

Abstract
Transcription-induced recombination has been reported in all organisms from bacteria to mammals. We have shown previously that the yeast genes HPR1 and THO2 may be keys to the understanding of transcription-associated recombination, as they both affect transcription elongation and hyper-recombination in a concerted manner. Using a yeast strain that has the wild-type THO2 gene replaced by one encoding a His6-HA-tagged version, we have isolated an oligomeric complex containing four proteins: Tho2, Hpr1, Mft1 and a novel protein that we have named Thp2. We have reciprocally identified a complex containing Hpr1, Tho2 and Mft1 using anti-Mft1 antibodies in immunoprecipitation experiments. The protein complex is mainly nuclear; therefore, Tho2 and Hpr1 are physically associated. Like hpr1Δ and tho2Δ cells, mft1Δ and thp2Δ cells show mitotic hyper- recombination and impaired transcription elongation, in particular, through the bacterial lacZ sequence. Hyper-recombination conferred by mft1Δ and thp2Δ is only observed in DNA regions under transcription conditions. We propose that this protein complex acts as a functional unit connecting transcription elongation with the incidence of mitotic recombination.
Keywords: HPR1/THO2/MFT1/THP2/mitotic recombination/THO protein complex/transcription-associated recombination/transcription elongation
Introduction
Transcription of DNA may have different consequences for other DNA transactions such as DNA replication, repair and recombination. The relationship between transcription and recombination is of particular interest, given the importance of recombination in genomic instability and its link with cancer. Different reports from bacteria to mammals have shown that transcription induces recombination. Since Ikeda and Matsumoto (1979) first showed that recombination of phage λ in Escherichia coli was stimulated by Rpo-mediated transcription, other cases have been reported in prokaryotes (Dul and Drexler, 1988; Vilette et al., 1992). In yeast, the finding that HOT1, a cis-acting hotspot of recombination, was dependent on RNA polymerase I (RNAPI)-driven transcription (Voelkel-Meiman et al., 1987; Stewart and Roeder, 1989) provided the first example of transcription-induced recombination. Subsequently, similar observations were made for RNA polymerase II (RNAPII)-driven transcription (Thomas and Rothstein, 1989; Grimm et al., 1991; Nevo-Caspi and Kupiec, 1994). In mammalian cells, RNAPII-driven transcription has been shown to stimulate homologous recombination (Nickoloff, 1992; Thyagarajan et al., 1995) and to positively control V(D)J recombination (Blackwell et al., 1986; Lauster et al., 1993; Oltz et al., 1993) and class switching (Daniels and Lieber, 1995).
The identification and analyses of the HPR1 and THO2 yeast genes have provided clues to understand transcription-associated recombination. HPR1 was identified by a mutation conferring hyper-recombination between DNA repeats (Aguilera and Klein, 1988) and THO2 as a high-copy suppressor of hpr1Δ (Piruat and Aguilera, 1998). Both null mutations confer strong stimulation of recombination (>3000 times the wild-type levels) (Aguilera and Klein, 1990; Piruat and Aguilera, 1998) that is dependent on RNAPII-driven transcription elongation, and increases in other forms of genetic instability such as plasmid and chromosome loss (Santos-Rosa and Aguilera, 1994; Chávez and Aguilera, 1997). Hyper-recombination is observed when RNAPII elongates through either the DNA repeats or the intervening regions flanked by the repeats. If transcription is impeded to proceed through such DNA regions by means of a transcription terminator or by turning off the promoter, hyper-recombination is abolished (Chávez and Aguilera, 1997; Prado et al., 1997; Piruat and Aguilera, 1998). It is probable that impairment of transcription elongation contributes in a major way to the generation of recombinagenic intermediates whose nature is yet to be determined.
Although the precise function of HPR1 and THO2 is not yet clear, their null mutations have consequences at transcription, suggesting that their functions may be related to RNAPII. This relationship is supported further by the identification of suppressors of the hyper-recombination or thermosensitive phenotypes of hpr1Δ as mutations in genes related to RNAPII holoenzyme, such as HRS1/PGD1 and SRB2 (Piruat and Aguilera, 1996; Santos-Rosa et al., 1996) or SOH1, RPB2 and SUA7 (Fan et al., 1996), respectively. Importantly, Hpr1 has been found in association with Cdc73, Paf1 and Ccr4 in an alternative form of the RNAPII holoenzyme (Chang et al., 1999).
In order to identify the functional roles of Hpr1 and Tho2 in yeast cells we need to define the structural and functional relationship between these proteins and other proteins with which they might physically interact. We have therefore undertaken a biochemical approach aimed at isolating a putative protein complex containing Tho2. We created yeast strains expressing a His6-HA-tagged form of THO2 functionally replacing the chromosomal copy. Using whole-cell extracts from this strain we have isolated a complex containing four proteins. The proteins were identified by western blotting, peptide microsequencing and mass spectrometry as Tho2, Hpr1, Mft1 (also called Mft52) and a new protein that we have named Thp2. Using anti-Mft1 monoclonal antibodies in immunoprecipitation experiments with whole cell extracts, we have indeed obtained both Hpr1 and Tho2. We show that, as previously reported for hpr1Δ and tho2Δ cells, the mft1Δ and thp2Δ mutations confer similar phenotypes of transcription impairment and transcription-dependent hyper-recombination. We believe that this novel multi-protein complex, termed the THO complex, acts as a functional unit affecting transcription elongation and recombination.
Results
Identification and purification of a THO protein complex containing Tho2, Hpr1, Mft1 and Thp2
We constructed a yeast strain, SChY73, carrying the His(6)-HA-THO2 gene fusion at the THO2 chromosomal locus. His6-HA-Tho2 was functional as deduced from its ability to confer wild-type frequencies of recombination in the chromosomal leu2-k::URA3::ADE2::leu2-k direct repeat recombination (data not shown). Whole-cell extracts from strain SChY73 was loaded onto Bio-Rex 70 and bound protein eluted with buffer containing 0.3, 0.6 and 1.2 M potassium acetate (Figure 1A). The majority of HA-Tho2 was found in the 1.2 M fraction, although a significant amount was also detected in the fraction eluted with 0.6 M potassium acetate. The 0.6 and 1.2 M fractions were loaded onto a column containing the monoclonal anti-HA antibody 12CA5 conjugated to protein G. Anti-HA-bound complexes were eluted with 1 mg/ml of HA peptide. As shown by silver staining after SDS–polyacrylamide gel electrophoresis (PAGE) (Figure 1B) the new fraction contained five polypeptides. The fraction eluting from the 12CA5 column was further purified by Ni-nitrilotriacetic acid (NTA) agarose chromatography. Fractionation of the eluted fraction by SDS–PAGE and subsequent silver staining revealed four polypeptides with apparent molecular sizes of ∼160, ∼90, ∼60 and ∼40 kDa (Figure 1B). Western blot analysis using anti-HA and anti-Hpr1 revealed that p160 and p90 corresponded to Tho2 and Hpr1, respectively.

Fig. 1. (A) Schematic diagram used for the purification of the THO complex. (B) SDS–polyacrylamide gels of the 1.2 M KOAc eluates from Bio-Rex70, the 2 mg/ml HA-peptide eluate from 12CA5-protein G–Sepharose fraction and the 0.4 M imidazole eluate from the Ni-NTA agarose mix. (C) SDS–PAGE of fractions 21–26 obtained after gel filtration through a Superose 6 PC3.2/30 column. (D) Western blots of the SDS–PAGE gels using anti-HA and Anti-Hpr1 antibodies (see text for details).
The protein fraction eluting from the Ni-NTA agarose column was subjected to gel filtration through Sepharose 6. All four polypeptides peaked in the same fractions (Figure 1C), indicating that they belong to a single protein complex. Based on the elution profile of size marker proteins from the same resin, we estimate the size of the THO complex to be ∼1400 kDa. This value is consistent with previous data indicating that Hpr1 was present in a complex of >1 MDa (Zhu et al., 1995).
To identify the subunits of the THO complex, we used two independent techniques; peptide mass fingerprinting using MALDI-reTOF mass spectrometry and SequenceTag/PepFrag database searching using limited amino acid sequence data obtained by ESI tandem mass spectrometry (Erdjument-Bromage et al., 1998). This revealed that p160 corresponds to Tho2 and p90 to Hpr1, consistent with our western blot results, whereas p60 corresponds to Mft1, a protein previously shown to be involved in mitochondrial protein import (Emr et al., 1986; Bedwell et al., 1987; Garrett et al., 1991; Beilharz et al., 1997). p40 corresponds to a novel protein encoded by the yeast gene YHR167w. We refer to the gene encoding p40 as THP2 (for Tho2/Hpr1 phenotype).
