

Abstract
XRCC4 is essential for carrying out non-homologous DNA end joining (NHEJ) in all eukaryotes and, in particular, V(D)J recombination in vertebrates. Xrcc4 protein forms a complex with DNA ligase IV that rejoins two DNA ends in the last step of V(D)J recombination and NHEJ to repair double strand breaks. XRCC4-defective cells are extremely sensitive to ionizing radiation, and disruption of the XRCC4 gene results in embryonic lethality in mice. Here we report the crystal structure of a functional fragment of Xrcc4 at 2.7 Å resolution. Xrcc4 protein forms a strikingly elongated dumb-bell-like tetramer. Each of the N-terminal globular head domains consists of a β-sandwich and a potentially DNA-binding helix– turn–helix motif. The C-terminal stalk comprising a single α-helix >120 Å in length is partly incorporated into a four-helix bundle in the Xrcc4 tetramer and partly involved in interacting with ligase IV. The Xrcc4 structure suggests a possible mode of coupling ligase IV association with DNA binding for effective ligation of DNA ends.
Keywords: DNA ligase IV/NHEJ/structure/tetramer/Xrcc4
Introduction
Double strand breakage of DNA presents cells with a serious challenge. If unrepaired, such breaks lead to cell death; if wrongly repaired, they can lead to gross genomic rearrangements (Gao et al., 2000). In eukaryotic cells, double strand breaks (DSBs) occur in the normal process of meiosis, and can also result from various types of specialized recombination, such as mating type switching in yeast or V(D)J recombination in vertebrate lymphoid cells. In addition, DSBs are common products of DNA damage by ionizing radiation or endogenous or exogenous oxidative agents. Efficient repair of DSBs is thus essential for maintaining viability and genomic integrity (Lieber, 1998).
In eukaryotes, DSBs are repaired by either homologous recombination or non-homologous end joining (NHEJ) (Jeggo et al., 1998). NHEJ is predominant in G0, G1 and early S phase of vertebrate cells, and is exclusively required for V(D)J recombination (Lieber, 1998). Genetic and biochemical studies have identified several proteins that have essential roles in NHEJ (Roth and Gellert et al., 2000). These include the Ku protein (Ku70–Ku80 heterodimer), the catalytic subunit of DNA-dependent protein kinase (DNA-PKCS), DNA ligase IV and Xrcc4. Mice lacking either of the subunits of Ku protein or DNA-PKCS exhibit sensitivity to radiation and are severely immunodeficient because they fail to complete V(D)J recombination (Lieber, 1998).
The Xrcc4 and ligase IV proteins form a complex in vivo (Critchlow et al., 1997; Grawunder et al., 1997). The presence of Xrcc4 is important for stability and activity of ligase IV (Grawunder et al., 1997; Bryans et al., 1999; Teo and Jackson et al., 2000). In vitro, Xrcc4 stimulates adenylation of ligase IV, the first chemical step in DNA ligation (Modesti et al., 1999). Xrcc4 may also bridge two DNA ends for ligation, perhaps by interacting with Ku protein already present at the ends (Chen et al., 2000; Nick McElhinny et al., 2000). In addition, purified Xrcc4 can bind cooperatively to DNA, with a preference for nicked or broken DNA, and a mutational study has suggested that DNA binding may be important for its biological activity (Modesti et al., 1999).
Disruption of the XRCC4 or ligase IV genes in mice leads to embryonic lethality, with a primary defect in neurogenesis and severe neuronal apoptosis (Barnes et al., 1998; Frank et al., 1998; Gao et al., 1998). This inspired speculations that a type of site-specific recombination might be involved in development of the central nervous system. However, additional inactivation of p53 allows these mice to survive with apparently normal brain development, suggesting instead that immature neuronal cells are hypersensitive to apoptosis when their DNA is damaged (Gao et al., 2000). Nevertheless, XRCC4–/– p53–/– mice develop B-cell lymphomas early in life, evidently due to an increased likelihood of chromosome translocations and gene amplifications resulting from broken ends in immunoglobulin loci (Gao et al., 2000). Thus, XRCC4 and the associated NHEJ factors appear to play a critical role not only in V(D)J recombination and maintaining the integrity of the genome, but also in guarding against cancer.
As the mode of action of Xrcc4 is not at all clear, it is particularly interesting to obtain structural information about the protein. We report here the atomic structure of a functional fragment of human Xrcc4 at 2.7 Å resolution and discuss implications for its function in NHEJ.
Results and discussion
Homologs of Xrcc4 have been found in human, mouse and yeast (LIF1), and perhaps in Drosophila and Arabidopsis (Figure 1). Full-length human Xrcc4 contains 336 residues (DDBJ/EMBL/GenBank accession No. AAD47298) (Li et al., 1995). However, deletional analysis revealed that the N-terminal 200 residues are sufficient to restore V(D)J recombination, and the N-terminal 250 residues confer resistance to ionizing radiation in XRCC4-defective cells (Mizuta et al., 1997; Grawunder et al., 1998a; Modesti et al., 1999). Based on protein stability analysis by partial proteolysis, we first purified and crystallized an Xrcc4 fragment comprising residues 1–265. The crystals belonged to space group C2 with two Xrcc4 subunits in each asymmetric unit, and diffracted X-rays to 2.9 Å resolution. The structure was phased by multiwavelength anomalous diffraction (MAD) with SeMet-substituted crystals. The refined model contains residues 1–203 of one subunit (L) and 1–178 in the second (S). The two protein subunits are associated by a pseudo 2-fold axis along the length of the molecule and the Xrcc4 dimer resembles a palm tree with a globular head and a long slender stem (Figure 2). A dumb-bell-like Xrcc4 tetramer results from a crystallographic 2-fold axis (Figure 3). Even though residues 204–265 are present, they are disordered in the crystal structure. We therefore cloned, expressed and purified another fragment containing only the first 203 residues of Xrcc4. This fragment crystallized isomorphously to the original C2 crystals, but offered improved diffraction to 2.7 Å. The structure of the 1–203 fragment, essentially identical to the structured part of the 1–265 fragment, has been refined to an R-value of 23.2% and an Rfree of 26.2% with standard geometry (Table I).

