

Abstract
Assembly of SNAREs (soluble N-ethylmaleimide- sensitive factor attachment protein receptors) mediates membrane fusions in all eukaryotic cells. The synaptic SNARE complex is represented by a twisted bundle of four α-helices. Leucine zipper-like layers extend through the length of the complex except for an asymmetric and ionic middle layer formed by three glutamines (Q) and one arginine (R). We have examined the functional consequences of Q–R exchanges in the conserved middle layer using the exocytotic SNAREs of yeast as a model. Exchanging Q for R in Sso2p drastically reduces cell growth and protein secretion. When a 3Q/1R ratio is restored by a mirror R→Q substitution in the R-SNARE Snc2p, wild-type functionality is observed. Secretion is near normal when all four helices contain Q, but defects become apparent when additional mutations are present in other layers. Using molecular dynamics free energy perturbation simulations, these findings are rationalized in structural and energetic terms. We conclude that the asymmetric arrangement of the polar amino acids in the central layer is essential for normal function of SNAREs in membrane fusion.
Keywords: exocytosis/membrane fusion/SNARE/yeast
Introduction
SNAREs (soluble N-ethylmaleimide-sensitive factor attachment protein receptors) represent families of small and mostly membrane-bound proteins that are located on the cytoplasmic surfaces of all membranes of the secretory pathway (Götte and Fischer von Mollard, 1998; Jahn and Südhof, 1999; Mayer, 1999; Pfeffer, 1999). Membranes destined to fuse contain matching sets of SNAREs. Based on their preferred localization, SNARE proteins were classified originally as either v-SNAREs (for SNAREs on trafficking vesicles) or t-SNAREs (for SNAREs on target membranes) (Söllner et al., 1993; Rothman, 1994). In vitro, appropriate sets of SNAREs spontaneously form tight core complexes. In vivo, complex formation probably bridges the opposing membranes (trans-complex) and ties the membranes closely together. Upon bilayer fusion, the complexes relax into the cis-orientation in which all membrane anchor domains are embedded in the same membrane (Hanson et al., 1997; Lin and Scheller, 1997; Weber et al., 1998; Ungermann et al., 1998). The complexes are then disassembled by the chaperone ATPase NSF (NEM-sensitive factor) in conjunction with cofactors termed SNAPs (soluble NSF attachment proteins) (Söllner et al., 1993).
The best characterized SNAREs are those involved in neuronal exocytosis. They include the vesicle protein synaptobrevin and the synaptic membrane proteins syntaxin 1 and SNAP-25. These proteins are present in a 1:1:1 stoichiometry when a core complex is formed. Deletion mutagenesis revealed that the interactions between these SNAREs are confined to a stretch of ∼60 amino acids. Syntaxin 1 and synaptobrevin each contain one such interacting region, whereas SNAP-25 contains two, one located on either side of its palmitoylated membrane anchor domain (Jahn and Südhof, 1999). Although the overall similarity between distantly related SNAREs is rather low, profile-based alignments of the interacting sequences reveal a conserved pattern of hydrophobic amino acids, in heptad repeats, referred to as the SNARE motif (Terrian and White, 1997; Weimbs et al., 1997, 1998). In the crystal structure of the neuronal complex, these amino acids form stacked leucine zipper-like layers in the core of the four-helix bundle (Sutton et al., 1998). SNARE mutations known to exhibit defects in membrane trafficking generally affect residues in the core layers, suggesting that an undisturbed layer structure is essential for function (Fasshauer et al., 1998).
The central layer of the helix bundle (‘0’ layer) is composed of three highly polar (glutamine) and one positively charged (arginine) amino acid side chain (Sutton et al., 1998). This layer is remarkable because: (i) it is the only polar/ionic layer of the entire complex that is shielded from water by surrounding hydrophobic groups; (ii) in contrast to most other layers, it is highly asymmetric; and (iii) the amino acids contributing to this layer are almost completely conserved in the entire SNARE superfamily. For these reasons, we have recently proposed the re-classification of SNAREs into Q- and R-categories. All known t-SNAREs are Q-SNAREs, and most v-SNAREs are R-SNAREs (Fasshauer et al., 1998). The remarkable conservation of the ionic layer and also most of the hydrophobic layers strongly suggests that the overall structure determined for the neuronal SNARE complex is common to all SNARE complexes. Thus, SNARE complexes may generally consist of four-helix bundles, with three helices derived from the Q-SNAREs and one helix derived from the R-SNAREs.
If formation of the helical bundle is decisive for membrane fusion, one would assume that the interactions between the amino acids in the ‘0’ layer are essential for SNARE function. Interestingly, the arrangement of the side chains in this layer displays rotational symmetry with respect to the plane of the layer. The central position of the three arginine nitrogens in the synaptic ‘0’ layer that participate in hydrogen bonding suggests that such a rotation is possible without incurring steric or electrostatic penalties (Sutton et al., 1998). In other words, there are no obvious structural reasons why the arginine needs to be contributed by synaptobrevin and its relatives instead of by one of the other constituents. Consequently, a switch of the arginine from synaptobrevin to syntaxin should affect neither the stability of the helical bundle nor the functioning of the SNAREs in fusion despite the high degree of conservation in these positions. Alternatively, however, the conserved glutamines and arginines may be essential for a function of the individual SNAREs unrelated to the formation of core complexes, e.g. in interactions with other proteins and/or in different conformations. In that case, an exchange of the arginine of synaptobrevin with the glutamine of one of the Q-SNAREs would be expected to impair the function of these SNAREs in membrane fusion.
In the present study, we have examined how conversion of the layer ‘0’ site of a Q-SNARE into an R and, conversely, of an R-SNARE into a Q affects SNARE function in membrane fusion. These experiments require that the endogenous SNAREs are replaced by the corresponding mutant versions. For these reasons, we used the exocytotic SNARE complex of the yeast Saccharomyces cerevisiae as a model since gene replacements can easily be performed in this organism. This SNARE complex is composed of the Q-SNAREs Sso1p/Sso2p (homologous to neuronal syntaxin) and Sec9p (homologous to neuronal SNAP-25), and the R-SNARE Snc1p/Snc2p (homologous to neuronal synaptobrevin) (Brennwald et al., 1994). Although the overall homology between the corresponding yeast and neuronal proteins is not high, the amino acids of the interacting layers are conserved to a much higher degree, with 50% being identical and most of the others being structurally very similar. Furthermore, the yeast proteins form core complexes in vitro that exhibit properties reminiscent of the neuronal proteins (Brennwald et al., 1994; Gerst, 1997; Rice et al., 1997; Katz et al., 1998; Fiebig et al., 1999), suggesting that the structures of the yeast and neuronal SNARE complexes are closely related. Moreover, structural distortions and changes in stability caused by the proposed mutations should be qualitatively similar to those of the neuronal SNARE complex. In the present work, we have, therefore, complemented the experiments with computer simulations of the mutations that were based on the crystal structure of the neuronal SNARE complex (Sutton et al., 1998).
Results
Effects of glutamine and arginine exchanges in SNC2 and SSO2
To examine the effects of Q/R exchanges in the ‘0’ layer, we constructed yeast strains carrying chromosomal deletions in four genes (SNC1, SNC2, SSO1 and SSO2). SNC1 and SNC2, as well as SSO1 and SSO2, are functionally redundant genes. They encode highly homologous proteins that are required for vesicle exocytosis and cell viability under standard conditions (Aalto et al., 1993; Protopopov et al., 1993). To allow normal growth and secretion, such a deletion strain expressed alleles of SNC2 and SSO2 from centromere-based vectors under the control of the cytochrome c1 promoter. In addition to these wild-type variants, two mutant alleles were constructed: SNC2R52Q and SSO2Q228R. To analyze the functional effects of these mutations, plasmids containing epitope-tagged versions of the mutant or the corresponding wild-type sequences were introduced into the deletion strain. The resulting combinations of expressed SNC2 and SSO2 alleles, encoding an arginine or glutamine residue in the ‘0’ layer of the SNARE proteins, respectively, are shown in Figure 1.
