

Abstract
Sphingosine-1-phosphate (S1P) is a pleiotropic bioactive lipid thought to be dysregulated in a variety of disease conditions. In this review, we discuss the roles of S1P in cancer and in wet age-related macular degeneration. We also explore potential treatment strategies for these disorders, including the utility of anti-S1P antibodies acting as molecular sponges to neutralize dysregulated S1P in relevant tissues.
Keywords: Sphingosine-1-phosphate, S1P, cancer, age-related macular degeneration, wet AMD, antibody therapeutics, ocular disease, choroidal neovascularization, retinal pigmented epithelial cells
Sphingosine-1-phosphate is an attractive target for drug discovery
Bioactive lipids are important signalling mediators that are becoming attractive targets for drug discovery because of their roles in cancer, inflammation and other pathological conditions. Examples of such bioactive lipids include: (i) eicosanoids (such as the thromboxanes and leukotrienes); (ii) phospholipids and their lysophospholipid derivatives such as platelet activating factor (PAF) and lysophosphatidic acid; and (iii) sphingolipids such as sphingosine-1-phosphate (S1P). In recent years, receptors and other targets have been discovered for many of these bioactive lipids, suggesting extracellular signalling roles for these lipid mediators and growth factors (Im, 2009). One difficulty in targeting proteins that are responsible for their dysregulation in disease is that there are commonly several biosynthetic pathways for a particular bioactive lipid. Equally challenging is that there are commonly several receptors, ion channels or other proteins responsible for the action of a particular bioactive lipid and, in some cases, not all of the receptors that have been elucidated as novel receptors continue to appear in the literature (Im, 2009).
Of the 1000 or so bioactive lipids, sphingolipids are recognized as important intercellular and intracellular signalling molecules participating in physiological and pathological processes associated with cellular survival, proliferation, differentiation and adhesion function (Moolenaar, 1999; Goetzl et al., 2002; Birgbauer and Chun, 2006; Gardell et al., 2006). The key elements of the sphingomyelin-associated signalling pathway include the bioactive lipid mediators, ceramide (CER), sphingosine (SPH) and S1P.
The most celebrated sphingolipid mediator is S1P whose well-documented pleiotropic biological activities are mediated via a family of G protein-coupled cell surface receptors (GPCRs) belonging to the family of endothelial differentiation genes (EDG). These high-affinity receptors are S1P1-5/EDG-1,3,5,6 and 8 and are coupled to heterotrimeric G-proteins (Gi/o, Gq-, Gi-, G12-13) and small GTPases of the Rho family (Anliker and Chun, 2004). Most of the growth-promoting actions of S1P described in multiple organ systems, including immune, inflammation and cardiovascular, are exerted by S1P's action on its cognate GPCRs.
The major source of S1P is that produced from SPH through the action of sphingosine kinase (SphK) (Taha et al., 2006). Two isoforms of the kinase have been identified, SphK1 and SphK2. Sphingosine kinase controls a ‘ceramide-S1P rheostat’ that decides whether a cell is sent into the death pathway (via CER or SPH) or is protected from apoptosis by S1P (Cuvillier et al., 1996). For example, increasing ceramide/S1P ratios makes cancer and other hyperproliferative cells more sensitive to apoptosis. Similarly, decreasing the S1P level, either by inhibiting SphK or removing S1P, diminishes cell proliferation and tumour-associated angiogenesis and makes cancer cells more susceptible to apoptotic cell death (Anliker and Chun, 2004; Colombaioni and Garcia-Gil, 2004; Hla, 2004; Hait et al., 2006).
There is a reservoir of S1P stored in and potentially released from red blood cells, platelets and mast cells to create a local pulse of free S1P sufficient enough to exceed the Kd of the S1PRs (e.g. Kd for S1P1= 20 nM), whereby wound healing and inflammation are promoted (Murata et al., 2000a). Under normal conditions, the total S1P in the human plasma is high (300–500 nM), far exceeding the Kd for the receptors. However, it has been hypothesized that most S1P may be ‘buffered’ by serum proteins, particularly lipoproteins [e.g. high-density lipoprotein (HDL) > low-density-lipoprotein (LDL) > very low-density lipoprotein] and albumin, so that the bio-available S1P (1–2% of total) is not sufficient to activate S1P receptors (Murata et al., 2000a,b;). If this were not the case, inappropriate cardiovascular effects, angiogenesis and inflammation would result as suggested by others (Murata et al., 2000b; Yatomi, 2006).
While we and others have initially focused on the importance of plasma S1P levels as mirroring what might be happening in the tumour microenvironment, it is clear from the work with FTY-720 and lyase inhibitors that interstitial fluid tissue levels of S1P might be the culprit. For example, SphK1 knockout mice have increased vascular permeability while experiencing no alterations in plasma S1P levels (Li et al., 2008b). One view is that plasma S1P is mostly ‘buffered’ and not biologically active unless S1P-binding proteins such as HDLs are able to present S1P to S1P receptors, possibly via ligand passing via Srb1-S1PR contact (Lucke and Levkau, 2010). It is also not known whether or not substantial amounts of S1P are associated with cell membranes other than RBCs (Bode et al., 2010) or in tissue lipid microenvironments. While SphK is ubiquitously expressed in tissues, the concentration of biologically active S1P in the interstitial compartment in local tissues is not known. It is generally believed that bioactive or free S1P can be dysregulated in cancer and inflammatory disorders such that the tissue levels of biologically active S1P can then exert autocrine and paracrine actions. In such cases, S1P can become harmful and can contribute to disease processes such as cancer and inappropriate angiogenesis (such as in exudative age-related macular degeneration or AMD). The next two sections of this review will concentrate on the role of dysregulated S1P in cancer and AMD as examples. Further, the merits of using anti-S1P antibodies to neutralize dysregulated extracellular S1P will be addressed for these two diseases, as anti-S1P antibodies are currently being tested in clinical trials.
S1P and cancer
Sphingosine kinase and the S1P receptors: roles in cancer
Because of the important role of sphingolipids in cancer progression, it has been argued that sphingolipid-based therapeutics will be the next generation of cancer treatments (Milstien and Spiegel, 2006; Fyrst and Saba, 2010; Pyne and Pyne, 2010). This view comes from findings that cancer cells exploit the sphingolipid rheostat by promoting conditions that favour the production of S1P through an up-regulation of SphK1, the isoform that is thought to be responsible for the release of S1P into the extracellular compartment (see comprehensive review by Pyne and Pyne, 2010). The ability of cancer cells to release S1P into the tumour microenvironment promotes the infiltration of platelets, fibroblasts, mast cells, endothelial cells (ECs) and neutrophils, resulting in an inflammatory response and tumour angiogenesis. The infiltrating cells cause further release of S1P into the tumour microenvironment, with the resulting manifestation of tumourigenic and pro-angiogenic effects of S1P.
The S1P produced by SphK1 has been established as a general growth-like factor and a potent protector against apoptosis caused by cytotoxics. The S1P produced promotes cellular proliferation, migration and protection from apoptosis (An et al., 1998; Maceyka et al., 2002; Radeff-Huang et al., 2004). S1P is also able to promote angiogenesis, the formation of new blood vessels, which are presumed to feed the growing tumour (Sabbadini, 2006; Teicher, 2011). Furthermore, it has been shown that the sphk1 gene is over-expressed in several tumour types and, as a result, directs attention to the kinase as a protein target for anti-cancer drug discovery (Milstien and Spiegel, 2006; Shida et al., 2008b).
NIH-3T3 fibroblasts and HEK 293 cells transfected with the kinase exhibited enhanced cell proliferation and protection from apoptosis accompanied by increased cellular S1P production (Olivera et al., 1999). In addition, the SphK1 over-expressers escaped contact inhibition, a property commonly exhibited by transformed cells. This observation is consistent with recent reports showing that S1P enhances metastatic potential of selected human cancer cell lines (Takuwa, 2002). Moreover, the SphK1 transfectants produced tumours when injected subcutaneously into non-obese diabetic/severe combined immune deficiency (SCID) mice (Xia et al., 2000). These results were recently confirmed in two studies showing that small molecule inhibitors of SphK1 could reduce tumour volumes in SCID mice grafted with either subcutaneous injections of JC mammary adenocarcinoma cells (French et al., 2003) or of human histocytic leukaemia U937 cells (Paugh et al., 2008). In addition, it has been demonstrated that several human tumour-derived cell lines became apoptotic when treated with the SphK1 small molecule inhibitors, and that their effectiveness could be accounted for by their abilities to reduce S1P levels (Bektas et al., 2005a). SphK1 has been shown to be over-expressed in many solid tumours, such as those of the breast, colon, lung, ovary, stomach, uterus, kidney and rectum (French et al., 2003; Johnson et al., 2005; Kawamori et al., 2006; Shida et al., 2008a). Increased expression of the SphK1 in tumour samples has been correlated with a significant decrease in survival rate in patients with several forms of cancer (Van Brocklyn et al., 2005; Ruckhaberle et al., 2008; Li et al., 2008a; 2009; Facchinetti et al., 2010; Long et al., 2010). However, the situation is predictably more complicated in breast cancer in that the expression and/or activity or SphK1 and its correlation with patient survival can depend on ER/HER status such that SphK1 can reverse its role and become protective (Long et al., 2010). For example, when one stratifies breast cancer patients, one finds that ER+ patients who have low HER1-3/SphK1 expression ratios, survive longer. This is in contrast to the inverse correlation between SphK1 and survival in unstratified breast cancer patient populations (Long et al., 2010). Certainly, there are other factors, including a potential role for SphK2, which conspire to influence time to disease progression.
SphK1 can confer resistance to cytotoxic agents and other therapies
It is widely appreciated that cancer cells are particularly successful in escaping therapy by adapting themselves to the tumour microenvironment and by mutating and evolving such that they can become resistant to cytotoxics or anti-angiogenic agents. The up-regulation of the oncogene, sphk1, and the resulting release of S1P into the tumour microenvironment could represent an important way cancer cells become resistance to treatment (recently reviewed by Raguz and Yague, 2008; Cuvillier et al., 2010). There is a growing consensus that individual patient genomics/proteomics profiling and biomarker evaluation will eventually identify which drug resistance mechanism is responsible for a patient's tumour progression. Thus, it may eventually be possible to identify some patients whose resistance can be attributed to an overproduction of S1P. While over-expression of SphK1 has been demonstrated in many cancer types and is correlated with patient survival (see above), a direct connection between SphK1 up-regulation and drug resistance has only recently been established, as reviewed by Cuvillier et al. (2010).
One potential mechanism explaining how chemoresistance can be established is the case of the S1P/hypoxia-inducible transcription factor (HIF) axis where the response of cancer cells to hypoxia involves the up-regulation of the S1P/SphK system. The S1P/HIF axis was the subject of a recent review by Ader et al. (2009).
