

Abstract
BACKGROUND AND PURPOSE
Betulinic acid (BA) is a naturally occurring triterpenoid widely distributed throughout the plant kingdom. We previously reported that BA inhibits lipopolysaccharide (LPS)-induced interleukin-6 production through modulation of nuclear factor κB (NF-κB) in human peripheral blood mononuclear cells (hPBMCs). This study attempted to identify other mechanisms through which BA modulates LPS signalling in mononuclear cells. The effects of BA on signalling pathways downstream were focused on in this study.
EXPERIMENTAL APPROACH
We determined the ability of BA to interfere with p38 and extracellular regulated kinase (ERK) phosphorylation as well as Akt phosphorylation and nuclear factor-κB activation using LPS-activated hPBMCs as an in vitro model. LPS-induced endotoxin shock in mice was the in vivo model employed.
KEY RESULTS
BA inhibited LPS-induced COX-2 protein expression and prostaglandin E2 production and also attenuated LPS-induced ERK and Akt phosphorylation, but not p38 in hPBMCs. BA abolished LPS-induced IκBα phosphorylation and thus normalized the levels of IκBα in cytosol. BA also inhibited LPS-induced reactive oxygen species formation and lactate dehydrogenase release. Interestingly, BA improved the life span of mice in endotoxin shock and also inhibited PGE2 production and myeloperoxidase activity in vivo.
CONCLUSIONS AND IMPLICATIONS
BA modulates LPS-induced COX-2 expression in hPBMCs by inhibiting ERK and Akt pathways as well as by modulating IκBα phosphorylation. At the same time, no cell toxicity was observed. The effect of the drug was confirmed through in vivo experiments. The study gives an insight into the molecular mechanisms of BA.
Keywords: betulinic acid, NF-κB, Akt, ERK, prostaglandin E2, cyclooxygenase, IκBα, inflammatory mediators, lipopolysaccharide, mononuclear cell
Introduction
Inflammation, which is characterized by oedema, pain, redness and heat, is one of the most crucial elements of host defence mechanisms against invading pathogens. However, dysregulated activation of inflammation and oxidative stress has been recognized as one of the principal causes of inflammatory diseases (Balkwill and Mantovani, 2001; Shacter and Weitzman, 2002).
One of the key enzymes involved in inflammation is COX, the rate-limiting enzyme involved in the biosynthesis of prostaglandins (PG). COX exists as two isoforms: the constitutively expressed COX-1 and the regulated isoform COX-2, which performs a crucial function in prostaglandin E2 (PGE2) production. During inflammation, the activation of critical mediators like COX-2 involves the triggering of a variety of transcription factors and cellular signalling pathways (Schachter, 2003). Nuclear factor-κB (NF-κB) is a transcription factor that performs a vital function in the regulation of the expression of genes responsible for a variety of cellular processes, including inflammatory responses, innate and adaptive immunity, and the pathways involved in cell death and proliferation (Baud and Karin, 2009). In unstimulated cells, NF-κB is sequestered in the cytosol in a latent form bound to inhibitory proteins, the inhibitor of κBs (IκB). The exposure of cells to a variety of extracellular stimuli results in the stimulation of a series of signalling events, which ultimately converge in the activation of one or more redox-sensitive kinases. These kinases exclusively phosphorylate IκBs, thereby resulting in the polyubiquitination of the proteins and their subsequent degradation by the 26S proteasome (Karin and Ben-Neriah, 2001). Free NF-κB is then translocated to the nucleus and stimulates the transcription of genes encoding for pro-inflammatory enzymes such as COX-2 (Lee et al., 2009).
Akt is a serine/threonine protein kinase also known as protein kinase B or Rac (Bellacosa et al., 1991; Jones et al., 1991). Akt is an inactive cytosolic protein recruited to the plasma membrane, and activated by phosphorylation at threonine 308 and serine 473 in response to growth factors or cytokines (Stephens et al., 1998; Meng et al., 2002; Germain et al., 2004) via the product of PI3-K, phosphatidylinositol 3, 4, 5 – triphosphate. The phosphoinositide 3-kinase pathway has been implicated in the activation of NF-κB. It has been demonstrated that both the regulatory and the catalytic subunit of phosphatidylinositol 3-kinase (PI3-K) play a role in NF-κB activation by the tyrosine phosphorylation-dependent pathway (Beraud et al., 1999). Akt is a downstream target of NF-κB (Meng et al., 2010). Akt also regulates COX-2 gene and protein expressions and is also demonstrated to be involved in IKK phosphorylation resulting in NF-κB activation (Wang et al., 2004).
A number of studies have suggested that certain bioactive chemicals present in plants may protect against inflammation (Plummer et al., 1999; Murakami and Ohigashi, 2007). Bacopa monniera (L.) Wettst (family: Scrophulariaceae family) is a renowned Ayurvedic plant reported to possess memory-enhancing (Mukherjee and Dey, 1996), cognitive (Vohora et al., 2000), antioxidant (Chowdhuri et al., 2002), anti-fertility (Singh and Singh, 2009), hepatoprotective (Vijayan and Helen, 2007), anti-cancer (Rohini and Devi, 2008), anti-ulcer (Sairam et al., 2001) and anti-inflammatory (Viji and Helen, 2008) properties. The plant is enriched with many phytochemicals of which betulinic acid (BA), a triterpenoid belonging to lupane series, was recently explored to its topoisomerase inhibitory potential (Chowdhury et al., 2002). BA has a wide range of other pharmacological properties like anti-cancer (Ren et al., 2010), anti-malarial (Santos et al., 2009), anti-retroviral and anti-inflammatory properties (Chowdhury et al., 2002). To the best of our admittedly limited knowledge, the effect of BA on inflammation has yet to be elucidated in detail. This study was aimed to elucidate the signalling pathways involved in the anti-inflammatory effect of BA.
Methods
Reagents
RPMI-1640, Histopaque-1077, fetal bovine serum, LY294002, SB203580, PD98059 and SP600125, penicillin, lipopolysaccharide (Escherichia coli serotype O127:B8), streptomycin, phenylmethylsulphonyl fluoride (PMSF), leupeptin, 4-nitroblue tetrazolium chloride/5-bromo-4-chloro-3-indolyl-phosphate stock solution were obtained from Sigma-Aldrich (St Louis, MO, USA). Antibodies against COX-2 (dilution 1:1000), IκBα (1:1000), IκBα-P (1:500), Akt (1:1000), Akt-P (1:250), extracellular regulated kinase (ERK) 1/2 (1:1000), ERK1/2-P (1:1000), β-actin, were obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA). PGE2 immunoassay kit was obtained from Cayman Chemical Co. (Ann Arbor, MI, USA).