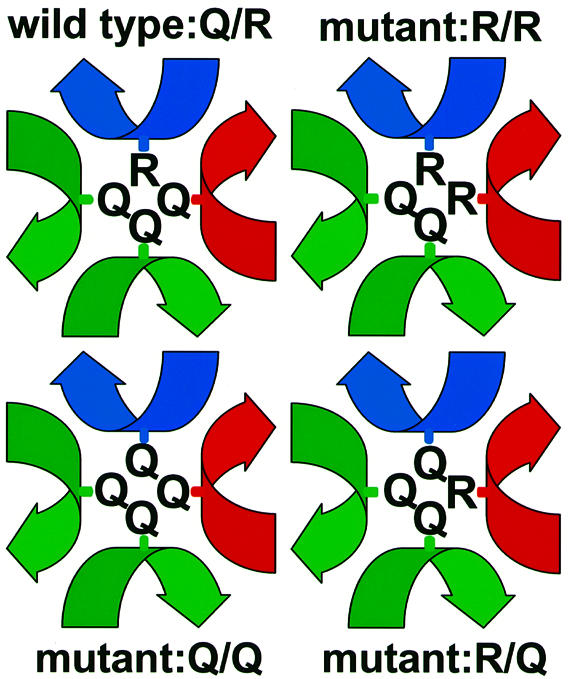
Fig. 1. Schematic illustration of the arrangement of amino acid side chains involved in the ‘0’ layers of wild-type and mutant SNARE complexes used in this study. Local helical axes of the SNARE proteins are displayed in blue for Snc2p, red for Sso2p and green for Sec9p.
Yeast transformants containing all four possible allele combinations were shown to be viable. However, expression of SSO2Q228R in the presence of wild-type SNC2, placing two arginine residues in the ‘0’ layer, results in significantly retarded growth at 25°C and growth arrest at 37°C (R/R mutant in Figure 2). The efficiency of protein secretion was analyzed by measuring the release of invertase. In these experiments, expression of endogenous invertase was derepressed during a 1 h incubation in low glucose concentration medium. As shown in Figure 3, the growth defect is mirrored by a serious defect in invertase secretion. The R/R mutant secreted only ∼40% of total invertase at 25°C, and even less at 37°C. Although both cellular growth and secretion defects are aggravated at elevated temperature, the severe functional impairment at 25°C clearly distinguishes this phenotype from typical temperature-sensitive (Ts–) defects caused by mutations such as sso2-1 (Figure 3). To exclude the possibility that the secretion defect is confined to the invertase-containing subpopulation of vesicles (David et al., 1998), we also monitored the pattern of proteins secreted into the medium after pulse-labeling followed by precipitation and SDS–PAGE analysis (Figure 4). The pattern of secreted proteins was similar in all four strains. However, a significant reduction in the amount of media proteins was observed with the R/R mutant, which was reduced further after shifting the cells to 37°C (Figure 4). To exclude that the differences between the strains are due to differences in the expression levels of the Sso2p and Snc2p variants, the protein levels were measured in each strain by immunoblotting (not shown). In all four strains, there was no significant difference in the levels of either Snc2p or Sso2p variants. However, the expression levels under the moderate cytochrome c1 promoter were somewhat higher than expression rates of SSO and SNC genes in wild-type strains (Snc2p, ∼3-fold; Sso2p, 4- to 5-fold; at both 25 and 37°C).
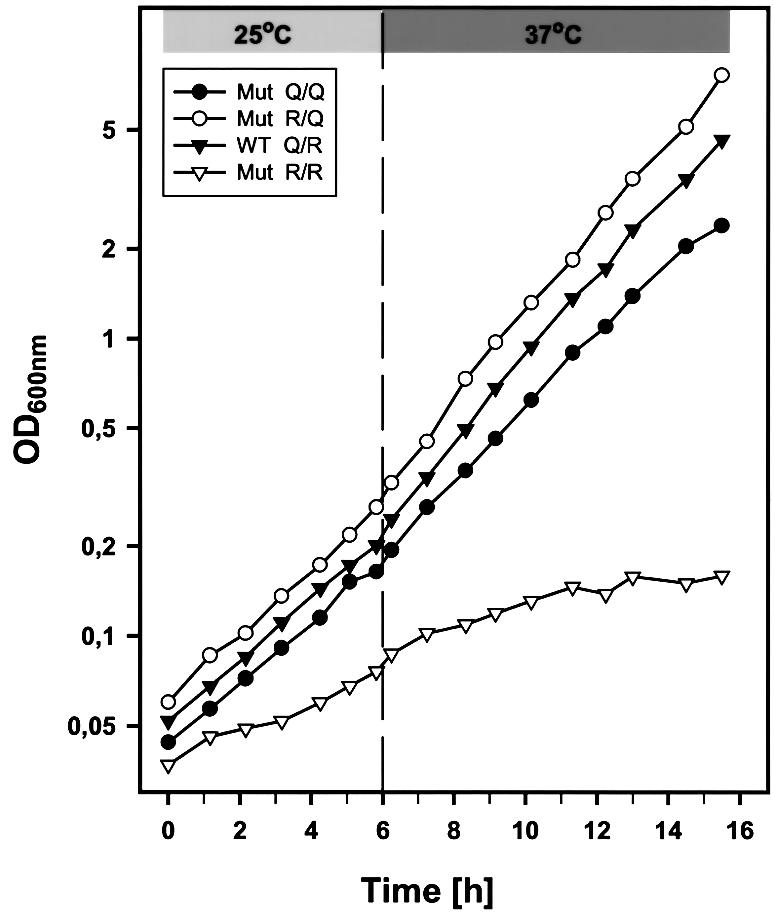
Fig. 2. Growth curves of yeast cells expressing different sets of wild-type and mutated SNAREs. Cells of strain Δ4-2D [sso1::URA3, sso2::LEU2, snc1::URA3, snc2::ADE8] were transformed with sets of plasmids as indicated: WT Q/R (filled inverted triangles), pRS314-SSO2wt/pRS313-SNC2wt; Mut R/R (open inverted triangles), pRS314-SSO2Q228R/pRS313-SNC2wt; Mut R/Q (open circles), pRS314-SSO2Q228R/pRS313-SNC2R52Q; Mut Q/Q (filled circles), pRS314-SSO2wt/pRS313-SNC2R52Q. Cell growth was monitored by measuring the optical density at 600 nm during incubation at 25°C and following a temperature shift to 37°C (after 6 h) as described in Materials and methods.

Fig. 3. Invertase secretion from yeast cells (strain Δ4-2D) expressing different pairs of SSO2 and SNC2 alleles from centromere-based plasmids as indicated in Figure 2. Transformed yeast cells were derepressed for invertase and incubated at 25°C (light gray bars) or 37°C (dark gray bars). The secreted invertase activity of the different transformants is shown as a percentage of the total enzyme activity. Each bar represents the mean value of four independent experiments (mean values ± SEM). Note that the untransformed strain H603 (sso2-1) exhibits a temperature-sensitive secretion defect, showing a significant decrease in invertase release only at the non-permissive temperature.
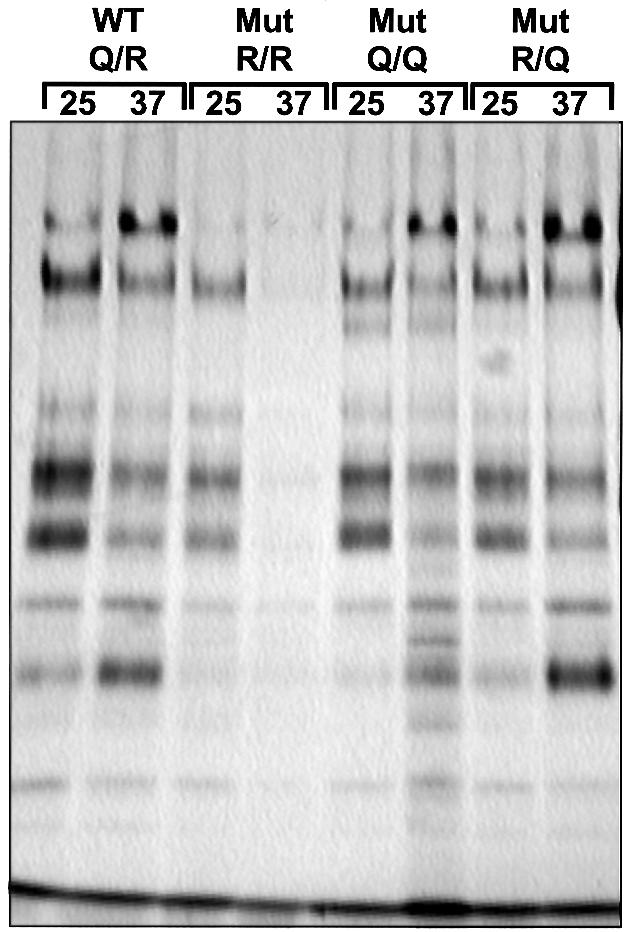
Fig. 4. Secretory defects of mutants expressing the R/R combination in the ‘0’ layer of the exocytotic SNARE complex. For a total secretion assay, yeast transformants containing SSO2 and SNC2 variants as shown in Figure 2 were radiolabeled, and secreted proteins were precipitated from the growth medium as described in Materials and methods. After separation by SDS–PAGE, radiolabeled proteins were detected by fluorography. The figure shows a representative example of several experiments. We also determined incorporation of radioactivity into total cell protein that was roughly comparable in all strains. Although the pattern of secreted proteins is similar for all strains, the R/R mutant obviously displays a reduced secretion efficiency exhibited by the reduced amount of media proteins.