Targeting extracellular S1P might be an optimum strategy for cancer
The overwhelming body of evidence so far implicates the type 1 isoform of SphK in chemoresistance and in the enhanced tumourigenicity adaptations to hypoxia, all of which correlate with poor patient outcomes in several cancer types. Thus, SphK1 is a very attractive target for drug discovery. Along the path towards the full validation of SphK1 as a cancer target, several papers have recently appeared demonstrating the efficacy of various SphK inhibitors in delaying the progression of xenografted human tumours in mice. For example, the kinase inhibitor, SK1-II, showed significant retardation of tumour growth of JC mammary adenocarcinoma cell allografts (French et al., 2003); the SK1-I (BML-258) SphK1-specific inhibitor reduced tumour growth and improved survival in glioblastoma xenograft models employing orthotopically placed LN229-H2B-GFP cells (Kapitonov et al., 2009) and in AML xenograft models using human U-297 cells (Paugh et al., 2008); the SphK1 inhibitor, B-5354c, reduced tumour progression in orthotopically implanted hormone-resistant PC3-GFP cells (Pchejetski et al., 2008). SphK1 is thought to be translocated to the surface membrane and to produce/release the extracellular S1P that is tumourigenic and angiogenic. There is also evidence that targeting SphK2 with a small molecule inhibitor, ABC294640, specific for this isoform can delay tumour progression in xenograft models of human renal cell cancer using A498 cells implanted subcutaneously in SCID mice (Beljanski et al., 2010) and in JC mammary allografts (French et al., 2006). The latter two findings with SphK2 inhibitors complicate the otherwise simple story that pointed to SphK1 as the single appropriate cancer target (Maceyka et al., 2005).
It has been argued that the two isoforms, SphK1 and SphK2, may have opposite actions with regard to cancer cell growth (Maceyka et al., 2005). If this is the case, an SphK-targeted anti-cancer therapeutic would have to selectively inhibit SphK1 and not SphK2. This may prove to be difficult, as both enzymes have five highly conserved domains and high-percent identity in their amino acid sequences. In addition, several splice variants of both isoforms have been characterized. Additionally, an acylglycerol kinase with homology to SphK1 has been recently identified (Bektas et al., 2005b), thus further complicating target selectivity of SphK-directed agents. Furthermore, other SphK-independent sources of S1P have been suggested. One such player is the ectoenzyme, autotaxin (ATX), which is capable of producing S1P from sphingosylphosphoryl choline (Clair et al., 2003). Potential additional mechanisms to generate S1P, like ATX or SphK2, may explain why sphk1-null mice exhibited no significant changes in tissue S1P levels even though tissue SphK1 activity was nearly eliminated (Allende et al., 2004). Consequently, selectively blocking only SphK1 activity may not mitigate S1P production by other routes.
Another intervention point in the sphingolipid pathway is to prevent S1P from interacting with its G-protein coupled receptors. The majority of pro-tumourigenic, angiogenic and metastatic actions of S1P are mediated through these receptors; however, intracellular roles for S1P have recently been demonstrated such as histone deacetylases 1 and 2 (Hait et al., 2009) as well as TRAF2 (Alvarez et al., 2010). There are five isoforms of the S1P receptor named S1P1-5 (Sanchez and Hla, 2004). S1P1-3 are usually expressed ubiquitously, while S1P4 and S1P5 are mainly expressed in the lymphatic system and the central nervous system respectively (Sanchez and Hla, 2004). Treatment with the Novartis compound, FTY720 (fingolimod), a functional antagonist (i.e. reverse agonist) of S1P receptors, reduces tumour growth and tumour-associated angiogenesis in a variety of animal models of human cancer (Ho et al., 2005; Schmid et al., 2005; Neviani et al., 2007; Liu et al., 2008; Lucas da Silva et al., 2008; Silva, 2008). It is believed that FTY720 acts as an antagonist of S1P1, S1P3, S1P4 and S1P5 but not S1P2. In particular, FTY720 causes internalization of the receptors, thus acting as reverse agonists/functional antagonist. As such, FTY720 has been shown to induce apoptosis in multiple myeloma cells (Yasui and Palade, 1995), inhibit angiogenesis and reduce tumour vascularization (Lamontagne et al., 2006).
It must also be appreciated that some S1P receptors have opposite actions. For example, S1P1 and S1P3 are primarily responsible for the pro-migratory and angiogenic effects of S1P (Chae et al., 2004; Langlois et al., 2004). Conversely, S1P2 has been demonstrated to have the opposite effects on cell migration and angiogenesis (Goparaju et al., 2005; Skoura and Hla, 2009). In the B16-F10 melanoma cell line, which exclusively expresses S1P2, S1P stimulation has no effect on proliferation but inhibits cell migration in vitro (Arikawa et al., 2003). S1P-mediated inhibition of migration by U118 and U138 cells correlated with S1P2 expression (Lepley et al., 2005). The over-expression of S1P2 in these cell lines further enhanced the suppression of migration upon S1P stimulus, while a down-regulation of S1P2 by RNA interference reversed this inhibitory effect (Lepley et al., 2005). The literature shows that each cancer cell lineage has a unique pattern of S1PR expression that varies inconsistently not only among cancer types but also among patients within a cancer type. So, targeting an individual S1P receptor becomes problematic and is not likely to be a viable anti-S1P drug discovery strategy for cancer unless all receptors are blocked.
A more direct approach, which avoids all of the said limitations, is the prevention of ligand binding to all cognate receptors using an anti-S1P monoclonal antibody (mAb). Preclinical data with the murine variant of the anti-S1P mAb (Sphingomab™ or LT1002) demonstrate that this approach deprives growing tumour cells of important growth and survival factors and largely prevents tumour angiogenesis (Visentin et al., 2006). Considerable experimental data suggest that preventing the action of extracellular S1P could be an effective therapeutic approach for targeting tumour cells and tumour-associated vasculature.
It remains to be determined whether or not absorbing extracellular S1P with a mAb can influence intracellular pools such that the mAb can pull S1P from the cell. This is unlikely as aggressive dosing of both murine and humanized mAbs in good laboratory practice (GLP) toxicology studies failed to elicit serious adverse events such as those one might expect to see if all sources of S1P (including intracellular) were neutralized by the mAb. If that had happened, mice and monkeys would have experienced serious adverse events stemming from increased vascular permeability. Mice with conditional deletion of both SphK1 and SphK2 [Sphk1fl/–:Sphk2–/–:Mx1Cre+ (pS1Pless mice)] experience profound increases in vascular permeability (and death) after PAF and histamine challenge (Camerer et al., 2009).
It is not known what role, if any, membrane-bound S1P might have in the tumour microenvironment or in other disorders where S1P is dysregulated. It is clear that the mAbs are able to extract/release membrane-associated S1P from RBCs in vitro (Bode et al., 2010) as they do from plasma proteins such as HDLs, LDLs and serum albumin (R. Sabbadini, unpubl. obs.). Using surface plasmon resonance, the affinities of full-length anti-S1P mAbs were found to be high, in the 0.03–0.06 nM range (O'Brien et al., 2009) or 1.1 nM for the Fab'(Wojciak et al., 2009), while HDLs LDLs and albumin were not measurable under identical conditions (R. Sabbadini, unpubl. obs.).
The anti-S1P approach has distinct advantages over ‘singly-targeted’ therapeutics in that S1P not only has its own dual effects (tumorigenic and angiogenic) which should be neutralized, but is also permissive in promoting the actions and/or release of other important growth factors [e.g. IL-8, IL-6, matrix metalloproteinases, platelet-derived growth factor (PDGF), transforming growth factor β (TGFβ), vascular endothelial growth factor (VEGF), etc.]. An additional advantage of targeting S1P is that, unlike protein targets that can exist in multiple isoforms and/or splice variants, S1P has a single molecular structure that is conserved among species, making it both an attractive therapeutic target and one in which animal data may be more translatable to human diseases.
Anti-S1P antibody as a potential therapeutic
The murine anti-S1P, Sphingomab, was humanized for clinical development by grafting the complementarity determining regions onto a human IgG1κ framework and was then optimized to retain the specificity and affinity characteristics of the murine mAb (O'Brien et al., 2009). This humanized, optimized antibody is referred to as sonepcizumab ('S-one-P' cizumab) or LT1009. The crystal structure of the Fab' fragment was resolved to 1.9 Angstrom resolution, demonstrating that the hypervariable domains of the Fab' interact with the ligand, S1P, in a manner predicted by site-directed mutagenesis studies (Wojciak et al., 2009).
In anticipation of the Phase I clinical studies, the strength and nature of the binding of sonepcizumab to S1P was determined in order to determine the performance characteristics of the antibody and to evaluate its safety and efficacy in humans. Sonepcizumab exhibited favourable characteristics with respect to kinetic, stoichiometric and binding specificity, as well as thermal stability, making it suitable as a clinical candidate. Binding kinetics of sonepcizumab to S1P as measured by BiaCore showed that sonepcizumab bound well to the S1P-tethered surfaces. The kinetics on the lowest density surface fit well to a simple 1:1 interaction model and yielded an affinity of <0.1 nM under these conditions (Wojciak et al., 2009).
The specificity of anti-S1P binding was tested in a competitive ELISA for cross-reactivity against over 60 bioactive lipids and other molecules of interest (O'Brien et al., 2009). Sonepcizumab did not recognize lipids if the phosphate group of the polar head was absent or substituted (S1P vs. sphingosine and d-galactosyl-sphingosine); it did not recognize lipid structures if a fatty acid was added to the amino group on the sphingoid base (S1P vs. ceramide-1-phosphate); sonepcizumab recognized a form of S1P with a reduction of the double bond in the sphingoid base (dihydro S1P); and phosphate ester group added to the sphingoid base forming sphingosylphosphoryl choline. Epitope mapping revealed that sonepcizumab recognized preferentially the phosphate group and the amino-alcohol carried by the polar head of the sphingosine base. This structure has been confirmed by X-ray diffraction of the crystallized Fab' of sonepcizumab and was recently published (Wojciak et al., 2009). The binding of sonepcizumab and Sphingomab towards an extracellular bioactive lipid target such as S1P affords a potential advantage for therapeutic efficacy in vivo, as S1P is highly conserved across species and therefore not subject to the drug-resistant mutations in response to therapy in the same manner as protein targets.
Thermal stability of sonepcizumab was determined to be greater than the murine mAb, Sphingomab: the thermal unfolding transitions (Tm) of sonepcizumab is 73 ± 2°C compared with 55 ± 2°C for Sphingomab (O'Brien et al., 2009).
Thereby, sonepcizumab displayed performance characteristics that made it suitable as a clinical candidate. Sonepcizumab was formulated into two separate drug candidates: (i) ASONEP™, the oncology formulation which was investigated in a recently completed Phase I trial in cancer patients (see next for details); and (ii) iSONEP™, the ocular formulation of which was also investigated in a recently completed Phase I trial for wet-AMD patients (see next for details).
Sonepcizumab is possibly the first humanized monoclonal antibody against a bioactive lysolipid, and it is certainly the first to be advanced into the clinic.
Preclinical pharmacology and efficacy
Preclinical studies with sonepcizumab and its murine counterpart, Sphingomab, administered every 2–3 days at doses of 10–80 mg·kg−1, demonstrated the ability of anti-S1P mAbs to reduce tumour volumes and metastatic potential, probably the result of inhibiting tumour-associated angiogenesis. In mouse models of murine and human cancer, the murine anti-S1P mAbs significantly retarded the progression of several orthotopic and subcutaneous human tumours implanted in nude mice (Visentin et al., 2006), including breast MDA MB 231 and 468, ovarian SKOV3 and lung A549, as well as melanoma B16/F10 allograft tumours. The anti-S1P mAbs inhibited S1P-induced tumour cell migration, proliferation and protection from apoptosis induced by chemotherapeutic agents (Visentin et al., 2006).