Preparation of BA
BA was isolated from B. monniera herbs as described by Chatterji et al. (1963) and Chowdhury et al. (2002). BA was purified by chromatography over silica gel (60–120 mesh) and by HPLC (Shimadzu, Kyoto, Japan) using C18 (250 × 4.8 mm) column. BA obtained by this method was greater than 90% pure. The presence of BA was confirmed by proton nuclear magnetic resonance spectroscopy. BA: colourless solid; melting point, 295–298°C. H1 NMR (400 MHz) spectrum taken in CHCl3 d6 showed the presence of six tertiary methyl groups resonated at δ 0.652, 0.749, 0.900, 0.958, 0.977 and 1.649 as singlets. The H1 NMR data is as follows: [δH: 0.652, 0.749, 0.900, 0.966, 0.977 and 1.649], vinyl methyl [δH: 1.671 (br d, J = 0.5 Hz)], a secondary carbinol [δH: 3.158 (dd, J = 9.5, 6.0 Hz)] and [δH: 2.947 (ddd, J = 9.5, 6.0 Hz, 0.5 Hz)], an exo-methylene group [δH: 4.557 (H1 d, δH: J = 0.4 Hz)], [δH: 4.659 (1h, d, J = 0.4 Hz)]. This H1 NMR data (CDCl3) and TLC analysis were in agreement with the data obtained for BA (C30H48O3) previously (Haque et al., 2006). The chemical structure of BA is given in Figure 1. The stock solution of BA was prepared in and further diluted with RPMI-1640 culture medium.
Figure 1.
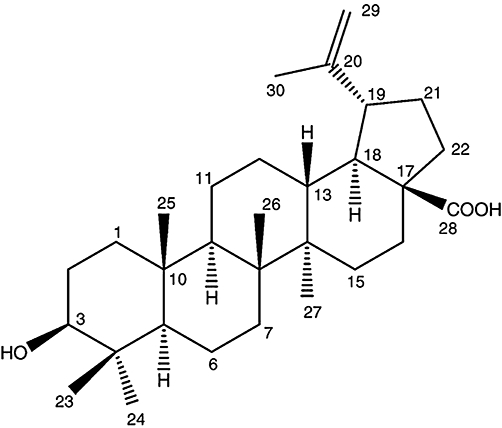
Chemical structure of betulinic acid [3-beta-hydroxy-20(29)-lupaene-28-oic acid] C30H48O3 (MW 456.7).
Ethical clearance for the conduct of experiments
All the experiments conducted on the human blood samples were with the donor's consent and in agreement with the institutional guidelines. Experimental procedures conducted on male Swiss mice were reviewed and approved by the Animal Experiment Committee (218/CPC-SEA) according to the Government of India's accepted principles for laboratory animal use and care (No. KU-12/2005-06) and in accordance with the British Pharmacological Society's Ethics Committee.
Test system
The human peripheral blood mononuclear cells (hPBMCs) were isolated from heparin-treated blood obtained from healthy individuals (age group 27 ± 2) by density gradient centrifugation using the method of Huch et al. (1996). Briefly, heparin-treated blood was overlaid on Histopaque-1077 (Sigma-Aldrich; 1.077 g·mL−1) and spun at 400×g for 30 min. The mononuclear cells at the interphase of phosphate-buffered saline (PBS)/Histopaque were collected and washed with PBS. The cells were re-suspended in RPMI-1640 medium supplemented with 100 U·mL−1 penicillin and 100 µg·mL−1 streptomycin and adjusted to the desired cell count. The viability of the cells was assayed by trypan blue exclusion assay. The hPBMCs were enriched for monocytes by adherence on type I collagen (50 µg·mL−1) coated plates under 37°C and 5% CO2 humidified atmosphere for 4 h. The non-adherent cells were removed by vigorous washing with RPMI-1640. Adherent cells were cultured in RPMI-1640 supplemented with 10% heat-inactivated autologous serum and antibiotics at a density of 5 × 106 cells·mL−1 (NF-κB experiments) or 1 × 106 cells·mL−1 (PGE2 experiments) overnight, and the monolayer was washed with PBS before induction with LPS. More than 85% of cells purified by this technique were determined to be monocytes. Cell viability determined by trypan blue exclusion was 94%. For experiments using BA, cells were incubated with varying concentrations of BA for 45 min prior to stimulation with lipopolysaccharide (LPS) (1 µg·mL−1 culture medium).
PGE2 assay
Cells were pretreated with BA or inhibitors prior to LPS induction and the conditioned media at 16th hour was taken for the assay of PGE2 (Cayman Chemical Co.). The concentration of PGE2 was measured according to manufacturer's instructions.
Cytotoxicity assay test
Cytotoxic assay test was conducted by measuring lactate dehydrogenase secreted in the medium using an lactate dehydrogenase (LDH) assay kit from Erba Transasia Biomedicals Ltd. (Daman, India) according to the manufacturer's protocol.
Measurement of ROS production in hPBMCs
The measurement of reactive oxygen species (ROS) production in the hPBMCs was performed as described previously (Furukawa et al., 2004). ROS production was measured by nitroblue tetrazolium (NBT) reduction. Briefly, hPBMCs were incubated for 60 min in PBS (137 mM NaCl, 8.1 mM Na2PO4, 2.68 mM KCl and 1.47 mM KH2PO4) containing 0.2% NBT. Formazan was dissolved in 50% acetic acid, and the absorbance was monitored at 560 nm using a spectrophotometer (Shimadzu).
Immunoblot analysis
The hPBMCs were cultured in 35 mm plate in the presence or absence of LPS or BA. Cells were lysed in lysis buffer [25 mM HEPES (pH 7.5), 300 mM NaCl, 1.5 mM MgCl2, 0.2 mM EDTA, 0.1% Triton X-100, 0.5 mM dithiothreitol, 20 mM β-glycerophosphate, 0.1 mM Na3VO4, 2 µg·mL−1 leupeptin and 1 mM PMSF] followed by sonication on ice for 20 s. The lysate was centrifuged at 14 000×g for 10 min at 4°C and the protein content of the supernatant was measured using Bradford assay (Bio-Rad, Hercules, CA, USA). The soluble lysate was mixed in 5x sample buffer and heated for 5 min at 95°C. Samples (60 µg) were loaded per lane and separated by SDS-PAGE using 4 and 10% acrylamide for stacking and separating gels respectively. Protein was transferred to nitrocellulose membrane (pore size: 0.45 µm) and the membrane was treated with 5% non-fat milk overnight at 4°C to block non-specific binding. The membrane was probed with a specific monoclonal or polyclonal primary antibody, then stripped and probed with a corresponding secondary antibody against total protein. Bands were visualized using enhanced chemiluminescence (Amersham Pharmacia Biotech, Piscataway, NJ, USA) and quantified by densitometry using Quantityone software (Bio-Rad).
Preparation of nuclear extract and electrophoretic mobility shift assay
Cells were pretreated with different concentrations of BA prior to LPS (1 µg·mL−1 culture medium) induction and incubated at 37°C for 24 h. After treatment, cells were spun at 300×g for 5 min at 4°C. The supernatant was discarded and the cell pellet obtained was re-suspended in ice-cold PBS/phosphate inhibitor solution (250 µL PIS containing 1 M sodium fluoride (NaF), 0.05 M β-glycerophosphate and 0.05 M sodium orthovanadate, and 10 mL 1x PBS) and spun at 300×g for 5 min at 4°C. The supernatant was discarded and the cells were re-suspended in ice-cold hypotonic buffer (20 mM HEPES, 5 mM NaF, 10 mM sodium molybdate, 0.1 mM EDTA, pH 7.5). The cell suspension was mixed and transferred to pre-chilled microfuge tubes on ice for 15 min to allow cells to swell. To this 50 µL 10% Igepal was added, mixed gently and pulse spun for 30 s at 4°C. The supernatant containing cytosolic fraction was collected to fresh tubes and stored at −80°C. The pellet was re-suspended in ice-cold extraction buffer (10 mM HEPES; pH 7.9) containing 0.1 mM EDTA, 1.5 mM MgCl2, 420 mM NaCl, 0.5 mM dithiothreitol, 0.5 mM PMSF, 1 µg·mL−1 pepstatin A, 1 µg·mL−1 leupeptin, 10 µg·mL−1 aproptinin, 20 mM NaF, 1 mM β-glycerophosphate, 10 mM sodium orthovanadate and 25% glycerol, and vortexed for 15 min at the highest setting followed by gentle rocking on ice for 15 min. This process was repeated and the suspension was spun at 14 000×g for 10 min at 4°C. The supernatant containing the nuclear fraction was flash frozen and quantified for protein content by the Bradford assay (Bio-Rad).
