

Abstract
Positive genetic toxicity data suggest carcinogenic hazard, and this can stop a candidate pharmaceutical reaching the clinic. However, during the last decade, it has become clear that many non-carcinogens produce misleading positive results in one or other of the regulatory genotoxicity assays. These doubtful conclusions cost a lot of time and money, as they trigger additional testing of apparently genotoxic candidates, both in vitro and in animals, to discover whether the suggested hazard is genuine. This in turn means that clinical trials can be put on hold. This review describes the current approaches to the ‘misleading positive’ problem as well as efforts to reduce the use of animals in genotoxicity assessment. The following issues are then addressed: the application of genotoxicity testing screens earlier in development; the search for new or improved in vitro genotoxicity tests; proposed changes to the International Committee on Harmonisation guidance on genotoxicity testing [S2(R1)]. Together, developments in all these areas offer good prospects of a more rapid and cost-effective way to understand genetic toxicity concerns.
Keywords: genotoxicity testing, safety assessment, ICH S2, screening, GADD45a
Chemical carcinogenicity and genetic toxicology
Children working as chimney sweeps often died from cancers. It was subsequently discovered that a chemical in soot, benzo[a]pyrene, causes animal cancers, as well as mutations in bacteria (reviewed by Phillips, 1983). Thus, the concept of chemical carcinogenicity and the discipline of genetic toxicology were born. Difficulties soon emerged. First, it was recognized that not all animal carcinogens are bacterial mutagens (McCann et al., 1975). This was thought to reflect the differences between animals and bacteria, including xenobiotic metabolism in the liver and other organs. So, mammalian cell lines were developed, and liver extracts were included in test protocols. This combination of bacterial and mammalian cell tests produces the ‘desired’ positive results for about nine out of 10 carcinogens (Kirkland et al., 2005). This is not, however, as good a result as it seems, as this same combination also produces positive results for as many as nine out of 10 non-carcinogens (Kirkland et al., 2005). Finally, it was recognized that many carcinogens do not actually have a genotoxic mode of action (Shaw and Jones, 1994). Thus, the unanticipated consequence of a focus on sensitivity during genotoxicity assay development was that many of the compounds now producing positive results are either not carcinogens, or carcinogens that do not have a genotoxic mode of action. This means that there are many compounds carrying positive genotoxicity information that is not relevant to the assessment of cancer risk. Some of these are potentially useful pharmaceuticals. It is therefore important to distinguish the true positives from the false, or more accurately misleading, positives.
For the purposes of this review, a genotoxic mechanism of carcinogenicity is one in which a chemical triggers an event, or in the short term a series of events, which can lead to a permanent change in the genome. Such events include direct damage to DNA, as well damage to or interference with the enzymes and systems required for DNA replication, repair and chromosome segregation. The repair of such damage can be part of the process which fixes permanent change. Such changes might or might not be lethal at the cellular level. While carcinogenicity is the exemplar of genotoxic hazard, there can be other equally serious health consequences of genome alteration, including reproductive impairment, developmental anomalies and genetic diseases. For brevity, these are not considered further here.
The human genome comprises 46 recognizable chromosomes, 23 from each parent, and with the possible exception of identical twins, each person's genome is unique. At a gross level, genome change might be identified as a change in chromosome number (aneugenesis) or other large chromosome rearrangements (karyotype changes), or a change in the sequence of a particular gene (the ‘genotype’). The former can be recognized by microscopic examination, as can the identification of ‘chromosome aberrations’ (abbreviated CA). The latter can be recognized by phenotypic examination, and the identification of mutants. Bacterial test systems detect mutations whereas eukaryotic test systems detect mutations and CA.
The definition of genotoxic carcinogenicity used here is deliberately precise. Every round of genome replication that precedes cell division is subject to the statistically unavoidable introduction of mutations: the high-fidelity DNA replication process has a net error rate of less than 1 in a million base pairs, but the genome has around three thousand million base pairs. Thus, chemicals that allow cells to escape the normal restrictions on cell division inevitably increase the chance of mutations arising in an individual. Such chemicals might therefore be carcinogenic by a non-genotoxic mechanism. For example, they might cause epigenetic changes (LeBarona et al., 2010), where chromatin remodelling, rather than mutation, leads to a loss of restriction of cell division. In this case, new mutations arise as a consequence of tumorigenicity rather than its cause. ‘Non-genotoxic’ carcinogenicity is not discussed further in this review, although interested readers are directed elsewhere (Hernández et al., 2009). There is currently no simple in vitro method that distinguishes genotoxic from non-genotoxic carcinogens. It should be noted that even with animal data available, the distinction between genotoxic and non-genotoxic carcinogens is neither readily made nor universally agreed. Indeed, such classifications require the review of available data and complex weight of evidence arguments by panels of experts.
It is clear that there are highly evolved, complex cellular systems which detect genome damage. These can both modulate the activity of repair enzymes and adjust cell cycle timings to ensure that repair is completed before mitosis. If repair capacity is exceeded, cell suicide (apoptosis) and/or necrosis mechanisms are triggered. In animals, this functionally altruistic response at the cellular level also protects against cancers. This coordinated response to DNA damage is regulated at the level of gene expression, as well as post-translationally, but it is the former mechanism that is being exploited by a new generation of assays to predict genotoxicity from the transcriptional response to genome damage. These are discussed in a later section.
Misleading in vitro data
A carcinogen for which observed tumorigenicity is clearly due to a genotoxic mode of action is classified as a ‘genotoxic carcinogen’. A carcinogen for which tumorigenicity is not due to a recognizably genotoxic mode of action is classified as a non-genotoxic carcinogen. Positive in vitro genotoxicity data produced from either non-genotoxic carcinogens or non-carcinogens are therefore irrelevant to the development of tumours in animals. These data are sometimes called ‘false’, although if reproducible they are of course true for the in vitro test concerned, so some authors prefer to use the terms ‘misleading’ or ‘irrelevant’ (Kirkland et al., 2007).