Fig. 1. Sequence alignment of Xrcc4 homologs of human, mouse and Saccharomyce cerevisiae. The related genes found in Drosophila and Arabidopsis, which are included in this alignment, are based on the sequence similarity, but their function in NHEJ has not been tested. Secondary structures found in human Xrcc4 are indicated by yellow arrows (β-strands) and green ribbons (α-helices). Regions involved in Xrcc4 dimer formation are underlined with gray lines ending in filled circles, tetramer formation with a black line ending in diamonds, and both tetramer and dimer formation with a line ending in a circle and diamond. Conserved sequences are highlighted in yellow for hydrophobic residues important for tertiary and quarternary structure formation, green for polar residues making structurally critical hydrogen bonds and salt bridges, blue for those potentially interacting with DNA, and red for those currently exposed to solvent but potentially interacting with ligase IV.

Fig. 2. Structure of an Xrcc4 dimer. (A) A ribbon diagram of the Xrcc4 dimer. The L subunit is shown in dark green and the S subunit in lighter green. (B) A stereo view of L (dark green) and S (light green) subunits superimposed. (C and D) Two orthogonal views of the S subunit in ribbon diagram. β-strands are in yellow and α-helices in aqua green. (E) Superposition of the αA and αB helices of Xrcc4 and the DNA recognition HTH motif of γδ-resolvase (PDB accession No. 1GDT). The r.m.s. deviation of the 88 main chain atoms within the HTH motifs is 0.9 Å. (F) Topology diagram of Xrcc4. The β-strands are represented by arrows and α-helices by rectangular boxes.

Fig. 3. Structure of the Xrcc4 tetramer. (A) Two orthogonal views of an Xrcc4 tetramer. The crystallographic dyad axis is marked. The two dimers are colored in green and red; a darker color for the L subunit and a lighter color for the S subunit. (B) A view that shows the interdigitation of the two Xrcc4 dimers. (C) Stereo drawings of the conserved residues between 170 and 203 in one L subunit of the Xrcc4 tetramer and (D) two layers of repeated salt bridges in the four-helix bundle region. Both are viewed in the same orientation as in (B).
Table I. Data collection and refinement.
| XRCC4 (amino acids 1–265) |
XRCC4 (1–203 aa) |
||||
|---|---|---|---|---|---|
| SeMet-substituted | Native 1 | Native 2 | |||
| Data collection | |||||
| unit cell (C2) (a, b, c, β) | 164.7 Å, 73.6 Å 103.8 Å, 103.8° | 165.5 Å, 73.7 Å, 87.2 Å, 103.5° | 164.9 Å, 74.8 Å, 87.3 Å, 104.0° | ||
| solvent content (%) | 71.0 | 71.0 | 77.8 | ||
| wavelength | 0.97892 | 0.97853 | 0.96859 | 0.9789 | 0.979 |
| resolution (Å)a | 3.2 (3.3–3.2) | 3.2 (3.3–3.2) | 3.2 (3.3–3.2) | 2.9 (3.0–2.9) | 2.6 (2.7–2.6) |
| completeness (%)a | 94.2 (76.6) | 94.1 (75.9) | 93.5 (74.3) | 90.3 (80.2) | 98.1 (95.9) |
| Rmerge (%) | 7.4 (33.5) | 7.8 (34.1) | 8.3 (35.5) | 5.5 (32.4) | 5.0 (40.8) |
| I/σ(I)a | 12.3 (1.5) | 12.1 (1.5) | 11.5 (1.3) | 13.2 (1.2) | 21.0 (1.2) |
| Refinement | |||||
| resolution (Å) | 30–2.7 | ||||
| refl. working set | 25 319 | ||||
| refl. test set | 2791 | ||||
| protein atoms | 3107 | ||||
| water and ion atoms | 77 | ||||
| R (Rfree) (%) | 23.2 (26.2) | ||||
| r.m.s.d. in bonds (Å) | 0.007 | ||||
| r.m.s.d. in angles (°) | 1.17 | ||||
aData for the highest resolution shell are shown in parentheses.
Xrcc4
An Xrcc4 protomer consists of an N-terminal head domain (amino acids 1–115) and a long helical C-terminal stalk (amino acids 119–203 of the L subunit and 119–178 of the S subunit) (Figure 2B). The head domain contains a β-sandwich, in which two antiparallel sets of strands 1, 2, 3 and 4, and 5, 6 and 7 form two β-sheets, and two α-helices (A and B) forming a typical helix–turn–helix (HTH) structure (Wintjens and Rooman et al., 1996) (Figure 2C, D and E). At one end of the β-sandwich, the two β-sheets are tied together between the antiparallel strands 4 and 7 and between parallel strands 1 and 5 by three and one hydrogen bonds, respectively. At the other end, the two helices between strands 4 and 5 insert the second helix (αB) between the β-sheets and make them flare out (Figure 2C and D). The helical stalk of Xrcc4 consists of a single but kinked α-helix (αC) that extends ∼120 Å from the head domain. The head domain and the stalk are held together by van der Waals contacts (R3, I5 and T35 of the first β-sheet, and V122, I123, L126 and Y129 of αC) and a pair of salt bridges between R3 and E125. The folding topology of the head domain is not similar to that of any known structures and is thus considered a new fold (Figure 2F).
A crystal asymmetric unit contains two Xrcc4 subunits, whose head domains are nearly identical. However, the two helical stalks differ in the length and curvature of their helical axes (Figure 2B). The L subunit with 203 residues is kinked first at G160 and again at K190, while the S subunit is relatively straight to residue 178, after which it is disordered. The interactions between the two subunits are quite extensive, burying a total of ∼2300 Å2 of molecular surface. The helical stalks, which form a pair of parallel α-helices with a single left-handed crossover, are central to the dimer interface. They interact reciprocally from residue 119 to 155 and could form an inter-chain disulfide bond between two C130 residues under non-reducing conditions, as previously observed (Leber et al., 1998). In addition, each helical stalk from residues 119 to 135 is in close contact with strands 1, 2 and 3 of the head domain that belong to the partner subunit, thus rigidifying the connection between the head domain and the stalk in the Xrcc4 dimer. The dimer interface is predominantly hydrophobic (Figure 1) and is stabilized and kept in register by one salt bridge between side chains of D132 and R7, and two hydrogen bonds, between R124 and the carbonyl oxygen of D38, and between N148 and N148. The hydrophobic nature, the extensiveness of the interface and the conservation of residues at the dimer interface from yeast to human (Figure 1) all suggest that this crystal dimer persists in solution.