The data described so far demonstrate that the replacement of the glutamine with an arginine in the Q-SNARE Sso2p, resulting in a 2Q/2R ‘0’ layer, severely disrupts the function of the SNARE complex. We then examined whether normal function can be restored when the reciprocal R→Q exchange is introduced in Snc2p. This complementary mutation (SNC2R52Q) re-establishes the 3Q/1R ratio of the original complex but effectively ‘rotates’ the arginine in the ‘0’ layer from the Snc2p helix to the neighboring Sso2p helix. Indeed, we found that both growth rate and protein secretion of this strain were indistinguishable from those of the strain expressing only wild-type proteins, even at the critical temperature of 37°C (Figures 2, 3 and 4). Moreover, the defects caused by the SSO2Q228R allele could also be rescued by expressing SNC2R52Q in yeast cells still harboring the chromosomal SNC wild-type genes (not shown). This result illustrates that the presence of two arginine residues in the ‘0’ layer severely disrupts SNARE function; nevertheless, it does not induce the formation of a ‘dead-end’ complex, which would result in a dominant-negative phenotype. Together, these findings show that the arginine can be swapped to a different helix in the ‘0’ layer without affecting the function of the complex.
Yeast cells containing the Q/Q combination (see Figure 1) expressed SNC2R52Q in the presence of wild-type Q-SNAREs, leading to only glutamines in the ‘0’ layer of the SNARE complex. This investigation was encouraged further by reports suggesting the involvement of SNARE complexes that contain only Q-SNAREs in certain intracellular fusion steps (Patel et al., 1998; Nichols et al., 1997; Rabouille et al., 1998). Surpris ingly, both growth rate and protein secretion of these cells (Figures 2, 3 and 4) were almost indistinguishable from those of the strain expressing the wild-type genes. The slight differences when compared with the wild-type and the R/Q strain [evident, for instance, by the morphological appearance (see below) and by the slightly reduced growth rate (Figure 2)] were difficult to establish unequivocally. We therefore investigated whether these minor defects become more evident when analyzed in conjunction with defects in other layers of the SNARE complex. To verify whether SNC2R52Q is functionally different from its wild-type counterpart, we first tested its ability to suppress other secretion defects. For this purpose, we selected a sec9-4 mutation strain containing a glycine to aspartic acid substitution in the –3 layer of the SNAP-25 homolog Sec9p (Brennwald et al., 1994) and a sso2-1 strain that carries an arginine to lysine substitution in the –8 layer. As already observed previously (Couve and Gerst, 1994; Gerst, 1997), the temperature-sensitive growth defects of these two mutant strains could be partially compensated by overexpression of wild-type SNC2. In addition, we used a strain expressing a Ts– mutation in SEC1. This strain is also rescued at restrictive temperature by overexpression of wild-type SNC2 (Couve and Gerst, 1994). Unlike SSO2 and SEC9, SEC1 encodes an essential SNARE-interacting protein that is not part of the SNARE core complex. Thus, the sec1-1 mutation can serve as control for a functional rescue that does not involve SNARE interactions in the core complex.
We observed that, unlike overexpression of wild-type SNC2, high level expression of SNC2R52Q was unable to restore growth of both sso2-1 and sec9-4 cells at the restrictive temperature of 34°C. In contrast, expression of both alleles was found to be equally effective in rescuing sec1-1 yeast cells (Figure 5, upper panel). More importantly, a synergistic negative effect of SNC2R52Q in conjunction with sso2-1 was observed at temperatures >30°C, i.e. a temperature range that still permits cellular growth of the sso2-1 strain. Overexpression of SNC2R52Q strongly inhibited cellular growth even in the presence of wild-type SNC genes (Figure 5, lower panel). With sec9-4, such a synergistic negative effect could not be observed. However, by using two different approaches, we were unable to obtain sec9-4 mutant cells that express the SNC2R52Q allele in the absence of wild-type SNC genes, strongly suggesting that SNC2R52Q is synthetically lethal in combination with the sec9-4 mutation. Thus, SNARE complexes with 4Q in the ‘0’ layer are capable of mediating membrane fusion at an efficiency close to that of 3Q/1R complexes. However, they do contain a structural defect that becomes more evident when combined with defects in other layers of the four-helix bundle.

Fig. 5. Genetic interaction between two SNC2 alleles and three mutations affecting the function of the exocytic SNARE complex in yeast. Upper panel: the two different SNC2 alleles (SNC2wt and SNC2R52Q) were tested for suppression of particular conditional defects affecting vesicle fusion at the plasma membrane. Snc2p (wild-type) or Snc2R52Q protein was overproduced in cells carrying Ts– defects in either the SEC1 gene (sec1-1), the SEC9 gene encoding the yeast SNAP-25 homolog (sec9-4) or the SSO2 gene (sso1::URA3; sso2-1). ↑, improvement of growth relative to transformants containing the pure vector; →, no change in growth rate; ↓, inhibition of growth. Cells transformed with multicopy vector pRS323 (URA3, 2µ), pRS323-SNC2wt or pRS323-SNC2R52Q were plated on synthetic minimal medium lacking uracil. Growth was scored after 3 or 4 days of incubation at 30, 32.5 or 35°C. The latter two temperatures are restrictive for growth of sec1-1, sec9-4 and sso2-1 mutants. Note that in contrast to the wild-type allele, the SNC2R52Q allele was unable to suppress the growth defect of sec9-4 mutants and even aggravated the growth defect caused by the sso2-1 mutation. However, overexpression of SNC2R52Q did sustain the growth of sec1-1 cells as efficiently as wild-type SNC2. Lower panel: sso2-1 mutants incubated at 32.5°C for 3 days. The mutants contained either pRS323 without insert (lower third of the plate), pRS323-SNC2 (upper left) or pRS323-SNC2R52Q (upper right). sso2-1 yeast transformants overexpressing SNC2R52Q showed a synergistic negative effect.
For further characterization of our mutants, we analyzed the morphology of the four strains by light and electron microscopy. When monitored by differential interference contrast (DIC) microscopy, the R/R mutant had a clearly abnormal appearance (Figure 6). Many cells were unusually large, and cell groups that apparently had not separated after division were evident. Furthermore, cells of this strain lack the vacuolar structures that were visible as indentations within wild-type cells. At the electron microscopic level, the R/R mutant was characterized by an accumulation of numerous vesicles with diameters of 80–100 nm that correspond to the size of post-Golgi vesicles (Figure 6). This phenotype is indicative of a defect in the final step of the secretory pathway. In contrast, the R/Q ‘swap’ mutant was morphologically indistinguishable from the strain expressing the wild-type combination. The Q/Q mutant also looked normal except that the cell size distribution was slightly more heterogeneous and the vacuole often had a more fragmented appearance.