How frequently are misleading genotoxicity data produced? The first systematic study to provide an answer to this came from a review of the 1999 Physicians' Desk Reference. Snyder and Green (2001) reported that about 50% of non-carcinogenic marketed pharmaceuticals had positive results in at least one of the regulatory in vitro genotoxicity tests. This figure is all the more remarkable because chemotherapeutics and nucleoside analogues, which are often genotoxic by design, were excluded from their analysis. A more recent report from Brambilla and Martelli (2009) reviewed data from 838 marketed pharmaceuticals. The majority had been licensed before standardizing of testing guidelines. However, of 315 compounds with some genotoxicity and carcinogenicity data, 50 of 166 non-carcinogens (30%) had positive genotoxicity data from at least one genotoxicity assay. With this high prevalence of misleading in vitro results, it is clear that either a great deal of work is being expended mitigating genetic toxicity problems, or in some cases potentially useful pharmaceuticals might be needlessly discarded.
All in vitro assays produce misleading positive results, but some produce more than others. The performance of in vitro genotoxicity assays is commonly described in terms of sensitivity and specificity (Cooper et al., 1979). Sensitivity is the proportion of genotoxic carcinogens that produces positive results in a given test, and specificity is the proportion of non-carcinogens (and ideally non-genotoxic carcinogens) that produces negative results. A low specificity figure therefore identifies a test that produces a high proportion of misleading positive results. The accuracy of both sensitivity and specificity figures depends on the reliability with which the mechanism of carcinogenicity within the carcinogen test set can be defined, but this is not a simple task. The actual figures can also depend on the way in which the in vitro test was performed. However, with those important caveats, Table 1 lists some reported figures for a selection of regulatory and non-regulatory in vitro genotoxicity assays.
Table 1.
Published performance parameters for a selection of in vitro genotoxicity assays
| Test name | Sensitivity (%) | Specificity (%) | References |
|---|---|---|---|
| 1.Regulatory | |||
| Bacterial reversion (Ames) | 60 | 77 | Kirkland et al., 2005 |
| Chromosome aberrations | 70 | 55 | Kirkland et al., 2005 |
| Mammalian mutation | 81 | 48 | Kirkland et al., 2005 |
| 2.Screening | |||
| Bacterial | |||
| SOS Umu C | 62 | 72 | Reifferscheid and Heil, 1996 |
| Ames MPF | 58 | 63a | Kamber et al., 2009 |
| Yeast | |||
| RAD54-GFP | 39 | 82 | Knight et al., 2007 |
| DEL | 86 | 80a | Brennan and Schiestl, 2004 |
| Mammalian | |||
| MNT | 81 | 54 | Kirkland et al., 2005 |
| GADD45a-GFPb | 87 | 95 | Hastwell et al., 2009 |
Limited dataset of 10 compounds.
GADD45a-GFP data refer specifically to genotoxic carcinogens.
GFP, green fluorescent protein; MNT, micronucleus test.
It is clear that test performance varies quite widely. Within the regulatory battery, the Ames test exhibits reasonable specificity, but poor sensitivity. The mammalian cell assays have better sensitivity, but poorer specificity. This reflects the observation that it is the latter which produce the majority of misleading positive results. Many new screening tests have been described, but the examples chosen here have been selected on the basis of wide use beyond the originating laboratory. They include tests developed to reproduce the results of the regulatory in vitro assays, and new tests developed to predict genotoxic carcinogenicity, largely genome damage response biomarker reporter assays. These are described and discussed in a later section.
The regulatory in vitro tests are briefly described later in this review and well described elsewhere. The reader is referred to regulatory guideline documents for further information: International Committee on Harmonisation (ICH) S2B (ICH, 1997) and the proposed, ICH S2(R1) (ICH, 2010; summarized in Table 2). While this review is principally concerned with in vitro testing, it should be noted that the in vivo tests produce their own anomalies. There are examples of compounds, including urethane and benzene, which generate negative or weak in vitro genotoxicity data that produce positive results in in vivo studies. There can be a variety of reasons for this, including the specific metabolism of compounds in the rodent species and pharmacological interference in metabolism (Tweats et al., 2007a). Compound-induced hypo- or hyperthermia in rodents can also lead to increases in micronucleated cells in the bone marrow (Tweats et al., 2007b).
Table 2.
An outline summary of the current ICH guidelines for the genotoxicity testing of pharmaceuticals compared with the proposed two-option revised guideline, ICH S2(R1)
| ICH S2 guidelines (A&B) | ICH S2(R1) – proposed revisions to S2 | |
|---|---|---|
| Current | Option 1 | Option 2 |
| Bacterial mutation (Ames) | Bacterial mutation (Ames) | Bacterial mutation (Ames) |
| With repeat test | One full test | One full test |
| In vitro mammalian cell test (to 10 mM) | In vitro mammalian cell test (to 1 mM) | |
| CA | CA | No requirement |
| or | or | |
| Mutation (e.g. tk) | Mutation (e.g. tk) | |
| or | ||
| MNT | ||
| In vivo cytogenetic assay | In vivo cytogenetic assay | In vivo assay |
| Integrated into 28-day acute toxicity study where possible | Two endpoints, integrated into 28-day acute toxicity study where possible | |
CA, chromosome aberrations; ICH, International Committee on Harmonisation; MNT, micronucleus test.
The application of genotoxicity testing early in development
How often do positive in vitro genotoxicity results arise in a pharmaceutical collection? Ignoring for the moment that many of these will be misleading positives, the answer to this question is probably between 10% and 15%. This is based on testing results from the collections at Pfizer (Aubrecht et al., 2007) and GlaxoSmithKline (GSK) (D. Tweats, pers. comm.). These large companies have been active in all therapeutic areas, so this range is probably representative. It is clear, however, that some pharmacologies are more likely to produce positive results than others, although detailed data from proprietary collections are, unsurprisingly, difficult to find. With due apologies for anecdote, there seems to be some consensus that neurological/psychiatric/central nervous system discovery programmes are particularly prone to producing genotoxic candidates. This may relate to specific core chemistries used in neuroscience, such as the tricyclic structure in many antidepressants which is hypothesized to cause damage through non-covalent interaction with DNA (Snyder et al., 2006).