Two Xrcc4 dimers related by a crystallographic 2-fold symmetry are packed tail-to-tail in the stalk region to form a tetramer (Figure 3A). Two pairs of helical stalks that run antiparallel toward each other are assembled into an antiparallel four-helix bundle between residues 159 and 173 (Figure 3B and C). Since the S subunit is disordered after residue 178, the tetramer interface is tapered into a three-helix bundle with residues 180–203 from one L subunit and residues 140–163 of the opposite dimer. Residues 175–190 of the L subunit are in lattice contact with a neighboring Xrcc4 molecule, which may stabilize the C-terminal helical conformation of that subunit. However, there is no lattice contact that would prevent the S subunit from continuing after residue 178 to complete a four-helix bundle structure in the Xrcc4 tetramer. Both C-termini of the S subunits are located on the inside of a concave surface, while those of the L subunits are on the outside (Figure 3A). We surmise that the C-terminal region of the S subunit is intrinsically mobile in an Xrcc4 tetramer and that the crystal structure captures rather than induces this asymmetric nature.
The tetramer interface buries ∼3300 Å2 of molecular surface between the two dimers and is ∼50% more extensive than the dimer interface. Residues 180–200 are the most conserved in the entire sequence among Xrcc4 homologs (Figure 1). Among these, many of the conserved hydrophobic residues in the L subunit are buried in the tetramer interface (Figure 3D). Interestingly, the same conserved region in the S subunit is disordered. In addition to the conserved hydrophobic interactions, the tetramer interface comprises a number of charged or polar residues. A single salt bridge between side chains of K169 and E173 is repeated four times to form two layers of interactions in the middle of the four-helix bundle (Figure 3E). These polar interactions probably hold the tetramer in register. Three additional pairs of polar interactions, K187(L1)– D155(S2), K188(L1)–N156(L2) and H195(L1)–E147(S2), in the three-helix bundle region on each side of the central four-helix bundle further strengthen the Xrcc4 tetramer.
Xrcc4 was first reported to be a dimer based on disulfide cross-linking (Leber et al., 1998) and later suggested to be an oligomer in solution based on the apparent mol. wt of ∼240 kDa estimated by size exclusion chromatography (Modesti et al., 1999). The crystal structure suggests that Xrcc4 forms a tetramer. Owing to the large radius of gyration of the elongated Xrcc4 and a similar overall molecular shape whether it is a dimer or tetramer, size exclusion chromatography cannot be definitive in characterizing the oligomeric state of Xrcc4. To resolve this question, we carried out protein cross-linking and equilibrium ultracentrifugation experiments (Figure 4), both of which support the existence of a tetrameric structure for the 1–265 fragment as well as the intact Xrcc4 in solution, as in the crystal. The dissociation constants of tetramer to dimer in 0.1 M KCl, thus approximating physiological conditions, are 3.0 and 22 µM for the Xrcc4 fragment and the full-length protein, respectively. When the KCl concentration is increased to 0.2 M, dissociation constants of 13 and 22 µM are measured for the Xrcc4 fragment and the full-length protein, respectively. The relatively high Kd values indicate that inside cells the dimeric and tetrameric Xrcc4 could exist in an equilibrium, which may be altered upon binding of ligase IV and DNA.
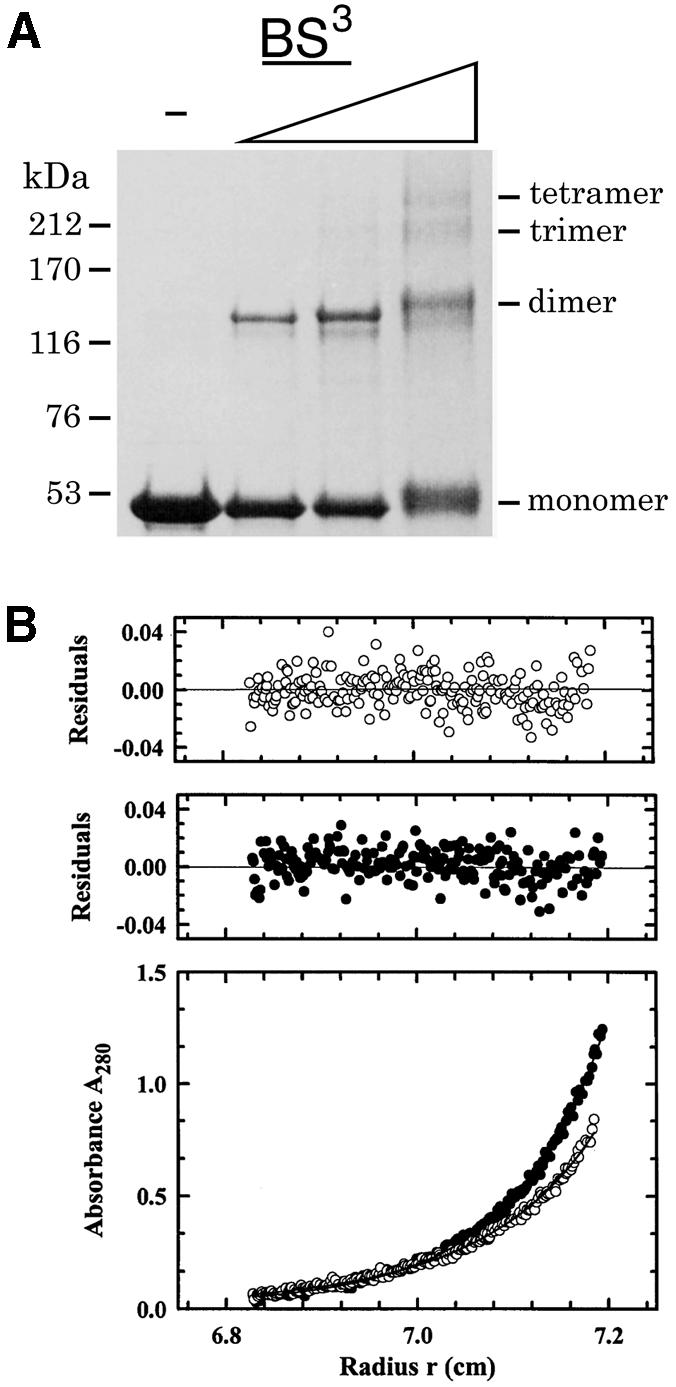