Fig. 6. Morphology of cells expressing different ‘0’ layer Q/R combinations analyzed by light microscopy (A, B, C and D) and electron microscopy (E, F, H and I, bar = 1 µm; G, bar = 370 nm; J, bar = 100 nm). Transformants of strain Δ4-2D were used as follows: (A), (E) and (J) WT Q/R, pRS314-SSO2wt/pRS313-SNC2wt; (B), (F), (G) and (K) Mut R/R, pRS314-SSO2Q228R/pRS313-SNC2wt; (C) and (H) Mut R/Q, pRS314-SSO2Q228R/pRS313-SNC2R52Q; (D) and (I) Mut Q/Q, pRS314-SSO2wt/pRS313-SNC2R52Q. Cells were grown and treated at 25°C before light microscopic inspection with DIC or incubated at 37°C for 45 min before fixation for electron microscopy. (J and K) Higher magnifications of the WT Q/R and Mut R/R strains that were obtained from spheroblasts using a different fixation and staining protocol. Note the aberrant cell shape and the numerous vesicles that are visible in the mutant containing two arginines in the ‘0’ layer of the exocytotic SNARE complex.
We also performed immunofluorescence microscopy in order to localize the Snc2p and Sso2p variants in all four strains. As shown in Figure 7, the plasma membrane localization of the Sso2Q228R protein was found to be indistinguishable from that of wild-type Sso2p regardless of whether the R or Q variant of Snc2p was expressed. These observations suggest that the defect in exocytosis in the R/R mutant is not due to missorting or serious degradation of the mutated protein. Interestingly, the R/R mutant showed strong and diffuse staining for Snc1p that was often enriched in the daughter cells (Figure 7). This probably reflects the accumulation of small Snc1p-containing transport vesicles as observed in the EM analysis (Figure 6).

Fig. 7. Indirect immunofluorescence microscopy to confirm that plasma membrane localization of the Sso2Q228R protein is indistinguishable from that of wild-type Sso2p. Yeast transformants of strain Δ4-2D containing different pairs of either wild-type (WT Q/R) or mutant (Mut R/R; Mut R/Q; Mut Q/Q) SSO2 and SNC2 alleles from centromeric plasmids (indicated in Figure 2) were fixed and prepared for immunocytochemistry. HA and c-Myc epitope-specific anibodies were used to detect the epitope-tagged SNARE proteins. Cy2-conjugated antibodies track Sso2wt or Sso2Q228R proteins (shown in green), while Cy3-conjugated antibodies labeled Snc2wt or Snc2R52Q proteins (shown in red). DAPI staining of DNA was used to localize the nucleus (blue).
To exclude the possibility that other intracellular trafficking pathways are affected in any of the mutant strains (e.g. defects in vacuolar sorting as suggested by the microscopic appearance), we examined the processing of the vacuolar enzyme carboxypeptidase Y (CPY) after pulse-labeling. After translocation, this protein is core-glycosylated in the endoplasmic reticulum (p1 form), further glycosylated in the Golgi apparatus (p2 form) and proteolytically processed upon arrival in the vacuole (mature form) (Stevens et al., 1982). As shown in Figure 8, all four strains exhibited a normal pattern of CPY processing, with the R/R strain being slightly retarded (probably secondary to the overall growth defect). These findings confirm that neither endoplasmic reticulum (ER)-to-Golgi nor Golgi-to-vacuole trafficking was impaired.

Fig. 8. Maturation of carboxypeptidase Y was monitored in order to follow intracellular transport in strain Δ4-2D cells expressing different pairs of SSO2 and SCN2 alleles. Transformants containing SSO2 and SNC2 variants as indicated in Figure 2 were grown in synthetic medium at 25°C and pulse–chased with 35S-labeled cysteine and methionine for 5 min at 37°C. Aliquots were removed at 0, 7.5 and 15 min of chase time. Cells were separated from the medium, lysed, and lysates and medium samples were subjected to immunoprecipitation with CPY-specific antibodies. The positions of the ER precursor (p1), the Golgi-modified precursor (p2) and the mature vacuolar enzyme (m) are indicated.
Calculation of free energy changes for Q/R substitutions in neuronal SNAREs using molecular dynamics simulations
The phenotypes described above suggest that an interference with the side chain interactions of the ‘0’ layer affects SNARE function in membrane fusion. However, it remains unclear to what extent the amino acid substitutions affect the structure of the ‘0’ layer region and the overall stability of the complex. We have therefore performed five molecular dynamics free energy calculation studies to gain more insight into the structural and energetic consequences of these mutations. It should be understood that these calculations can only provide a qualitative picture, since a crystal structure (as required as input for the simulations) is only available from the neuronal SNARE complex (Sutton et al., 1998). In particular, we have simulated the following transitions: synaptobrevin R56Q (WT→Q/Q), syntaxinQ226R (WT→ R/R), synaptobrevinR56Q plus syntaxinQ226R (WT→R/Q), as well as the subsequent transitions Q/Q→R/R and R/Q→R/R (see Figure 9).
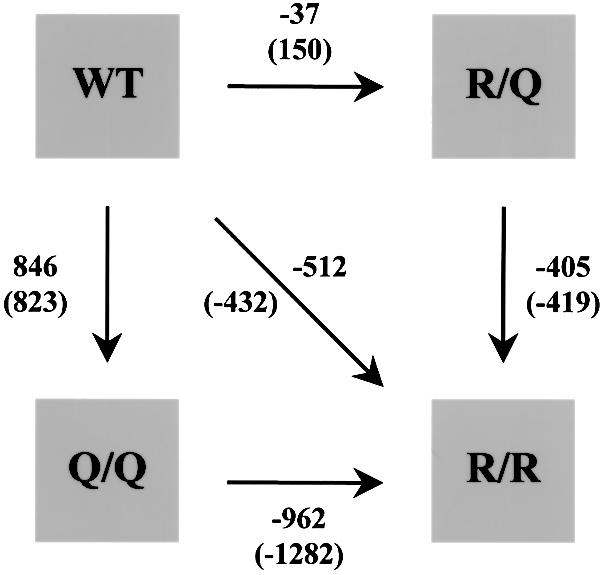
Fig. 9. Computed differences between the free energies of folding (in kJ/mol) for the studied mutations as obtained from 1 ns (numbers) and 100 ps simulations (numbers in parentheses). All calculations are based on the crystal structure of the neuronal SNARE complex (Sutton et al., 1998). Note that there are three paths connecting WT to R/R, which should all have an identical free energy difference. For further details see Materials and methods.
The calculated differences between the free energies of folding are shown in Figure 9 both for 1 ns simulations and, as a check for convergence, for 100 ps simulations (numbers in parentheses). As a further check of convergence, the respective back-transitions were also simulated and compared with the forward transitions (results not shown). In all cases, no significant differences in the free energies were observed except for the transitions involving the R/R mutant.
As can be seen in Figure 10A, the structure of the wild-type SNARE complex remains essentially intact during the 1 ns equilibration run in comparison with the crystal structure (cf. Figure 3 of Sutton et al., 1998). Especially in the central region around synaptobrevinR56, the structural features of the crystal structure are unaltered. In particular, all hydrogen bonds of the experimental structure remain intact. In the synaptobrevin Arg56 to Gln (Q/Q) mutation (Figure 10C), the created glutamine cannot form a comparably strong hydrogen-bonding network to the wild-type arginine. Moreover, the stabilizing effect of the positively charged wild-type arginine is lost. As a result, the Q/Q mutant is estimated to be significantly less stable (∼900 kJ/mol) than the wild-type SNARE complex. Nevertheless, the overall structure of the complex is only slightly altered by this mutation.

Fig. 10. Structures of the central ionic ‘0’ layer (snapshots) as obtained from 1 ns free energy perturbation simulations. The backbones of synaptobrevin, syntaxin and SNAP-25 are shown as blue, red and green ribbons, respectively; the four side groups of the ‘0’ layer are shown as ball-and-stick models. Hydrogen bonds are drawn as dashed lines. Shown are (A) WT structure after equilibration, as well as the calculated structures (B) of the R/Q double mutant, (C) the Q/Q mutant and (D) the R/R mutant. All plots were generated using Bobscript (Kraulis, 1991; Esnouf, 1997) and Raster3D (Merritt and Bacon, 1997).
From the 1 ns simulation, the R/Q double mutant (Figure 10B) appears to be marginally more stable (40 kJ/mol) than the wild-type SNARE complex. This indicates that the stabilizing role of the positively charged arginine can be taken over by syntaxin, although, in the final simulation structure, the complex lacks one hydrogen bond compared with the wild-type complex (Figure 10A). The overall structure of the complex is maintained, however, and, sterically, formation of this third hydrogen bond also appears possible.