The Good Laboratory Practice (GLP) format genotoxicity assays prescribed by the ICH guidelines (S2B; ICH, 1997) require relatively large (gram) quantities of compound, and are not simple to perform. As a consequence of this, their application has historically been part of the preclinical safety assessment phase of drug development (see Figure 1). Typically, such testing is restricted to a lead candidate and a back-up from the same chemical series. An additional candidate from a different chemical series might also be included. The bacterial mutation assay (typically the Ames test/Salmonella typhimurium reversion) and an in vitro mammalian assay for either CA (clastogenicity and aneugenesis) or mutation are followed by an in vivo genotoxicity assessment (generally CA or micronucleus induction). If positive in vitro data are produced, then additional in vivo testing is required, and in some circumstances this also triggers a 2-year rodent carcinogenicity study. Thus, positive in vitro data can lead to a lot of extra work, or terminate a programme, at a stage when the choice of compounds has become very limited. In either case, a lot of money has been spent, and a lot more can be spent. These issues have driven the interest in earlier testing, which initially focused on the development of ‘cut-down’ versions of the regulatory safety assessment stage tests. The notion behind this focus was to obtain an earlier preview of the regulatory tests outside the onerous GLP testing environment and with much lower compound usage. They were applied to the slightly larger and chemically diverse group of compounds from which the candidates were selected (‘lead or candidate selection’). In theory, this increases the likelihood of finding candidates without genotoxic liability. However, the best time to find liabilities is when there is still active chemistry in progress on the target, as this allows chemists to actively search for modifications that remove the genotoxic liability while maintaining the useful pharmacology.
Figure 1.
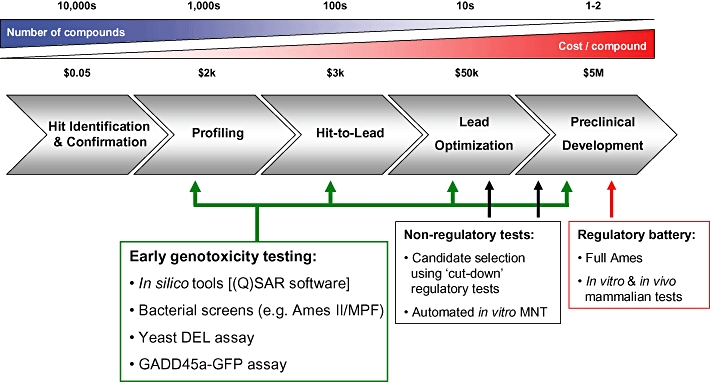
A cartoon of the drug discovery ‘pipeline’ and the relative stages at which various genotoxicity assays are or can be utilized. The indicative shaded bars above the pipeline represent (L–R) decreasing numbers of compounds and increasing cost per compound through the stages of the process. GFP, green fluorescent protein; MNT, micronucleus test.
The application of in vitro genotoxicity screening at pre-candidate selection at GSK has reduced the incidence of positive genotoxicity data in GLP testing to around 4% (P. Hastwell, pers. comm.). The figure is probably not zero for two reasons. First, screening is usually carried out at lower concentrations (sub-millimolar) than the ICH regulatory requirement of 10 mM (or 5 mg·mL−1 or 5 mg per plate; S2A; ICH, 1995). The testing concentration constraint encountered during the discovery phase is caused by the limited amount of compound available. The second reason is that there are compounds that only produce positive results in vivo. Overall, however, reducing attrition from 15% to 4% represents a significant efficiency saving. Screening out an in vitro genotoxic liability does not of course get rid of the misleading positive problem itself and inevitably limits the chemistry in lead compounds to those that produce neither true nor misleading positives. Of course, the latter might be useful drugs, so it is an expedient, but still a wasteful approach. It would be better to screen with tests that did not produce too many misleading positives. The next few paragraphs describe progress in that endeavour.
Improving in vitro genotoxicity testing
The European Centre for the Validation of Alternative Methods (ECVAM) sponsored a workshop in 2006 (Kirkland et al., 2007), to ‘discuss whether there exist cells and test systems that have a reduced tendency to false positive results, to review potential modifications to existing protocols and cell systems that might result in improved specificity, and to review the performance of some new test systems that show promise of improved specificity without sacrificing sensitivity’. Their findings and proposals are summarized below. Some of these are now integrated into proposed new testing guidance.
Cell lines and p53 status
The commonly used cell lines in in vitro genetic toxicology are often deficient in DNA repair, p53 function or metabolic competency, and many derive from malignancies in rodents. As noted in the introduction, chemical interference with systems evolved to protect the genome can be genotoxic, so one might anticipate that cell lines with mutations which affect those systems would be made particularly sensitive to challenges that might normally be met without adverse effect. P53 mutations are common in cell lines, not least because these mutations often arise during the development of malignancy, and it is from such malignancies that many cell lines were established. P53 has been called ‘the guardian of the genome’ as a consequence of its functions in the maintenance of genome stability (Lane, 1992). The singular importance of genome stability was exemplified by a study from Kirkland's group (described in Kirkland et al., 2007). They demonstrated that 51 passages with the commonly used Chinese Hamster Ovary cell line were sufficient to affect sensitivity to two known clastogens, and to introduce karyotypic changes affecting both the number and range of identifiable chromosomes. Fortunately, there are cell lines that are derived from malignancies that still retain p53 proficiency. Two examples of such cell lines that already have some limited usage in genotoxicity testing are the human hepatoma HepG2 cells and the human B-cell-derived TK6. Studies are ongoing to determine if p53-competent cell lines such as these might offer advantages for accurate testing, and whether benefit is gained by using human origin cell lines as opposed to rodent.