Fig. 4. The equilibrium of Xrcc4 dimer and tetramer. (A) The full-length Xrcc4 is cross-linked by BS3 and analyzed by SDS–PAGE. The Xrcc4 concentration is fixed at 50 µg/ml and concentrations of BS3 are increased from 0.6 to 5 mM, as indicated by the ascending triangle. The molecular weights are marked at the side of the figure. Similar analysis and results were obtained with Xrcc4 (1–265) (data not shown). (B) Sedimentation equilibrium profiles at 280 nm and 4.0°C for Xrcc4 (1–265) at a loading concentration of 0.4 mg/ml. Data were collected at 10 000 r.p.m. and analyzed in terms of a reversible dimer–tetramer equilibrium. The best fit is shown as a line through the experimental points, and the corresponding distributions of the residuals are shown above the plot. Filled circles correspond to data collected in 0.1 M KCl, whereas open circles correspond to data collected in 0.2 M KCl. A similar analysis was performed with full-length Xrcc4 (see text and Materials and methods).
Similarity of Xrcc4 to SMC proteins
The overall structure of the Xrcc4 tetramer, which resembles an elongated dumb-bell, is reminiscent of the structures of SMC protein observed by electron microscopy (Connelly et al., 1998; Melby et al., 1998). Although the SMC family differs from Xrcc4 in possessing an ATPase activity in the head domain, SMC proteins are predicted to contain a similar long helical stalk (Hirano et al., 1995). In SMC proteins, an ATPase domain is com posed of N- and C-terminal regions separated by hundreds of angstroms because of the intervening α-helical stalk. SMC molecules have been suggested to form a dimer with a head-to-tail configuration so that the N- and C-terminal regions are in suitable proximity to form the ATPase domain (Hirano et al., 1995). The recently reported crystal structure of the ATPase domain of Rad50, a member of the SMC family, confirms this model and provides further insight (Hopfner et al., 2000). Not only is each ATPase structural domain composed of N- and C-terminal polypeptide chains, a functional Rad50 also requires two of these ATPase domains and thus four polypeptide chains to associate. It is possible that such a tetrameric ATPase structure is the result of folding a dimeric SMC protein in the middle and bringing together the two globular ends. However, an SMC protein could also be assembled into a tetramer using the kinked helical stalks, as is observed in Xrcc4, with active ATPase sites at each end of the dumb-bell-like structure.
Modeling of bound DNA
Xrcc4 protein has been shown to bind DNA (Modesti et al., 1999; Chen et al., 2000; Nick McElhinny et al., 2000). However, with a calculated pI of 4.7, Xrcc4 is predicted to be quite negatively charged at neutral pH. Our structure reveals that the molecular surface of Xrcc4 is in fact almost entirely covered with negative electrostatic potential, which makes it unattractive to DNA. One DNA-binding region is likely to be within the N-terminal 203 residues, because the 1–203 fragment binds DNA, as does the full-length protein (Modesti et al., 1999). In the Xrcc4 structure, the one notable region that shows positive electrostatic potential is at the base of the head domain, surrounding the second helix (αB) of the HTH motif. HTH is a well known motif in sequence-specific DNA binding (Wintjens and Rooman et al., 1996). Recently, it was also reported to bind the phosphate–sugar backbone of DNA (Luger et al., 1997). A short piece of B-DNA can easily be modeled into this region based on charge and shape complementarity (Figure 5A). The following residues within the head domain are predicted to be important for DNA binding: K4, K26, K65, R71, K72, K99 and K102. In addition, the six-residue connection between the αB helix and β5 strand contains two glycine and two alanine residues, which make it potentially flexible. Comparison of the L and S subunits reveals that the structure of this region is indeed variable (Figure 2B). It is possible that the two β-sheets in the head domain close toward each other and αB moves outwards from the β-sandwich in order to make a broader contact with the DNA.