The murine anti-S1P mAb dramatically reduced tumour-associated angiogenesis in subcutaneous murine melanoma B16-F10 allograft, human lung A549 and ovarian SKOV3 xenograft models (Visentin et al., 2006). Both murine and humanized antibodies neutralized bFGF- and VEGF-induced angiogenesis in the murine Matrigel plug assay (Visentin et al., 2006; O'Brien et al., 2009). Consistent with in vivo anti-angiogenic properties, the antibodies neutralized S1P-induced EC tube formation, migration and protection from cell death in various in vitro assays (Visentin et al., 2006). S1P-induced release of pro-angiogenic growth factors such as IL-8, IL-6 and VEGF from tumour cells was also demonstrated in vitro and in vivo (Visentin et al., 2006).
In completely different preclinical animal models of ocular angiogenesis, Sphingomab and sonepcizumab blocked choroidal and retinal neovascularizations (Caballero et al., 2009; O'Brien et al., 2009; Xie et al., 2009).
The efficacy in animal xenograft and angiogenesis studies with the anti-S1P mAbs was demonstrable despite the fact that plasma S1P levels are ∼threefold higher in mice compared with humans (He et al., 2009) such that, in mice, the antibody molecular sponge has more antigen to neutralize. The efficacy of anti-S1P mAbs in these models suggests that treatment with an anti-S1P antibody may provide an innovative and useful approach to cancer treatment in humans. Antibody-mediated neutralization of extracellular S1P could result in a marked decrease in cancer progression in humans as a result of inhibition of new blood vessel formation with concomitant loss of the nutrients and oxygen needed to support tumour growth. In addition, the remaining blood vessels would be expected to be normalized to allow for more efficient delivery of cytotoxic drugs. In fact, many angiogenesis inhibitors may also act as anti-invasive and anti-metastatic compounds, which could also aid in the mitigation of the spread of cancer to sites distant from the initial tumour (Teicher, 2011).
Preclinical safety and toxicity
GLP-quality safety/toxicology evaluations in mice and non-human primates (NHPs) were performed for sonepcizumab. A battery of preclinical studies with anti-S1P mAb complying with the International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use guidelines was performed to support the initial clinical development of the antibody in the proposed clinical indication showing no dose-limiting toxicities. Two GLP toxicology studies with sonepcizumab in NHPs were completed to support the initial clinical development of sonepcizumab. In addition, three tissue cross-reactivity studies were conducted with sonepcizumab and three mouse tissue cross-reactivity studies were completed with the murine antibody, Sphingomab. Neither the murine nor the humanized mAbs recognized any protein epitopes in the more than 30 tissues studied.
C57BL6 mice were generally tolerant of large, single doses of Sphingomab, with no evidence of toxicity up to 240 mg·kg−1 administered intravenously. The administration of this drug to mice over 28 days produced a decrease in absolute lymphocyte counts. At 30 mg·kg−1 (cumulative dose over 28 days of 840 mg·kg−1), the lymphopoenia observed in mice was more pronounced with Sphingomab than the very mild, reversible decrease in lymphocyte counts (but not lymphopoenia) in monkeys treated with a similar 100 mg·kg−1 dose of sonepcizumab (cumulative dose of 1000 mg·kg−1). Decreased lymphocytes were an expected pharmacological response to systemic anti-S1P mAb treatment as S1P has been shown to control trafficking of lymphocytes out of primary and secondary lymphoid tissues into the peripheral circulation (Cyster, 2004; Graler and Goetzl, 2004). In NHP studies, lymphocyte counts were reduced in a dose-dependent fashion after neutralization of systemic S1P by sonepcizumab in agreement with published accounts of mice treated with Sphingomab (O'Brien et al., 2009) as well as the GLP toxicology studies performed in mice cited previously. As a result, decreased lymphocyte counts were used as a surrogate marker of anti-S1P pharmacological activity in vivo for a Phase I sonepcizumab trial in cancer patients (see next). While this signal may not necessarily represent an effect of sonepcizumab on tumour progression per se, it does demonstrate pharmacological activity of the antibody. Additionally, the ability of anti-S1P mAbs to reduce tumour progression in xenografted nude mice that are not immunocompetent suggests that anti-S1P actions are independent of effects on lymphocyte trafficking.
Besides the mild decreases in absolute lymphocyte counts shown in the mice and monkey studies, no consistent adverse events were noted in either species. For both sonepcizumab and Sphingomab, no effects were seen in animal mortality, body weight, clinical pathology (haematology except for lymphocytes, clinical chemistry, coagulation and urinalysis) and lymphocyte phenotyping. Mild histopathological changes were noted in mice only in the spleen. No histopathological changes were noted in monkeys dosed with sonepcizumab.
A battery of other safety and pharmacological studies were performed with both sonepcizumab and Sphingomab. The findings, with respect to vascular permeability and wound healing, suggest little effect of the antibody treatment on these systems. No toxicologically relevant effects of sonepcizumab administration were observed in cytomologous monkeys in GLP ophthalmological studies (intravitreal administration, including measurement of electroretinogram), or in cardiovascular function as measured by ECG. The data also indicated that sonepcizumab does not induce antibody-dependent cellular cytotoxicity, cytokine release or blood haemolysis.
Taken together, the safety and pharmacology studies supported an Investigational New Drug application with the Food and Drug Administration and warranted moving sonepcizumab into clinical development with the oncology formulation, ASONEP.
Phase I clinical trial with sonepcizumab
Lpath has recently conducted a Phase I clinical trial in cancer. The Phase I study was a multi-centre, open-label, single-arm, Phase I dose escalation study of sonepcizumab administered as a single agent weekly to subjects with refractory advanced solid tumours. The objectives of this study were to characterize the safety, tolerability and dose-limiting toxicities (DLTs) for sonepcizumab. Doses between 1 and 24 mg·kg−1 were tested. Other than infusion-related reactions observed at the highest dose of 24 mg·kg−1, sonepcizumab was well tolerated across the range of doses that was tested, and no DLTs were observed.
Of the 21 patients who completed the initial four-treatment evaluation period, 12 showed stable disease at the end of the first cycle and 11 had stable disease for 2 months or longer. One patient with an extremely aggressive metastatic melanoma showed stable disease through 8 months; another patient with an adenoid cystic tumour was treated for over a year without disease progression. Yet another patient – with carcinoid tumour – is still being treated (as of the writing of this article) with sonepcizumab 26 months after the initial dose, without disease progression. This patient has also shown significant symptomatic improvement (near elimination of the diarrhoea and flushing that makes carcinoid so debilitating). These data were presented at a recent ASCO meeting (Gordon et al., 2010).
Because sonepcizumab was well tolerated by cancer patients in the Phase I clinical study and because of the potential to see an efficacy signal in cancer patients in first or second-line treatment settings, sonepcizumab is being considered for Phase II clinical trials.
S1P and exudative AMD
Exudative (i.e. wet) AMD is characterized by choroidal neovascular (CNV) lesions that eventually lead to the degeneration of the macula, the area of the retina responsible for central vision. Macular damage is caused collectively by: (i) new and leaky blood-vessel growth from the choroid layer into the retinal region; (ii) sub-retinal fibrosis; and (iii) inflammation in the retinal area. The most widely used drugs for wet AMD are Lucentis® and off-label use of Avastin®, both of which target the protein, VEGF, a well-established promoter of the vascular permeability experienced by wet-AMD patients. These drugs exert most of their beneficial effect via an anti-permeability action that results in the resolution of intra- and sub-retinal oedema, and reduces the progression of further neovascularization. Although anti-VEGF drugs improve vision in about a third of patients, the majority do not experience visual improvement over the long term. Anti-VEGF therapies do not cause regression of already-established lesions and the treatments have only a modest effect in reducing pigmented epithelial detachments (PEDs) in patients experiencing this common complication of wet AMD (Ach et al., 2010). Thus, despite the impressive clinical success of anti-VEGF therapies, there is room for a second generation of agents to treat AMD.
Growing evidence suggests that S1P modulates exudative-AMD-associated neovascularization, inflammation and fibrosis. S1P's effects on cell migration, proliferation and protection from cell death have been observed in multiple cell types including fibroblasts, ECs, pericytes and inflammatory cells, all implicated in the pathogenesis of exudative AMD. S1P is also implicated in the trans-activation and production of VEGF, FGF, PDGF and other growth factors that play a major role in the pathogenesis of CNV and are targets of other CNV therapeutics (Spiegel and Milstien, 2003). As the next paragraphs will demonstrate, considerable experimental data suggest that preventing the action of S1P could be an effective therapeutic approach for exudative AMD with distinct non-overlapping mechanisms of action vis a vis current anti-VEGF based therapies.
S1P and pathological neovascularization and vascular permeability in the retina
Several lines of evidence suggest that S1P, along with its complement of receptors, plays a major regulatory role in the angiogenic process (Argraves et al., 2004). S1P stimulates DNA synthesis and chemotactic motility of local and bone marrow-derived vascular EC to sites of vascularization and induces differentiation of multicellular structures consistent with early blood vessel formation (Lee et al., 1999b). Also, S1P stimulates the formation and maintenance of vascular EC assembly and integrity by activating S1P1, S1P3 and S1P-induced EC adherens junction assembly mediated by Rho and Rac GTPases (Lee et al., 1999a; Paik et al., 2001). Interestingly, S1P promotes N-cadherin junction formation between ECs and mural cells (Paik et al., 2004). In these studies, cocultures of 10T1/2 pericytes with ECs produced junctions which were eliminated when EC S1P1 receptors were knocked down with siRNA. Normal connections were restored upon expression of the S1P1 receptor. All of these effects were completely independent of VEGF and angiopoietin 1 and 2.
Importantly, S1P induces significant capillary tube formation and is thought to be at least as pro-angiogenic as basic fibroblast growth factor and VEGF in promoting the development of vascular networks in vivo (Lee et al., 1999a,b; Visentin et al., 2006). In addition to the direct angiogenic effects of S1P, there is some cross-talk between S1P and other pro-angiogenic growth factors. For example, S1P transactivates epidermal growth factor and VEGF2 receptors (Tanimoto et al., 2002) and VEGF up-regulates S1P1 receptors (Igarashi et al., 2003). S1P as well as VEGF independently activate the endothelial isoform of nitric oxide synthase (eNOS), which has essential roles in angiogenesis (Igarashi et al., 2003).
A pivotal paper implicating a role of S1P in ocular angiogenesis was published by Skoura et al. (2007). Using a S1P2 knockout mouse and a retinopathy of prematurity (ROP) model, they demonstrated that the retinal neovascularization characteristic of this model could be retarded by the genetic deletion of S1P2. Unfortunately, very few studies have directly demonstrated the presence of S1P in the retina. One study reported a biochemical determination of S1P that revealed substantial levels of S1P in rabbit vitreous fluid (Xie et al., 2009), and it has also been demonstrated that S1P can be localized by immunohistochemistry in retinal tissue (Caballero et al., 2009). These data are supported by findings of SphK expression in relevant primary human retinal cell types (Swaney et al., 2008) or by cell lines (Zhu et al., 2009).