Highest testing concentration
Current regulations require in vitro genotoxicity tests to be carried out up to a top concentration of 10 mM, unless limited by toxicity (see below) or solubility. This is well in excess of the Michaelis constant for physiological biochemical reactions, which rarely exceeds 100 µM, and is often unreachable in vitro and lethal in vivo. Where these levels are reached, multiple systems can be affected. This makes the reason for a positive genotoxicity result difficult to identify, and probably irrelevant at intended dose levels. This leads to an obvious conclusion that the maximum test concentration should be reduced, but this must be supported by evidence to ensure that high concentration in vitro positive compounds with a genuine liability at lower doses in animals would not go undetected. Poor specificity is principally a problem with mammalian cell tests, so a study sponsored by ECVAM identified 24 carcinogens that tested negative or equivocal in the Ames bacterial mutation test, yet produced positive results in in vitro mammalian cells at concentrations of 1–10 mM. Of these, almost half were concluded not to be mechanistically genotoxic carcinogens and some were only carcinogens at excessive doses in animals, which under current animal testing guidance would not have produced carcinogenesis (Parry et al., 2010). The rest produced negative results up to 10 mM or positive results at or below 2 mM – using revised in vitro genotoxicity test protocols (Kirkland and Fowler, 2010: see below). However, at the time of writing, amendments to the guideline requirements for maximum testing concentration (e.g. a reduction to 1 mM) were still being keenly debated and consensus had not been reached.
Toxicity is also used to limit top dose. This a rather vague limit, as there are numerous different endpoints used to measure toxicity, and they are not equivalent. A systematic study of different cytotoxicity assessment methods in the micronucleus test, through both theoretical modelling and practical comparisons, concluded that measurement of relative cell counts (RCC; the final cell count of the treated culture expressed as a percentage of the final cell count of the control culture) underestimated toxicity compared with measures of proliferation such as relative increase in cell counts (RICC) or relative population doubling (RPD; Fellows et al. 2008; Lorge et al. 2008). Both RICC and RPD factor in the change in cell number during the treatment period, which enables them to reflect types of cytotoxicity that are effectively masked using RCC. The apparent toxicity underestimation with RCC means that compounds can be tested to higher concentrations. Selection of these potentially excessive concentrations may lead to an increase in the generation of irrelevant, toxicity-related positive results.
Using the currently recommended protocols, with measures of toxicity based on proliferation and a reduction in top test dose to 1 mM, the incidence of misleading positives is reduced without detriment to the sensitivity to genotoxic carcinogens. Thus, the ECVAM exercise was successful in improving the performance of currently used tests. Finally, the ECVAM group recognized that in order to discover whether a new test provided an improvement on the current regulatory tests, then a reference set of chemicals should be compiled which included examples of both expected positives and expected negatives. This was subsequently made available and has already been used in several studies (Kirkland et al. 2008; Birrell et al. 2010; Westerink et al. 2010).
New genotoxicity tests
The International Life Sciences Institute's Health and Environmental Sciences Institute project committee on ‘the relevance and follow-up of positive results in in vitro genetic toxicity testing’ established an Emerging Technologies and New Strategies Workgroup to review the current state of the art in genetic toxicology testing. This group focused on the identification of promising new technologies, including mature as well as maturing approaches that are not yet integrated into regulatory frameworks. In the mature category, three technologies were reviewed (Lynch et al. 2010). They are briefly described below, but well described elsewhere and reader is referred in the text to appropriate reviews.
The ‘in silico’ methods are based on computationally derived structural similarities between new compounds and known genotoxins. There are several methods in use at present, and some authors have suggested that there is some merit in combining methods in order to gain the maximum benefit (Yang et al. 2008). In some systems, the statistical approach is supplemented by reference to expert knowledge, gleaned from peer-reviewed studies, or proprietary compound studies. While widely used, and mature in that sense, these methods are only as good as the information they use to construct the predictive models. This leads to a lack of sensitivity to novel chemistries or complex biologically derived materials, and a tendency to misclassify as positive, compounds similar to genotoxins, but without genotoxic properties. Their use as an early pre-screen can, however, flag up compounds for early in vitro testing where an alert is produced. The regulatory agencies clearly have access to a great deal more proprietary compound data than other in silico model developers, and as a consequence their models can generate new alerts for a new drug application which is without alerting structures in other software.
The in vivo Comet assay (also known as the single cell gel electrophoresis assay) simply and rapidly allows an assessment of DNA double-stranded and single-stranded breaks in individual cells derived from different tissues (Brendler-Schwaab et al. 2005). While there has been no regulatory requirement for the in vivo Comet assay, in recent years it has often been used as the second in vivo assay in following up positive in vitro genotoxicity data. Some common recommendations for its performance have been developed (e.g. Tice et al. 2000; Hartmann et al. 2003; Burlinson et al. 2007), but there is still a need for greater standardization of methodology and interpretation. Efforts to achieve this uniformity are currently underway and are coordinated by the Japanese Centre for the Validation of Alternative Methods. The principle of the assay is quite simple (reviewed by Collins et al. 2008): if a direct current electric field is established through an appropriate aqueous gel matrix, DNA fragments migrate towards the anode faster than whole chromosomes. Following staining, these have the appearance of extraterrestrial comets, although in the assay the tail runs ahead of the main body. The most common version of the assay is performed in alkaline conditions to facilitate the detection of abasic sites and single-stranded breaks.