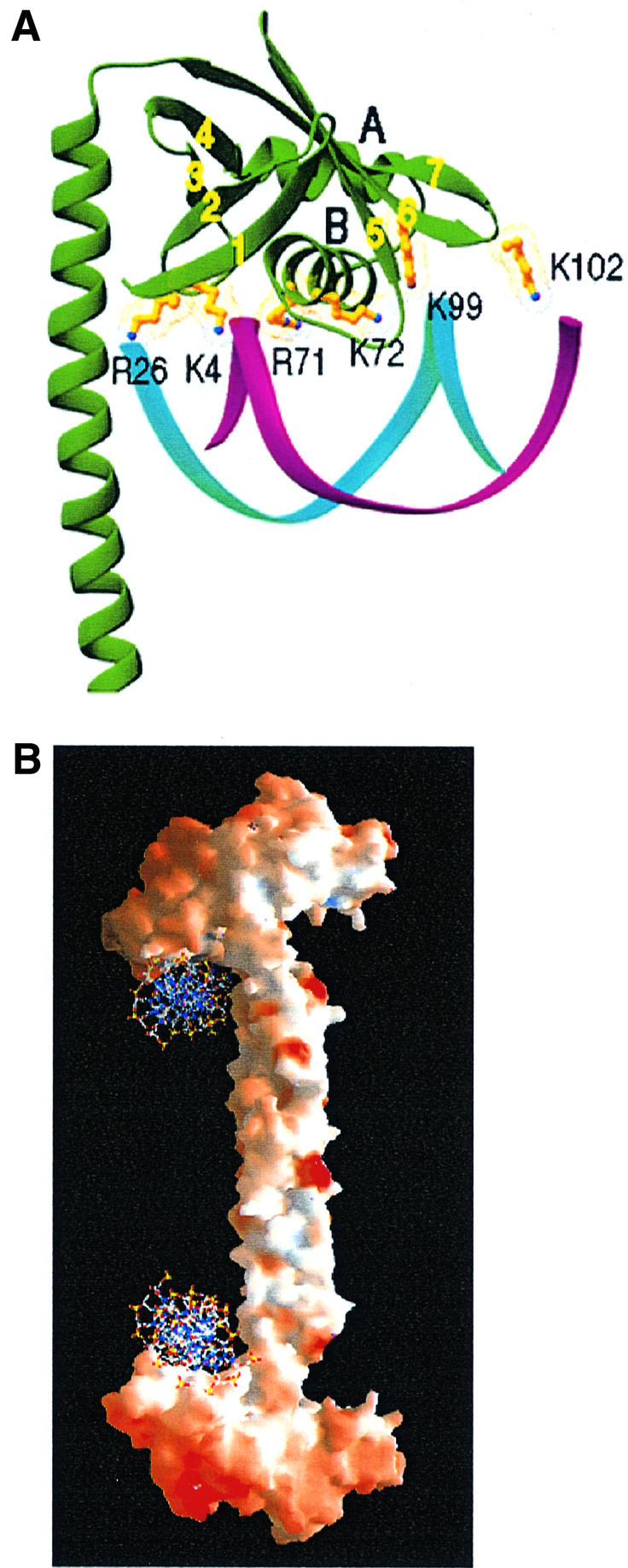
Fig. 5. A model of DNA binding by Xrcc4. (A) Basic residues of Xrcc4, which are potentially involved in DNA binding, are shown together with a B-DNA. The αB helix of the HTH motif faces the major groove and interacts with the DNA backbone. (B) A molecular surface presentation of Xrcc4 shown with positive (blue) and negative (red) electrostatic potential. Two DNA molecules are modeled to bind the S subunits as shown in (A) at the base of the head domain, as one of many possible ways in which the protein and DNA can associate with one another.
Xrcc4 can bind cooperatively to DNA (Modesti et al., 1999), suggesting that a functional Xrcc4 molecule contains multiple DNA-binding sites. As modeled here, there is one DNA-binding surface available in each Xrcc4 subunit. By forming a tetramer, Xrcc4 can thus simultaneously bind as many as four DNA duplexes. Xrcc4 is suggested to align DNA ends for ligation by ligase IV (Li et al., 1995; Leber et al., 1998). As one example of a number of possible arrangements of DNA among the Xrcc4 subunits, two DNA ends have been modeled to bind a pair of equivalent subunits in the tetramer of Xrcc4 (Figure 5B).
Interaction with DNA ligase IV
Xrcc4 forms a complex with DNA ligase IV that is stable enough to resist challenge by 1 M salt in vitro (Critchlow et al., 1997; Modesti et al., 1999). Previous analysis has implicated residues 162–231 of Xrcc4 as being important for interaction with residues 767–783 in ligase IV, which lie between two BRCT domains (Mizuta et al., 1997; Grawunder et al., 1998a,b; Modesti et al., 1999; Teo and Jackson et al., 2000). Residues 180–198 within this region of Xrcc4 are the most conserved among Xrcc4 homologs (Figure 1). Interestingly, in the crystal structure, these conserved residues are found to be either involved in Xrcc4 tetramerization (in the L subunit) or disordered (in the S subunit). These residues may be conserved because they interact directly with ligase IV, or they may be important for tetramerization of Xrcc4, which in turn is required for its association with ligase IV.
Amino acid composition analysis of Xrcc4 and ligase IV separated after co-purification as a two-protein complex indicates that the molar ratio of Xrcc4 protomers to ligase IV is 2:1 (M.S.Junop, unpublished results). If Xrcc4 retains the arrangement seen in the crystal structure when in solution, either the L or S subunits may interact with ligase IV. The two L subunits that utilize their hydrophobic residues to form the Xrcc4 tetramer still contain a number of conserved yet exposed charged residues between 162 and 200 (Figure 3D), which potentially may interact with ligase IV. The region of ligase IV that was mapped to interact with Xrcc4 is predicted to have a high propensity to form an amphipathic α-helix, which may interact with the three-helix bundle region of the Xrcc4 tetramer interface to form a four-helix bundle (Figure 6). The question then would be what role the disordered region in the S subunit plays. Another possibility is that residues after 178 in the S subunits, which are disordered in the crystal structure, interact directly with ligase IV and thus become ordered (Figure 6). This model is attractive since the conserved residues would then be utilized in both Xrcc4 tetramer formation (L) and Xrcc4–ligase IV interaction (S).

Fig. 6. Four possible modes of Xrcc4 interaction with ligase IV. If Xrcc4 remains as a tetramer, ligase IV is shown as binding either to the helical stem of the L subunits (upper panel) or to the corresponding disordered region of the S subunits (lower panel). If Xrcc4 dissociates to become a dimer, ligase IV interacts with either one or both helical stems of the two Xrcc4 subunits. The proposed DNA-binding sites are labeled as black asterisks.
Alternatively, binding of ligase IV may dissociate Xrcc4 from tetramer to dimer, which leads to two additional possible models for the Xrcc4–ligase IV complex (Figure 6). The ends of the αC helices of the Xrcc4 dimer may interact with the predicted helix of ligase IV in a manner similar to the three-helix bundle found in the Xrcc4 tetramer. It is also possible that the C-terminal end of the L subunit may fold back to form a three-helix bundle within an Xrcc4 dimer, with the tail of the S subunit interacting with ligase IV in a manner similar to that proposed for the Xrcc4 tetramer.