SDS–PAGE analysis of the different fractions alongside gel filtration experiments (Figure 1C) also revealed two additional weak bands with apparent molecular masses of ∼200 and ∼92 kDa, whose intensities varied from one experiment to another. Western blots of the SDS–PAGE (Figure 1D) and peptide mass fingerprinting unambiguously identified these as alternative forms of Tho2 and Hpr1, respectively.
In a parallel series of experiments, in which we were trying to isolate proteins that might form a complex with Mft1, we raised a monoclonal antibody to the C-terminal domain of Mft1. Both western blotting and immunoprecipitation analysis confirmed the antibody to be specific for an epitope contained within this domain (T.Beilharz and T.Lithgow, unpublished data). When total spheroplast extracts of strain JK9-3da were subjected to immunoprecipitation with the anti-Mft1, two proteins with apparent molecular sizes of 170 and 90 kDa were co-immunoprecipitated (Figure 2). Also present in the precipitated complex was a protein of 35–40 kDa that could not be peptide sequenced. Several additional yeast proteins precipitated non-specifically, being present in the precipitate from mft1Δ cells (Figure 2) as well as in the absence of the antibody (data not shown).
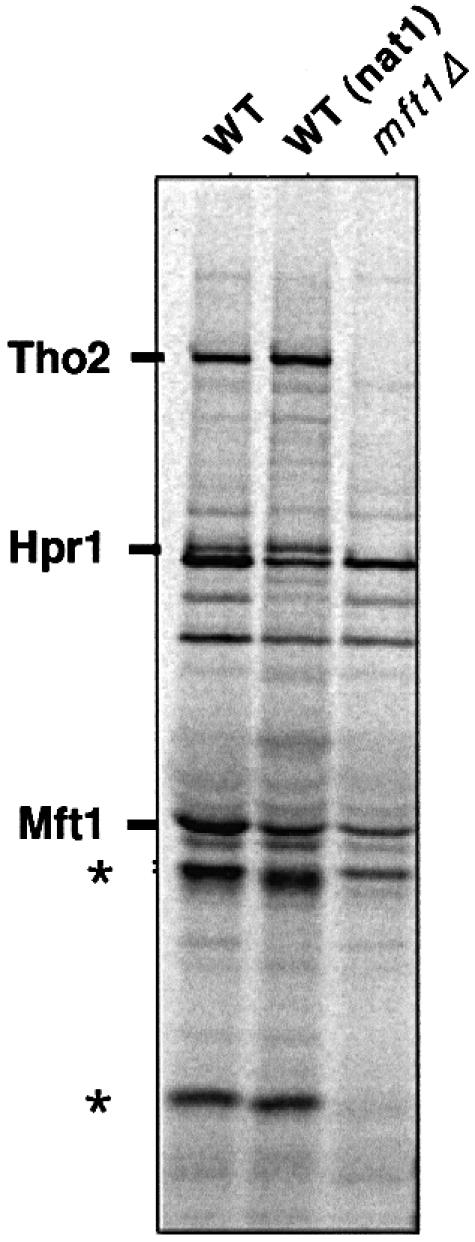
Fig. 2. Tho2 and Hpr1 are co-immunoprecipitated with a monoclonal anti-Mft1. Cell extracts were prepared from either wild-type (JK9-3da and nat1) or mft1Δ strains of yeast metabolically labeled with [35S]methionine, and then subjected to immunoprecipitation with the monoclonal antibody P1A12H8. Immunoprecipitated proteins were analyzed by SDS–PAGE and phosphoimaging. After transfer to PVDF membranes, the bands indicated on the gels were identified as Tho2 and Hpr1. Mft1 migrates close to the IgG heavy chain and is compressed together with several minor endogeous proteins by the immunoglobulin heavy chains during SDS–PAGE. The minor endogenous proteins are still visible in the mft1Δ lane. Mft1 is subject to some proteolytic degradation during the course of the experiment. Proteolytic fragments of Mft1 (indicated with an asterisk) have been characterized previously (Cartwright et al., 1997).
Attempts to peptide sequence the partner proteins precipitated from the wild-type extracts were initially unsuccessful. However, using the nat1Δ cells, which lack the cytoplasmic N-acetyltransferase responsible for co-translationally modifying nascent polypeptides, the sequence from the precipitated proteins unambiguously identified the larger protein subunit as Tho2 and the other subunit as Hpr1. During the course of the experiment, proteolytic fragments of Mft1 ranging from 35 to 55 kDa, corresponding to the first 283 amino acids of the protein (Cartwright et al., 1997), were usually observed (Figure 1, asterisks) and may mask the presence of Thp2 in the precipitates.
Physical interactions between Mft1, Hpr1 and Tho2
As Hpr1 and Tho2 were identified in genetic studies that revealed a strong and similar effect on transcription, recombination and genetic instability, it was not unexpected to find both proteins in the same protein complex. Mft1, however, was identified by a genetic screen designed to identify genes involved in mitochondrial protein import pathway (Emr et al., 1986; Garrett et al., 1991; Beilharz et al., 1997). Consequently, we decided to define the subunit interactions between Mft1, Hpr1 and Tho2. We constructed a series of glutathione S-transferase (GST) fusion proteins for expression in vivo, consisting of GST plus either the entire Mft1 (GST–Mft1), the 188 Mft1 N-terminal residues [GST–Mft1(Δ189–392)] or the 203 C-terminal residues [GST–Mft1(Δ1–188)]. Whereas the two first constructs complemented the temperature-sensitive defect of mft1Δ cells, the latter did not, suggesting an important function for the N-terminal domain of Mft1 (data not shown).
Both GST–Mft1(Δ1–188) and GST–Mft1(Δ189–392) were expressed at high levels in yeast, but only the GST–Mft1(Δ189–392) fusion protein is incorporated into the THO complex. After sucrose density gradient centrifugation of extracts containing either the intact Mft1 (data not shown) or GST–Mft1(Δ189–392), a proportion of the over-expressed GST–Mft1 protein thus migrates through the gradient almost at the position of the 60S ribosomal marker protein L3 (Figure 3). This is consistent with the chromatography of the particle containing Mft1 during gel filtration, where the protein behaves as if it were >1 MDa (Cartwright et al., 1997). In contrast, GST–Mft1(Δ1–188) remains at the top of the gradient (data not shown), indicating that the protein is not incorporated into the THO complex and suggesting that the N-terminal domain of Mft1 is required for interaction with other proteins of the complex. Indeed, migration of Mft1 as a large entity is dependent on association with Tho2, since in extracts prepared from tho2Δ cells, GST–Mft1(Δ189–392) is found exclusively at the top of the sucrose gradients (Figure 3). To address independently whether the N-terminal domain of Mft1 interacts with Tho2, GST fusion proteins were precipitated with glutathione–agarose in a classical pull-down experiment from yeast extracts. Both Tho2 and Hpr1 were precipitated on glutathione–agarose loaded with extract from the cells expressing the full-length GST–Mft1 and the GST–Mft1(Δ189–329), but not from the extract of GST–Mft1(Δ1–188) cells (data not shown).

Fig. 3. The N-terminal domain of Mft1 is required to form an oligomeric particle. Extracts were prepared from yeast cells expressing GST–Mft1(Δ189–392) and subjected to sucrose density gradient centrifugation. Fractions were collected from the sucrose density gradients and analyzed by SDS–PAGE and immunoblotting with antibodies recognizing Mft1 and ribosomal protein L3. Also shown are equivalent fractions of sucrose density gradient analysis from extracts of tho2Δ yeast cells expressing the GST–Mft1(Δ189–392) fusion protein.
Subcellular localization of the proteins of the THO complex
To determine the subcellular localization of the THO complex, we made green fluorescent protein (GFP) fusion proteins of three of the polypeptides: Hpr1, Mft1 and Thp2. All GFP fusion constructs are functional, as they complement either the temperature-sensitive defect or the hyper-recombination phenotype (see later) caused by the respective deletion mutations (data not shown). All fusion proteins, Mft1–GFP, Hpr1–GFP and Thp2–GFP, expressed from the MET25 promoter, were found concentrated in the nucleus of living yeast cells (Figure 4). A nuclear localization has also been observed for Tho2–GFP (M.Gallardo and A.Aguilera, unpublished results). When the plasmid system was used to express GFP alone, fluorescence was seen uniformly throughout the cytoplasm. As expected, the nuclear mRNA export protein Npl3 (Lee et al., 1996) fused to GFP and expressed under the control of the same promoter was localized in the nucleus (Figure 4). Therefore, even though we can also detect weak signals from the respective overexpressed GFP fusion proteins in the cytoplasm, we conclude that the THO complex is concentrated in the nucleus.