In contrast to the mutations considered above, in the simulation of the syntaxin Q226R mutation (R/R) the overall structure of the SNARE complex is significantly distorted. This structural distortion is initiated by a conformational transition of the newly created arginine in syntaxin, which in the course of the simulation folds outwards due to electrostatic repulsion with the neighboring synaptobrevin wild-type arginine. This distortion of the syntaxin backbone initially is local (Figure 10D) but subsequently also affects adjacent SNAP-25 residues, thereby severely disrupting the whole coiled-coil. The calculated free energy difference of approximately –400 kJ/mol compared with the wild-type SNARE complex is dominated by favorable electrostatic interactions of the formed arginine with the numerous nearby negative charges on the protein (the wild-type SNARE complex has a net negative charge of –16). This free energy value, however, has been computed from a simulation, which has not yet reached equilibrium but rather provides only an intermediate of an ongoing and presumably drastic conformational change. Therefore, it cannot be used to estimate the stability of the R/R SNARE complex.
Discussion
Our results demonstrate that the interactions in the ionic ‘0’ layer, a hallmark of the crystal structure of the neuronal SNARE complex, are crucial for the biological activity of SNAREs. They show that structural predictions based on the crystal structure can be tested by precisely planned sequence changes followed by functional analyses in vivo. We reasoned that the conversion of a glutamine into an arginine, resulting in two arginines in the ‘0’ layer, would be highly disruptive for the stability of the complex due to steric and electrostatic hindrances. This consideration was clearly confirmed by our findings. More importantly, the data prove that the ionic layer as identified in the crystal structure of the neuronal complex exists as a structural entity of functional relevance in the distantly related yeast complex: the fact that rotation of the arginine to a different position in the ‘0’ layer restores normal function cannot be explained in any other way.
Given the high degree of evolutionary conservation of the amino acids in the ‘0’ layer, the crystal structure of the neuronal SNARE complex is likely to be paradigmatic for SNARE complexes in general. Consequently, all SNARE complexes are predicted to contain three Q-SNAREs and one R-SNARE. Unfortunately, so far, there are only a few such complexes whose composition has been elucidated beyond doubt. Since it is now well established that some SNAREs participate in more than one complex, an unambiguous identification of all SNAREs cooperating in a given complex is far from trivial. For instance, more than four SNARE proteins have been found to be associated in immunoprecipitation assays (Ungermann et al., 1999). However, no rigorous structural characterization of these complexes has been performed. Further more, SNARE complexes containing only Q-SNAREs have been implicated in some intracellular fusion events such as homotypic fusion of cisternae of the ER, of vacuoles, and fusion of Golgi stacks after mitosis (Patel et al., 1998; Nichols et al., 1997; Rabouille et al., 1998). Since our data show that four glutamines in the ‘0’ layer can combine to form a functional complex, it is possible that biologically relevant complexes consisting of four Q-SNAREs do exist in specialized pathways. It should be kept in mind, however, that in none of these cases has the participation of a hitherto unidentified R-SNARE been excluded.
The structural distortions derived from the simulation of the mutations correlate well with the observed phenotypes. In particular, no structural distortions of the helical backbones were observed in the R/Q double mutant although the arrangement of the side chains is less regular and the hydrogen bond network is weaker than in the wild-type structure. Furthermore, the stability, as estimated from the calculated folding free energy, is close to that of the wild-type. Also, no major structural changes are observed in the simulation of the Q/Q mutation. However, in contrast to the R/Q double mutant, this complex appears to be considerably less stable as estimated from the large difference in free energy. The most surprising simulation result is the severe structural distortion induced by the R/R mutation. The unexpected outward bending of one arginine residue is, however, in perfect agreement with the highly disruptive phenotype of the corresponding yeast mutant. It should be borne in mind that this structure represents a short-time ‘intermediate’ of an ongoing distortion process that could not be investigated further. Also, for this reason, the free energy value obtained for this mutant does not allow conclusions to be drawn about the stability of the mutant.
Generally, the calculated differences of folding free energies appear rather large. It is well known that free energy perturbation calculations can be inaccurate if larger changes in the distribution of charges are involved, as is the case for the Q/Q and R/R calculations presented here. In particular, the peculiar electrostatic environment of the ‘0’ layer, involving a large number of negative charges, might reduce the accuracy of our calculations. Since, additionally, our heptapeptide model of the unfolded state is rather crude, we would expect the absolute values of the free energies to be generally smaller than those calculated from the simulations. Finally, the number of negative charges in the vicinity of the ‘0’ layer is slightly smaller in the yeast than in the neuronal system. For these reasons, we have used these values only as a qualitative assessment for which we consider them sufficiently reliable.
Other mutations of amino acids of the ‘0’ layer have been reported previously. It was shown recently that exocytosis of permeabilized PC12 cells in which one helix of SNAP-25 was proteolyzed by botulinum neurotoxin E could be restored by external addition of the fragment corresponding to this helix (Chen et al., 1999). Interest ingly, fragments carrying Q→A and Q→I exchanges within the ‘0’ layer restored function in a way similar to the unmodified version, whereas exchanges in the neighboring hydrophobic layers were less well tolerated. However, minor to moderate differences in function may have escaped detection. More recently, Wei et al. (2000) have overexpressed a ‘0’ layer Q→L exchange mutant of SNAP-25 in chromaffin cells and found that sustained release was compromised whereas the exocytotic burst remained unchanged. More severe defects were observed in yeast secretory mutants containing ‘0’ layer exchanges such as sec22-3 (R→G) (Novick et al., 1980; Sacher et al., 1997) and vti1-12 (Q→R) (Fischer von Mollard and Stevens, 1998). The latter mutant is particularly interesting since it corresponds to one of the mutations analyzed here. However, the interpretation of defects exhibited by this mutant is compromised by the presence of a second mutation in one of the neighboring layers (Fischer von Mollard and Stevens, 1998).
The role of the ‘0’ layer during SNARE assembly and disassembly remains to be established. First, the interactions within this layer may contribute significantly to the overall stability of the SNARE complex. The shielding of the polar groups from the surrounding water may result in an increase of the bonding energies due to the lack of dielectric weakening of the electrostatic interactions (Sutton et al., 1998). Secondly, the asymmetry of the ionic layer may contribute to correct alignment of the four helices during assembly. Together with other asymmetric layers, this may ensure that the long helices are kept ‘in register’ during complex formation. Since SNARE function is not only dependent on the integrity of the ‘0’ layer, the integrity of other layers of the four-helix bundle must also be taken into account. According to the emerging picture, relatively benign substitutions in any of the layers (including the ‘0’ layer) can be tolerated but are synergistic with defects in surrounding layers. In contrast, substitutions in single layers that are predicted to be structurally disruptive suffice to cause severe functional defects. All these observations lend support to the model described above, which involves a zipper-like assembly of SNAREs as an essential step in membrane fusion (Hanson et al., 1997; Lin and Scheller, 1997; Weber et al., 1998; Ungermann et al., 1998).
Finally, our observation that ‘transforming a Q- into an R-SNARE’ causes severe disruption and that this can be rescued by the converse R→Q substitution in the corresponding R-SNARE provides a powerful tool to identify cognate SNARE pairs in systems that can be manipulated genetically. Thus, rescue should only be observed by mutating R-SNAREs that can form functional complexes with the mutated Q-SNARE. This is of particular importance since in vitro interactions between SNAREs are promiscuous and thus cannot be used to distinguish cognate from non-cognate SNARE complexes (Fasshauer et al., 1999; Yang et al., 1999). Furthermore, it is possible that such mutagenesis can also be used to clarify whether complexes consisting of only Q-SNAREs operate in certain fusion steps as suggested (see above): thus, a single Q→R substitution in such a complex might be tolerated, whereas it is predicted to be highly disruptive if the complex does contain an R-SNARE.