Electrophoretic mobility shift assay (EMSA) was conduced as described previously. Briefly, nuclear extracts prepared from control and treated cells were incubated with γ32P-end-labeled, 45mer, double stranded NF-κB oligonucleotide (15 µg protein with 16 fmol DNA) from HIV long terminal repeat. The DNA protein complex formed was freed from oligonucleotide on 6.6% native PAGE. A double-stranded DNA probe for the consensus sequence of NF-κB (5′-ATG TGA GGG GAC TTT CCC AGG C-3′) was employed for examining the specificity of binding of NF-κB to DNA. The specificity of binding was also examined by competition with unlabelled oligonucleotide. The gels were dried and visualized and quantified by Imagequant software.
To control any selective differences between normal and experimental groups in protein levels or in the EMSAs, internal standard Oct-1 was used. The binding of Oct-1 to DNA was determined by incubating 20 µg nuclear extracts with 16 fmol 32P-end-labeled with the octamer-binding protein (Oct-1) consensus oligonucleotide (5′-TGTCGAATGCAAATCACTAGAA-3′; Promega, Madison, WI, USA) (boldface indicates Oct-1 binding site) for 30 min at 37°C and then analysed using 5% native polyacrylamide gel. Using these internal standards, we compared NF-κB binding normalized by protein levels with the data normalized by Oct-1 binding. Nuclear fraction was quantified for protein content and also used for p65 NF-κB assay according to the manufacturer's instructions (Cayman Chemical Co.).
Mice endotoxin shock model
A total of 120 Swiss mice (24–26 g) were used for this experiment. Mice were bred and reared in the department animal house and maintained on normal laboratory diet (Amruth Rat Feed Ltd., Maharashtra, India). They were housed in polypropylene cages in room with temperature maintained at 25 ± 1°C and 12 h light and dark cycles. Water was provided ad libitum.
Mice were pretreated with BA (20 mg·kg−1 in 0.1% DMSO i.p.) or vehicle (0.1% DMSO intraperitoneally) three times (days 1, 2 and 3) before LPS challenge (E. coli O55:B5 in sterilized physiological saline; 32 mg·kg−1 i.p.) on the 3rd day (Zuchermann and Bendele, 1989). Injections were made in a total volume of 100 µL. On the day of LPS induction, BA was administered 3 h before LPS injection. Animals were observed for 7 days. The total duration of experiment was 10 days. After experiment, the remaining animals were killed by cervical dislocation. The lungs and livers were removed immediately by thoracotomy and laparotomy and stored at −80°C for the assay of PGE2 and myeloperoxidase (MPO) in the tissue samples.
PGE2 assay was done using an express kit from Cayman Chemical Co. Briefly tissues washed in phosphate buffer containing 1 mM EDTA and 10 µM indomethacin. Tissues were homogenized in 10% methanol and allowed to precipitate. The samples were spun and the supernatant was added to extraction C18 SPE extraction column (Waters Corporation, USA) pre-conditioned with methanol and water. The column was washed sequentially with water, 10% methanol and petroleum ether. The eicosanoid was eluted with methyl formate, evaporated under nitrogen and reconstituted in enzyme immunoassay (EIA) buffer for assay according to manufacturer's instruction.
Assay of MPO
The MPO activity was assayed by the method of Bradley et al. (1982). MPO activity was analysed as an index of neutrophil infiltration. Tissues were first homogenized in a solution containing 50 mM potassium phosphate buffer, pH 7.0 containing 0.5% hexadecyltrimethylammonium bromide. This was freeze thawed three times and then centrifuged at 20 000×g for 30 min at 4°C. An aliquot of the supernatant was allowed to react with a solution of o-dianisidine dihydrochloride (0.167 mg·mL−1) and 0.0005% hydrogen peroxide. MPO activity has been defined as the concentration of enzyme degrading 1 µM of peroxide min−1 at 37°C and was expressed as U·mg−1 protein.
Histopathological studies
After treatment, lungs and livers were removed immediately by thoracotomy and laparotomy. The right lungs were used for histological studies. The tissue specimens were fixed overnight in 4% buffered formaldehyde, processed by standard methods, and stained for hematoxylin and eosin (H&E). Semi-quantitative analysis of tissues was performed by one observer in a blinded fashion.
Data analysis and statistics
Data are presented as ± SEM of indicated number of observations. The results were analysed using a statistical program SPSS/PC+, version 11.0 (SPSS Inc., Chicago, IL, USA). One-way anova was employed for comparisons between the groups. Pair-fed comparisons between the groups were made by Duncan's multiple range tests. P < 0.05 was considered to be significant.
Results
Effect of BA on LPS-induced PGE2 release by hPBMCs
We recently demonstrated the expression of IL-6 after treatment with BA in LPS-activated cells and found that BA inhibited its release at 8th hour post-LPS induction. To the best of our knowledge, no information about cell signal transduction pathways controlling COX-2 expression and PGE2 production in normal cells is currently available. We addressed this issue here and in order to study the effect of BA on LPS-induced production of PGE2 by hPBMCs, isolated cells were maintained in culture and pretreated with different concentrations of BA (0.1, 0.5, 1, 2, 5 and 10 µg·mL−1 culture medium) for 30 min prior to activation with LPS (1 µg·mL−1 culture medium). The cultures were maintained for 16 h and PGE2 levels were measured in culture supernatants by EIA. Normal hPBMCs produced very low levels of PGE2 but the production significantly increased following LPS stimulation (Figure 2A). Treatment with BA significantly decreased LPS-induced PGE2 production; with an increase in concentration of BA, there was a progressive decrease in PGE2 production. Treatment of cells with BA alone did not elicit any PGE2 production.
Figure 2.
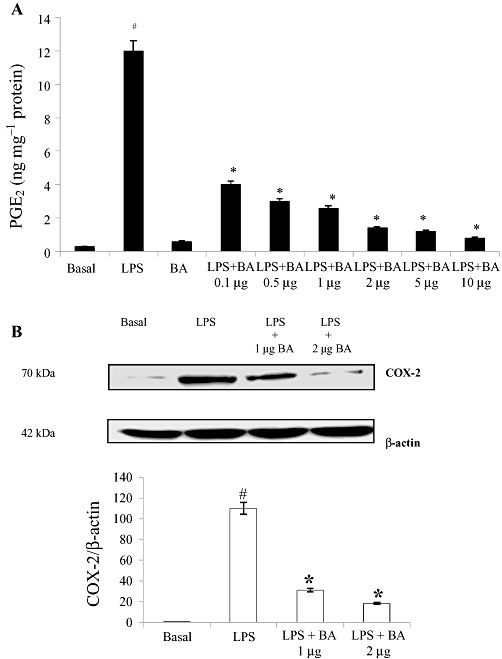
Effect of BA on LPS-induced PGE2 production and COX-2 protein expression in hPBMCs. (A) BA reduced LPS-induced PGE2 production in hPBMCs. Cells were pretreated with BA (0.1–10 µg·mL−1 culture medium) 30 min prior to LPS (1 µg·mL−1 culture medium) stimulation. The hPBMCs were cultured for 16 h. After 16 h, PGE2 was measured in the culture medium as described in the Methods. (B) Cell lysates were subjected to Western blotting with an anti-COX-2 or β-actin antibody. The relative abundance of each band to its own β-actin was quantified, and the LPS control levels (1 µg LPS + 0 µg BA) were set to 100%. Results are expressed as mean ± SEM; n = 6. #Significantly different from basal values (P < 0.05). *Significantly different from LPS control (P < 0.05). BA, betulinic acid; hPBMCs, human peripheral blood mononuclear cells; LPS, lipopolysaccharide; PGE2, prostaglandin E2.