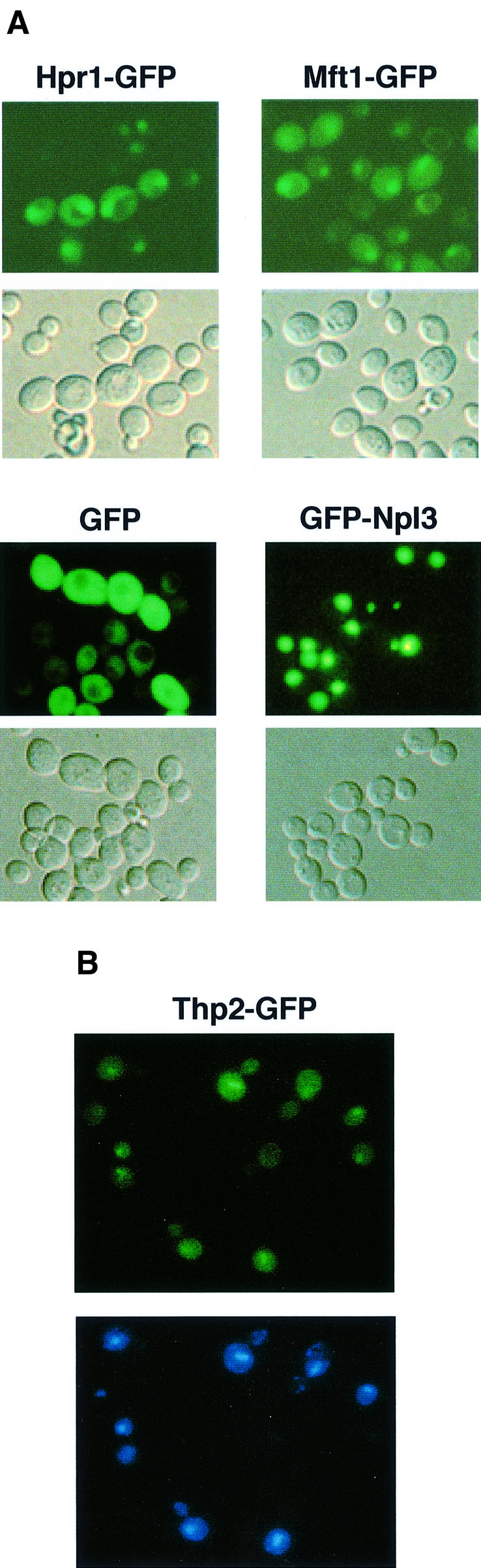
Fig. 4. Subcellular colocalization of Mft1, Hpr1 and Thp2. (A) Diploid JK9 cells were transformed with plasmids encoding Mft1–GFP, Hpr1–GFP, GFP and GFP–Npl3, and analyzed by fluorescence microscopy to visualize GFP fusions (top) and with Nomarski optics to visualize whole cells (bottom). (B) Haploid strain BY-HR167 was transformed with plasmids encoding Thp2-GFP and analyzed by fluorescence microscopy to visualize either GFP (top) and nuclei (bottom) after DAPI staining. Although only localization of Thp2–GFP expressed from plasmid pUG23 is shown, identical results were obtained for the GFP–Thp2 fusion expressed from pUG34 (data not shown).
The mft1Δ and thp2Δ mutations confer the same phenotypes of thermosensitivity, gene expression and recombination as hpr1Δ and tho2Δ
In order to show that the THO complex identified in this study acts as a functional unit, we performed a genetic and molecular analysis of transcription and recombination in mft1Δ and thp2Δ mutants. As reported previously for hpr1Δ, tho2Δ and mft1Δ, thp2Δ cells are thermosensitive for viability at 37°C. In addition, expression of the bacterial lacZ sequence under the control of the GAL1 promoter, as determined by β-galactosidase activity, is significantly reduced to 21.6 and 17% of wild-type levels, values that are close to those of hpr1Δ and tho2Δ cells (11.2 and 5.6%, respectively) (Figure 5A). In contrast, expression of the yeast PHO5 coding region from the same GAL1 promoter, as determined by acid phosphatase activity, was only slightly affected in both mft1Δ (82.8% of wild-type levels) and thp2Δ cells (1.7-fold increase) (Figure 5B). These results indicate that both mutants are capable of activating transcription driven from the GAL1 promoter. Therefore, the reduction in lacZ expression probably reflects an inability of the mutants to express lacZ.

Fig. 5. Effect of the mft1Δ and thp2Δ mutations on gene expression and recombination. β-galactosidase (A) and acid phosphatase (B) activities of wild-type (W303–1A) and mutant strains mft1Δ (WMK-1A), and thp2Δ (BY-HR167) transformed with centromeric plasmid p416GAL1-lacZ and pSCh202 containing lacZ and PHO5 under the GAL1 promoter, respectively. Average value and standard deviation of two different transformants are shown for each strain. Only data of induced expression are given (2% galactose). Under repressed conditions (2% glucose), values were 1–2 U and 4–8 mU for β-galactosidase and acid phosphatase, respectively. (C) Frequency of recombination of the chromosomal direct repeat system leu2-k::ADE2-URA3::leu2-k wild type (wt) and mutant strains mft1Δ (YTW-13C) and thp2Δ (HRW167–18D). Data from hpr1Δ and tho2Δ strains are taken from Piruat and Aguilera (1998) and correspond to W303 isogenic strains. Similar results for recombination were obtained for strains SEW-2C and SEW-14B (wild type), SEW-2D (mft1-ts), YTW-5A (mft1Δ) or HRW167–12D and HRW167–16A (thp2Δ).
Finally, recombination between the 2.16-kb LEU2 genes of the chromosomal leu2-k::URA3-ADE2::leu2-k repeat was clearly increased in mft1Δ and thp2Δ mutants (180 and 121 times wild-type levels) (Figure 5C). Therefore, despite minor quantitative differences observed in hpr1Δ, tho2Δ, mft1Δ and thp2Δ, our results indicate that all four mutations confer similar phenotypes.
Our gene expression analysis suggests that the incapacity of mft1Δ and thp2Δ strains to express lacZ is not caused by a defect in activation of the GAL1 promoter, but by an inability of mft1Δ and thp2Δ cells to transcribe lacZ sequences. In order to confirm this at the mRNA level we determined the kinetics of mRNA accumulation in the GAL1-lacZ and GAL1-PHO5 constructs. Whereas the kinetics of accumulation of full-length lacZ mRNA is strongly reduced in both mft1Δ and thp2Δ mutants, accumulation of full-length PHO5 mRNA was not affected in mft1Δ cells or it occurred at a slightly faster rate in thp2Δ cells (Figures 6 and 7), consistent with the enzymatic assays (see Figure 5). Similar experiments were performed for the analysis of expression of the endogenous chromosomal GAL1 gene. The kinetics of accumulation of GAL1 mRNA are similar to wild type in mft1Δ and thp2Δ cells (Figures 6 and 7). Again, and as observed for PHO5, accumulation of GAL1 mRNA occurs at higher rate in thp2Δ versus wild-type cells. These results indicate that the transcription defect of mft1Δ and thp2Δ is not at the level of promoter activation. Transcription is, indeed, activated in both mutants, being more proficient in thp2Δ cells. However, whereas transcription proceeds through PHO5 or GAL1, it cannot traverse the lacZ sequences properly, as shown previously for hpr1Δ and tho2Δ cells (Chàvez and Aguilera, 1997; Piruat and Aguilera, 1998).