In all cases, ligase IV will interact with the region C-terminal to the head domain of Xrcc4, where DNA is proposed to bind. Therefore, the binding regions for DNA and ligase IV are non-overlapping in Xrcc4, supporting the notion that Xrcc4 can bind ligase IV and DNA simultaneously, and possibly align DNA ends for ligase IV to join them. In addition, the known interaction of Ku protein with Xrcc4–ligase IV could be used to bring the complex to Ku-bound DNA ends. If an NHEJ complex is composed of an Xrcc4 dimer and one ligase IV, the two DNA ends have to bind to the two head domains of Xrcc4 in a side-by-side configuration. This arrangement would require either bending of the DNA molecules or rearrangement of the Xrcc4 dimer in order for DNAs to be aligned properly for ligation. Alternatively, if a Xrcc4 tetramer and two ligase IVs form a complex, the two DNA ends can be placed at the opposite ends of the Xrcc4 tetramer as depicted in Figure 5B. This configuration provides DNA ends and ligase IV with ample space and flexibility, which is demonstrated in the asymmetric tetramer interface and the disordered part of the helical stalk, to align and assemble for the chemical reaction.
Conclusion
The crystal structure of Xrcc4 offers the first glimpse of the NHEJ machinery. The dumb-bell-like Xrcc4 tetramer provides a structural basis for multivalent DNA binding and coordinated association with ligase IV at the same time. The structure of Xrcc4 is also reminiscent of the overall shape of SMC proteins. The resemblance to the SMC family is probably more than a mere coincidence. Future experiments are needed to test the specific models of Xrcc4 binding to ligase IV and DNA, and the predicted assembly of these three components in NHEJ.
Materials and methods
Cloning and purification
PCR products containing the T7 promoter region and coding sequence for a His6 tag and amino acids 1–265 or 1–203 of human XRCC4 were generated using pBMM42 (Modesti et al., 1999) as template, digested with BglII–XhoI and ligated into the BamHI–SalI-digested pACYC154. The resulting plasmids were named pWY1139 (1–265) and pWY1190 (1–203).
Xrcc4 was expressed in BL21(DE3) cells (Novagen) induced with 0.5 mM isopropyl-β-d-thiogalactopyranoside (IPTG) for 3 h at 20°C. Cell pellets were resuspended in 20 mM Tris pH 8.5, 0.5 M KCl, 2 mM β-mercaptoethanol, 10 mM imidazole, 10% glycerol, 0.03% lauryldimethylamine oxide (LDAO), and lysed by passage though a French pressure cell at 8000 p.s.i. After clarification by centrifugation at 30 000 g for 40 min, Xrcc4 was purified by nickel affinity chromatography (Pharmacia). The column was washed and the sample was eluted with a gradient from 40 to 300 mM imidazole. Pooled fractions were treated with 500 U of TEV protease (Life Technologies) per milligram of of protein for 90 min at 20°C to remove the His tag, and loaded onto a MonoQ column (Pharmacia) equilibrated with 20 mM Tris pH 8.0, 10 mM dithiothreitol (DTT), 10% glycerol, 150 mM KCl. Proteins were eluted with a linear gradient from 150 to 400 mM KCl. Peak fractions were concentrated and buffer exchanged for 20 mM Tris pH 7.0, 10 mM DTT, 150 KCl, 1 mM EDTA using a Centriprep 30K (Amicon). SeMet-substituted Xrcc4 was expressed as described (Hendrickson et al., 1990) and purified in the same way as the native protein.
Protein cross-linking and sedimentation equilibrium ultracentrifugation
Purified samples of 1–265 and full-length Xrcc4 were dialyzed exhaustively against buffer containing 0.1 M KCl, 0.02 M Tris pH 8.0, 0.025 M 2-mercaptoethanol, 1 mM EDTA prior to both cross-linking and sedimentation equilibrium studies.
Cross-linking was performed by diluting Xrcc4 protein to 50 µg/ml in 1 ml of buffer containing 0.02 M HEPES pH 7.5, 0.2 M KCl, 0.01 M DTT, 1 mM EDTA, bis[sulfosuccinimidyl]suberate (BS3). BS3 concentrations ranged from 0.6 to 5 mM. Reactions were quenched after a 45 min incubation at room temperature by addition of 0.05 ml of buffer containing 1 M Tris pH 8.0 and 1 M glycine. Protein was precipitated by addition of 0.115 ml of 100% trichloroacetic acid (TCA). Protein was collected by centrifugation at 10 000 g for 5 min, washed once with 1 ml of acetone, air dried, resuspended in SDS–PAGE load dye and analyzed by SDS–PAGE using 4–20% polyacrylamide gels (Novex).
Sedimentation equilibrium experiments were conducted at 4.0°C on a Beckman Optima XL-A analytical ultracentrifuge. Xrcc4 (1–265) and full-length samples, at loading concentrations of 0.2 and 0.4 mg/ml, were studied at different rotor speeds, ranging from 6000 to 12 000 r.p.m. For samples in 0.1 M KCl, data were collected at 6000, 8000 and 10 000 r.p.m. Samples in 0.2 M KCl were studied at 8000, 10 000 and 12 000 r.p.m. In all cases, data were acquired as an average of eight absorbance measurements at a radial spacing of 0.001 cm and collected at a nominal wavelength of 280 nm. Equilibrium, monitored for by successive scans at 6 h intervals, was achieved within 36 h. Data analysis in terms of a single ideal solute led to weight-average molecular masses that were larger than that expected for the Xrcc4 dimer, yet smaller than that calculated for a tetramer.
Accordingly, the sedimentation equilibrium data were analyzed in terms of reversible dimer–tetramer equilibrium. Data collected at three different rotor speeds were analyzed simultaneously by mathematical modeling using SigmaPlot 2000 (SPSS Inc.) operating on a Windows 95, Pentium II 233 MHz platform. Simultaneous weighted non-linear least squares fitting of the data sets for each loading concentration was performed using a mathematical model of the following form (Perkins, 1986):
Ar = Ao exp[2HM(1 – vρ)(r2 – r2o)] + Ao2 exp[ln(k2,4) + 4HM(1 – vρ)(r2 – r2o)] + E
where Ao is the absorbance at a reference point ro, Ar is the absorbance at a given radial position r, H represents ω2/2RT, ω the angular speed in rad/s, R is the gas constant, T is the absolute temperature, k2,4 is the equilibrium dimer–tetramer association constant on an absorbance (A280) scale and E a small baseline correction. Values of the buoyant molecular masses, M (1 – vρ), for the 1–265 and full-length Xrcc4 were calculated. Densities, ρ, at 4.0°C were obtained from standard tables. Values of the partial specific molar volumes, v, were calculated based on the amino acid composition using the consensus data for the partial specific molar volumes of amino acids published by Perkins (1986).