LPS induction also caused increased COX-2 protein expression at 16th hour post-LPS induction. Treatment with BA (1 µg and 2 µg·mL−1 culture medium) significantly reduced LPS-induced COX-2 up-regulation by 3.6- and sixfold respectively (Figure 2B).
Effect of MAPK, PI3-K inhibitors and BA on LPS-induced PGE2 release
To address the role of MAPK and PI3-K pathways on LPS-induced PGE2 release in hPBMCs, the cells were pretreated with pharmacological inhibitors of MAPK pathway viz. SB203580 (p38 inhibitor; 25 µM), PD98059 (ERK inhibitor; 10 µM) and SP600125 (JNK inhibitor; 10 µM) (Figure 3). PD98059 brought about a 15-fold decrease in LPS-induced PGE2 production whereas SP600125, a JNK inhibitor, or SB203580, a p38 inhibitor, did not exert significant effects. The addition of BA into the medium caused a significant reduction in PGE2 production up to ninefold. The addition of the PI3-K inhibitor, LY294002 (10 µM), into the medium also caused a substantial decrease in PGE2 production of up to 20-fold. Taken together, these data indicate that the down-regulation of LPS-induced PGE2 production is mediated by a LY294002- and PD98059-sensitive pathway.
Figure 3.
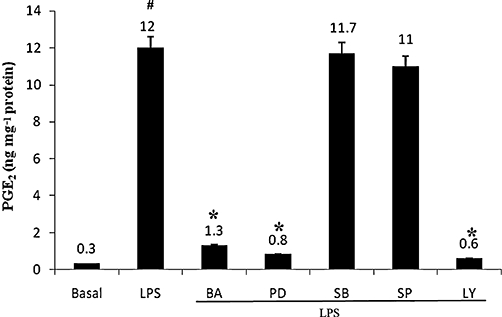
Effects of MAPK and Akt inhibitors and BA on LPS-induced PGE2 production in hPBMCs. Cells were pretreated with pharmacological inhibitors extracellular regulated kinase inhibitor (PD98059; 10 µM), p38 inhibitor (SB203580; 25 µM), JNK inhibitor (SP600125; 10 µM), PI-3 K inhibitor (LY294002; 10 µM) or BA (2 µg·mL−1 culture medium) 30 min prior to stimulation. hPBMCs were stimulated with LPS (1 µg·mL−1 culture medium) for 16 h. After 16 h, PGE2 was measured in the culture supernatants as described in Methods. Data are presented as mean ± SEM; n = 6. #Significantly different from basal values (P < 0.05). *Significantly different from LPS control (P < 0.05). BA, betulinic acid; hPBMCs, human peripheral blood mononuclear cells; LPS, lipopolysaccharide; PGE2, prostaglandin E2.
BA inhibits LPS-induced phosphorylation of Akt and ERK1/2 in hPBMCs
The administration of LPS to hPBMCs dramatically activated Akt and the MAPK family proteins ERK1/2 and p38 MAPK (Figure 4). The addition of BA significantly inhibited LPS-induced phosphorylation of ERK1/2 and Akt (Figure 4A,C), whereas the phosphorylation of p38 MAPK was unaffected by BA pretreatment (Figure 4B). BA alone (0.1–2 µg·mL−1 culture medium) did not elicit Akt nor ERK 1/2 phosphorylation (Figure 5).
Figure 4.
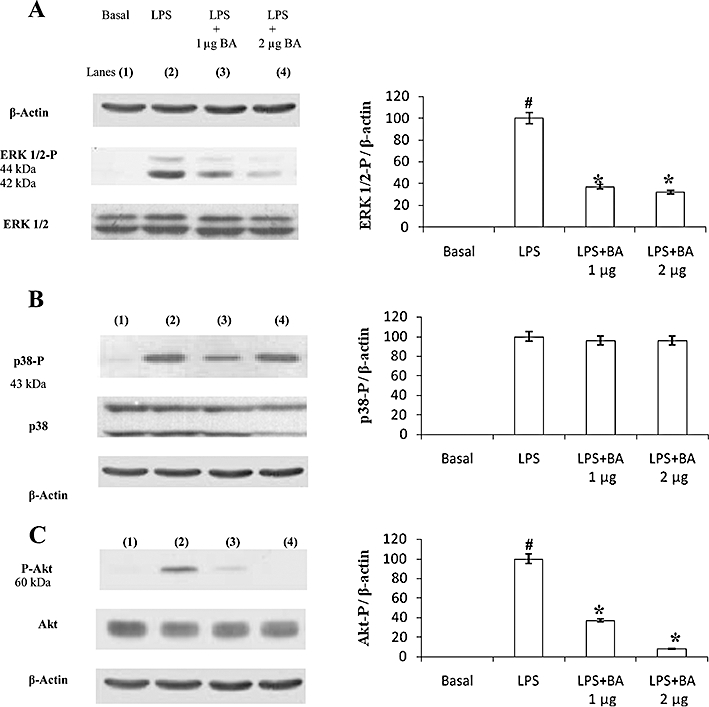
BA inhibits LPS-induced phosphorylation of extracellular regulated kinase (ERK) 1/2 (A) and Akt (C) but not p38 (B) in hPBMCs. hPBMCs were pretreated with BA (1–2 µg·mL−1 culture medium) for 30 min in RPMI-1640 containing 5% autologous serum. LPS (1 µg·mL−1 culture medium) was then added and incubated for an additional 30 min. Cell lysates were subjected to Western blotting with their relevant antibodies. Photographs of chemiluminescent detection of the blots, which were representative of three independent experiments, are shown. The relative abundance of each band to its own β-actin was quantified. Data are presented as mean ± SEM; n = 6. #Significantly different from basal values (P < 0.05). *Significantly different from LPS control (P < 0.05). BA, betulinic acid; hPBMCs, human peripheral blood mononuclear cells; LPS, lipopolysaccharide.
Figure 5.
