G, Lenaerts T, Remacle C, Collen D, Vermylen J, Hoylaerts MF. Circulating activated platelets assist THP-1 monocytoid/endothelial cell interaction under shear stress. Blood. 1999;94:2725–2734. [PubMed] [Google Scholar]
- 58.da Costa Martins P, van den Berk N, Ulfman LH, Koenderman L, Hordijk PL, Zwaginga JJ. Platelet-monocyte complexes support monocyte adhesion to endothelium by enhancing secondary tethering and cluster formation. Arterioscler Thromb Vasc Biol. 2004;24:193–199. doi: 10.1161/01.ATV.0000106320.40933.E5. [DOI] [PubMed] [Google Scholar]
- 59.da Costa Martins PA, van Gils JM, Mol A, Hordijk PL, Zwaginga JJ. Platelet binding to monocytes increases the adhesive properties of monocytes by up-regulating the expression and functionality of beta1 and beta2 integrins. Journal of Leukocyte Biology. 2006;79:499–507. doi: 10.1189/jlb.0605318. [DOI] [PubMed] [Google Scholar]
- 60.Singbartl K, Forlow SB, Ley K. Platelet, but not endothelial, P-selectin is critical for neutrophil-mediated acute postischemic renal failure. Faseb J. 2001;15:2337–2344. doi: 10.1096/fj.01-0199com. [DOI] [PubMed] [Google Scholar]
- 61.Pitchford SC, Yano H, Lever R, Riffo-Vasquez Y, Ciferri S, Rose MJ, Giannini S, Momi S, Spina D, O’Connor B, Gresele P, Page CP. Platelets are essential for leukocyte recruitment in allergic inflammation. J Allergy Clin Immunol. 2003;112:109–118. doi: 10.1067/mai.2003.1514. [DOI] [PubMed] [Google Scholar]
- 62.Semple MJ, Al-Hasani SF, Kioy P, Savidge GF. A double-blind trial of ticlopidine in sickle cell disease. Thromb Haemost. 1984;51:303–306. [PubMed] [Google Scholar]
- 63.Greenberg J, Ohene-Frempong K, Halus J, Way C, Schwartz E. Trial of low doses of aspirin as prophylaxis in sickle cell disease. J Pediatr. 1983;102:781–784. doi: 10.1016/s0022-3476(83)80258-3. [DOI] [PubMed] [Google Scholar]
- 64.Chaplin H, Jr, Monroe MC, Malecek AC, Morgan LK, Michael J, Murphy WA. Preliminary trial of minidose heparin prophylaxis for painful sickle cell crises. East Afr Med J. 1989;66:574–584. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.