Consistent with a role for S1P in ocular angiogenesis and vascular permeability, a recent report in the rat steptozotocin-induced diabetic retinopathy model showed that the inhibition of S1P production using a SphK inhibitor, SK1-II, attenuated VEGF-induced retinal EC proliferation and migration and reduced retinal vascular leakage (Maines et al., 2006). The systemic administration of SK1-II also inhibited subcutaneous VEGF-induced vascular permeability in the Miles assay, suggesting that leakage from the peripheral vascular bed can be reduced by anti-S1P interventions.
Recently, two papers demonstrated the efficacy of anti-S1P mAbs in preventing choroidal and retinal angiogenesis in standard murine CNV and ROP models. Xie et al. (2009) found that single intravitreal injection of sonepcizumab showed a dose-dependent decrease in retinal neovascularization and leakiness in the ROP model of ischaemia-induced angiogenesis. These findings were consistent with role of S1P2 in promoting angiogenesis in ROP mice (Skoura et al., 2007). It was also demonstrated that both murine and humanized anti-S1P mAbs substantially reduced choroidal neovascularization after laser disruption of Bruch's membrane (the CNV model of wet AMD) (Caballero et al., 2009; O'Brien et al., 2009). Importantly, immunostaining of CNV lesions revealed a dramatic up-regulation of S1P levels in what appeared to be produced by the retinal pigmented epithelium (RPE) layer after laser-induced injury (Caballero et al., 2009). This is consistent with the finding that human RPE cells express both isoforms of SphK, including SphK1 (Swaney et al., 2008; Zhu et al., 2009), the isoform thought to be responsible for the extracellular release of S1P. Significantly, both human choroidal and retinal ECs also express SphKs and S1P receptors (Maines et al., 2006; Swaney et al., 2008). The S1P receptors expressed by endothelia are probably the targets for extracellular S1P's actions in promoting angiogenesis and vascular permeability.
S1P and fibrosis
S1P is increasingly being recognized for its ability to promote pro-fibrotic function by several cell types. S1P promotes the transformation of human lung fibroblasts and human dermal fibroblasts to the profibrotic, myofibroblast phenotype (Keller et al., 2007). Also TGFβ, a well-known profibrotic factor, stimulates the expression of SphK1 (Yamanaka et al., 2004). Recent reports have also shown that S1P exhibits cross-talk with other profibrotic signalling pathways such as TGFβ (Xin et al., 2004) and PDGF (Alderton et al., 2001; Hobson et al., 2001), and S1P stimulates the expression of connective tissue growth factor (Katsuma et al., 2005), a protein implicated in numerous fibrotic disorders as well as in wet AMD (He et al., 2003). A fibrogenic role for S1P is also suggested by findings that transgenic mice over-expressing SphK1 exhibit profound cardiac remodelling associated with myocardial fibrosis (Takuwa et al., 2009).
Using primarily S1P2 and Rho signalling, we demonstrated that S1P stimulates the expression of collagen in primary fibroblasts coincident with promoting differentiation of fibroblasts to the myofibroblast phenotype (Gellings Lowe et al., 2009). Substantial cross-talk between S1P and TGFβ was also demonstrated in the cardiac fibroblast in that study. Importantly, the murine anti-S1P mAb, Sphingomab, blocked the profibrotic effects of S1P in murine primary cardiac fibroblasts (Gellings Lowe et al., 2009). Of particular relevance to ocular fibrosis, we recently showed that human primary retinal RPE cells express S1P receptors and that S1P's action on these GPCRs promotes the differentiation of RPE cells into a myofibroblast phenotype capable of expressing collagen as well as other fibrotic mediators such as PAI-1 and HSP-47 (Swaney et al., 2008). We also have published work demonstrating a role of S1P in ocular and other fibrotic processes, including data showing the efficacy of anti-S1P mAbs in mitigating fibrotic responses after laser-induced injury of Bruch's membrane (Caballero et al., 2009).
Combined, these data suggest that S1P may serve as a novel mediator of fibrosis, and in particular, the ocular fibrosis (e.g. disciform scarring) associated with advanced stages of wet AMD. Anti-S1P antibodies may interfere with extracellular S1P actions and this might be a useful therapeutic strategy in disorders such as wet AMD where dysregulated fibrosis and/or scarring plays a role in the pathogenesis or progression of disease.
S1P and inflammation
There is growing evidence that S1P is an important mediator of inflammatory events (Olivera and Rivera, 2005), contributing to both wet and dry AMD. Activated platelets and mast cells serve as rich sources of S1P during coagulation and inflammation (Yatomi et al., 2000). S1P receptors are expressed by macrophages from a variety of sources and these GPCRs mediate S1P-dependent activation of macrophage function and protection against cell death, as well as having a stimulating effect on macrophage trafficking (Gude et al., 2008; Weigert et al., 2009). One notable exception is a recent paper showing increased macrophage trafficking in a S1P2 KO mouse model (Michaud et al., 2010).
Macrophages are important mediators of the dysfunctional inflammatory response associated with AMD. The presence of macrophages in histological studies of CNV implicates these cells as a contributing factor in the progression of wet AMD (Tsutsumi et al., 2003). In addition, generalized macrophage depletion has been shown to reduce the size and leakage of laser-induced CNV (Sakurai et al., 2003). Because mast cells, neutrophils, platelets and macrophages are important components in the inflammatory response and tissue loss, S1P may regulate these inflammatory events via the control of inflammatory cell function in ocular disorders such as wet and dry AMD as well as diabetic retinopathy.
As an initial demonstration that ocular S1P may be involved in retinal inflammation, Xie et al. (2009) have published work demonstrating that intravitreal injection of Sphingomab could substantially reduce the infiltration of F4/80 positive macrophages in a murine model of ROP. It has been argued that this effect may have been due to reduced survival of macrophages caused by antibody neutralization of the anti-apoptotic S1P, rather than a direct effect on macrophage trafficking (Weigert et al., 2009).
The alternative complement pathway has also been implicated in CNV based on the finding that inhibitors of the alternate complement pathway reduce the size of CNV lesions (Bora et al., 2006; Rohrer et al., 2009). Dysregulated complement has also been implicated in the pathogenesis of dry AMD (Petrukhin, 2007). Recently, a connection between complement and S1P in a C5a/S1P axis of inflammation has been established. Specifically, the activation of macrophages by the alternative complement pathway has been attributed to SphK1 where S1P is implicated as the major downstream mediator of C5a action (Melendez et al., 2000; Melendez and Ibrahim, 2004; Vlasenko and Melendez, 2005; Pushparaj et al., 2008; Puneet et al., 2010). Collectively, these papers demonstrate that the inflammatory action of complement C5a on the relevant GPCR expressed on macrophages is mediated by an up-regulation of SphK1 (Melendez and Ibrahim, 2004), thus proving support for the C5a/S1P axis mechanism. S1P-dependent C5a actions include degranulation, migration and release of TNFα, IL-8 and IL-6. Interestingly, it is well established that C5a receptors are also expressed by RPE cells of the eye (Fukuoka and Medof, 2001). Thus, one may speculate that the alternative complement pathway could be a stimulus for S1P production and release by RPE cells, as well as leading to C5a-dependent macrophage activation, recruitment and cytokine release in the posterior segment of the eye. The C5a/S1P axis could involve a positive feedback system for promoting an exaggerated inflammatory cascaded in the eye through an interaction between RPE cells and macrophages (see Figure 1).
Figure 1.
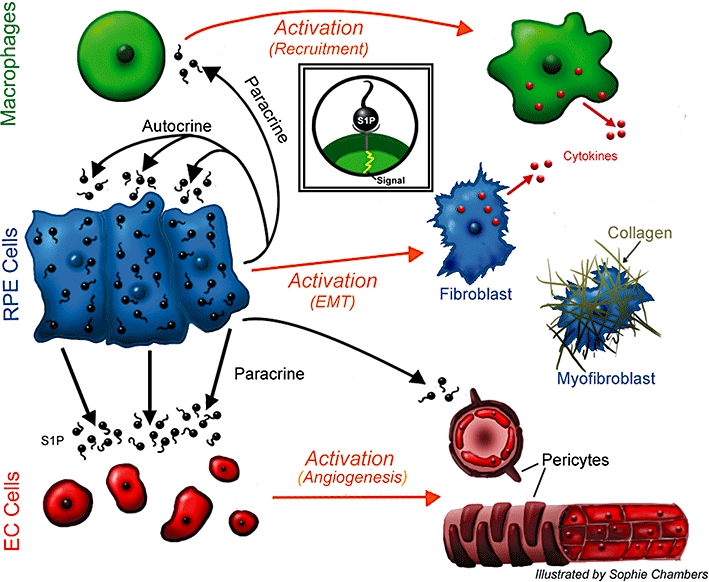
Proposed central role of RPE cells in wet AMD. We speculate that the RPE cells are a major source of S1P in the posterior segment of the eye and that the S1P stored and released from RPEs is responsible for the pathological angiogenesis, vascular permeability, fibrosis and inflammatory responses associated with wet AMD. A positive feedback loop is proposed whereby S1P released from the RPE layer acts in an autocrine fashion to further activate RPE cells and promote their differentiation to a myofibroblast, pro-fibrotic phenotype capable of expressing collagen. Released S1P also serves a paracrine function to promote choroidal endothelial cells and pericytes to form new blood vessels that will eventually create a CNV. S1P also promotes the inflammatory component of wet AMD by either directly activating macrophages or indirectly promoting their survival. RPE, retinal pigmented epithelium; AMD, age-related macular degeneration; S1P, sphingosine-1-phosphate; CNV, choroidal neovascularization.
Central role for RPE cells in ocular pathology
We hypothesize that RPE cells play a central role in the angiogenic, inflammatory and fibrotic responses to injury or in wet AMD (Figure 1). While human choroidal and retinal ECs express SphK1 as a potential source for S1P in the posterior segment of the eye, we propose that the RPE layer could be the major source of S1P in injured eyes and that the release of S1P from the RPE cells acts in a paracrine manner to stimulate choroidal ECs to participate in pathological neovascularization. Additionally, the postulated local pulse of S1P may serve to activate choroidal fibroblasts and promote the infiltration and activity of macrophages, and other inflammatory cells as discussed previously. Similarly, we propose that the S1P pulse could serve in an autocrine fashion to promote the epithelial-to-mesenchymal transition (EMT) of RPE cells towards the hypercontracile myofibroblast phenotype, an effect that has been described as one of S1P's actions on human RPE cells in vitro (Swaney et al., 2008).
RPEs play an important role in wet AMD as many of these patients experience PEDs where the RPE layer is detached from the basement membrane. Sub-retinal oedema is often the cause but one must consider that the hyper-contractile nature of the RPE cells could also be responsible for the detachment. This was elegantly demonstrated by Agrawal et al. (2007) who demonstrated in a model of peripheral vitreoretinopathy (PVR) that human RPE cells injected into rabbit vitreous promoted retinal detachment. We speculate that PED could be due to the effect of S1P in promoting the EMT and the resultant hyper-contractile state of the RPE layer (Figure 1).