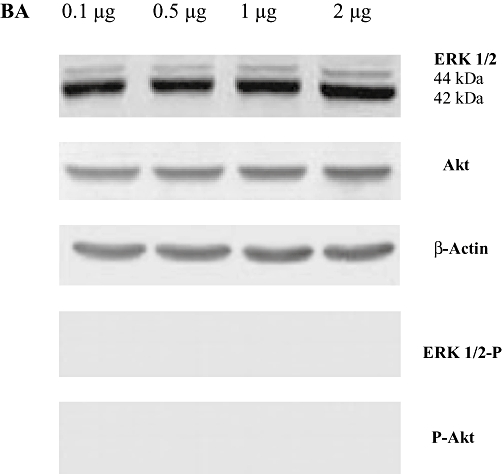
BA alone does not induce phosphorylation of extracellular regulated kinase (ERK) 1/2 or Akt in normal cells. hPBMCs were pretreated with different concentrations of BA (0.1–2 µg·mL−1 culture medium) for 30 min in RPMI-1640 containing 5% autologous serum. LPS (1 µg·mL−1 culture medium) was then added and incubated for an additional 30 min. Cell lysates were subjected to Western blotting with their relevant antibodies. Photographs of chemiluminescent detection of the blots, which were representative of three independent experiments, are shown. The relative abundance of each band to its own β-actin was quantified. Data are presented as mean ± SEM; n = 6. BA, betulinic acid; hPBMCs, human peripheral blood mononuclear cells; LPS, lipopolysaccharide.
BA inhibits LPS-induced NF-κB signalling in hPBMCs
In order to determine whether IκBα stabilization by BA was indeed due to its inhibitory effects on IκBα phosphorylation, Western blot analysis was conducted. LPS increased the levels of phospho-IκBα and the administration of BA to cells significantly prevented the LPS-induced increases in phospho-IκBα levels (Figure 6A). Using EMSA, it was demonstrated that BA significantly prevented LPS-induced NF-κB nuclear translocation (Figure 6B).
Figure 6.
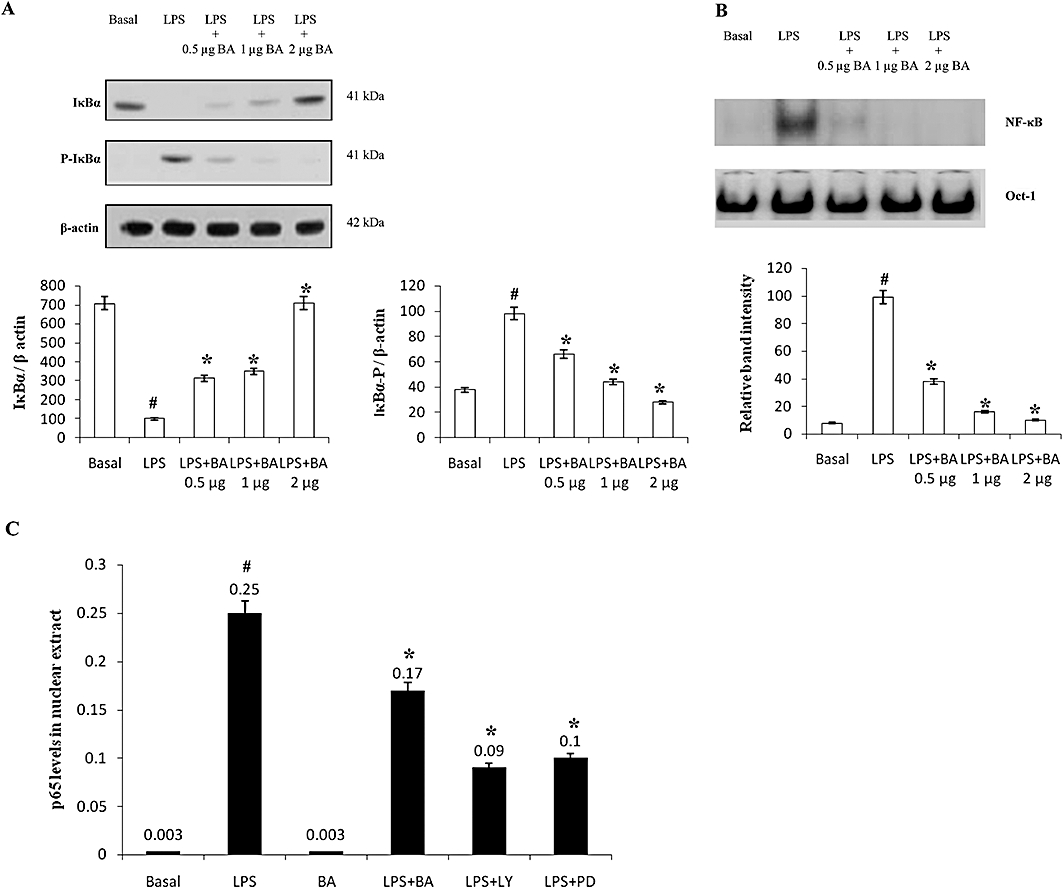
BA inhibits LPS-induced NF-κB signalling in hPBMCs. hPBMCs were pretreated with BA (0.5–2 µg·mL−1 culture medium) for 30 min in RPMI-1640 containing 5% autologous serum. LPS (1 µg·mL−1 culture medium) was then added and incubated for an additional 30 min. (A) Western blots showing that BA inhibits LPS-induced phosphorylation of IκBα. (B) Electrophoretic mobility shift assay showing that BA inhibits LPS-induced NF-κB nuclear translocation. Oct-1 electrophoretic mobility shift assay served as a loading control. (C) Effect of BA (2 µg·mL−1 culture medium), PD98059 (25 µM) and LY294002 (10 µM) on LPS-induced p65 nuclear translocation. Data are presented as mean ± SEM; n = 6. #Significantly different from basal values (P < 0.05). *Significantly different from LPS control (P < 0.05). BA, betulinic acid; hPBMCs, human peripheral blood mononuclear cells; LPS, lipopolysaccharide; NF-κB, nuclear factor κB.
To illustrate the modulatory roles of MAPK and PI3-K on LPS-induced NF-κB nuclear translocation, cells were pretreated with PD98059, LY294002 or BA, and the level of p65 in the nucleus was assayed at the 16th hour (time point when maximum LPS-induced PGE2 production was observed). It was observed that LPS induction caused significant nuclear translocation of p65 NF-κB subunit while BA alone did not induce any p65 NF-κB nuclear translocation. In LPS-induced cell cultures, BA caused a 1.4-fold inhibition of p65 NF-κB nuclear translocation whereas inhibitors like PD98059 and LY294002 caused 2.7- and 2.5-fold inhibition of p65 NF-κB nuclear translocation (Figure 6C).
Effect of BA on the cytotoxicity and ROS production in hPBMCs
To determine if BA exerted any cytotoxicity, LDH was assayed. LDH release markedly increased when cells were cultured in medium containing LPS and this release was inhibited by BA (Figure 7A). BA alone did not induce any LDH secretion.
Figure 7.