Fig. 6. Transcription analysis of GAL1-lacZ, GAL1-PHO5 and endogenous GAL1 gene in wild-type and mft1Δ cells. Northern blot analyses of lacZ and PHO5 mRNAs driven from the GAL1 promoter in the strains W303–1A (WT) and WMK-1A (mft1Δ) transformed with plasmids p416GAL1lacZ and pSCh202 are shown. Mid-log phase cells were diluted in 3% glycerol–2% lactate SC-ura and diluted into identical fresh media to an OD600 of 0.5 and incubated for 16 h. Galactose (Gal) was then added and samples were taken for northern blot analysis at different times, as specified. DNA probes used were the 3 kb BamHI–BamHI 5′ end fragment of lacZ, a 0.9 kb EcoRV–EcoRV PHO5 internal fragment, a 0.75 kb PvuII–AvaI GAL1 internal fragment and a 589 bp 28S rRNA internal fragment obtained by PCR (rRNA). The kinetics of induction of mRNAs as determined by quantification of northern blots in a Fuji FLA3000 are shown. The mRNA values are given in arbitrary units (AU) with respect to rRNA levels.

Fig. 7. Transcription analysis of GAL1-lacZ, GAL1-PHO5 and the endogenous GAL1 gene in wild-type (BY4741) and thp2Δ cells (BY-HR167). Other details are as in the legend to Figure 6.
Hyper-recombination conferred by mft1Δ and thp2Δ is linked to transcription-elongation impairment
In order to determine whether the transcriptional defect of mft1Δ and thp2Δ is linked to their hyper-recombination phenotypes, we analyzed the recombination levels of such mutants in a set of direct repeat constructs based on the same 0.6 kb repeat sequence (an internal LEU2 fragment) and containing different sequences flanked by the repeats. In all cases transcription is controlled by one promoter located outside of the repeat contruct. All repeat systems lie on the centromeric plasmid pRS314. We first used either the LY or LYΔNS repeat systems, in which transcription initiated from the external LEU2 promoter traverses the first leu2 repeat and enters the pBR322 sequences located between the repeats. As reported previously for hpr1Δ cells, transcription through pBR322 is impaired and leads to hyper-recombination (Prado et al., 1997). As shown in Figure 8, both mft1Δ and thp2Δ mutations induced recombination 30- and 11-fold greater than wild-type levels in these repeat systems. However, in a similar system not containing intervening sequence (system L), no significant hyper-recombination phenotype was observed for any of the mutations. Similarly, mft1Δ and thp2Δ induce recombination 8- and 3-fold greater than wild-type levels in the LNA system, in which the pBR322 sequences of the intervening region are also transcribed, but no induction was observed in the LNAT system, identical to LNA but with a CYC1 transcription terminator that impedes transcription entering the pBR322 sequence.

Fig. 8. Recombination analysis of direct repeat systems in wild-type (WT), mft1Δ and thp2Δ strains. (A) Scheme of the deletion resulting from recombination between the direct repeats used. (B) Recombination frequencies were determined in strains SEY2102 (WT) and Mft1(m6) (mft1) transformed with plasmid pRS314-LYΔNS (LYΔNS) or pRS314-L (L); strains BY4741 (WT) and BY-HR167 (thp2) transformed with pRS314-LY (LY) or pRS314-L (L); strains SEW-14D (WT) and SEW-14C (mft1) or HRW167–18Bu (WT) and HRW167-16Au (thp2) transformed with pRS314-LNA (LNA) or pRS314-LNAT (LNAT); and strains W303-1A (WT) and WMK-1A (mft1) or HRW167-18Bu (WT) and HRW167-18Du (thp2) transformed with pSCh204 (L-lacZ) or pSCh206 (L-PHO5). In the scheme of each repeat system, the transcripts driven from the external LEU2 promoter are indicated by arrows. For fluctuation tests, colonies were obtained from SC-Trp with 2% glucose and recombinants selected in SC-Leu-Trp. The median frequency of 6–12 cultures is given in each case.
Although the hyper-recombination phenotype of LY and LNA systems in mft1Δ and thp2Δ is lower than in hpr1Δ cells (Prado et al., 1997), it is only observed when transcription proceeds through DNA sequences known to impair transcription-elongation in hpr1Δ cells. This suggests that the hyper-rec phenotype of mft1Δ and thp2Δ cells is also associated with transcription-elongation impairment. This was confirmed with the analysis of the L-lacZ and L-PHO5 systems containing lacZ and PHO5 as intervening sequences. As can be seen in Figure 8, L-lacZ, through which transcription is impaired in mft1Δ and thp2Δ (Figures 6 and 7), has frequencies of recombination 91 and 7.5 times the wild-type level for each mutant, respectively. In constrast, L-PHO5, through which transcription is not impaired in the mutants, shows frequencies of recombination close to the wild-type levels in mft1Δ and thp2Δ cells, respectively.
Therefore, the major hyper-recombination phenotype of mft1Δ and thp2Δ is transcription dependent. It is observed when RNAPII proceeds through DNA sequences that require a functional THO complex for transcription elongation.
Discussion
We have constructed a yeast strain with its wild-type THO2 gene replaced by a fully functional His6-HA-tagged copy. Starting with whole-cell extracts prepared from this strain, we have purified a novel protein complex composed of four different proteins: Tho2 and Hpr1, both previously shown to be involved in transcription-associated recombination, Mft1, previously reported to be involved in mitochondrial protein targeting, and a protein whose function was previously unknown, Thp2. Reciprocally, using anti-Mft1 monoclonal antibodies, we have shown in immunoprecipitation experiments that Mft1 indeed forms a complex with Hpr1 and Tho2.
Therefore, Hpr1 and Tho2, two proteins that by genetic means were shown to functionally interact and to participate in the same biological process are present in the same protein complex in vivo. In addition, we show in this study that mft1Δ and thp2Δ strains show phenotypes of hyper-recombination and transcription impairment similar to hpr1Δ and tho2Δ. They confer very high levels of recombination between DNA repeats (>100-fold greater than wild-type levels) and an inability to transcribe the lacZ sequence properly. We propose that this novel protein complex acts as a functional unit connecting transcription elongation and the incidence of mitotic recombination.
The THO complex as a structural and functional unit
HPR1 was identified by mutations conferring hyper-recombination between direct repeats (Aguilera and Klein, 1988). THO2 was identified as a multicopy suppressor of hpr1Δ mutants (Piruat and Aguilera, 1998). The finding that Hpr1 and Tho2 act in the same protein complex provides a probable explanation for the ability of Tho2 overexpression to suppress certain hpr1Δ phenotypes. An assembly of an at least partially functional complex in the absence of a Hpr1 might require a large amount of Tho2 protein. This would suggest that Tho2 may be the most important ‘effector’ protein in the THO complex. Indeed, the tho2Δ mutation confers the strongest phenotypes of hyper-recombination and transcription of all four null mutations of the complex (Piruat and Aguilera, 1998; Figure 5). Among the four proteins, Tho2 is the only protein that consistently has obvious structural homologs (>25% identity) in higher eukaryotes (Schizosaccharomyces pombe, Caenorhabditis elegans, Drosophila melanogaster, Mus musculus and Homo sapiens).
The identification of Mft1 as a subunit of the THO complex is, at first glance, somewhat surprising. Based on two lines of evidence, it was previously suggested that the particle containing Mft1 might be involved in protein targeting to the mitochondria (Beilharz et al., 1997; Cartwright et al., 1997). The MFT1 gene was identified first in a genetic screen for genes that affect the mitochondrial accumulation of a lacZ fusion protein, and secondly based on in vitro assays measuring binding of mitochondrial precursor proteins. There is also a functional similarity, since the purified GST–Mft(Δ1–188) protein can bind mitochondrial precursors in vitro (Cartwright et al., 1997). Heterologous reporter proteins have proved useful in the identification of components of the protein import machinery of mitochondria (Kübrich et al., 1995). In yeast cells lacking nascent polypeptide-associated complex (NAC), proteins including F1β-LacZ fail to accumulate in the mitochondria due to defects in protein delivery to the organelle (George et al., 1998), and the three other MFT genes identified in the same screen as MFT1 may well define further components of the protein import pathway (Garrett et al., 1991). However, the finding of Mft1 as a subunit of the THO complex and the fact that mft1Δ cells show hyper-recombination and transcriptional phenotypes similar to hpr1Δ and tho2Δ suggests that Mft1 is involved in transcription and genetic stability. Thus, the genetic screen that identified Mft1 as a protein involved in mitochondrial import was based on the reduction in the levels of β-galactosidase from a F1β-LacZ fusion protein. Since one of the hallmark phenotypes of cells lacking a gene encoding a THO subunit is their inability to transcribe lacZ, this genetic screen, in the case of mft1, have actually picked up a mutation impairing transcription through lacZ.