The profound pro-fibrotic effects of S1P on cells from multiple regions of the eye and the anti-fibrosis data in mice suggest that sonepcizumab or other anti-S1P therapies could be efficacious in disorders such as PED, PVR and various anterior-segment diseases, including intraocular pressure disorders such as glaucoma. Regarding glaucoma, S1P has been shown recently to reduce outflow facility in ex vivo perfused human eyes probably through actions on trabelcular cells and inner wall Schlemm's canal cells expressing S1P1 and S1P3 receptors (Stamer et al., 2009). Thus, anti-S1P-based therapy could correct S1P-mediated intraocular hypertension in glaucoma. As discussed previously, the systemic administration of the SphK inhibitor, SK1-II, attenuated retinal vascular leakage in the STZ rat model of diabetic retinopathy (Maines et al., 2006), suggesting that S1P may play a role in this ocular disorder as well.
Sonepcizumab as a potential treatment for wet AMD
Taken together, the data suggest that inhibiting the action of S1P with anti-S1P mAbs could be an effective and novel therapeutic treatment for wet AMD as well as other ocular disorders. As a consequence, the anti-S1P mAbs were shown to markedly reduce CNV lesion volume, sub-retinal fibrosis and pericyte recruitment in a murine model of laser-induced rupture of Bruch's membrane. These findings were the first demonstration that a non-protein (specifically, a lipid) is a biological mediator of CNV formation. In addition, S1P is present in vitreous fluids and several ocular cell types express S1P receptors and SphK isoforms. In preclinical animal studies, anti-S1P mAbs exhibited a favourable safety and pharmacokinetic profile following both systemic and intravitreal administrations.
It is therefore possible that iSONEP, the ocular formulation of sonepcizumab, could deprive fibroblasts, pericytes, endothelial and immune cells of important growth factors. The ability of sonepcizumab/iSONEP to neutralize S1P-mediated trans-activation of VEGF and PDGF could prove effective in mitigating macular oedema associated with these growth factors (Vinores et al., 2000; Sanchez et al., 2003). Pericytes play a critical role in the development and maintenance of vascular tissue, and their presence seems to confer a resistance to anti-VEGF agents and compromise their ability to inhibit CNV (Ishibashi et al., 1995; Yamagishi and Imaizumi, 2005). S1P promotes adherens junction formation between pericytes and ECs, and promotes maturation of blood vessels during angiogenesis (Paik et al., 2004). By interfering with pericyte signalling, sonepcizumab could strip pericytes from existing lesions and could promote lesion regression by depriving CNV lesions of supportive mural cells. Finally, S1P produced locally by ischaemic/damaged cells could, in part, be responsible for the maladaptive wound healing associated with remodelling and scar formation. By inhibiting S1P, sonepcizumab could diminish the degree of fibroblast infiltration and collagen deposition associated with remodelling and scar formation. A therapeutic agent like sonepcizumab that simultaneously targets the vascular and extravascular components of exudative AMD has the potential to be a more effective treatment than ‘singly-targeted’ therapies such as anti-VEGF agents. Importantly, the success of Lucentis and Avastin in the treatment of wet AMD has demonstrated that antibodies have long half-lives, biodistribution and stability characteristics suited for intravitreal injection.
Thus, considerable experimental data have been generated to support the hypothesis that inhibiting the action of S1P could be an effective therapeutic approach for treating wet AMD, and this approach may have distinct non-overlapping mechanisms of action compared with current anti-VEGF therapies that solely target one vascular component of wet AMD. Because of the pleiotropic nature of S1P's actions in inflammation, angiogenesis and fibrosis, it is possible that anti-S1P treatment in wet AMD could have beneficial long-term outcomes including lesion regression and prevention of RPE detachments (PED or pigmented epithelial detachments). In fact, preliminary anecdotal findings from our Phase I clinical trial supports this contention (see next).
Phase I clinical trial in wet AMD with sonepcizumab
A multi-centre, open-label, single-arm, Phase I, dose escalation study of sonepcizumab administered as an intravitreal injection to subjects with CNV secondary to AMD was initiated. Five dose levels were evaluated: 0.2, 0.6, 1.0, 1.4 and 1.8 mg per eye. Subjects received a single intravitreal injection of sonepcizumab in one eye. The objectives were to determine the safety, tolerability, maximum tolerated dose (MTD) and DLT of sonepcizumab, and to characterize the systemic pharmacokinetics of sonepcizumab, determine doses for future clinical studies and investigate preliminary efficacy on retinal lesion thickness by optical coherence tomography, size and extent of CNV and lesion area and visual acuity. Results of this study were presented at a recent ophthalmics meeting (Stoller et al., 2010).
The patients in the cohort were largely those who were refractory to previous anti-VEGF treatments. The most significant benefit observed was with the five patients diagnosed with some form of occult CNV (vs. classic CNV). All of these five patients exhibited an apparent strong biological response. Specifically, three of the four occult patients with an active lesion exhibited a significant regression (>75%) in their CNV lesion, which is the underlying cause of the disease that eventually leads to the degeneration of the macula, the area of the retina responsible for central vision. This type of clinical benefit is not typical, as the published data (Heier et al., 2006) suggest that, even with repeated Lucentis dosing, the total physical size of CNV lesion does not show substantial reduction, especially with a single dose. Another distinctive benefit for the patients with occult disease was the resolution of RPE detachment (PED or pigmented epithelial detachment), a potentially serious condition that is often a part of the pathology of wet AMD. Of the two occult patients diagnosed with RPE detachment in the Phase I trial, both experienced complete or near-complete resolution of the condition. Neither Lucentis nor Avastin commonly produces this type of clinical benefit with a single dose (Ach et al., 2010). Because of these anecdotal signals of potential efficacy combined with strong safety data, sonepcizumab is now being advanced into Phase II clinical trials.
Conclusions
When considering the roles of dysregulated S1P levels in the pathogenesis of such diverse diseases as cancer and wet AMD, one is struck by the many commonalities these two disorders share. For example, both pathologies suffer from exaggerated and pathogenic elements of the inflammatory and wound-healing responses. Dysfunctional angiogenesis is also common to cancer (i.e. tumour angiogenesis) and to wet AMD (i.e. CNV). In addition, both are hyperproliferative disorders; the cancer being one of transformed, mitotically active primary cells and AMD being complicated by hyperproliferative ECs. Serendipitously, the pleiotropic nature of S1P in promoting angiogenesis, inflammation, wound healing and cell survival explain the reason why anti-S1P therapy could be useful in controlling both of these disorders. The anti-S1P antibody molecular sponge approach is one of several therapeutic strategies that are being explored but holds promise as a novel, first-in-class treatment.
Acknowledgments
This work was supported by Award Number R43EYO18020 from the National Eye Institute (NEI) and 5R44CA110298 from the National Cancer Institute (NCI). The content is solely the responsibility of the author and does not necessarily represent the official views of the NEI, NCI or the National Institutes of Health. The author has a potential conflict of interest associated with being a consultant for Lpath, Inc., having stock in Lpath and as an inventor on key patents that are held by Lpath. Appreciation goes to Sophie Chambers for the illustration in Figure 1.
Glossary
Abbreviations
- AMD
age-related macular degeneration
- CNV
choroidal neovascularization
- GLP
good laboratory practices
- NHP
non-human primate
- PED
pigmented epithelial detachment
- RPE
retinal pigmented epithelium
- S1P
sphingosine-1-phosphate
- SphK
sphingosine kinase
Supplementary material
Supporting Information: Teaching Materials; Fig 1 as PowerPoint slide.
References
- Ach T, Hoeh AE, Ruppenstein M, Kretz FT, Dithmar S. Intravitreal bevacizumab in vascular pigment epithelium detachment as a result of subfoveal occult choroidal neovascularization in age-related macular degeneration. Retina. 2010;30:1420–1425. doi: 10.1097/IAE.0b013e3181d87e97. [DOI] [PubMed] [Google Scholar]
- Ader I, Malavaud B, Cuvillier O. When the sphingosine kinase 1/sphingosine 1-phosphate pathway meets hypoxia signaling: new targets for cancer therapy. Cancer Res. 2009;68:8635–8642. doi: 10.1158/0008-5472.CAN-09-0389. [DOI] [PubMed] [Google Scholar]
- Agrawal RN, He S, Spee C, Cui JZ, Ryan SJ, Hinton DR. In vivo models of proliferative vitreoretinopathy. Nat Protoc. 2007;2:67–77. doi: 10.1038/nprot.2007.4. [DOI] [PubMed] [Google Scholar]
- Alderton F, Rakhit S, Kong KC, Palmer T, Sambi B, Pyne S, et al. Tethering of the platelet-derived growth factor beta receptor to G-protein-coupled receptors. A novel platform for integrative signaling by these receptor classes in mammalian cells. J Biol Chem. 2001;276:28578–28585. doi: 10.1074/jbc.M102771200. [DOI] [PubMed] [Google Scholar]
- Allende ML, Sasaki T, Kawai H, Olivera A, Mi Y, van Echten-Deckert G, et al. Mice deficient in sphingosine kinase 1 are rendered lymphopenic by FTY720. J Biol Chem. 2004;279:52487–52492. doi: 10.1074/jbc.M406512200. [DOI] [PubMed] [Google Scholar]