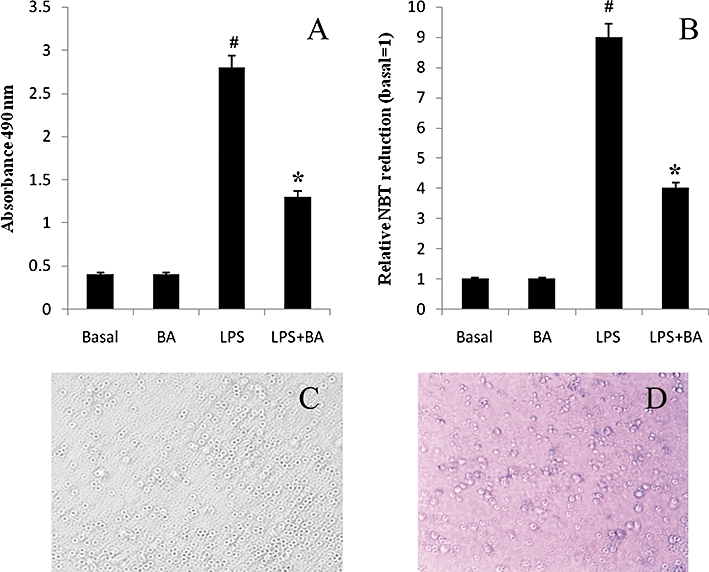
Effect of BA on LPS-induced lactate dehydrogenase release, ROS generation and cell viability (A) BA is not cytotoxic to hPBMCs. (B) BA (2 µg·mL−1 culture medium) inhibited LPS-induced ROS production. ROS production was measured by nitroblue tetrazolium reduction. (C) Normal mononuclear cells. (D) Trypan blue assay showing that BA is non-cytotoxic to normal cells. Data are presented as mean ± SEM; n = 6. #Significantly different from basal values (P < 0.05). *Significantly different from LPS control (P < 0.05). BA, betulinic acid; hPBMCs, human peripheral blood mononuclear cells; LPS, lipopolysaccharide; ROS, reactive oxygen species.
We investigated the antioxidant effects of betulinic acid by measuring NBT reduction. Betulinic acid significantly decreased the levels of LPS-induced ROS in hPBMCs (Figure 7B). Through trypan blue assay, it was evident that BA did not induce any cell death (Figure 7C,D).
Effect of BA on survival in a mouse model of sepsis and its effect on PGE2 production and MPO activity
To test if BA can increase survival rate in a mouse model of sepsis, Swiss mice were injected with vehicle (0.1% DMSO) or BA (20 mg·kg−1 i.p.) before LPS (32 mg·kg−1 i.p.). The dose of LPS used was close to LD90 and the dose of BA chosen for lethality studies was the one that showed most promise in pilot experiments (results not shown). Mice receiving DMSO or BA did not die, whereas only 8% of the mice receiving LPS survived after 7 days. Pretreatment with BA increased survival rate throughout the observation period, with 40% of the mice pretreated with BA remaining alive after 7 days (Figure 8).
Figure 8.
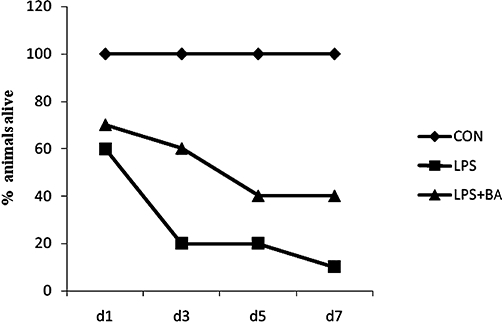
BA protects against LPS-induced toxicity. Mice were pretreated with (i) vehicle [DMSO; control (CON; n = 6]; (ii) LPS (32 mg·kg−1 i.p.; n = 50); and (iii) BA followed by LPS (LPS = 32 mg·kg−1; BA = 20 mg·kg−1 i.p.; n = 60). Mice were observed for 7 days. BA, betulinic acid; hPBMCs, human peripheral blood mononuclear cells; LPS, lipopolysaccharide.
We have previously shown that BA inhibits IL-6 production in rat blood mononuclear cells in response to E. coli LPS (serotype 0B26). Here we demonstrated that LPS augments PGE2 production in liver and lungs of mice and this increase was prevented by BA pretreatment. Results shown in Figure 9 demonstrate that pretreatment with BA significantly inhibited LPS-induced PGE2 level in liver and lungs. BA treatment alone did not elicit any significant amounts of PGE2 in the organs studied.
Figure 9.
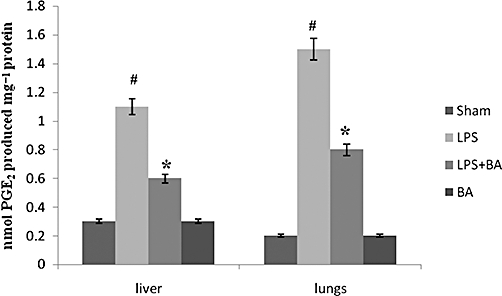
Effect of BA on LPS-induced endotoxic shock and PGE2 production in mice. Mice were pretreated with BA (20 mg·kg−1 in 0.1% DMSO), injected with LPS and then monitored for a period of 7 days. On day 7, mice (which survived) were killed; liver and lungs were removed as described in Methods. PGE2 was extracted from tissue homogenates using SPE columns and the elutes obtained were used for PGE2 quantification. Results are expressed as mean ± SEM (n = 6–8 observations). #Significantly different from normal control (P < 0.05). *Significantly different from LPS control (P < 0.05). BA, betulinic acid; LPS, lipopolysaccharide; PGE2, prostaglandin E2.
To test the functional relevance of the reduced PGE2 production in the face of BA pretreatment, we studied the inflammatory infiltration in organs that constitute major targets of LPS. MPO, an index of neutrophil infiltration, was found significantly increased in the liver and lungs of LPS-induced mice (Figure 10). Histopathological analysis of H&E-stained tissue sections from LPS-treated mice revealed a marked peribronchiolar and perivascular infiltrations of leucocytes in the lung and around portal or central spaces in the liver (Figure 11B,F). Pretreatment with BA abolished the accumulation of leucocytes in both organs, as evidenced by decreased MPO activity and histopathological analysis (Figures 10 and 11B,F). However, BA did not prevent more subtle changes in tissue architecture caused by LPS, such as the thickening of alveolar septa and the hyperplasia of bronchial epithelium in the lung or the hyperplasia of vascular endothelium in the liver, as well as hyperplasia of the epithelium in the biliary ducts (Figure 11C,G). Treatment with BA alone did not induce any change in tissue structure, indicating its non-toxic effects (Figure 11D,H).
Figure 10.
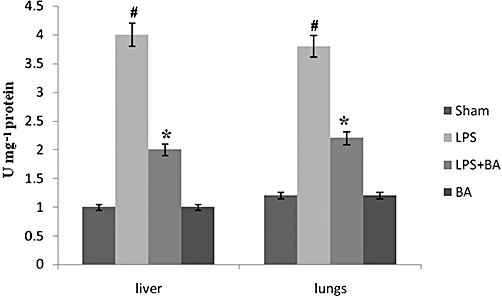
BA reduces myeloperoxidase activity in liver and lungs of LPS-induced mice. Mice were pretreated with BA (20 mg·kg−1 in 0.1% DMSO), injected with LPS and then monitored for a period of 7 days. On day 7, mice (which survived) were killed; liver and lungs were removed as described in Methods. Results are expressed as mean ± SEM (n = 6–8 observations). #Significantly different from normal control (P < 0.05). *Significantly different from LPS control (P < 0.05). BA, betulinic acid; LPS, lipopolysaccharide.
Figure 11.