Previously, chromatography of total yeast cell extracts by gel filtration and analysis of the fractions collected by immunoblotting suggested that Hrp1p behaves as if it were part of a complex >1 MDa in size (Zhu et al., 1995). The identification of Hpr1 as the 90 kDa subunit of the THO complex is consistent with this finding. Our gel filtration experiments show that purified THO complex also behaves as a complex >1 MDa in size (Figures 1 and 2). This apparent size of the THO complex is somewhat surprising, given the molecular weight of the complex is predicted to be ∼350 kDa. The apparent molecular size might be due to the complex having an elongated shape, or that the complex is formed by more than one subunit of each of the four proteins.
A novel form of RNAPII distinct from the Mediator/Srb-containing holoenzyme, but containing a discrete set of proteins that include Paf1, Cdc73, Hpr1, Ccr4 and at least 10 other subunits has been identified in yeast (Shi et al., 1997; Chang et al., 1999). Our discovery of Hpr1 as a component of the THO complex presents different plausible scenarios for the connection to the Paf1-Cdc73 RNAPII holoenzyme. First, Hpr1 might have distinct roles as a component of both complexes. Alternatively, the entire THO complex might form part of the Cdc73-Paf1 RNAPII holoenzyme or it might interact with RNAPII independently of Paf1 and Cdc73. It is worth noting in this connection that whereas a major fraction of cellular Paf1 and Cdc73 was found associated with RNAPII (Shi et al., 1997), the fraction of Hpr1 colocalizing with Paf1 and Cdc73 is small (Chang et al., 1999). One possible interpretation of this result would be that a fraction of the THO complex co-purifies with the Cdc73-Paf1 RNAPII holoenzyme due to independent interactions with RNAPII. Our observed in vivo effect of the THO complex in transcriptional elongation by RNAPII would be consistent with such interactions. Preliminary evidence indicates that purified THO complex does not form a strong complex with unphosphorylated RNAPII in vitro (data not shown).
The THO complex defines a structural and functional unit, as deletion of all subunits lead to identical phenotypes of transcription and recombination. Deletion of the genes encoding subunits of the Paf1/Cdc73 RNAPII holoenzyme leads to a variety of phenotypes. Thus, deletion of CCR4 does not cause hyper-recombination, and although paf1Δ and cdc73Δ display hyper-recombination between repeats, it is not yet known whether this phenotype is transcription-dependent. In addition, whereas double mutant combinations of tho2Δ and hpr1Δ are not lethal but show epistatic relationships (as expected for proteins belonging to the same functional complex), the double paf1Δ hpr1Δ and ccr4Δ hpr1Δ mutants are lethal (Chang et al., 1999). Moreover, hpr1Δ has not been found to confer the cell wall-integrity phenotypes typical of paf1Δ and cdc73Δ strains (Chang et al., 1999). Finally, it is unlikely that the THO complex is involved in transcription of a subset of genes downstream of the protein kinase C (PKC) signaling pathway, as proposed for the Cdc73-Paf1 RNAPII holoenzyme (Chang et al., 1999), as cells lacking Tho2 are affected in transcription of a variety of genes not related with PKC, such as GAL1 or ACT1 (Piruat and Aguilera, 1998). These results strongly suggest that at least some of the proteins of the Paf1/Cdc73 RNAPII holoenzyme play roles different to those of the THO complex.
The recent finding that hpr1Δ cells do not export poly(A+) RNA at 37°C (Schneiter et al., 1999) could imply a wider relationship of Hpr1, and possibly the THO complex, with mRNA metabolism. However, it is not yet clear whether such a relationship is indirect, particularly since hpr1Δ cells do not grow at 37°C.
Biological role of the THO complex
Cells lacking HPR1, THO2, MFT1 or THP2 have the same transcription and recombination phenotypes and the encoded proteins comprise the subunits of the same protein complex. This data make it reasonable to conclude that the four proteins form a structural and functional unit that affects both transcription elongation and the incidence of recombination. The four proteins are consistently predominantly found in the nucleus (Figure 4; M.Gallardo and A.Aguilera, unpublished data). Nevertheless, the biochemical function of the THO complex is yet to be elucidated.
It is important to emphasize the results indicating that the THO complex affects the incidence of recombination associated with transcription elongation. First, the transcriptional defects observed in cells lacking a THO subunit are not observed at the level of promoter activation. Whereas the GAL1 promoter can be activated in all mutant backgrounds, as determined by analysis of the transcripts of a fused PHO5 gene or the endogenous GAL1 gene, transcription of lacZ fused to GAL1 promoter is strongly reduced (Figures 5–7) (Chávez and Aguilera, 1997; Piruat and Aguilera, 1998). This result suggests that the main transcriptional defect observed in cells that are compromised for THO complex function is a dramatically reduced ability of RNAPII to transcribe certain coding regions.
Finally, a recombination event between direct repeats has to initiate in the repeats or in the intervening region, whereas transcription can be initiated outside of the recombinagenic region. Since, as we have shown here, hyper-recombination is completely dependent on transcription, this dependency must occur at the level of transcriptional elongation. Taken together, our results indicate that mft1Δ and thp2Δ, as well as hpr1Δ and tho2Δ, impair transcription elongation causing a recombinogenic intermediate. Whether this intermediate is caused by a collision between replication forks and stalled RNAPII, by negatively supercoiled DNA allowed to accumulate during RNAPII elongation, by an opening of the chromatin around the stalled RNAPII, or by a DNA–RNA hybrid are possibilities that can now be addressed with a combination of genetic and biochemical approaches.
Materials and methods
Yeast strains and plasmids
Yeast strains used in this study are listed in Table I. All plasmids used to determine recombination frequencies, enzymatic activities and mRNA expression levels have been described previously (Piruat and Aguilera, 1998).
Table I. Yeast strains.
| Strain | Genotype | Source |
|---|---|---|
| W303–1A | MATa ade2-1 can1-100 his3-11,15 leu2-3,112 trp1-1 ura3-1 | R.Rothstein |
| AYW3-1B | MATα ade2 his3 trp1 ura3 leu2-k::ADE2-URA3::leu2-k | Piruat and Aguilera (1996) |
| WRK5-3C | MATa ade2 can1-100 his3 leu2 trp1 ura3 leu2-k::ADE2-URA3::leu2-k | Piruat and Aguilera (1998) |
| SEY2102 | MATα gal2 his4 leu2 suc2 ura3 | S.Emr |
| Mft1(m6) | MATα gal2 his4 leu2 suc2 ura3 mft1-ts | J.Garret |
| nat1 | MATa his3 leu2 trp1 ura3 ade2 can1 nat1::LEU2 | Mullen et al. (1989) |
| BJ5457 | MATα ura3-52 trp1 lys2-801 leu2Δ1 his3Δ200 pep4::his3 prb1Δ1.6R can1 | E.Jones |
| BY4741 | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Delta;0 | EUROSCARFa |
| BY-HR167 | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 thp2::KAN | EUROSCARFa |
| JK9-3da | MATa leu2 ura3 his4 trp1 | M.Hall |
| YHTB2 | MATα his4 leu2 trp1 ura3 mft1Δ::LEU2 | this study |
| YHTB5 | MATa his3 leu2 trp1 ura3 ade2 tho2::spHIS5 | this study |
| SChY73 | MATα ura3-52 trp1 lys2-801 leu2Δ1 his3Δ200 pep4::HIS3 prb1Δ1.6R can1 THO2-6(His)-HA | this study |
| SEW-2C | MATa ade2 trp1 ura3 leu2-k::ADE2-URA3::leu2-k | This study |
| SEW-14B | MATa ade2 his3 ura3 leu2-k::ADE2-URA3::leu2-k | this study |
| SEW-2D | MATα ade2 can1-100 ura3 leu2-k::ADE2-URA3::leu2-k mft1-ts | this study |
| SEW-14C | MATα gal2 can1-100 his3 leu2 trp1 ura3 mft1-ts | this study |
| SEW-14D | MATα gal2 his4 leu2 trp1 ura3 | this study |
| YTW-3A | MATa ade2 gal2 ura3 leu2-k::ADE2-URA3::leu2-k | this study |
| YTW-5A | MATa ade2 his3 ura3 leu2-k::ADE2-URA3::leu2-k mft1ΔLEU2 | this study |
| YTW-13C | MATa ade2 can1-100 his3 ura3 leu2-k::ADE2-URA3::leu2-k mft1ΔLEU2 | this study |
| WMK-1A | MATa ade2-1 can1-100 his3-11 leu2-3,112 trp1-1 ura3-1 mft1::KAN | this study |
| HRW167–12A | MATα ade2 his3 met15Δ0 ura3 leu2-k::ADE2-URA3::leu2-k | this study |
| HRW167–12D | MATα ade2 his3 ura3 leu2-k::ADE2-URA3::leu2-k thp2::KAN | this study |
| HRW167–16A | MATα ade2 his3 met15Δ0 trp1 ura3 leu2-k::ADE2-URA3::leu2-k thp2::KAN | this study |
| HRW167–18D | MATa ade2 his3 met15Δ0 trp1 ura3 leu2-k::ADE2-URA3::leu2-k thp2::KAN | this study |
| HRW167–12C | MATa his3 leu2 met15Δ0 trp1 ura3 | this study |
| HRW167–18Bu | MATα ade2 his3 leu2-k trp1 ura3 | this study |
| HRW167–18Du | MATa ade2 his3 leu2-k met15Δ0 trp1 ura3 thp2::KAN | this study |
| HRW167–16Au | MATα ade2 his3 leu2-k met15Δ0 trp1 ura3 thp2::KAN | this study |
aFrankfurt, Germany.