The values of ln(k2,4) obtained experimentally were converted to ln(Ka) values, Ka now being the association constant on a molar scale, using the extinction coefficients (ε) calculated for the dimeric forms of 1–265 and full-length Xrcc4. It can be shown (Mach et al., 1992) that for a given path length l:
ln(Ka) = ln(k2,4) + ln(εl/2)
Crystallization and data collection
Crystals were grown at 20°C by the hanging drop vapor diffusion method. Protein (8 mg/ml) was mixed with an equal volume of well solution containing 100 mM sodium cacodylate pH 6.5, 10 mM DTT, 1.2 M ammonium sulfate, 10 mM magnesium acetate. Crystals were soaked in cryo-protectant buffer (100 mM Na cacodylate pH 6.5, 10 mM DTT, 10 mM magnesium acetate, 14% PEG 6000, 25% ethylene glycol) and frozen in liquid propane.
Diffraction data for SeMet-based MAD phasing were collected at the inflection point (0.97892 Å), absorption peak (0.97853 Å) and one remote wavelength at 0.96859 Å from a single cryo-cooled SeMet-Xrcc4 crystal (amino acids 1–265) at the X9B beam line of the National Synchrotron Light Source Brookhaven National Laboratory (Table I). Native data from a cryo-cooled crystal (amino acids 1–203) to 2.7 Å resolution were also collected at the X9B beam line. All data sets were indexed, integrated and scaled using programs HKL (Otwinowski and Minor, 1997) (Table I).
Structure determination and refinement
Six out of eight possible Se sites were found and refined at 3.2 Å resolution by SOLVE (Terwilliger et al., 1997). Both missing Se atoms were in the disordered region between residues 204 and 265. Solvent flipping by SOLOMON (CCP4, 1994) yielded an interpretable map with clear electron density for both Xrcc4 subunits. Residues 1–175 of both subunits could be easily traced. Residues 176–203 of the L subunit could be traced after iterative cycles of manual rebuilding and refinement using O (Jones et al., 1991) and CNS (Brünger et al., 1998).
All four cysteine residues, which are C93 exposed, C128 and C130 at the dimer interface, and C165 at the tetramer interface, were modified to S-dimethylarsenic cysteine by cacodylate. Modification of cysteine residues seems not to have changed the tetrameric nature of Xrcc4 in solution, and Xrcc4 crystals could be grown in the absence of cacodylate, but they were much smaller.
The current refined atomic model includes the S and L subunits of Xrcc4, 61 water molecules and four acetates (Table I). Ninety-two percent of the main chain torsion angles lie in the most favored regions in the Ramachandran plot, and the rest are inside the ‘additional allowed’ regions.
Figures 2, 3 and 5A were made using RIBBONS (Carson et al., 1987), and Figure 5B using GRASP (Nicholls et al., 1991).
The coordinates of Xrcc4 have been deposited with the Protein Data Bank (PDB). The accession code is 1FU1.
Acknowledgments
Acknowledgements
We thank Dr Z.Dauter for synchrotron beamline support and Dr C.Ban for assistance in X-ray data collection.
References
- Barnes D.E., Stamp,G., Rosewell,I., Denzel,A. and Lindahl,T. (1998) Targeted disruption of the gene encoding DNA ligase IV leads to lethality in embryonic mice. Curr. Biol., 8, 1395–1398. [DOI] [PubMed] [Google Scholar]
- Brünger A.T. et al. (1998) Crystallography and NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr. D, 54, 905–921. [DOI] [PubMed] [Google Scholar]
- Bryans M., Valenzano,M.C. and Stamato,T.D. (1999) Absence of DNA ligase IV protein in XR-1 cells: evidence for stabilization by XRCC4. Mutat. Res., 433, 53–58. [DOI] [PubMed] [Google Scholar]
- Carson M. (1987) Ribbon models of macromolecules. J. Mol. Graph., 5, 103–106. [Google Scholar]
- CCP4 (1994) The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D, 50, 760–763. [DOI] [PubMed] [Google Scholar]
- Chen L., Trujillo,K., Sung,P. and Tomkinson,A.E. (2000) Interactions of the DNA ligase IV–Xrcc4 complex with DNA ends and the DNA-dependent protein kinase. J. Biol. Chem., 275, 26196–26205. [DOI] [PubMed] [Google Scholar]
- Connelly J.C., Kirkham,L.A. and Leach,R.F. (1998) The sbcCD nuclease of Escherichia coli is a structural maintenance of chromosomes (SMC) family protein that cleaves hairpin DNA. Proc. Natl Acad. Sci. USA, 95, 7969–7974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Critchlow S.E., Bowater,R.P. and Jackson,S.P. (1997) Mammalian DNA double-strand break repair protein XRCC4 interacts with DNA ligase IV. Curr. Biol., 7, 588–598. [DOI] [PubMed] [Google Scholar]
- Frank K.M., Sekiguchi,J.M., Seidl,K.J., Swat,W., Rathbun,G.A., Cheng,H.L., Davidson,L., Kangaloo,L. and Alt,F.W. (1998) Late embryonic lethality and impaired V(D)J recombination in mice lacking DNA ligase IV. Nature, 396, 173–177. [DOI] [PubMed] [Google Scholar]
- Gao Y. et al. (1998) A critical role for DNA end-joining proteins in both lymphogenesis and neurogenesis. Cell, 95, 891–902. [DOI] [PubMed] [Google Scholar]
- Gao Y. et al. (2000) Interplay of p53 and DNA-repair protein XRCC4 in tumorigenesis, genomic stability and development. Nature, 404, 897–900. [DOI] [PubMed] [Google Scholar]