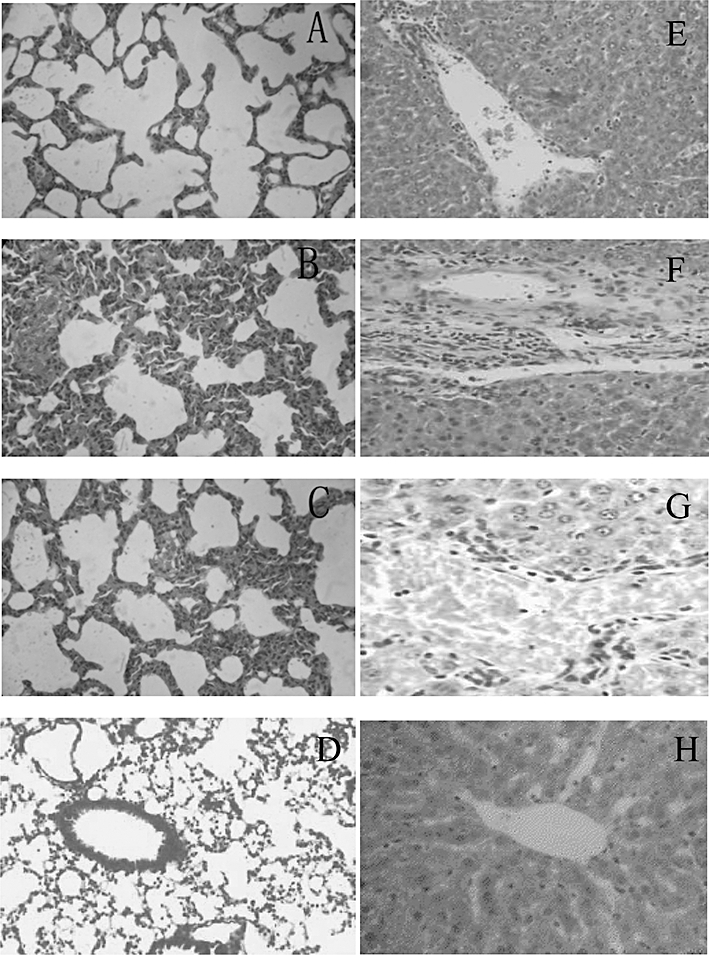
Betulinic acid abolishes leucocyte infiltration in the lung and liver of LPS-treated animals. Representative photomicrographs of hematoxylin and eosin-stained sections from control (A, E), LPS (B, F), or betulinic acid + LPS (C, G)-treated mice. A, B, C and D are lung sections, and E, F and G are liver sections. LPS, lipopolysaccharide.
Discussion and conclusions
For centuries, dietary and medicinal plants have been employed as anti-inflammatory remedies, and recently identifications of their active components and investigations of their mechanisms of action have been conducted by multiple investigative groups. As the dysregulated activation of inflammation has been shown to stimulate the initiation and promotion of various diseases, the primary purpose of the present study was to determine the cell signalling mechanisms through which BA, a potent anti-inflammatory agent, inhibits LPS-induced pro-inflammatory mediators like COX-2 and PGE2, employing activated hPBMCs and endotoxin-induced mouse as model systems.
The control of COX-2 transcription and thereby PGE2 production is mediated by NF-κB, which is an important proximal mechanism for the production of inflammatory mediators in LPS-activated mononuclear cells (Janssen-Heininger et al., 2000). NF-κB is maintained in a latent form in the cytoplasm, where it exists in complex with IκBs (Barnes and Karin, 1997; Li and Verma, 2001). On IκB kinase-dependent phosphorylation and subsequent ubiquitination and degradation, free NF-κB is translocated to the nucleus where it binds to the consensus sequence of pro-inflammatory genes and evokes its expression (Janssen-Heininger et al., 2000). Earlier works by Takada and Aggarwal (2003) demonstrated that BA prevents carcinogen-induced NF-κB activation through the inhibition of IκBα kinase and p65 phosphorylation, and abrogates COX-2 expression in carcinoma cell lines. It was recently reported that BA effectively attenuated TNF-α-induced NF-κB activation in human umbilical vein endothelial cells (Yoon et al., 2010). Another previous report stated that BA suppresses the expression of COX-2 in LPS-stimulated RAW 264.7 cells (Yun et al., 2003), but no information regarding the cell signalling cascades were reported. In this work, we demonstrated that BA inhibits COX-2 and PGE2 production through NF-κB, ERK MAPK and Akt pathways. Here we present results that BA caused dose-dependent attenuation of LPS-induced phosphorylation of IκBα in normal cells. By the inhibition of IκBα, BA prevents LPS-induced nuclear translocation of NF-κB (EMSA) as shown in Figure 6B, which was also confirmed earlier by p65 NF-κB EIA analysis (Viji et al., 2010).
Most of the works on BA have been targeted to its effect on carcinoma cell lines and very few reports point out the effect of BA in countering oxidative stress or inflammatory responses in normal cells. We report here that BA inhibits LPS-induced NF-κB nuclear translocation, in a similar manner to that observed in cancer cells. However, the effect of this triterpenoid on other cell signalling cascades differs in normal cells than that observed in cancer cells. Previous studies demonstrated that BA activates p38, JNK MAPKs and Akt to induce programmed cell death in human melanoma cells and leaves the anti-apoptotic ERK MAPK cascade unaffected (Sezler et al., 2002; Tan et al., 2003). However, in this study, normal cells like hPBMCs have responded differently. BA dampened LPS-induced ERK1/2 and Akt phosphorylation in hPBMCs without modulating p38. At the same time, BA treatment alone did not elicit the phosphorylation of ERK, p38 or Akt in hPBMCs. It has been noted that the activation of MAPK phosphorylation transduces signals for the activation of the transcription of NF-κB-mediated pro-inflammatory mediator production (Ajizian et al., 1999; Lee and Lim, 2008). Here in the present study, the incubation of cells with PD98059 and LY29004 significantly inhibited PGE2 production as well as decreased p65 NF-κB nuclear translocation, indicating that ERK MAPK and PI3K/Akt cascades operate with NF-κB in order to inhibit LPS-induced PGE2 production. The extent of p65 NF-κB nuclear translocation was less in the presence of PD98059 and LY29004, indicating that ERK1/2 and Akt signalling have predominant roles in reinforcing NF-κB-mediated PGE2 production.
Akt has been shown to enhance the degradation of IκB, which results in the activation of NF-κB in Jurkat T cells (Kane et al., 1999). Again, the transcription factor CCAAT/enhancer-binding protein beta, which is involved downstream Akt activation pathway, is essential for COX-2 gene regulation (Reddy et al., 2000; Wang et al., 2000; Gorgoni et al., 2001; Piwien-Pilipuk et al., 2001). Akt is a downstream target of NF-κB as p65 overexpression results in higher Akt phosphorylation (Meng et al., 2010). The same was observed in the present study. LPS-induced phosphorylation of Akt coincided with IκB phosphorylation of NF-κB at the 16th hour post-endotoxin stimulation, which was significantly inhibited by BA. Taken together, the inhibition of ERK1/2 and Akt activation contributes to the inhibition of NF-κB signalling. Here, BA countered the inflammatory response to LPS by inhibiting COX-2 up-regulation and PGE2 production, and preventing the phosphorylation of IκB and ERK and Akt pathways (anti-apoptotic pathways) and thereby improving the survival rate of cells. Further in-depth study will be required to establish the role of other signalling molecules involved in the anti-inflammatory effects of BA in normal cells as opposing mechanisms tend to operate in malignant cells (Sezler et al., 2002; Tan et al., 2003).