- Alvarez SE, Harikumar KB, Hait NC, Allegood J, Strub GM, Kim EY, et al. Sphingosine-1-phosphate is a missing cofactor for the E3 ubiquitin ligase TRAF2. Nature. 2010;465:1084–1088. doi: 10.1038/nature09128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- An S, Goetzl EJ, Lee H. Signaling mechanisms and molecular characteristics of G protein-coupled receptors for lysophosphatidic acid and sphingosine 1-phosphate. J Cell Biochem Suppl. 1998;30-31:147–157. [PubMed] [Google Scholar]
- Anliker B, Chun J. Cell surface receptors in lysophospholipid signaling. Semin Cell Dev Biol. 2004;15:457–465. doi: 10.1016/j.semcdb.2004.05.005. [DOI] [PubMed] [Google Scholar]
- Argraves KM, Wilkerson BA, Argraves WS, Fleming PA, Obeid LM, Drake CJ. Sphingosine-1-phosphate signaling promotes critical migratory events in vasculogenesis. J Biol Chem. 2004;279:50580–50590. doi: 10.1074/jbc.M404432200. [DOI] [PubMed] [Google Scholar]
- Arikawa K, Takuwa N, Yamaguchi H, Sugimoto N, Kitayama J, Nagawa H, et al. Ligand-dependent inhibition of B16 melanoma cell migration and invasion via endogenous S1P2 G protein-coupled receptor. Requirement of inhibition of cellular RAC activity. J Biol Chem. 2003;278:32841–32851. doi: 10.1074/jbc.M305024200. [DOI] [PubMed] [Google Scholar]
- Bektas M, Jolly PS, Muller C, Eberle J, Spiegel S, Geilen CC. Sphingosine kinase activity counteracts ceramide-mediated cell death in human melanoma cells: role of Bcl-2 expression. Oncogene. 2005a;24:178–187. doi: 10.1038/sj.onc.1208019. [DOI] [PubMed] [Google Scholar]
- Bektas M, Payne SG, Liu H, Goparaju S, Milstien S, Spiegel S. A novel acylglycerol kinase that produces lysophosphatidic acid modulates cross talk with EGFR in prostate cancer cells. J Cell Biol. 2005b;169:801–811. doi: 10.1083/jcb.200407123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beljanski V, Knaak C, Smith CD. A novel sphingosine kinase inhibitor induces autophagy in tumor cells. J Pharmacol Exp Ther. 2010;333:454–464. doi: 10.1124/jpet.109.163337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birgbauer E, Chun J. New developments in the biological functions of lysophospholipids. Cell Mol Life Sci. 2006;63:2695–2701. doi: 10.1007/s00018-006-6155-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bode C, Sensken SC, Peest U, Beutel G, Thol F, Levkau B, et al. Erythrocytes serve as a reservoir for cellular and extracellular sphingosine 1-phosphate. J Cell Biochem. 2010;109:1232–1243. doi: 10.1002/jcb.22507. [DOI] [PubMed] [Google Scholar]
- Bora NS, Kaliappan S, Jha P, Xu Q, Sohn JH, Dhaulakhandi DB, et al. Complement activation via alternative pathway is critical in the development of laser-induced choroidal neovascularization: role of factor B and factor H. J Immunol. 2006;177:1872–1878. doi: 10.4049/jimmunol.177.3.1872. [DOI] [PubMed] [Google Scholar]
- Caballero S, Swaney J, Moreno K, Afzal A, Kielczewski J, Stoller G, et al. Anti-sphingosine-1-phosphate monoclonal antibodies inhibit angiogenesis and sub-retinal fibrosis in a murine model of laser-induced choroidal neovascularization. Exp Eye Res. 2009;88:367–377. doi: 10.1016/j.exer.2008.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camerer E, Regard JB, Cornelissen I, Srinivasan Y, Duong DN, Palmer D, et al. Sphingosine-1-phosphate in the plasma compartment regulates basal and inflammation-induced vascular leak in mice. J Clin Invest. 2009;119:1871–1879. doi: 10.1172/JCI38575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chae SS, Paik JH, Furneaux H, Hla T. Requirement for sphingosine 1-phosphate receptor-1 in tumor angiogenesis demonstrated by in vivo RNA interference. J Clin Invest. 2004;114:1082–1089. doi: 10.1172/JCI22716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clair T, Aoki J, Koh E, Bandle RW, Nam SW, Ptaszynska MM, et al. Autotaxin hydrolyzes sphingosylphosphorylcholine to produce the regulator of migration, sphingosine-1-phosphate. Cancer Res. 2003;63:5446–5453. [PubMed] [Google Scholar]
- Colombaioni L, Garcia-Gil M. Sphingolipid metabolites in neural signalling and function. Brain Res Brain Res Rev. 2004;46:328–355. doi: 10.1016/j.brainresrev.2004.07.014. [DOI] [PubMed] [Google Scholar]
- Cuvillier O, Pirianov G, Kleuser B, Vanek PG, Coso OA, Gutkind S, et al. Suppression of ceramide-mediated programmed cell death by sphingosine-1-phosphate. Nature. 1996;381:800–803. doi: 10.1038/381800a0. [DOI] [PubMed] [Google Scholar]
- Cuvillier O, Ader I, Bouquerel P, Brizuela L, Malavaud B, Mazerolles C, et al. Activation of sphingosine kinase-1 in cancer: implications for therapeutic targeting. Curr Mol Pharmacol. 2010;3:53–65. doi: 10.2174/1874467211003020053. [DOI] [PubMed] [Google Scholar]
- Cyster JG. Chemokines, sphingosine-1-phosphate, and cell migration in secondary lymphoid organs. Annu Rev Immunol. 2004;23:127–159. doi: 10.1146/annurev.immunol.23.021704.115628. [DOI] [PubMed] [Google Scholar]
- Facchinetti MM, Gandini NA, Fermento ME, Sterin-Speziale NB, Ji Y, Patel V, et al. The expression of sphingosine kinase-1 in head and neck carcinoma. Cells Tissues Organs. 2010;192:314–324. doi: 10.1159/000318173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- French KJ, Schrecengost RS, Lee BD, Zhuang Y, Smith SN, Eberly JL, et al. Discovery and evaluation of inhibitors of human sphingosine kinase. Cancer Res. 2003;63:5962–5969. [PubMed] [Google Scholar]
- French KJ, Upson JJ, Keller SN, Zhuang Y, Yun JK, Smith CD. Antitumor activity of sphingosine kinase inhibitors. J Pharmacol Exp Ther. 2006;318:596–603. doi: 10.1124/jpet.106.101345. [DOI] [PubMed] [Google Scholar]
- Fukuoka Y, Medof EM. C5a receptor-mediated production of IL-8 by the human retinal pigment epithelial cell line, ARPE-19. Curr Eye Res. 2001;23:320–325. doi: 10.1076/ceyr.23.5.320.5437. [DOI] [PubMed] [Google Scholar]
- Fyrst H, Saba JD. An update on sphingosine-1-phosphate and other sphingolipid mediators. Nat Chem Biol. 2010;6:489–497. doi: 10.1038/nchembio.392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardell SE, Dubin AE, Chun J. Emerging medicinal roles for lysophospholipid signaling. Trends Mol Med. 2006;12:65–75. doi: 10.1016/j.molmed.2005.12.001. [DOI] [PubMed] [Google Scholar]
- Gellings Lowe N, Swaney JS, Moreno KM, Sabbadini RA. Sphingosine-1-phosphate and sphingosine kinase are critical for transforming growth factor-beta-stimulated collagen production by cardiac fibroblasts. Cardiovasc Res. 2009;82:303–312. doi: 10.1093/cvr/cvp056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goetzl EJ, Graeler M, Huang MC, Shankar G. Lysophospholipid growth factors and their G protein-coupled receptors in immunity, coronary artery disease, and cancer. ScientificWorldJournal. 2002;2:324–338. doi: 10.1100/tsw.2002.124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goparaju SK, Jolly PS, Watterson KR, Bektas M, Alvarez S, Sarkar S, et al. The S1P2 receptor negatively regulates platelet-derived growth factor-induced motility and proliferation. Mol Cell Biol. 2005;25:4237–4249. doi: 10.1128/MCB.25.10.4237-4249.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon MS, Just R, Rosen LS, Dorr A. A phase 1 study of sonepcizumab (S), a humanized monoclonal antibody to sphingosine-1-phosphate (S1P), in patients with advanced solid tumors. J Clin Oncol. 2010;28:219s. No 15S Part 1 of 2. [Google Scholar]
- Graler MH, Goetzl EJ. The immunosuppressant FTY720 down-regulates sphingosine 1-phosphate G-protein-coupled receptors. FASEB J. 2004;18:551–553. doi: 10.1096/fj.03-0910fje. [DOI] [PubMed] [Google Scholar]
- Gude DR, Alvarez SE, Paugh SW, Mitra P, Yu J, Griffiths R, et al. Apoptosis induces expression of sphingosine kinase 1 to release sphingosine-1-phosphate as a ‘come-and-get-me’ signal. FASEB J. 2008;22:2629–2638. doi: 10.1096/fj.08-107169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hait NC, Oskeritzian CA, Paugh SW, Milstien S, Spiegel S. Sphingosine kinases, sphingosine 1-phosphate, apoptosis and diseases. Biochim Biophys Acta. 2006;1758:2016–2026. doi: 10.1016/j.bbamem.2006.08.007. [DOI] [PubMed] [Google Scholar]
- Hait NC, Allegood J, Maceyka M, Strub GM, Harikumar KB, Singh SK, et al. Regulation of histone acetylation in the nucleus by sphingosine-1-phosphate. Science. 2009;325:1254–1257. doi: 10.1126/science.1176709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He S, Jin ML, Worpel V, Hinton DR. A role for connective tissue growth factor in the pathogenesis of choroidal neovascularization. Arch Ophthalmol. 2003;121:1283–1288. doi: 10.1001/archopht.121.9.1283. [DOI] [PubMed] [Google Scholar]
- He X, Huang CL, Schuchman EH. Quantitative analysis of sphingosine-1-phosphate by HPLC after napthalene-2,3-dicarboxaldehyde (NDA) derivatization. J Chromatogr B Analyt Technol Biomed Life Sci. 2009;877:983–990. doi: 10.1016/j.jchromb.2009.02.048. [DOI] [PubMed] [Google Scholar]
- Heier JS, Antoszyk AN, Pavan PR, Leff SR, Rosenfeld PJ, Ciulla TA, et al. Ranibizumab for treatment of neovascular age-related macular degeneration: a phase I/II multicenter, controlled, multidose study. Ophthalmology. 2006;113:633, e1–e4. doi: 10.1016/j.ophtha.2005.10.052. [DOI] [PubMed] [Google Scholar]
- Hla T. Physiological and pathological actions of sphingosine 1-phosphate. Semin Cell Dev Biol. 2004;15:513–520. doi: 10.1016/j.semcdb.2004.05.002. [DOI] [PubMed] [Google Scholar]
- Ho JW, Man K, Sun CK, Lee TK, Poon RT, Fan ST. Effects of a novel immunomodulating agent, FTY720, on tumor growth and angiogenesis in hepatocellular carcinoma. Mol Cancer Ther. 2005;4:1430–1438. doi: 10.1158/1535-7163.MCT-05-0021. [DOI] [PubMed] [Google Scholar]
- Hobson JP, Rosenfeldt HM, Barak LS, Olivera A, Poulton S, Caron MG, et al. Role of the sphingosine-1-phosphate receptor EDG-1 in PDGF-induced cell motility. Science. 2001;291:1800–1803. doi: 10.1126/science.1057559. [DOI] [PubMed] [Google Scholar]
- Igarashi J, Erwin PA, Dantas AP, Chen H, Michel T. VEGF induces S1P1 receptors in endothelial cells: implications for cross-talk between sphingolipid and growth factor receptors. Proc Natl Acad Sci USA. 2003;100:10664–10669. doi: 10.1073/pnas.1934494100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Im DS. New intercellular lipid mediators and their GPCRs: an update. Prostaglandins Other Lipid Mediat. 2009;89:53–56. doi: 10.1016/j.prostaglandins.2009.01.002. [DOI] [PubMed] [Google Scholar]