Materials and methods
Yeast strains and genetic techniques
Saccharomyces cerevisiae strain Δ4-2D (MATa ura3 leu2 his3 trp1 ade8 sso1::URA3 sso2::LEU2 snc1::URA3 snc2::ADE8) was obtained by a series of crosses from the following strains: H404 (MATα ura3 leu2 his3 trp1 ade2 sso2::LEU), H403 (MATa ura3 leu2 his3 trp1 ade2 sso1::URA3) (Aalto et al., 1993), JG8 T15:85 (MATa ura3 leu2 his3 trp1 ade8 snc1::URA3 snc2::ADE8 pTGAL-SNC1) (Protopopov et al., 1993) and H603 (MATa sso2-1 sso1::HIS3 ura3 leu2 his3 trp1 can1 ade2); NY782 (MATa sec9-4 leu2 ura3) was from P.Novick and MEY51 (MATa sec1-1 ura3 leu2 his4 lys2 bar1) was from H.Riezman. The relevant SNC and SSO genotypes were confirmed by complementation analysis and PCR. Genetic analysis was performed as described (Rose et al., 1990).
Strain Δ4-2D transformed with a set of plasmids is described as follows: wild-type Q/R, pRS314-SSO2wt/pRS313-SNC2wt; mutant R/R, pRS314-SSO2Q228R/pRS313-SNC2wt; mutant R/Q, pRS314-SSO2Q228R/pRS313-SNC2R52Q; and mutant Q/Q, pRS314-SSO2wt/pRS313-SNC2R52Q.
The sso2-1 mutation was isolated in a screen for temperature-sensitive SSO2 alleles, and will be described elsewhere (J.Jäntti, M.K.Aalto, M.Öyen, L.Sundquist, S.Keränen and H.Ronne, submitted). The mutant protein has an R200K substitution.
Nucleic acid techniques
DNA manipulations were performed according to Sambrook et al. (1989). Plasmids were constructed as follows: the CYC1 promoter [1.1 kb SalI–BamHI fragment from pSCCZ100 (Boehm et al., 1994)], the 3′-non-translated region of YPT1 (BssHII–MluI), and a 61 bp oligonucleotide (BamHI–NotI) to introduce an N-terminal c-Myc epitope were inserted into centromere-based vector pRS314 (Sikorski and Hieter, 1989). SSO2Q228R was obtained as two fragments by PCR; YEpSSO2 (Aalto et al., 1993) was used as template. PCR products were inserted as PstI–SpeI and SpeI–SacI fragments into the vector. In addition to silent mutations (to introduce an SpeI and a SalI site), the SSO2Q228R reading frame contains a mutated codon in position 228 (AGA coding for arginine). To construct an equivalent vector encoding the c-Myc-tagged wild-type Sso2p (pRS314-SSO2wt), a short SalI–SpeI fragment was replaced with an oligonucleotide containing the wild-type codon 228 (CAA).
SNC2R52Q was cloned as two PCR fragments (PstI–ClaI and ClaI–BssHII) using a vector containing genomic SNC2 derived from a multicopy library. The PCR products were ligated into the pRS314-based vector described above. The PstI–ClaI fragment that contains a mutated codon 52 in the reading frame of SNC2 (CAA coding for glutamine) was replaced by an equivalent PCR fragment with the wild-type codon (CGT) to construct pRS314-SNC2wt. SNC2 sequences were shifted as a PvuII fragment into pRS313 or 2µ-based high copy number vector pRS323 (Strom et al., 1993). Hemagglutinin (HA) epitope-tagged versions were constructed by replacing the HindIII–BamHI fragment of pRS314-SSO2Q228R and pRS314-SSO2wt (encoding the c-Myc epitope) with an oligonucleotide coding for the HA epitope and were used for the growth assays and immunofluorescence studies.
Growth assays
Overnight yeast cultures grown to the logarithmic phase at 25°C in YEPD were harvested and diluted into fresh YEPD (4% glucose) to an OD600 of ∼0.05. Cells were incubated at 25°C using a waterbath shaking at 150 r.p.m. After 6 h, the cells were shifted to 37°C and the incubation was continued under constant shaking. Samples were taken frequently to measure OD600. To prolong the exponential phase of growth, cultures were diluted ∼40-fold into fresh YEPD medium (4% glucose) when they had reached an OD600 of ∼2. The precise dilution factor was calculated from parallel OD600 determinations of the original and diluted cultures.
Secretion assays, protein labelling and immunoprecipitation
To assay secretion of invertase Δ4-2D, transformants were grown to the mid-logarithmic phase in YEPD medium at 25°C. Equivalents of one OD600 of cells were harvested. Expression of invertase was derepressed by shifting to low-glucose (0.1%) YEPD and incubation at either 37 or 25°C. After 1 h of incubation, cells were harvested and split into two equal aliquots for quantitation of secreted (intact cells) and total (lysed cells) invertase activity as described previously (Boehm et al., 1994).
Anterograde protein transport of the yeast transformants was tested by a total secretion assay. Yeast were grown in minimal medium at 25°C and pulsed with Tran35S-label (ICN) for 10 min. Samples were split, and cells were collected by a brief centrifugation, resuspended in pre-warmed YEPD medium and chased for 30 min at 25 or 37°C. Secreted media proteins were precipitated from supernatants using 10% trichloroacetic acid (TCA), and proteins were separated by 8% SDS–PAGE.
CPY maturation was assayed essentially as described by Benli et al. (1996). Cells growing in minimal medium were pulse-labeled for 5 min with Tran35S-label (ICN) at 37°C and chased with cold methionine and cysteine. Sample were taken immediately after the pulse period and after 30 min of chase at 37°C. Cells were broken by using 0.2 g of glass beads in 100 µl of phosphate-buffered saline (PBS) containing 1% SDS. Extracts were diluted, beads and cells debris were discarded and labeled proteins were immunoprecipitated using specific antibodies and separated by SDS–PAGE. After incubating the gel with Amplify (Amersham) for 30–45 min, the proteins were detected by exposing the gels to X-Omat AR (Kodak) at –80°C
Yeast cell morphology and indirect immunofluorescence
Yeast cells were grown overnight in YEPD at 25°C to mid-logarithmic phase, harvested, washed three times in PBS and incubated in PBS for 15 min in a shaker at room temperature. Aliquots were plated on concanavalin A-coated coverslips. After 5 min, the coverslips were rinsed carefully with PBS to wash out non-adherent cells. Cells were imaged using a Zeiss Axiphot II microscope with DIC optics and a slow-scan CCD camera (Princeton Instruments TE/CCD1317K).
For indirect immunofluorescence, yeast cells were fixed by adding formaldehyde solution to a final concentration of 4% directly to the growing cells. Preparation for immunostaining was performed essentially as described by Kilmartin and Adams (1984). Fixed spheroplasts were immobilized on polylysine-coated slides and labeled with monoclonal mouse c-Myc epitope-specific antibodies (9E10) and rabbit anti-HA eptiope antibodies (Roche). For secondary labeling, Cy3-conjugated goat anti-mouse and Cy2-conjugated goat anti-rabbit (Dianova) IgGs were used. For nuclear staining, the mounting medium contained 0.3 µg/ml 4′,6-diamidino-2-phenylindole (DAPI).
For electron microscopic inspection, yeast cells were grown overnight in YEPD at 25°C to logarithmic phase, diluted into fresh YEPD to an OD600 of 1 and incubated for another 45 min at 25 or 37°C. Two different protocols were used. First (Figure 6E–I), 40 ml of cell suspension were harvested, washed once in water at the indicated temperature and fixed immediately in 1.5% KMnO4. After fixation at room temperature for 2 h, cells were washed three times with distilled water, and the final pellet was resuspended in 2% agarose. Secondly (Figure 4J and K), spheroplasts were prepared and fixed in glutaraldehyde as described (Salminen and Novick, 1987). Small agarose blocks were prepared, dehydrated consecutively by increasing ethanol concentration and embedded in Embed 812 (Serva) (Luft, 1961; Kaiser and Schekman, 1990). Ultrathin sections were stained with 1% aqueous uranyl acetate and Reynold’s lead citrate and examined in a Philips CM 120 electron microscope.
Molecular dynamics free energy calculations
As a qualitative measure of stability of the mutants with respect to the wild type, we have calculated the differences between the respective free energies of folding (Gmutfolded – Gmutunfolded) – (Gwtfolded – Gwtunfolded). Since these two free energies of folding cannot be calculated directly, we have used the usual rearrangement (Gmutfolded – Gwtfolded) – (Gmutunfolded – Gwtunfolded), which gives the same result, but requires only the calculation of ‘mutation free energies’ in the folded and unfolded state, respectively. As a crude model for the unfolded state, we have used a heptapeptide comprising residues 53–59 of synaptobrevin solvated in SPC water. The free energy difference of the R→Q mutation in this ‘unfolded’ system was calculated to be +58 kJ/mol.