Construction of GFP- and GST-fusion genes
To generate GFP-tagged proteins the full-length open reading frame (ORF) encoding Mft1 (excluding the stop codon) was subcloned into the centromeric plasmid p416MET25 upstream of GFPS65T (J.Hegemann, Düsseldorf, Germany). The full-length HPR1 gene was PCR amplified replacing the stop codon with a BamHI restriction site.
THP2–GFP and GFP–THP2 fusions were constructed in centromeric plasmids pUG34 and pUG23 (J.Hegemann, Düsseldorf, Germany) carrying yEGFP (yeast enhanced GFP) under the MET25 promoter. The full-length THP2 ORF was PCR amplified adding an EcoRI site immediately upstream of the gene and a SalI site immediately downstream for cloning into both pUG34 and pUG23. The only difference was that for the cloning into pUG23, the THP2 stop codon was excluded from the downstream primer.
GST fusions were constructed by subcloning either the full-length ORF, the N-terminal domain of Mft1 (amino acids 1–188) utilising the natural EcoRV site, or the C-terminal domain (amino acids 189–392) again using EcoRV into the pYEX series of vectors (AMRAD Biotech, Melbourne, Australia).
Construction of a yeast strain expressing His6-HA-tagged Tho2
To construct a His6-HA-tagged Tho2, we first inserted a double-stranded oligonucleotide coding for His6 and the HA epitope into an integrative plasmid derived from pRS306, harboring 1.2 kb of the THO2 5′UTR and 1.5 kb of the 5′ part of its ORF. We annealed the oligos SC1 and SC2 (Table II) and inserted the PCR-derived products between the PstI and NcoI restriction sites inmediately after the THO2 start codon. The resulting pSCh239 plasmid was then used to replace the endogenous THO2 in the protease-deficient strain BJ5457. Replacement was performed in two steps: first, the linearized plasmid was integrated into the THO2 locus, and subsequently, excission of the THO2 wild-type copy was selected on 5-FOA.
Table II. Primers.
| Primer | DNA sequencea |
|---|---|
| SC1 | TCACCATCACCATCACTATCCATATGATGTTCCTGACTATGC |
| SC2 | CATGGCATAGTCAGGAACATCATATGGATAGTGATGGTGATGGTGATGCA |
| AR1 | GAACTAAAGCCAAAGGAGACTAACTCACAATGCCTCTGTCCAGCTGAAGCTTCGTACGCT |
| AR2 | TGGCTCTTCTATCACTCCAGTTTCTTTGTCTGTTTCCTCACTAGTGGATCTGATATCATC |
| TB1 | AATAGGGTGAAACGGTTTCAGTTGATACATATTCGCACCAGTATACATTTTCAGGACTTTCGAGCTCGTTTAAACTGG |
| TB2 | CCCACATGGCCTGGTTAACACCATCAAATTGAACAGCAGGCCTGTTATAAGTTAAGCGTACCTCGAGGTCGACGGTATC |
aAll sequences are given from 5′ to 3′.
Construction of mft1Δ and tho2Δ strains
The mft1Δ::LEU2 deletion was made in JK9-3da cells using previously described DNA constructs (Beilharz et al., 1997). MFT1 was also deleted in strain W303–1A with the Kan-MX4 cassette by the PCR-mediated SFH (short flanking homology) method (Wach et al., 1994) using the primers AR1 and AR2 (Table II). Correct chromosomal deletions were confirmed by Southern blot, PCR and/or tetrad analysis.
PCR-mediated disruption was also used to disrupt the THO2 gene in JK9-3da cells with the heterologous HIS5 gene from S.pombe. The oligonucleotide primers TB1 and TB2 (Table II) were specifically designed to leave a THO2 1 kbp internal ORF intact in tho2Δ mutant cells.
Production of polyclonal anti-Hpr1 and monoclonal anti-Mft1 antibodies
To produce rabbit anti-Hpr1 antibodies, a synthetic peptide encoding the last 20 amino acids of the predicted Hpr1 sequence was coupled to the C-terminal of keyhole limpet haemocyanin (Calbiochem) as described (Hancock et al., 1998), and the fusion protein used to inoculate rabbits.
Anti-Mft1 monoclonal antibodies were produced from mice immunized with purified His6-tagged Mft1 (Cartwright et al., 1997). Hybridoma supernatants were screened for the presence of antibodies by immunoblot analysis against recombinant forms of intact Mft1 and the 35 kDa N-terminal fragment, and subsequently rescreened against the GST–Mft(Δ1–188) fusion protein. The hybridoma line P1A12H8 is clonal and secretes an antibody specifically recognizing the C-terminal domain of Mft1.
Purification of a THO protein complex using His6-HA-tagged Tho2
Yeast whole-cell extracts (WCE), up to the ultracentrifugation step, were prepared as described (Sayre et al., 1992; Otero et al., 1999). Soluble fractions prepared from 300–500 ml of WCE were collected, pooled and diluted with 3 vols of buffer A [20% glycerol, 40 mM HEPES pH 7.6, 1 mM EDTA, 1 mM dithiothreitol (DTT)]. The resulting fraction, containing 15–30 mg/ml protein, was loaded onto 200 ml Bio-Rex 70 resin (Bio-Rad) pre-equilibrated in buffer A-150 (150 mM KAc). The column was washed with 1 column volume (CV) of buffer A-150 and stepwise eluted with 2 CV A-300, A-600 and A-1200. Tho2 and Hpr1 co-eluted in A-600 (20%) and A-1200 (80%).
A-600 and A-1200 eluates from Bio-Rex 70 were diluted with buffer A, adjusting the glycerol to 10% and ammonium acetate to 600 mM (A*-600), and independently subjected to inmunoaffinity purification using 12CA5-protein G–Sepharose. To prepare 12CA5 beads, 60 mg of monoclonal anti-HA 12CA5 were coupled to 12 ml protein G– Sepharose beads in phosphate-buffered saline (PBS) buffer for 30 min before washing with PBS. The two fractions were incubated for 4 h with 6 ml of 12CA5 beads, transferred to columns and washed with 100 ml A*-600 and 80 ml of A-600 buffer not containing EDTA and DTT, but including 20 mM 2-mercaptoethanol and 5 mM imidazole (A-600β). The column was then stepwise eluted with 10 ml 1 mg/ml and 2 mg/ml HA-peptide in A-600β. More than 90% of Tho2 and Hpr1 co-eluted in 1 mg/ml HA-peptide, both in the preparations coming from Bio-Rex A-600 and A-1200.
The eluate from 12CA5-immunoaffinity beads was mixed with 1.5 ml Ni-NTA agarose beads (Qiagen) equilibrated in A600β, washed with 5 ml A600β and stepwise eluated with A600β containing 50, 100 and 400 mM imidazole (0.5 ml). More than 70% of Tho2 and Hpr1 co-eluted in the 400 mM imidazole fraction. Size was determined by gel filtration through a Superose 6 PC3.2/30 column (Pharmacia) equilibrated in 20 mM Tris–HCl pH 7.5, 0.01% Nonidet P-40 (NP-40) and 200 mM NaCl, using the SMART system (Pharmacia). Gel filtration standards (thyroglobulin, bovine-γ-globulin, chicken ovalbumin, equine myoglobin) were from Bio-Rad. Protease inhibitors [leupeptin, pepstatin A, phenylmethylsulfonyl fluoride (PMSF) and benzamidine] were included in all buffers used throughout purification.