- Ishibashi T, Inomata H, Sakamoto T, Ryan SJ. Pericytes of newly formed vessels in experimental subretinal neovascularization. Arch Ophthalmol. 1995;113:227–231. doi: 10.1001/archopht.1995.01100020111041. [DOI] [PubMed] [Google Scholar]
- Johnson KR, Johnson KY, Crellin HG, Ogretmen B, Boylan AM, Harley RA, et al. Immunohistochemical distribution of sphingosine kinase 1 in normal and tumor lung tissue. J Histochem Cytochem. 2005;59:1159–1166. doi: 10.1369/jhc.4A6606.2005. [DOI] [PubMed] [Google Scholar]
- Kapitonov D, Allegood JC, Mitchell C, Hait NC, Almenara JA, Adams JK, et al. Targeting sphingosine kinase 1 inhibits Akt signaling, induces apoptosis, and suppresses growth of human glioblastoma cells and xenografts. Cancer Res. 2009;69:6915–6923. doi: 10.1158/0008-5472.CAN-09-0664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katsuma S, Ruike Y, Yano T, Kimura M, Hirasawa A, Tsujimoto G. Transcriptional regulation of connective tissue growth factor by sphingosine 1-phosphate in rat cultured mesangial cells. FEBS Lett. 2005;579:2576–2582. doi: 10.1016/j.febslet.2005.03.073. [DOI] [PubMed] [Google Scholar]
- Kawamori T, Osta W, Johnson KR, Pettus BJ, Bielawski J, Tanaka T, et al. Sphingosine kinase 1 is up-regulated in colon carcinogenesis. FASEB J. 2006;20:386–388. doi: 10.1096/fj.05-4331fje. [DOI] [PubMed] [Google Scholar]
- Keller CD, Rivera Gil P, Tolle M, van der Giet M, Chun J, Radeke HH, et al. Immunomodulator FTY720 induces myofibroblast differentiation via the lysophospholipid receptor S1P3 and Smad3 signaling. Am J Pathol. 2007;170:281–292. doi: 10.2353/ajpath.2007.060485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamontagne K, Littlewood-Evans A, Schnell C, O'Reilly T, Wyder L, Sanchez T, et al. Antagonism of sphingosine-1-phosphate receptors by FTY720 inhibits angiogenesis and tumor vascularization. Cancer Res. 2006;66:221–231. doi: 10.1158/0008-5472.CAN-05-2001. [DOI] [PubMed] [Google Scholar]
- Langlois S, Gingras D, Beliveau R. Membrane type 1-matrix metalloproteinase (MT1-MMP) cooperates with sphingosine 1-phosphate to induce endothelial cell migration and morphogenic differentiation. Blood. 2004;103:3020–3028. doi: 10.1182/blood-2003-08-2968. [DOI] [PubMed] [Google Scholar]
- Lee MJ, Thangada S, Claffey KP, Ancellin N, Liu CH, Kluk M, et al. Vascular endothelial cell adherens junction assembly and morphogenesis induced by sphingosine-1-phosphate. Cell. 1999a;99:301–312. doi: 10.1016/s0092-8674(00)81661-x. [DOI] [PubMed] [Google Scholar]
- Lee OH, Kim YM, Lee YM, Moon EJ, Lee DJ, Kim JH, et al. Sphingosine 1-phosphate induces angiogenesis: its angiogenic action and signaling mechanism in human umbilical vein endothelial cells. Biochem Biophys Res Commun. 1999b;264:743–750. doi: 10.1006/bbrc.1999.1586. [DOI] [PubMed] [Google Scholar]
- Lepley D, Paik JH, Hla T, Ferrer F. The G protein-coupled receptor S1P2 regulates Rho/Rho kinase pathway to inhibit tumor cell migration. Cancer Res. 2005;65:3788–3795. doi: 10.1158/0008-5472.CAN-04-2311. [DOI] [PubMed] [Google Scholar]
- Li JY, Wang H, May S, Song X, Fueyo J, Fuller GN. Constitutive activation of c-Jun N-terminal kinase correlates with histologic grade and EGFR expression in diffuse gliomas. J Neurooncol. 2008a;88:11–17. doi: 10.1007/s11060-008-9529-1. [DOI] [PubMed] [Google Scholar]
- Li X, Stankovic M, Bonder CS, Hahn CN, Parsons M, Pitson SM, et al. Basal and angiopoietin-1-mediated endothelial permeability is regulated by sphingosine kinase-1. Blood. 2008b;111:3489–3497. doi: 10.1182/blood-2007-05-092148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Yu CP, Xia JT, Zhang L, Weng GX, Zheng HQ, et al. Sphingosine kinase 1 is associated with gastric cancer progression and poor survival of patients. Clin Cancer Res. 2009;15:1393–1399. doi: 10.1158/1078-0432.CCR-08-1158. [DOI] [PubMed] [Google Scholar]
- Liu Q, Zhao X, Frissora F, Ma Y, Santhanam R, Jarjoura D, et al. FTY720 demonstrates promising preclinical activity for chronic lymphocytic leukemia and lymphoblastic leukemia/lymphoma. Blood. 2008;111:275–284. doi: 10.1182/blood-2006-10-053884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long JS, Edwards J, Watson C, Tovey S, Mair KM, Schiff R, et al. Sphingosine kinase 1 induces tolerance to human epidermal growth factor receptor 2 and prevents formation of a migratory phenotype in response to sphingosine 1-phosphate in estrogen receptor-positive breast cancer cells. Mol Cell Biol. 2010;30:3827–3841. doi: 10.1128/MCB.01133-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucas da Silva LB, Ribeiro DA, Cury PM, Cordeiro JA, Bueno V. FTY720 treatment in experimentally urethane-induced lung tumors. J Exp Ther Oncol. 2008;7:9–15. [PubMed] [Google Scholar]
- Lucke S, Levkau B. Endothelial functions of sphingosine-1-phosphate. Cell Physiol Biochem. 2010;26:87–96. doi: 10.1159/000315109. [DOI] [PubMed] [Google Scholar]
- Maceyka M, Payne SG, Milstien S, Spiegel S. Sphingosine kinase, sphingosine-1-phosphate, and apoptosis. Biochim Biophys Acta. 2002;1585:193–201. doi: 10.1016/s1388-1981(02)00341-4. [DOI] [PubMed] [Google Scholar]
- Maceyka M, Milstien S, Spiegel S. Sphingosine kinases, sphingosine-1-phosphate and sphingolipidomics. Prostaglandins Other Lipid Mediat. 2005;77:15–22. doi: 10.1016/j.prostaglandins.2004.09.010. [DOI] [PubMed] [Google Scholar]
- Maines LW, French KJ, Wolpert EB, Antonetti DA, Smith CD. Pharmacologic manipulation of sphingosine kinase in retinal endothelial cells: implications for angiogenic ocular diseases. Invest Ophthalmol Vis Sci. 2006;47:5022–5031. doi: 10.1167/iovs.05-1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melendez AJ, Ibrahim FB. Antisense knockdown of sphingosine kinase 1 in human macrophages inhibits C5a receptor-dependent signal transduction, Ca2+ signals, enzyme release, cytokine production, and chemotaxis. J Immunol. 2004;173:1596–1603. doi: 10.4049/jimmunol.173.3.1596. [DOI] [PubMed] [Google Scholar]
- Melendez AJ, Carlos-Dias E, Gosink M, Allen JM, Takacs L. Human sphingosine kinase: molecular cloning, functional characterization and tissue distribution. Gene. 2000;251:19–26. doi: 10.1016/s0378-1119(00)00205-5. [DOI] [PubMed] [Google Scholar]
- Michaud J, Im DS, Hla T. Inhibitory role of sphingosine 1-phosphate receptor 2 in macrophage recruitment during inflammation. J Immunol. 2010;184:1475–1483. doi: 10.4049/jimmunol.0901586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milstien S, Spiegel S. Targeting sphingosine-1-phosphate: a novel avenue for cancer therapeutics. Cancer Cell. 2006;9:148–150. doi: 10.1016/j.ccr.2006.02.025. [DOI] [PubMed] [Google Scholar]
- Moolenaar WH. Bioactive lysophospholipids and their G protein-coupled receptors. Exp Cell Res. 1999;253:230–238. doi: 10.1006/excr.1999.4702. [DOI] [PubMed] [Google Scholar]
- Murata N, Sato K, Kon J, Tomura H, Okajima F. Quantitative measurement of sphingosine 1-phosphate by radioreceptor-binding assay. Anal Biochem. 2000a;282:115–120. doi: 10.1006/abio.2000.4580. [DOI] [PubMed] [Google Scholar]
- Murata N, Sato K, Kon J, Tomura H, Yanagita M, Kuwabara A, et al. Interaction of sphingosine 1-phosphate with plasma components, including lipoproteins, regulates the lipid receptor-mediated actions. Biochem J. 2000b;352(Pt 3):809–815. [PMC free article] [PubMed] [Google Scholar]
- Neviani P, Santhanam R, Oaks JJ, Eiring AM, Notari M, Blaser BW, et al. FTY720, a new alternative for treating blast crisis chronic myelogenous leukemia and Philadelphia chromosome-positive acute lymphocytic leukemia. J Clin Invest. 2007;117:2408–2421. doi: 10.1172/JCI31095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Brien N, Jones ST, Williams DG, Cunningham HB, Moreno K, Visentin B, et al. Production and characterization of monoclonal anti-sphingosine-1-phosphate antibodies. J Lipid Res. 2009;50:2245–2257. doi: 10.1194/jlr.M900048-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olivera A, Rivera J. Sphingolipids and the balancing of immune cell function: lessons from the mast cell. J Immunol. 2005;174:1153–1158. doi: 10.4049/jimmunol.174.3.1153. [DOI] [PubMed] [Google Scholar]
- Olivera A, Kohama T, Edsall L, Nava V, Cuvillier O, Poulton S, et al. Sphingosine kinase expression increases intracellular sphingosine-1-phosphate and promotes cell growth and survival. J Cell Biol. 1999;147:545–558. doi: 10.1083/jcb.147.3.545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paik JH, Chae S, Lee MJ, Thangada S, Hla T. Sphingosine 1-phosphate-induced endothelial cell migration requires the expression of EDG-1 and EDG-3 receptors and Rho-dependent activation of alpha vbeta3- and beta1-containing integrins. J Biol Chem. 2001;276:11830–11837. doi: 10.1074/jbc.M009422200. [DOI] [PubMed] [Google Scholar]
- Paik JH, Skoura A, Chae SS, Cowan AE, Han DK, Proia RL, et al. Sphingosine 1-phosphate receptor regulation of N-cadherin mediates vascular stabilization. Genes Dev. 2004;18:2392–2403. doi: 10.1101/gad.1227804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paugh SW, Paugh BS, Rahmani M, Kapitonov D, Almenara JA, Kordula T, et al. A selective sphingosine kinase 1 inhibitor integrates multiple molecular therapeutic targets in human leukemia. Blood. 2008;112:1382–1391. doi: 10.1182/blood-2008-02-138958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pchejetski D, Doumerc N, Golzio M, Naymark M, Teissie J, Kohama T, et al. Chemosensitizing effects of sphingosine kinase-1 inhibition in prostate cancer cell and animal models. Mol Cancer Ther. 2008;7:1836–1845. doi: 10.1158/1535-7163.MCT-07-2322. [DOI] [PubMed] [Google Scholar]
- Petrukhin K. New therapeutic targets in atrophic age-related macular degeneration. Expert Opin Ther Targets. 2007;11:625–639. doi: 10.1517/14728222.11.5.625. [DOI] [PubMed] [Google Scholar]
- Puneet P, Yap CT, Wong L, Lam Y, Koh DR, Moochhala S, et al. SphK1 regulates proinflammatory responses associated with endotoxin and polymicrobial sepsis. Science. 2010;328:1290–1294. doi: 10.1126/science.1188635. [DOI] [PubMed] [Google Scholar]