All free energy calculations were carried out with the Gromacs simulation package (Van der Spoel et al., 1995), using the free energy perturbation (FEP) scheme (McQuarrie, 1973; Resat and Mezel, 1993). A soft-core potential as defined in Beutler et al. (1994) was applied, using an α value of 1.0. The gromacs forcefield was applied, which is the gromos 87 forcefield (Van Gunsteren and Berendsen, 1987) with slight modifications (Van Buuren et al., 1993) and explicit hydrogens on aromatic residues. Weak coupling to an external temperature and pressure bath was applied to ensure constant temperature and pressure, respectively (Berendsen et al., 1984). Shake (Ryckaert et al., 1977) was applied to all bond lengths to allow a time step of 2 fs. A cut-off radius of 10 and 14 Å was employed for the calculation of van der Waals and electrostatic forces, respectively. As a starting structure, the crystal structure of the neuronal SNARE complex was taken [chains A–D of PDB entry 1SFC (Sutton et al., 1998)]. The structure was solvated in a rectangular box containing 8277 SPC (Berendsen et al., 1981) water molecules. The resulting simulation system comprised 27 832 atoms. After energy minimization, the 16 water molecules with the most favorable electrostatic potential were replaced by sodium ions to compensate the net negative charge of the complex. The system was then equilibrated for 1 ns. The obtained structure was used as the starting structure for all FEP simulations.
All presented FEP simulations had a duration of 1 ns, during which the perturbation parameter λ was slightly modified at every integration step. FEP simulations of 100 ps duration of both forward and back transitions were carried out as a control. Five mutations were studied using the scheme described above (see also Figure 9), introducing the following abbreviations: WT, wild-type; Q/Q, synaptobrevin Arg56→Gln; R/R, syntaxin Gln226→Arg; Q/R, double mutant with synaptobrevin Arg56→Gln and syntaxin Gln226→Arg. For the ‘mutations’ of arginine to glutamine, a ‘dummy particle’ without charge and Lennard–Jones interactions was covalently connected to the δ carbon. During the FEP simulation, this ‘dummy’ was slowly perturbed into the ε oxygen of glutamine. Simultaneously, the η nitrogen atoms and the attached protons of arginine were converted into ‘dummies‘, and the ζ carbon of arginine was converted into the second ε proton of glutamine. All bond parameters were perturbed accordingly. The inverse glutamine to arginine mutations were performed in exactly the same way, backwards.
Acknowledgments
Acknowledgements
We thank Berk Hess and Ronen Zangi for their assistance on using the gromacs FEP implementation. This work was supported in part by a grant from the VW foundation to H.G. and R.J., by EU Biotechnology grant BIO4-CT98-0024 to B.d.G., and a grant of the Academy of Finland (grant No. 42160) to S.K.
References
- Aalto M.K., Ronne,H. and Keränen,S. (1993) Yeast syntaxins Sso1p and Sso2p belong to a family of related membrane-proteins that function in vesicular transport. EMBO J., 12, 4095–4104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benli M., Döring,F., Robinson,D.G., Yang,X. and Gallwitz,D. (1996) Two GTPase isoforms, Ypt31p and Ypt32p, are essential for Golgi function in yeast. EMBO J., 15, 6460–6475. [PMC free article] [PubMed] [Google Scholar]
- Berendsen H.J.C., Postma,J.P.M., Van Gunsteren,W.F. and Hermans,J. (1981) Interaction models for water in relation to protein hydration. In Pullman,B. (ed.), Intermolecular Forces. D.Reidel Publishing Company, Dordrecht, The Netherlands, pp. 331–342. [Google Scholar]
- Berendsen H.J.C., Postma,J.P.M., DiNola,A. and Haak,J.R. (1984) Molecular dynamics with coupling to an external bath. J. Chem. Phys., 81, 3684–3690. [Google Scholar]
- Beutler T.C., Mark,A.E., van Schaik,R.C., Gerber,P.R. and van Gunsteren,W.F. (1994) Avoiding singularities and numerical instabilities in free-energy calculations based on molecular simulations. Chem. Phys. Lett., 222, 529–539. [Google Scholar]
- Boehm J., Ulrich,H.D., Ossig,R. and Schmitt,H.D. (1994) Kex2-dependent invertase secretion as a tool to study the targeting of transmembrane proteins which are involved in ER–Golgi transport in yeast. EMBO J., 13, 3696–3710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennwald P., Kearns,B., Champion,K., Keränen,S., Bankaitis,V. and Novick,P. (1994) Sec9 is a SNAP-25-like component of a yeast SNARE complex that may be the effector of Sec4 function in exocytosis. Cell, 79, 245–258. [DOI] [PubMed] [Google Scholar]
- Chen Y.A., Scales,S.J., Patel,S.M., Doung,Y.C. and Scheller,R.H. (1999) SNARE complex formation is triggered by Ca2+ and drives membrane fusion. Cell, 97, 165–174. [DOI] [PubMed] [Google Scholar]
- Couve A. and Gerst,J.E. (1994) Yeast Snc proteins complex with Sec9. Functional interactions between putative SNARE proteins. J. Biol. Chem., 269, 23391–23394. [PubMed] [Google Scholar]
- David D., Sundarababu,S. and Gerst,J.E. (1998) Involvement of long-chain fatty-acid elongation in the trafficking of secretory vesicles in yeast. J. Cell Biol., 143, 1167–1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esnouf R.M. (1997) An extensively modified version of MolScript that includes greatly enhanced coloring capabilities. J. Mol. Graph. Model., 15, 112–113, 132–134. [DOI] [PubMed] [Google Scholar]
- Fasshauer D., Sutton,R.B., Brunger,A.T. and Jahn,R. (1998) Conserved structural features of the synaptic fusion complex: SNARE proteins reclassified as Q- and R-SNAREs. Proc. Natl Acad. Sci. USA, 95, 15781–15786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fasshauer D., Antonin,W., Margittai,M., Pabst,S. and Jahn,R. (1999) Mixed and non-cognate SNARE complexes—characterization of assembly and biophysical properties. J. Biol. Chem., 274, 15440–15446. [DOI] [PubMed] [Google Scholar]
- Fiebig K.M., Rice,L.M., Pollock,E. and Brünger,A.T. (1999) Folding intermediates of SNARE complex assembly. Nature Struct. Biol., 6, 117–123. [DOI] [PubMed] [Google Scholar]
- Fischer von Mollard G. and Stevens,T.H. (1998) A human homolog can functionally replace the yeast vesicle-associated SNARE Vti1p in two vesicle transport pathways. J. Biol. Chem., 273, 2624–2630. [DOI] [PubMed] [Google Scholar]
- Gerst J.E. (1997) Conserved α-helical segments on yeast homologs of the synaptobrevin/VAMP family of v-SNAREs mediate exocytic function. J. Biol. Chem., 272, 16591–16598. [DOI] [PubMed] [Google Scholar]
- Götte M. and Fischer von Mollard,G. (1998) A new beat for the SNARE drum. Trends Cell Biol., 8, 215–218. [DOI] [PubMed] [Google Scholar]
- Hanson P.I., Roth,R., Morisaki,H., Jahn,R. and Heuser,J.E. (1997) Structure and conformational changes in NSF and its membrane receptor complexes visualized by quick-freeze/deep-etch electron microscopy. Cell, 90, 523–535. [DOI] [PubMed] [Google Scholar]
- Jahn R. and Südhof,T.C. (1999) Membrane fusion and exocytosis. Annu. Rev. Biochem., 68, 863–911. [DOI] [PubMed] [Google Scholar]
- Kaiser C.A. and Schekman,R. (1990) Distinct sets of SEC genes govern transport vesicle formation and fusion early in the secretory pathway. Cell, 61, 723–733. [DOI] [PubMed] [Google Scholar]