The flow cytometric assessment of micronucleus formation in peripheral red blood reticulocytes provides a very good prediction of the more arduous microscopic chromosomes aberration assays (Dertinger et al. 1996; Torous et al. 2003). As the name implies, micronuclei are effectively small nuclei. They can contain a whole chromosome or a chromosome fragment and there are several mechanisms by which they might appear. In a normal mitosis, there is a carefully ordered series of events that ensures the precise segregation of replicated sister chromatid pairs. This involves checkpoints that prevent anaphase until all replicated chromosomes have been captured at the centromere by spindle fibres and aligned at the metaphase plate. Following anaphase, the two clusters of chromosomes travel to opposing centrioles and are enveloped by nuclear membrane. A chromosome break generates one fragment with a centromere and one without: the latter can not be transported by the spindle mechanism, so some micronuclei contain chromosome fragments without centromeres. However, there are also micronuclei containing chromosomes with centromeres. These might be whole chromosomes or chromosome fragments, but in both cases, they have either failed to reach the nascent daughter nucleus or in some way left the nucleus during interphase. An agent that causes the appearance of micronuclei which all have a centromere is generally classified as an aneugen. The relatively high proportion of micronucleated cells in a normal population of cells (0.2–0.5%) suggests, perhaps, that chromosomes might spontaneously leave and rejoin the nucleus quite frequently. There have been a number of efforts to validate the flow cytometric methodology for the in vivo micronucleus test: for example, a method transferability study was performed across 14 laboratories for the enumeration of micronucleated reticulocytes (Torous et al. 2001). This study was then extended into an interlaboratory validation study to further assess the correspondence between the flow cytometry and traditional microscopy scoring methods, as well as between-laboratory reproducibility (Torous et al. 2005).
Four maturing tests were noted. Yeasts have for many years provided a readily manipulable model system for the development of simple assays which provide an insight into eukaryote-specific genotoxin targets. The yeast DEL assay detects DNA deletions that arise through homologous recombination events that restore histidine biosythetic capacity in engineered cells. Subsequent estimation of the frequency of these events, through growth on histidine deficient media in microwell plates, provides an indirect measure of the frequency of deletion events (Hontzeas et al. 2007). It was proposed that the DEL assay alongside a system's biology approach could replace the current regulatory framework (Ku et al. 2007), although as yet no transferability studies have been reported.
Mutagens, clastogens and aneugens all cause increased expression of the mammalian GADD45a gene. This has been exploited by the authors' laboratory in the development of a green fluorescent protein (GFP) reporter assay for the gene, in a human B lymphocyte-derived cell line (Hastwell et al. 2006). Validation studies have demonstrated the expected high sensitivity that comes from the comprehensive response to different genotoxin classes, as well as a higher specificity than the other in vitro mammalian assays (e.g. Hastwell et al. 2006; 2009; Birrell et al. 2010). International multi-laboratory ‘ring trials’ have demonstrated transferability of assay versions both with and without S9 metabolic activation (Billinton et al. 2008, 2010). This assay is performed in 96-well microplate format, and the consequent low compound requirement has already led to its adoption during lead optimization and selection by many biotech and pharmaceutical companies.
The assessment of micronucleus formation in vitro has now been validated as an alternative to in vitro CA tests (Corvi et al. 2008; Homiski et al. 2010), and an Organisation for Economic Co-operation and Development guideline (TG 487) has been approved. As with the in vivo assay, flow cytometry is proving a very popular technique for collecting data due to the statistical robustness that derives from the larger datasets that can be collected by automated scoring. Flow cytometry also provides a ready means to collect toxicity and cell cycle perturbation data – that is, it provides ‘high content’ screening (Avlasevich et al. 2006; Bryce et al. 2008).
The fourth ‘maturing’ technology considered was reconstructed three-dimensional skin models. These have application in the testing of topically applied compounds – both pharmaceutical and cosmetic. Under the 7th Amendment to the EC Cosmetics Directive (76/768/EEC), the use of animals for genotoxicity assessment is now banned in Europe and this has created a strong drive for developments in this field. In essence, a reconstructed skin model consists of human keratinocytes cultured in vitro. The top layers exposed to air, dry out and take on characteristics of the dermis. Cultured fibroblasts can be co-cultured beneath, and form an epidermal layer. The material clearly has utility for topically applied medicines, as well as those which might partition in the skin. Modified micronucleus (Hu et al. 2009; Mun et al. 2009) and comet assays (Flamand et al. 2006) are now being evaluated in the different systems.
Radical proposals to the International Committee on Harmonisation guidance on genotoxicity testing
An alternative and quite radical solution to the ‘false positives’ problem has been proposed at the regulatory level [S2(R1); ICH, 2010] and is summarized in Table 2. As an Ames positive result is almost certain to halt the development of a candidate pharmaceutical, and an Ames negative compound with positive in vitro mammalian data can often be ‘rescued’ by two in vivo studies, one obvious option is to remove the regulatory requirement for an in vitro mammalian study. There are consequences for both drug developers and animals in this proposal. The Ames test does not detect all carcinogens: that is why the in vitro mammalian tests were developed. However, regulatory testing is only applied at the preclinical stage, and drug developers would be at liberty to apply in vitro mammalian tests at whatever stage they chose to ensure that eukaryote-specific genotoxins are identified. An earlier section has already discussed how screening can be an effective approach to reducing attrition at preclinical safety assessment. Such testing need not be carried out to GLP, and without regulatory requirement to perform them, it will surely be tempting not to use the poorer specificity tests.
The second consequence of the ‘Ames only’ proposal is that more animals will be used because of the requirement for two animal tests. However, this was anticipated in the ICH proposals, which suggest that the two in vivo genotoxicity test endpoints should be collected from one group of dosed animals. Furthermore, samples for these tests should be integrated into 28-day repeat dose toxicity studies. The net results of this approach would actually reduce animal testing below the current level. A recent study (Dertinger et al. 2010) has now demonstrated proof of principle for the approach.
In the short term at least, it is likely that medicinal chemists will continue to produce putative drug candidates that are carcinogens. The current in vitro genotoxicity tests will continue to be effective in preventing pharmaceuticals with unanticipated carcinogenicity reaching the market. However, the refinement of existing methods and the development of new methods should now start to reduce the needless loss of new and effective drugs, which might at present be prevented from reaching patients because of misleading hazard alerts.
Acknowledgments
Andrew Billinton is thanked for his critical reading of the manuscript.
Glossary
Abbreviations
- CA
chromosome aberrations
- ECVAM
European Centre for the Validation of Alternative Methods
- GLP
Good Laboratory Practice
- ICH
International Committee on Harmonisation
- RCC
relative cell counts
- RICC
relative increase in cell counts
- RPD
relative population doubling
Conflict of interest
Richard Walmsley is the founder and a director of, and Nicholas Billinton is employed by Gentronix Ltd., which developed and markets the RAD54-GFP and GADD45a-GFP assays.