Immunoprecipitation analysis using anti-Mft1
Cells were grown in synthetic-complete media lacking methionine to an OD600 of 0.5–0.8/ml. Cells equivalent to 30 OD600 units were harvested, resuspended in fresh pre-warmed media with 10 µCi trans 35S-label per unit cells, and incubated with shaking for 15 min at 30°C. Cell pellets were washed once in spheroplasting buffer (1 M sorbitol, 20 mM HEPES, pH 7.4) before treatment with 2.5 mg Zymolyase T-20/g cells. Spheroplast pellets were resuspended in 1 ml/mg cells lysis buffer (20 mM HEPES pH 7.4, 500 mM KAc, 1% Triton X-100, 1 mM EDTA, 20% glycerol), RNase A and DNase I were added to 0.3 mg/ml, together with a protease inhibitor cocktail (1.25 µg/ml leupeptin, 0.75 µg/ml antipain, 0.25 µg/ml chymostatin, 5 µg/ml pepstatin, 0.5 mM PMSF), and incubated for 15 min at 4°C. After centrifugation to pellet cell debris, the supernatant was added to a slurry of antibody–protein A–Sepharose beads and incubated for 2–4 h at 4°C. After washing four times in buffer (1.0% Triton X-100, 150 mM NaCl, 0.2% SDS, 5 mM EDTA, 50 mM Tris–HCl pH 7.4), the samples were analyzed by SDS–PAGE and phosphoimaging.
Sucrose density gradient centrifugation
Cell extracts were prepared as described above, except that spheroplasts were resuspended in 5 ml/g cells of lysis buffer containing 10% glycerol. Cell extract (300 µl) was loaded onto a 15–45% continuous sucrose gradient made up in PBS with 0.1% Triton X-100 and centrifuged at 4°C in a Beckman SW40 rotor for 4 h at 38 000 r.p.m. Gradients were fractionated by bottom puncture and upward displacement using automated fractionator (Brandell). Seventeen fractions of 750 µl were collected, proteins were precipitated by TCA (10% w/v) and analyzed by SDS–PAGE and western blotting.
Protein identification by mass spectrometry
Two-hundred microliters of the 400 mM imidazole fraction were precipitated with TCA and loaded in a 8–15% discontinuous SDS–polyacrylamide gel. Stained bands were excised from the gels, digested with trypsin and processed for mass spectrometric fingerprinting as described previously (Erdjument-Bromage et al., 1998). The peptide mixture was passed over a Poros 50 R2 RP micro-tip, and analyzed by matrix-assisted laser-desorption/ionization reflectron time-of-flight mass spectrometry (MALDI-reTOF MS) using a Reflex III instrument (Brüker Franzen; Bremen, Germany) and by electrospray ionization (ESI) tandem MS on an API 300 triple quadrupole instrument (PE-SCIEX; Thornhill, Canada), modified with a custom-made fine ionization source, as described (Geromanos et al., 2000). Selected mass values from the MALDI-reTOF experiments were taken to search the NCBI protein database (NIH, Bethesda, MD) and MS/MS spectra from the ESI triple quadrupole analyses were inspected for y” ion series, and the resultant information used for SequenceTag/PepFrag database searching as described previously (Erdjument-Bromage et al., 1998). Any identification thus obtained was verified by comparing the computer-generated fragment ion series of the predicted tryptic peptide with the experimental MS/MS data.
Analysis of gene expression and recombination
For the analysis of GAL1-driven expression by enzymatic activity, mid-log phase cells were inoculated in 3% glycerol–2% lactate synthetic medium at a concentration of 1.5–2.0 × 107 cells/ml. After 16 h of incubation at 30°C, either 2% glucose or 2% galactose were added and incubation was continued for other 8 h at 30°C. β-galactosidase and acid phosphatase activities and mRNA levels were determined as described (Chávez and Aguilera, 1997).
Recombination frequencies were calculated as the median frequency of six independent cultures according to Prado and Aguilera (1995).
Miscellaneous
Published methods were used for SDS–PAGE and immunoblotting (Glick et al., 1992), protein sequencing from PVDF membranes (Beilharz et al., 1997), and DNA and RNA hybridization (Prado et al., 1997). DNA amplification was done by PCR with Expand-High-Fidelity Taq polymerase (Boehringer Mannheim).
Acknowledgments
Acknowledgements
We thank M.Hall, R.Sternglanz and P.Walsh for yeast strains, S.Gratzer, S.Emr and J.Hegemann for plasmids, J.Scoble for preparing the glutathione–agarose, R.Condron for protein sequencing, G.Newman and L.Lacomis for mass spectrometry analysis, and J.Hoogenraad for anti-Mft1 monoclonal antibody production. This project was supported by the Spanish Ministry of Science and Culture grants PB96–1350 and HB98–230 (to A.A.), the Human Frontiers Science Program grants RG0075/1999-M (to A.A.) and RG0203/1998-M (to T.L.), an Australian Research Council grant A09925005 (to T.L.), an NCI Cancer Center grant P30 CA08748 (to P.T.) and a grant from the ICRF (to J.S.). T.B. and A.G.R. were recipients of an Australian Postgraduate Research Award and a Spanish Predoctoral-Training Fellow from the Spanish Ministry of Science and Culture, respectively. S.C. was the recipient of an EMBO short-term fellowship.
References
- Aguilera A. and Klein,H.L. (1988) Genetic control of intrachromosomal recombination in Saccharomyces cerevisiae. I. Isolation and genetic characterization of hyper-recombination mutations. Genetics, 119, 779–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguilera A. and Klein,H.L. (1990) HPR1, a novel yeast gene that prevents intrachromosomal excision recombination, shows carboxy-terminal homology to the Saccharomyces cerevisiae TOP1 gene. Mol. Cell. Biol., 10, 1439–1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bedwell D.M., Klionsky,D.J. and Emr,S.D. (1987) The yeast F1-ATPase beta subunit precursor contains functionally redundant mitochondrial protein import information. Mol. Cell. Biol., 7, 4038–4047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beilharz T., Beddoe,T., Landl,K., Cartwright,P. and Lithgow,T. (1997) The protein encoded by the MFT1 gene is a targeting factor for mitochondrial precursor proteins and not a core ribosomal protein. FEBS Lett., 407, 220–224. [DOI] [PubMed] [Google Scholar]
- Blackwell T.K., Moore,M.W., Yancopoulos,G.D., Suh,H., Lutzker,S., Selsing,E. and Alt,F.W. (1986) Recombination between immunoglobulin variable region gene segments is enhanced by transcription. Nature, 324, 585–589. [DOI] [PubMed] [Google Scholar]
- Cartwright P., Beilharz,T., Hansen,P., Garrett,J. and Lithgow,T. (1997) Mft52, an acid-bristle protein in the cytosol that delivers precursor proteins to yeast mitochondria. J. Biol. Chem., 272, 5320–5325. [DOI] [PubMed] [Google Scholar]
- Chang M., French-Cornay,D., Fan,H.Y., Klein,H., Denis,C.L. and Jaehning,J.A. (1999) A complex containing RNA polymerase II, Paf1p, Cdc73p, Hpr1p and Ccr4p plays a role in protein kinase C signaling. Mol. Cell. Biol., 19, 1056–1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chávez S. and Aguilera,A. (1997) The yeast HPR1 gene has a functional role in transcriptional elongation that uncovers a novel source of genome instability. Genes Dev., 11, 3459–3470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniels G.A. and Lieber,M.R. (1995) RNA:DNA complex formation upon transcription of immunoglobulin switch regions: implications for the mechanism and regulation of class switch recombination. Nucleic Acids Res., 23, 5006–5011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dul J.L. and Drexler,H. (1988) Transcription stimulates recombination. I. Specialized transduction of Escherichia coli by λtrp phages. Virology, 162, 466–470. [DOI] [PubMed] [Google Scholar]
- Emr S.D., Vassarotti,A., Garrett,J., Geller,B.L., Takeda,M. and Douglas,M.G. (1986) The amino terminus of the yeast F1-ATPase beta-subunit precursor functions as a mitochondrial import signal. J. Cell Biol., 102, 523–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erdjument-Bromage H., Lui,M., Lacomis,L., Grewal,A., Annan,R.S., McNulty,D.E., Carr,S.A. and Tempst,P. (1998) Examination of micro-tip reversed-phase liquid chromatographic extraction of peptide pools for mass spectrometric analysis. J. Chromatogr. A, 826, 167–181. [DOI] [PubMed] [Google Scholar]