- Grawunder U., Wilm,M., Wu,X., Kulesza,P., Wilson,T.E., Mann,M. and Lieber,M.R. (1997) Activity of DNA ligase IV stimulated by complex formation with XRCC4 protein in mammalian cells. Nature, 388, 492–495. [DOI] [PubMed] [Google Scholar]
- Grawunder U., Zimmer,D., Kulesza,P. and Lieber,M.R. (1998a) Requirement for an interaction of XRCC4 with DNA ligase IV for wild-type V(D)J recombination and DNA double-strand break repair in vivo. J. Biol. Chem., 273, 24708–24714. [DOI] [PubMed] [Google Scholar]
- Grawunder U., Zimmer,D. and Lieber,M.R. (1998b) DNA ligase IV binds to XRCC4 via a motif located between rather than within its BRCT domains. Curr. Biol., 8, 873–876. [DOI] [PubMed] [Google Scholar]
- Hendrickson W.A., Horton,J.R. and LeMaster,D.M. (1990) Seleno methionyl proteins produced for analysis by multiwavelength anomalous diffraction (MAD): a vehicle for direct determination of three-dimensional structure. EMBO J., 9, 1665–1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirano T., Mitchison,T.J. and Swedlow,J.R. (1995) The SMC family: from chromosome condensation to dosage compensation. Curr. Opin. Cell Biol., 7, 329–336. [DOI] [PubMed] [Google Scholar]
- Hopfner K.-P., Karcher,A., Shin,D.S., Craig,L., Arthur,L.M., Carney,J.P. and Tainer,J.A. (2000) Structural biology of Rad50 ATPase: ATP-driven conformational control in DNA double-strand break repair and the ABC-ATPase superfamily. Cell, 101, 789–900. [DOI] [PubMed] [Google Scholar]
- Jeggo P.A. (1998) Identification of genes involved in repair of DNA double-strand breaks in mammalian cells. Radiat. Res., 150, S80–S91. [PubMed] [Google Scholar]
- Jones T.A., Zou,J.-Y. and Cowan,S.W. (1991) Improved methods for building models in electron density maps and the location of errors in these models. Acta Crystallogr. A, 47, 110–119. [DOI] [PubMed] [Google Scholar]
- Leber R., Wise,T.W., Mizuta,R. and Meek,K. (1998) The XRCC4 gene product is a target for and interacts with the DNA-dependent protein kinase. J. Biol. Chem., 273, 1794–1801. [DOI] [PubMed] [Google Scholar]
- Li Z., Otevrel,T., Gao,Y., Cheng,H.L., Seed,B., Stamato,T.D., Taccioli,G.E. and Alt,F.W. (1995) The XRCC4 gene encodes a novel protein involved in DNA double-strand break repair and V(D)J recombination. Cell, 83, 1079–1089. [DOI] [PubMed] [Google Scholar]
- Lieber M.R. (1998) Pathological and physiological double-strand breaks: roles in cancer, aging and the immune system. Am. J. Pathol., 153, 1323–1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luger K., Mader,A.W., Richmond,R.K., Sargent,D.F. and Richmond,T.J. (1997) Crystal structure of the nucleosome core particle at 2.8 Å resolution. Nature, 389, 251–260. [DOI] [PubMed] [Google Scholar]
- Mach H., Middaugh,C.R. and Lewis,R.V. (1992) Statistical determin ation of the average values of the extinction coefficients of tryptophan and tyrosine in native proteins. Anal. Biochem., 200, 74–80. [DOI] [PubMed] [Google Scholar]
- Melby T.E., Ciampaglio,C.N., Briscoe,G. and Erickson,H.P. (1998) The symmetrical structure of structural maintenance of chromosomes (SMC) and MukB proteins: long, antiparallel coiled coils, folded at a flexible hinge. J. Cell Biol., 142, 1595–1604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizuta R., Cheng,H.L., Gao,Y. and Alt,F.W. (1997) Molecular genetic characterization of XRCC4 function. Int. Immunol., 9, 1607–1613. [DOI] [PubMed] [Google Scholar]
- Modesti M., Hesse,J.E. and Gellert,M. (1999) DNA binding of Xrcc4 protein is associated with V(D)J recombination but not with stimulation of DNA ligase IV activity. EMBO J., 18, 2008–2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholls A., Sharp,K.A. and Honig,B. (1991) Protein folding and association: insights from the interfacial and thermodynamic properties of hydrocarbons. Proteins, 11, 281–296. [DOI] [PubMed] [Google Scholar]
- Nick McElhinny S.A., Snowden,C.M., McCarville,J. and Ramsden,D.A. (2000) Ku recruits the XRCC4–ligase IV complex to DNA ends. Mol. Cell. Biol., 20, 2996–3003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otwinowski Z. and Minor,W. (1997) Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol., 276, 307–326. [DOI] [PubMed] [Google Scholar]
- Perkins S.J. (1986) Protein volumes and hydration effects. The calculations of partial specific volumes, neutron scattering matchpoints and 280-nm absorption coefficients for proteins and glycoproteins from amino acid sequences. Eur. J. Biochem., 15, 169–180. [DOI] [PubMed] [Google Scholar]
- Roth D.B. and Gellert,M. (2000) New guardians of the genome. Nature, 404, 823–825. [DOI] [PubMed] [Google Scholar]
- Teo S.H. and Jackson,S.P. (2000) Lif1p targets the DNA ligase Lig4p to sites of DNA double-strand breaks. Curr. Biol., 10, 165–168. [DOI] [PubMed] [Google Scholar]
- Terwilliger T.C. (1997) SOLVE: An Automated Structure Solution for MAD and MIR. Edition 1.16 (www.solve.lanl.gov, Los Alamos National Laboratory, 1997). [Google Scholar]
- Wintjens R. and Rooman,M. (1996) Structural classification of HTH DNA-binding domains and protein–DNA interaction modes. J. Mol. Biol., 262, 294–313. [DOI] [PubMed] [Google Scholar]