- Pushparaj PN, H'Ng SC, Melendez AJ. Refining siRNA in vivo transfection: silencing SPHK1 reveals its key role in C5a-induced inflammation in vivo. Int J Biochem Cell Biol. 2008;40:1817–1825. doi: 10.1016/j.biocel.2008.01.015. [DOI] [PubMed] [Google Scholar]
- Pyne NJ, Pyne S. Sphingosine 1-phosphate and cancer. Nat Rev Cancer. 2010;10:489–503. doi: 10.1038/nrc2875. [DOI] [PubMed] [Google Scholar]
- Radeff-Huang J, Seasholtz TM, Matteo RG, Brown JH. G protein mediated signaling pathways in lysophospholipid induced cell proliferation and survival. J Cell Biochem. 2004;92:949–966. doi: 10.1002/jcb.20094. [DOI] [PubMed] [Google Scholar]
- Raguz S, Yague E. Resistance to chemotherapy: new treatments and novel insights into an old problem. Br J Cancer. 2008;99:387–391. doi: 10.1038/sj.bjc.6604510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohrer B, Long Q, Coughlin B, Wilson RB, Huang Y, Qiao F, et al. A targeted inhibitor of the alternative complement pathway reduces angiogenesis in a mouse model of age-related macular degeneration. Invest Ophthalmol Vis Sci. 2009;50:3056–3064. doi: 10.1167/iovs.08-2222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruckhaberle E, Rody A, Engels K, Gaetje R, von Minckwitz G, Schiffmann S, et al. Microarray analysis of altered sphingolipid metabolism reveals prognostic significance of sphingosine kinase 1 in breast cancer. Breast Cancer Res Treat. 2008;112:41–52. doi: 10.1007/s10549-007-9836-9. [DOI] [PubMed] [Google Scholar]
- Sabbadini RA. Targeting sphingosine-1-phosphate for cancer therapy. Br J Cancer. 2006;95:1131–1135. doi: 10.1038/sj.bjc.6603400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakurai E, Anand A, Ambati BK, van Rooijen N, Ambati J. Macrophage depletion inhibits experimental choroidal neovascularization. Invest Ophthalmol Vis Sci. 2003;44:3578–3585. doi: 10.1167/iovs.03-0097. [DOI] [PubMed] [Google Scholar]
- Sanchez T, Hla T. Structural and functional characteristics of S1P receptors. J Cell Biochem. 2004;92:913–922. doi: 10.1002/jcb.20127. [DOI] [PubMed] [Google Scholar]
- Sanchez T, Estrada-Hernandez T, Paik JH, Wu MT, Venkataraman K, Brinkmann V, et al. Phosphorylation and action of the immunomodulator FTY720 inhibits vascular endothelial cell growth factor-induced vascular permeability. J Biol Chem. 2003;278:47281–47290. doi: 10.1074/jbc.M306896200. [DOI] [PubMed] [Google Scholar]
- Schmid G, Guba M, Papyan A, Ischenko I, Bruckel M, Bruns CJ, et al. FTY720 inhibits tumor growth and angiogenesis. Transplant Proc. 2005;37:110–111. doi: 10.1016/j.transproceed.2004.12.278. [DOI] [PubMed] [Google Scholar]
- Shida D, Fang X, Kordula T, Takabe K, Lepine S, Alvarez SE, et al. Cross-talk between LPA1 and epidermal growth factor receptors mediates up-regulation of sphingosine kinase 1 to promote gastric cancer cell motility and invasion. Cancer Res. 2008a;68:6569–6577. doi: 10.1158/0008-5472.CAN-08-0411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shida D, Takabe K, Kapitonov D, Milstien S, Spiegel S. Targeting SphK1 as a new strategy against cancer. Curr Drug Targets. 2008b;9:662–673. doi: 10.2174/138945008785132402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucas da Silva LB, Ribeiro DA, Cury PM, Cordeiro JA, Bueno V. FTY720 treatment in experimentally urethane-induced lung tumors. J Exp Ther Oncol. 2008;7:9–15. [PubMed] [Google Scholar]
- Skoura A, Hla T. Regulation of vascular physiology and pathology by the S1P2 receptor subtype. Cardiovasc Res. 2009;82:221–228. doi: 10.1093/cvr/cvp088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skoura A, Sanchez T, Claffey K, Mandala SM, Proia RL, Hla T. Essential role of sphingosine 1-phosphate receptor 2 in pathological angiogenesis of the mouse retina. J Clin Invest. 2007;117:2506–2516. doi: 10.1172/JCI31123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spiegel S, Milstien S. Sphingosine-1-phosphate: an enigatic signalling lipid. Mol Cell Biol. 2003;4:397–407. doi: 10.1038/nrm1103. [DOI] [PubMed] [Google Scholar]
- Stamer WD, Read AT, Sumida GM, Ethier CR. Sphingosine-1-phosphate effects on the inner wall of Schlemm's canal and outflow facility in perfused human eyes. Exp Eye Res. 2009;89:980–988. doi: 10.1016/j.exer.2009.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoller GL, Lapierre-Holme F, Peterkin J, Garland W, Sabbadini R. iSONEP™, an anti-sphingosine-1-phosphate (Anti-S1P) monoclonal antibody for investigation in exudative AMD: results from a phase 1 prospective open-label dose-escalating multi-center study. Invest Ophthalmol Vis Sci. 2010;51 E-Abstract 1253. [Google Scholar]
- Swaney JS, Moreno KM, Gentile AM, Sabbadini RA, Stoller GL. Sphingosine-1-phosphate (S1P) is a novel fibrotic mediator in the eye. Exp Eye Res. 2008;87:367–375. doi: 10.1016/j.exer.2008.07.005. [DOI] [PubMed] [Google Scholar]
- Taha TA, Hannun YA, Obeid LM. Sphingosine kinase: biochemical and cellular regulation and role in disease. J Biochem Mol Biol. 2006;39:113–131. doi: 10.5483/bmbrep.2006.39.2.113. [DOI] [PubMed] [Google Scholar]
- Takuwa Y. Subtype-specific differential regulation of Rho family G proteins and cell migration by the Edg family sphingosine-1-phosphate receptors. Biochim Biophys Acta. 2002;1582:112–120. doi: 10.1016/s1388-1981(02)00145-2. [DOI] [PubMed] [Google Scholar]
- Takuwa N, Ohkura S, Takashima S, Ohtani K, Okamoto Y, Tanaka T, et al. S1P3-mediated cardiac fibrosis in sphingosine kinase 1 transgenic mice involves reactive oxygen species. Cardiovasc Res. 2009;85:484–493. doi: 10.1093/cvr/cvp312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanimoto T, Jin ZG, Berk BC. Transactivation of vascular endothelial growth factor (VEGF) receptor Flk-1/KDR is involved in sphingosine 1-phosphate-stimulated phosphorylation of Akt and endothelial nitric-oxide synthase (eNOS) J Biol Chem. 2002;277:42997–43001. doi: 10.1074/jbc.M204764200. [DOI] [PubMed] [Google Scholar]
- Teicher B. Antiangiogenic agents and targets: a perspective. Biochem Pharmacol. 2011;81:6–12. doi: 10.1016/j.bcp.2010.09.023. [DOI] [PubMed] [Google Scholar]
- Tsutsumi C, Sonoda KH, Egashira K, Qiao H, Hisatomi T, Nakao S, et al. The critical role of ocular-infiltrating macrophages in the development of choroidal neovascularization. J Leukoc Biol. 2003;74:25–32. doi: 10.1189/jlb.0902436. [DOI] [PubMed] [Google Scholar]
- Van Brocklyn JR, Jackson CA, Pearl DK, Kotur MS, Snyder PJ, Prior TW. Sphingosine kinase-1 expression correlates with poor survival of patients with glioblastoma multiforme: roles of sphingosine kinase isoforms in growth of glioblastoma cell lines. J Neuropathol Exp Neurol. 2005;64:695–705. doi: 10.1097/01.jnen.0000175329.59092.2c. [DOI] [PubMed] [Google Scholar]
- Vinores SA, Seo MS, Okamoto N, Ash JD, Wawrousek EF, Xiao WH, et al. Experimental models of growth factor-mediated angiogenesis and blood-retinal barrier breakdown. Gen Pharmacol. 2000;35:233–239. doi: 10.1016/s0306-3623(01)00117-3. [DOI] [PubMed] [Google Scholar]
- Visentin B, Vekich JA, Sibbald BJ, Cavalli AL, Moreno KM, Matteo RG, et al. Validation of an anti-sphingosine-1-phosphate antibody as a potential therapeutic in reducing growth, invasion, and angiogenesis in multiple tumor lineages. Cancer Cell. 2006;9:225–238. doi: 10.1016/j.ccr.2006.02.023. [DOI] [PubMed] [Google Scholar]
- Vlasenko LP, Melendez AJ. A critical role for sphingosine kinase in anaphylatoxin-induced neutropenia, peritonitis, and cytokine production in vivo. J Immunol. 2005;174:6456–6461. doi: 10.4049/jimmunol.174.10.6456. [DOI] [PubMed] [Google Scholar]
- Weigert A, Weis N, Brune B. Regulation of macrophage function by sphingosine-1-phosphate. Immunobiology. 2009;214:748–760. doi: 10.1016/j.imbio.2009.06.003. [DOI] [PubMed] [Google Scholar]
- Wojciak JM, Zhu N, Schuerenberg KT, Moreno K, Shestowsky WS, Hiraiwa M, et al. The crystal structure of sphingosine-1-phosphate in complex with a Fab fragment reveals metal bridging of an antibody and its antigen. Proc Natl Acad Sci USA. 2009;106:17717–17722. doi: 10.1073/pnas.0906153106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia P, Gamble J, Wang L, Pitson SM, Moretti PA, Wattenberg BW, et al. An oncogenic role of sphingosine kinase. Curr Biol. 2000;10:1527–1530. doi: 10.1016/s0960-9822(00)00834-4. [DOI] [PubMed] [Google Scholar]
- Xie B, Shen J, Dong A, Rashid A, Stoller G, Campochiaro PA. Blockade of sphingosine-1-phosphate reduces macrophage influx and retinal and choroidal neovascularization. J Cell Physiol. 2009;218:192–198. doi: 10.1002/jcp.21588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xin C, Ren S, Kleuser B, Shabahang S, Eberhardt W, Radeke H, et al. Sphingosine 1-phosphate cross-activates the Smad signaling cascade and mimics transforming growth factor-beta-induced cell responses. J Biol Chem. 2004;279:35255–35262. doi: 10.1074/jbc.M312091200. [DOI] [PubMed] [Google Scholar]
- Yamagishi S, Imaizumi T. Pericyte biology and diseases. Int J Tissue React. 2005;27:125–135. [PubMed] [Google Scholar]
- Yamanaka M, Shegogue D, Pei H, Bu S, Bielawska A, Bielawski J, et al. Sphingosine kinase 1 (SPHK1) is induced by transforming growth factor-beta and mediates TIMP-1 up-regulation. J Biol Chem. 2004;279:53994–54001. doi: 10.1074/jbc.M410144200. [DOI] [PubMed] [Google Scholar]
- Yasui K, Palade PT. Sphingolipid action on sodium and calcium currents of rat ventricular myocytes. Am J Physiol. 1995;270:C645–C649. doi: 10.1152/ajpcell.1996.270.2.C645. [DOI] [PubMed] [Google Scholar]
- Yatomi Y. Sphingosine 1-phosphate in vascular biology: possible therapeutic strategies to control vascular diseases. Curr Pharm Des. 2006;12:575–587. doi: 10.2174/138161206775474404. [DOI] [PubMed] [Google Scholar]
- Yatomi Y, Ohmori T, Rile G, Kazama F, Okamoto H, Sano T, et al. Sphingosine 1-phosphate as a major bioactive lysophospholipid that is released from platelets and interacts with endothelial cells. Blood. 2000;96:3431–3438. [PubMed] [Google Scholar]
- Zhu D, Sreekumar PG, Hinton DR, Kannan R. Expression and regulation of enzymes in the ceramide metabolic pathway in human retinal pigment epithelial cells and their relevance to retinal degeneration. Vision Res. 2009;50:643–651. doi: 10.1016/j.visres.2009.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.