- Fan H.Y., Cheng,K.K. and Klein,H.L. (1996) Mutations in the RNA polymerase II transcription machinery suppress the hyperrecombination mutant hpr1Δ of Saccharomyces cerevisiae. Genetics, 142, 749–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrett J.M., Singh,K.K., von der Haar,R.A. and Emr,S.D. (1991) Mitochondrial protein import: isolation and characterization of the Saccharomyces cerevisiae MFT1 gene. Mol. Gen. Genet., 225, 483–491. [DOI] [PubMed] [Google Scholar]
- George R., Beddoe,T., Landl,K. and Lithgow,T. (1998) The yeast nascent polypeptide-associated complex initiates protein targeting to mitochondria in vivo. Proc. Natl Acad. Sci. USA, 95, 2296–2301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geromanos S., Freckleton,G. and Tempst,P. (2000) Tuning of an electrospray ionization source for maximum peptide-ion transmission into a mass spectrometer. Anal. Chem., 72, 777–790. [DOI] [PubMed] [Google Scholar]
- Glick B.S., Brandt,A., Cunningham,K., Muller,S., Hallberg,R.L. and Schatz,G. (1992) Cytochromes c1 and b2 are sorted to the intermembrane space of yeast mitochondria by a stop-transfer mechanism. Cell, 69, 809–822. [DOI] [PubMed] [Google Scholar]
- Grimm C., Schaer,P., Munz,P. and Kohli,J. (1991) The strong ADH1 promoter stimulates mitotic and meiotic recombination at the ADE6 gene of Schizosaccharomyces pombe. Mol. Cell. Biol., 11, 289–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hancock D.C., O’Reilly,N.J. and Evan,G.I. (1998) Synthesis of peptides for use as immunogens. In Pound,J.D. (ed.), Immunochemical Protocols. Methods in Molecular Biology. Humana, Totowa, NJ, pp. 69–79. [DOI] [PubMed] [Google Scholar]
- Ikeda H. and Matsumoto,T. (1979) Transcription promotes recA-independent recombination mediated by DNA-dependent RNA polymerase in Escherichia coli. Proc. Natl Acad. Sci. USA, 76, 4571–4575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kübrich M., Dietmeier,K. and Pfanner,N. (1995) Genetic and biochemical dissection of the mitochondrial protein-import machinery. Curr. Genet., 27, 393–403. [DOI] [PubMed] [Google Scholar]
- Lauster R., Reynaud,C.A., Martensson,I.L., Peter,A., Bucchini,D., Jami,J. and Weill,J.C. (1993) Promoter, enhancer and silencer elements regulate rearrangement of an immunoglobulin transgene. EMBO J., 12, 4615–4623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee M.S., Henry,M. and Silver,P.A. (1996) A protein that shuttles between the nucleus and the cytoplasm is an important mediator of RNA export. Genes Dev., 10, 1233–1246. [DOI] [PubMed] [Google Scholar]
- Mullen J.R., Kayne,P.S., Moerschell,R.P., Tsunasawa,S., Gribskov,M., Colavito-Shepanski,M., Grunstein,M., Sherman,F. and Sternglanz,R. (1989) Identification and characterization of genes and mutants for an N-terminal acetyltransferase from yeast. EMBO J., 8, 2067–2075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nevo-Caspi Y. and Kupiec,M. (1994) Transcriptional induction of Ty recombination in yeast. Proc. Natl Acad. Sci. USA, 91, 12711–12715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nickoloff J.A. (1992) Transcription enhances intrachromosomal homologous recombination in mammalian cells. Mol. Cell. Biol., 12, 5311–5318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oltz E.M., Alt,F.W., Lin,W.C., Chen,J., Taccioli,G., Desiderio,S. and Rathbun,G. (1993) A V(D)J recombinase-inducible B-cell line: role of transcriptional enhancer elements in directing V(D)J recombination. Mol. Cell. Biol., 13, 6223–6230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otero G., Fellows,J., Li,Y., de Bizemont,T., Dirac,A.M., Gustafsson, C.M., Erdjument-Bromage,H., Tempst,P. and Svejstrup,J.Q. (1999) Elongator, a multisubunit component of a novel RNA polymerase II holoenzyme for transcriptional elongation. Mol. Cell, 3, 109–118. [DOI] [PubMed] [Google Scholar]
- Piruat J.I. and Aguilera,A. (1996) Mutations in the yeast SRB2 general transcription factor suppress hpr1-induced recombination and show defects in DNA repair. Genetics, 143, 1533–1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piruat J.I. and Aguilera,A. (1998) A novel yeast gene, THO2, is involved in RNA pol II transcription and provides new evidence for transcriptional elongation-associated recombination. EMBO J., 17, 4859–4872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prado F. and Aguilera,A. (1995) Role of reciprocal exchange, one-ended invasion crossover and single-strand annealing on inverted and direct repeat recombination in yeast: different requirements for the RAD1, RAD10 and RAD52 genes. Genetics, 139, 109–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prado F., Piruat,J.I. and Aguilera,A. (1997) Recombination between DNA repeats in yeast hpr1Δ cells is linked to transcription elongation. EMBO J., 16, 2826–2835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos-Rosa H. and Aguilera,A. (1994) Increase in incidence of chromosome instability and non-conservative recombination between repeats in Saccharomyces cerevisiae hpr1Δ strains. Mol. Gen. Genet., 245, 224–236. [DOI] [PubMed] [Google Scholar]
- Santos-Rosa H., Clever,B., Heyer,W.D. and Aguilera,A. (1996) The yeast HRS1 gene encodes a polyglutamine-rich nuclear protein required for spontaneous and hpr1-induced deletions between direct repeats. Genetics, 142, 705–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sayre M.H., Tschochner,H. and Kornberg,R.D. (1992) Purification and properties of Saccharomyces cerevisiae RNA polymerase II general initiation factor a. J. Biol. Chem., 267, 23383–23387. [PubMed] [Google Scholar]
- Schneiter R., Guerra,C.E., Lampl,M., Gogg,G., Kohlwein,S.D. and Klein,H.L. (1999) The Saccharomyces cerevisiae hyperrecombination mutant hpr1Δ is synthetically lethal with two conditional alleles of the acetyl coenzyme A carboxylase gene and causes a defect in nuclear export of polyadenylated RNA. Mol. Cell. Biol., 19, 3415–3422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi X., Chang,M., Wolf,A.J., Chang,C.H., Frazer-Abel,A.A., Wade,P.A., Burton,Z.F. and Jaehning,J.A. (1997) Cdc73 and Paf1 are found in a novel RNA polymerase II-containing complex distinct from the Srbp-containing holoenzyme. Mol. Cell. Biol., 17, 1160–1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart S.E. and Roeder,G.S. (1989) Transcription by RNA polymerase I stimulates mitotic recombination in Saccharomyces cerevisiae. Mol. Cell. Biol., 9, 3464–3472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas B.J. and Rothstein,R. (1989) Elevated recombination rates in transcriptionally active DNA. Cell, 56, 619–630. [DOI] [PubMed] [Google Scholar]
- Thyagarajan B., Johnson,B.L. and Campbell,C. (1995) The effect of target site transcription on gene targeting in human cells in vitro. Nucleic Acids Res., 23, 2784–2790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilette D., Uzest,M., Ehrlich,S.D. and Michel,B. (1992) DNA transcription and repressor binding affect deletion formation in Escherichia coli plasmids. EMBO J., 11, 3629–3634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voelkel-Meiman K., Keil,R.L. and Roeder,G.S. (1987) Recombination-stimulating sequences in yeast ribosomal DNA correspond to sequences regulating transcription by RNA polymerase I. Cell, 48, 1071–1079. [DOI] [PubMed] [Google Scholar]
- Wach A., Brachat,A., Pohlmann,R. and Philippsen,P. (1994) New heterologous modules for classical or PCR-based gene disruptions in Saccharomyces cerevisiae. Yeast, 10, 1793–1808. [DOI] [PubMed] [Google Scholar]
- Zhu Y., Peterson,C.L. and Christman,M.F. (1995) HPR1 encodes a global positive regulator of transcription in Saccharomyces cerevisiae. Mol. Cell. Biol., 15, 1698–1708. [DOI] [PMC free article] [PubMed] [Google Scholar]