Supplementary material
Supporting Information: Teaching Materials; Fig 1 as PowerPoint slide.
References
- Aubrecht J, Osowski JJ, Persaud P, Cheung JR, Ackerman J, Lopes SH, et al. Bioluminescent Salmonella reverse mutation assay: a screen for detecting mutagenicity with high throughput attributes. Mutagenesis. 2007;22:335–342. doi: 10.1093/mutage/gem022. [DOI] [PubMed] [Google Scholar]
- Avlasevich SL, Bryce SM, Cairns SE, Dertinger SD. In vitro micronucleus scoring by flow cytometry: differential staining of micronuclei versus apoptotic and necrotic chromatin enhances assay reliability. Environ Mol Mutagen. 2006;47:56–66. doi: 10.1002/em.20170. [DOI] [PubMed] [Google Scholar]
- Billinton N, Bruce S, Hansen JR, Hastwell PW, Jagger C, McComb C, et al. A pre-validation transferability study of the GreenScreen HC GADD45a-GFP assay with a metabolic activation system (S9) Mutat Res. 2010;700:44–50. doi: 10.1016/j.mrgentox.2010.05.001. [DOI] [PubMed] [Google Scholar]
- Billinton N, Hastwell PW, Beerens D, Birrell L, Ellis P, Maskell S, et al. Interlaboratory assessment of the GreenScreen HC GADD45a-GFP genotoxicity screening assay: an enabling study for independent validation as an alternative method. Mutat Res. 2008;653:23–33. doi: 10.1016/j.mrgentox.2008.02.011. [DOI] [PubMed] [Google Scholar]
- Birrell L, Cahill P, Hughes C, Tate M, Walmsley RM. GADD45a-GFP GreenScreen HC assay results for the ECVAM recommended lists of genotoxic and non-genotoxic chemicals for assessment of new genotoxicity tests. Mutat Res. 2010;695:87–95. doi: 10.1016/j.mrgentox.2009.12.008. [DOI] [PubMed] [Google Scholar]
- Brambilla G, Martelli A. Update on genotoxicity and carcinogenicity testing of 472 marketed pharmaceuticals. Mutat Res. 2009;681:209–229. doi: 10.1016/j.mrrev.2008.09.002. [DOI] [PubMed] [Google Scholar]
- Brendler-Schwaab S, Hartmann A, Pfuhler S, Speit G. The in vivo comet assay: use and status in genotoxicity testing. Mutagenesis. 2005;20:245–254. doi: 10.1093/mutage/gei033. [DOI] [PubMed] [Google Scholar]
- Brennan RJ, Schiestl RH. Detecting carcinogens with the yeast DEL assay. Methods Mol Biol. 2004;262:111–124. doi: 10.1385/1-59259-761-0:111. [DOI] [PubMed] [Google Scholar]
- Bryce SM, Avlasevich SL, Bemis JC, Lukamowicz M, Elhajouji A, Van Goetham F, et al. Interlaboratory evaluation of a flow cytometric, high content in vitro micronucleus assay. Mutat Res. 2008;650:181–195. doi: 10.1016/j.mrgentox.2007.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burlinson B, Tice RR, Speit G, Agurell E, Brendler-Schwaab SY, Collins AR, et al. Fourth international workgroup on genotoxicity testing: results of the in vivo comet assay workgroup. Mutat Res. 2007;627:31–35. doi: 10.1016/j.mrgentox.2006.08.011. [DOI] [PubMed] [Google Scholar]
- Collins AR, Azqueta Oscoz A, Brunborg G, Gaivão I, Giovannelli L, Kruszewski M, et al. The comet assay: topical issues. Mutagenesis. 2008;23:143–151. doi: 10.1093/mutage/gem051. [DOI] [PubMed] [Google Scholar]
- Cooper JA, Saracci R, Cole P. Describing the validity of carcinogen screening tests. Br J Cancer. 1979;39:87–89. doi: 10.1038/bjc.1979.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corvi R, Albertini S, Hartung T, Hoffmann S, Maurici D, Pfuhler S, et al. ECVAM retrospective validation of in vitro micronucleus test (MNT) Mutagenesis. 2008;23:271–283. doi: 10.1093/mutage/gen010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dertinger SD, Swaths P, Franklin D, Weller P, Torous DK, Bryce SM, et al. Integration of mutation and chromosomal damage endpoints into 28-day repeat dose toxicology studies. Tox Sci. 2010;115:401–411. doi: 10.1093/toxsci/kfq070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dertinger SD, Torous DK, Tometsko KR. Simple and reliable enumeration of micronucleated reticulocytes with a single-laser flow cytometer. Mutat Res. 1996;371:283–292. doi: 10.1016/s0165-1218(96)90117-2. [DOI] [PubMed] [Google Scholar]
- Fellows MD, O'Donovan MR, Lorge E, Kirkland D. Comparison of different methods for an accurate assessment of cytotoxicity in the in vitro micronucleus test II: practical aspects with toxic agents. Mutat Res. 2008;655:4–21. doi: 10.1016/j.mrgentox.2008.06.004. [DOI] [PubMed] [Google Scholar]
- Flamand N, Marrot L, Belaidi J-P, Bourouf L, Dourille E, Feltes M, et al. Development of genotoxicity test procedures with Episkin®, a reconstructed human skin model: towards new tools for in vitro risk assessment of dermally applied compounds? Mutat Res. 2006;606:39–51. doi: 10.1016/j.mrgentox.2006.02.009. [DOI] [PubMed] [Google Scholar]
- Hartmann A, Agurell E, Beevers C, Brendler-Schwaab S, Burlinson B, Clay P, et al. Recommendations for conducting the in vivo alkaline comet assay. Mutagenesis. 2003;18:45–51. doi: 10.1093/mutage/18.1.45. [DOI] [PubMed] [Google Scholar]