- Katz L., Hanson,P.I., Heuser,J.E. and Brennwald,P. (1998) Genetic and morphological analyses reveal a critical interaction between the C-termini of two SNARE proteins and a parallel four helical arrangement for the exocytic SNARE complex. EMBO J., 17, 6200–6209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilmartin J.V. and Adams,A.E. (1984) Structural rearrangements of tubulin and actin during the cell cycle of Saccharomyces. J. Cell Biol., 98, 922–933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraulis P.J. (1991) MOLSCRIPT: a program to produce both detailed and schematic plots of protein structures. J. Appl. Crystallogr., 24, 946–950. [Google Scholar]
- Lin R.C. and Scheller,R.H. (1997) Structural organization of the synaptic exocytosis core complex. Neuron, 19, 1087–1094. [DOI] [PubMed] [Google Scholar]
- Luft J.H. (1961) Improvements in epoxy resin embedding methods. J. Biophys. Biochem. Cyt., 9, 409–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer A. (1999) Intracellular membrane fusion: SNAREs only? Curr. Opin. Cell Biol., 11, 447–452. [DOI] [PubMed] [Google Scholar]
- McQuarrie D.A. (1973) Statistical Thermodynamics. Harper & Row, New York, NY. [Google Scholar]
- Merritt E.A. and Bacon,D.J. (1997) Raster3D: photorealistic molecular graphics. Methods Enzymol., 277, 505–524. [DOI] [PubMed] [Google Scholar]
- Nichols B.J., Ungermann,C., Pelham,H.R., Wickner,W.T. and Haas,A. (1997) Homotypic vacuolar fusion mediated by t- and v-SNAREs. Nature, 387, 199–202. [DOI] [PubMed] [Google Scholar]
- Novick P., Field,C. and Schekman,R. (1980) Identification of 23 complementation groups required for post-translational events in the yeast secretory pathway. Cell, 21, 205–215. [DOI] [PubMed] [Google Scholar]
- Patel S.K., Indig,F.E., Olivieri,N., Levine,N.D. and Latterich,M. (1998) Organelle membrane-fusion—a novel function for the syntaxin homolog Ufe1p in ER membrane-fusion. Cell, 92, 611–620. [DOI] [PubMed] [Google Scholar]
- Pfeffer S.R. (1999) Transport-vesicle targeting: tethers before SNAREs. Nature Cell Biol., 1, E17–E22. [DOI] [PubMed] [Google Scholar]
- Protopopov V., Govindan,B., Novick,P. and Gerst,J.E. (1993) Homologs of the synaptobrevin vamp family of synaptic vesicle proteins function on the late secretory pathway in Saccharomyces cerevisiae. Cell, 74, 855–861. [DOI] [PubMed] [Google Scholar]
- Rabouille C., Kondo,H., Newman,R., Hui,N., Freemont,P. and Warren,G. (1998) Syntaxin 5 is a common component of the NSF- and p97-mediated reassembly pathways of Golgi cisternae from mitotic Golgi fragments in vitro. Cell, 92, 603–610. [DOI] [PubMed] [Google Scholar]
- Resat I.L. and Mezel,M. (1993) Studies on free energy calculations. I. Thermodynamic integration using a polynomial path. J. Chem. Phys., 99, 6052–6061. [Google Scholar]
- Rice L.M., Brennwald,P. and Brünger,A.T. (1997) Formation of a yeast SNARE complex is accompanied by significant structural changes. FEBS Lett., 415, 49–55. [DOI] [PubMed] [Google Scholar]
- Rose M., Winston,F. and Hieter,P. (1990) Methods in Yeast Genetics: A Laboratory Course Manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- Rothman J.E. (1994) Mechanism of intracellular protein transport. Nature, 372, 55–63. [DOI] [PubMed] [Google Scholar]
- Ryckaert J.P., Ciccotti,G. and Berendsen,H.J.C. (1977) Numerical integration of the cartesian equations of motion of a system with constraints; molecular dynamics of n-alkanes. J. Comp. Phys., 23, 327–341. [Google Scholar]
- Sacher M., Stone,S. and Ferro-Novick,S. (1997) The synaptobrevin-related domains of Bos1p and Sec22p bind to the syntaxin-like region of Sed5p. J. Biol. Chem., 272, 17134–17138. [DOI] [PubMed] [Google Scholar]
- Salminen A. and Novick,P. (1987) A ras-like protein is required for a post-Golgi event in yeast secretion. Cell, 49, 527–538. [DOI] [PubMed] [Google Scholar]
- Sambrook J., Fritsch,E.F. and Maniatis,T. (1989) Molecular Cloning. A Laboratory Manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- Sikorski R.S. and Hieter,P. (1989) A system of yeast shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics, 122, 19–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Söllner T., Whitehart,S.W., Brunner,M., Erdjument-Bromage,H., Geromanos,S., Tempst,P. and Rothman,J.E. (1993) SNAP receptors implicated in vesicle targeting and fusion. Nature, 362, 318–324. [DOI] [PubMed] [Google Scholar]
- Stevens T., Esmon,B. and Schekman,R. (1982) Early stages in the yeast secretory pathway are required for transport of carboxypeptidase Y to the vacuole. Cell, 30, 439–448. [DOI] [PubMed] [Google Scholar]
- Strom M., Vollmer,P., Tan,T.J. and Gallwitz,D. (1993) A yeast GTPase-activating protein that interacts specifically with a member of the Ypt/Rab family. Nature, 361, 736–739. [DOI] [PubMed] [Google Scholar]
- Sutton R.B., Fasshauer,D., Jahn,R. and Brünger,A.T. (1998) Crystal structure of a SNARE complex involved in synaptic exocytosis at 2.4 Å resolution. Nature, 395, 347–353. [DOI] [PubMed] [Google Scholar]
- Terrian D.M. and White,M.K. (1997) Phylogenetic analysis of membrane trafficking proteins: a family reunion and secondary structure predictions. Eur. J. Cell Biol., 73, 198–204. [PubMed] [Google Scholar]
- Ungermann C., Sato,K. and Wickner,W. (1998) Defining the functions of trans-SNARE pairs. Nature, 396, 543–548. [DOI] [PubMed] [Google Scholar]
- Ungermann C., Fischer von Mollard,G., Jensen,O.N., Margolis,N., Stevens,T.H. and Wickner,W. (1999) Three v-SNAREs and two t-SNAREs, present in a pentameric cis-SNARE complex on isolated vacuoles, are essential for homotypic fusion. J. Cell Biol., 145, 1435–1442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Buuren A.R., Marrink,S.-J. and Berendsen,H.J.C. (1993) A molecular dynamics study of the decane/water interface. J. Phys. Chem., 97, 9206–9212. [Google Scholar]
- Van der Spoel D., Berendsen,H.J.C., Van Buuren,A.R., Apol,E., Meulenhoff,P.J., Sijbers,A.L.T.M. and Van Drunen,R. (1995) Gromacs User Manual. Groningen, The Netherlands. Electronic access: http://md.chem.rug.nl/~gmx [Google Scholar]
- Van Gunsteren W.F. and Berendsen,H.J.C. (1987) Gromos Manual. BIOMOS biomolecular software, Laboratory of Physical Chemistry, University of Groningen, The Netherlands. [Google Scholar]
- Weber T., Zemelman,B.V., Mcnew,J.A., Westermann,B., Gmachl,M., Parlati,F., Söllner,T.H. and Rothman,J.E. (1998) SNAREpins—minimal machinery for membrane-fusion. Cell, 92, 759–772. [DOI] [PubMed] [Google Scholar]
- Wei S., Xu,T., Ashery,U., Kollewe,A., Matti,U., Antonin,W., Rettig,J. and Neher,E. (2000) Exocytotic mechanism studied by truncated and zero layer mutants of the C-terminus of SNAP-25. EMBO J., 19, 1279–1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weimbs T., Low,S.H., Chapin,S.J., Mostov,K.E., Bucher,P. and Hofmann,K. (1997) A conserved domain is present in different families of vesicular fusion proteins—a new superfamily. Proc. Natl Acad. Sci. USA, 94, 3046–3051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weimbs T., Mostov,K., Low,S.H. and Hofmann,R. (1998) A model for structural similarity between different SNARE complexes based on sequence relationships. Trends Cell Biol., 8, 260–262. [DOI] [PubMed] [Google Scholar]
- Yang B., Gonzalez,L., Prekeris,R., Steegmaier,M., Advani,R.J. and Scheller,R.H. (1999) SNARE interactions are not selective—implications for membrane fusion specificity. J. Biol. Chem., 274, 5649–5653. [DOI] [PubMed] [Google Scholar]