- Hastwell PW, Chai L-L, Roberts KJ, Webster TW, Harvey JS, Rees RW, et al. High-specificity and high-sensitivity genotoxicity assessment in a human cell line: validation of the GreenScreen HC GADD45a-GFP genotoxicity assay. Mutat Res. 2006;607:160–175. doi: 10.1016/j.mrgentox.2006.04.011. [DOI] [PubMed] [Google Scholar]
- Hastwell PW, Webster TW, Tate M, Billinton N, Lynch AM, Harvey JS, et al. Analysis of 75 marketed pharmaceuticals using the GADD45a-GFP ‘GreenScreen HC’ genotoxicity assay. Mutagenesis. 2009;24:455–463. doi: 10.1093/mutage/gep029. [DOI] [PubMed] [Google Scholar]
- Hernández LG, van Steeg H, Luijtena M, van Benthem J. Mechanisms of non-genotoxic carcinogens and importance of a weight of evidence approach. Mutat Res. 2009;682:94–109. doi: 10.1016/j.mrrev.2009.07.002. [DOI] [PubMed] [Google Scholar]
- Homiski ML, Muehlbauer PA, Dobo KL, Schuler MJ, Aubrecht J. Concordance analysis of an in vitro micronucleus screening assay and the regulatory chromosome aberration assay using pharmaceutical drug candidates. Environ Mol Mutagen. 2010;51:39–47. doi: 10.1002/em.20507. [DOI] [PubMed] [Google Scholar]
- Hontzeas N, Hafer K, Schiestl RH. Development of a microtiter plate version of the yeast DEL assay amenable to high-throughput toxicity screening of chemical libraries. Mutat Res. 2007;634:228–234. doi: 10.1016/j.mrgentox.2007.07.001. [DOI] [PubMed] [Google Scholar]
- Hu T, Kaluzhny Y, Mun GC, Barnett B, Karetsky V, Wilt N, et al. Intralaboratory and interlaboratory evaluation of the EpiDerm™ 3D human reconstructed skin micronucleus (RSMN) assay. Mutat Res. 2009;673:100–108. doi: 10.1016/j.mrgentox.2008.12.003. [DOI] [PubMed] [Google Scholar]
- International Conference on Harmonisation. S2(R1) revised guidance on genotoxicity testing and data interpretation for pharmaceuticals intended for human use. 2010. http://www.ich.org/cache/compo/502-272-1.html. [PubMed]
- International Conference on Harmonisation. Topic S2A, Genotoxicity: guidance on Specific Aspects of Regulatory Genotoxicity Tests for Pharmaceuticals. 1995.
- International Conference on Harmonisation. Harmonised tripartite guideline, S2B, genotoxicity: a standard battery for genotoxicity testing of pharmaceuticals. 1997.
- Kamber M, Fluckiger-Isler S, Engelhardt G, Jaeckh R, Zeiger E. Comparison of the Ames II and traditional Ames test responses with respect to mutagenicity, strain specificities, need for metabolism and correlation with rodent carcinogenicity. Mutagenesis. 2009;24:359–366. doi: 10.1093/mutage/gep017. [DOI] [PubMed] [Google Scholar]
- Kirkland D, Aardema M, Henderson L, Müller L. Evaluation of the ability of a battery of three in vitro genotoxicity tests to discriminate rodent carcinogens and non-carcinogens. I. Sensitivity, specificity and relative predictivity. Mutat Res. 2005;584:1–256. doi: 10.1016/j.mrgentox.2005.02.004. [DOI] [PubMed] [Google Scholar]
- Kirkland D, Fowler P. Further analysis of Ames-negative rodent carcinogens that are only genotoxic in mammalian cells in vitro at concentrations exceeding 1 mM, including retesting of compounds of concern. Mutagenesis. 2010 doi: 10.1093/mutage/geq041. doi: 10.1093/mutage/geq041. [DOI] [PubMed] [Google Scholar]
- Kirkland D, Kasper P, Müller L, Corvi R, Speit G. Recommended lists of genotoxic and non-genotoxic chemicals for assessment of the performance of new or improved genotoxicity tests: a follow-up to an ECVAM workshop. Mutat Res. 2008;653:99–108. doi: 10.1016/j.mrgentox.2008.03.008. [DOI] [PubMed] [Google Scholar]
- Kirkland D, Pfuhler S, Tweats D, Aardema M, Corvi R, Darroudi F, et al. How to reduce false positive results when undertaking in vitro genotoxicity testing and thus avoid unnecessary follow-up animal tests: report of an ECVAM Workshop. Mutat Res. 2007;628:31–55. doi: 10.1016/j.mrgentox.2006.11.008. [DOI] [PubMed] [Google Scholar]
- Knight AW, Billinton N, Cahill PA, Scott A, Harvey JS, Roberts KJ, et al. An analysis of results from 305 compounds tested with the yeast RAD54-GFP genotoxicity assay (GreenScreen GC) – including relative predictivity of regulatory tests and rodent carcinogenesis and performance with autofluorescent and coloured compounds. Mutagenesis. 2007;22:409–416. doi: 10.1093/mutage/gem036. [DOI] [PubMed] [Google Scholar]
- Ku WW, Aubrecht J, Mauthe RJ, Schiestl RH, Fornace AJ., Jr Genetic toxicity assessment: employing the best science for human safety evaluation part VII: why not start with a single test: a transformational alternative to genotoxicity hazard and risk assessment. Toxicol Sci. 2007;99:20–25. doi: 10.1093/toxsci/kfm147. [DOI] [PubMed] [Google Scholar]
- Lane DP. P53, guardian of the genome. Nature. 1992;358:15–16. doi: 10.1038/358015a0. [DOI] [PubMed] [Google Scholar]
- LeBarona MJ, Rasoulpoura RJ, Klapacza J, Ellis-Hutchings RG, Hollnagel HM, Gollapudi BB. Epigenetics and chemical safety assessment. Mutat Res. 2010;705:83–95. doi: 10.1016/j.mrrev.2010.04.003. [DOI] [PubMed] [Google Scholar]
- Lorge E, Hayashi M, Albertini S, Kirkland D. Comparison of different methods for an accurate assessment of cytotoxicity in the in vitro micronucleus test I. Theoretical aspects. Mutat Res. 2008;655:1–3. doi: 10.1016/j.mrgentox.2008.06.003. [DOI] [PubMed] [Google Scholar]
- Lynch AM, Sasaki JC, Elespuru R, Jacobson-Kram D, Thybaud V, De Boeck M, et al. New and emerging technologies for genetic toxicity testing. Environ Mol Mutagen. 2010 doi: 10.1002/em.20614. doi: 10.1002/em.20614. [DOI] [PubMed] [Google Scholar]
- McCann J, Choi E, Yamaski E, Ames BN. Detection of carcinogens as mutagens in the Salmonella/microsome test. Part 1, assay of 300 chemicals. Proc Natl Acad Sci U S A. 1975;72:5135–5139. doi: 10.1073/pnas.72.12.5135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mun GC, Aardema MJ, Hu T, Barnett B, Kaluzhny Y, Klausner M, et al. Further development of the EpiDerm™ 3D reconstructed human skin micronucleus (RSMN) assay. Mutat Res. 2009;673:92–99. doi: 10.1016/j.mrgentox.2008.12.004. [DOI] [PubMed] [Google Scholar]
- Parry JM, Parry E, Phrakonkha P, Corvi R. Analysis of published data for top concentration considerations in mammalian cell genotoxicity testing. Mutagenesis. 2010 doi: 10.1093/mutage/geq046. doi: 10.1093/mutage/geq046. [DOI] [PubMed] [Google Scholar]
- Phillips DM. Fifty years of Benzo{a]pyrene. Nature. 1983;303:468–472. doi: 10.1038/303468a0. [DOI] [PubMed] [Google Scholar]
- Reifferscheid G, Heil J. Validation of the SOS/umu test using test results of 486 chemicals and comparison with the Ames test and carcinogenicity data. Mutat Res. 1996;369:129–145. doi: 10.1016/s0165-1218(96)90021-x. [DOI] [PubMed] [Google Scholar]
- Shaw IC, Jones HB. Mechanisms of non-genotoxic carcinogenesis. Trends Pharmacol Sci. 1994;15:89–93. doi: 10.1016/0165-6147(94)90284-4. [DOI] [PubMed] [Google Scholar]
- Snyder RD, Ewing D, Hendry LB. DNA intercalative potential of marketed drugs testing positive in in vitro cytogenetics assays. Mutat Res. 2006;609:47–59. doi: 10.1016/j.mrgentox.2006.06.001. [DOI] [PubMed] [Google Scholar]
- Snyder RD, Green JW. A review of the genotoxicity of marketed pharmaceuticals. Mutat Res. 2001;488:151–169. doi: 10.1016/s1383-5742(01)00055-2. [DOI] [PubMed] [Google Scholar]
- Tice RR, Agurell E, Anderson D, Burlinson B, Hartmann A, Kobayashi H, et al. Single cell gel/comet assay: guidelines for in vitro and in vivo genetic toxicity testing. Environ Mol Mutagen. 2000;35:206–221. doi: 10.1002/(sici)1098-2280(2000)35:3<206::aid-em8>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- Torous DK, Hall NE, Dertinger SD, Diehl MS, Illi-Love AH, Cederbrant K, et al. Flow cytometric enumeration of micronucleated reticulocytes: high transferability among 14 laboratories. Environ Mol Mutagen. 2001;38:59–68. doi: 10.1002/em.1051. [DOI] [PubMed] [Google Scholar]
- Torous DK, Hall NE, Illi-Love AH, Diehl MS, Cederbrant K, Sandelin K, et al. Interlaboratory validation of a CD71-based flow cytometric method (MicoFlow®) for the scoring of micronucleated reticulocytes in mouse peripheral blood. Environ Mol Mutagen. 2005;45:44–55. doi: 10.1002/em.20081. [DOI] [PubMed] [Google Scholar]
- Torous DK, Hall NE, Murante FE, Gleason SE, Tometsko CR, Dertinger SD. Comparative scoring of micronucleated reticulocytes in rat peripheral blood by flow cytometry and microscopy. Toxicol Sci. 2003;74:309–314. doi: 10.1093/toxsci/kfg143. [DOI] [PubMed] [Google Scholar]
- Tweats DJ, Blakey D, Heflich RH, Jacobs A, Jacobsen SD, Morita T, et al. Report of the IWGT working group on strategy/interpretation for regulatory in vivo tests II. Identification of in vivo-only positive compounds in the bone marrow micronucleus test. Mutat Res. 2007a;627:92–105. doi: 10.1016/j.mrgentox.2006.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tweats DJ, Blakey D, Heflich RH, Jacobs A, Jacobsen SD, Morita T, et al. Report of the IWGT working group on strategies and interpretation of regulatory in vivo tests I. Increases in micronucleated bone marrow cells in rodents that do not indicate genotoxic hazards. Mutat Res. 2007b;627:78–91. doi: 10.1016/j.mrgentox.2006.10.005. [DOI] [PubMed] [Google Scholar]
- Westerink WMA, Stevenson JCR, Horbach GJ, Schoonen WGEJ. The development of RAD51C, Cystatin A, p53 and Nrf2 luciferase-reporter assays in metabolically competent HepG2 cells for the assessment of mechanism-based genotoxicity and of oxidative stress in the early research phase of drug development. Mutat Res. 2010;696:21–40. doi: 10.1016/j.mrgentox.2009.12.007. [DOI] [PubMed] [Google Scholar]
- Yang C, Hasselgren CH, Boyer S, Arvidson K, Aveston S, Dierkes P, et al. Understanding genetic toxicity through data mining: the process of building knowledge by integrating multiple genetic toxicity databases. Toxicol Mech Methods. 2008;18:277–295. doi: 10.1080/15376510701857502. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.