Free radicals have long been implicated as mediators of inflammation and oxidative stress (Cnubben et al., 2001). In this study, the addition of LPS caused a significant increase in ROS production followed by the loss of cell viability, as evidenced by the increased LDH release into the medium. Addition of BA alone into the culture (at the specified concentration) did not have any impact on LDH release or ROS formation, while pretreatment with BA reversed LPS-induced LDH release and ROS formation. In contrast to the reports on BA eliciting ROS generation in human melanoma cells (Tan et al., 2003), BA did not induce cell death (as evidenced by Trypan blue assay) or ROS generation in our experiments.
The major finding of the present study is that the triterpenoid BA protects mice from LPS-induced lethal toxicity. Earlier, we demonstrated that BA significantly reduced LPS-induced IL-6 expression in rats in vivo (Viji et al., 2010). Here we show that BA protects mouse against a high dose of LPS (32 mg·kg−1) and at the same time inhibits LPS-induced mortality. To determine the anti-inflammatory effects of BA in vivo, we determined the ability of BA to decrease PGE2 production in mice challenged with LPS. Interestingly, BA prevented LPS-induced PGE2 production in the liver and lungs, two of the most susceptible organs affected by this endotoxin. Moreover a good correlation occurred between attenuation of PGE2 production and decreased ROS formation in hPBMCs indicating the potential of BA both as an antioxidant and as an anti-inflammatory agent. At the same time, to figure out the role of BA against neutrophil infiltration, MPO was assayed in the tissues. The activity of MPO, an indicator of neutrophil infiltration was significantly increased in LPS-induced mice. The immune reactivity of MPO in vivo after injection of LPS has already been demonstrated (Watanabe et al., 2002). In the current study, MPO activity was observed augmented in the liver and lung tissues of LPS induced mice. The increased neutrophil infiltration was also evident from histopathological data. Blood-borne monocytes are the facilitators of neutrophil recruitment and coordinated neutrophil and monocyte recruitment is a characteristic feature of inflammatory responses especially in the lungs (Maus et al., 2003). BA reduced infiltration of leucocytes as evidenced by the decreased MPO activity and also by reversal of cell structural changes in the lungs and around the portal vein in the liver. Histopathological data also indicate that BA improved the histology of lung and liver tissue as evidenced by the decreased cell infiltration, emphysema formation and cell degeneration. Perhaps the ability of BA to inhibit mononuclear extravasation may have contributed to decreased neutrophil infiltration, though studies to investigate the role of BA against neutrophils need to be done in detail before a conclusion can be drawn in this regard. The results, however, are in line with our results in vitro on the protective effects of BA and strengthen our hypothesis that triterpenoids like BA can be used to block cascades triggered by LPS in vivo.
In conclusion, the results of this study demonstrate that BA inhibits the inflammatory responses in LPS-stimulated hPBMCs. The anti-inflammatory effects of BA may be mediated by decreased COX-2 protein production and PGE2 formation via the modulation of Akt and ERK and the subsequent down-regulation of NF-κB signalling. In addition, BA markedly reduced LPS-induced PGE2 production in mouse liver and lungs. Thus, this study provides some of the experimental basis for the use of BA as a potential anti-inflammatory agent.
Acknowledgments
The authors express their sincere gratitude to Prof P.R. Sudhakaran (Former Head of the Department of Biochemistry, University of Kerala), Academic Coordinator and Controller of Examinations, Central University of Kerala, for his guidance. Help rendered from the Department of Applied Chemistry, Cochin University of Science Technology and Environment and Rajiv Gandhi Centre for Biotechnology, Thiruvananthapuram is gratefully acknowledged. Funding from Kerala State Council for Science, Technology and Environment, Thiruvananthapuram, Kerala, India is also acknowledged.
Glossary
Abbreviations
- COX
cyclooxygenase
- ERK
extracellular regulated kinase
- LDH
lactate dehydrogenase
- LPS
lipopolysaccharide
- NF-κB
nuclear factor κ B
- PGE2
prostaglandin E2
- ROS
reactive oxygen species
Conflict of interest
The authors declare that there are no conflicts of interest.
Supplementary material
Supporting Information: Teaching Materials; Figs 1–11 as PowerPoint slide.
References
- Ajizian SJ, English BK, Meals EA. Specific inhibitors of p38 and extracellular signal-regulated kinase mitogen-activated protein kinase pathways block inducible nitric oxide synthase and tumor necrosis factor accumulation in murine macrophages stimulated with lipopolysaccharide and interferon-γ. J Infect Dis. 1999;179:939–944. doi: 10.1086/314659. [DOI] [PubMed] [Google Scholar]
- Balkwill F, Mantovani A. Inflammation and cancer: back to Virchow? Lancet. 2001;357:539–545. doi: 10.1016/S0140-6736(00)04046-0. [DOI] [PubMed] [Google Scholar]
- Barnes PJ, Karin M. Nuclear factor-κB: a pivotal transcription factor in chronic inflammatory diseases. N Engl J Med. 1997;336:1066–1071. doi: 10.1056/NEJM199704103361506. [DOI] [PubMed] [Google Scholar]
- Baud V, Karin M. Is NF-κB a good target for cancer therapy? Hopes and pitfalls. Nat Rev Drug Discov. 2009;8:33–40. doi: 10.1038/nrd2781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellacosa A, Testa JR, Staal SP, Tsichlis PN. A retroviral oncogene, Akt, encoding a serine-threonine kinase containing an SH2-like region. Science. 1991;254:274–277. doi: 10.1126/science.254.5029.274. [DOI] [PubMed] [Google Scholar]
- Beraud C, Henzel WJ, Baeuerle PA. Involvement of regulatory and catalytic subunits of phosphoinositide 3-kinase in NF-κB activation. Proc Natl Acad Sci USA. 1999;96:429–434. doi: 10.1073/pnas.96.2.429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradley PP, Priebat DA, Christensen RD, Rothstein G. Measurement of cutaneous inflammation: estimation of neutrophil content with an enzyme marker. J Invest Dermatol. 1982;78:206–209. doi: 10.1111/1523-1747.ep12506462. [DOI] [PubMed] [Google Scholar]
- Chatterji N, Rastogi RP, Dhar ML. Chemical examination of Bacopa monniera Wettst: part I-isolation of chemical constituents. Indian J Chem. 1963;1:212–217. [Google Scholar]
- Chowdhuri DK, Pannar D, Kakkar P. Anti-stress effects of bacosides of Bacopa monnieri: modulation of Hsp70 expression, superoxide dismutase and cytochrome P450 activity in rat brain. Phytother Res. 2002;16:639–645. doi: 10.1002/ptr.1023. [DOI] [PubMed] [Google Scholar]
- Chowdhury AR, Mandal S, Mittra B, Sharma S, Mukhopadhyay S, Majumder HK. Betulinic acid, a potent inhibitor of eukaryotic topoisomerase I: identification of the inhibitory step, the major functional group responsible and development of more potent derivatives. Med Sci Monit. 2002;8:254–265. [PubMed] [Google Scholar]
- Cnubben NHP, Rietjens IMCM, Wortelboer H, Van-Zanden J, Van Bladeren PJ. The interplay of glutathione related processes in antioxidant defense. Environ Toxicol Pharmacol. 2001;10:141–152. doi: 10.1016/s1382-6689(01)00077-1. [DOI] [PubMed] [Google Scholar]
- Furukawa S, Fujita T, Shimabukuro M, Iwaki M, Yamada Y, Nakajima Y. Increased oxidative stress in obesity and its impact on metabolic syndrome. J Clin Invest. 2004;114:1752–1761. doi: 10.1172/JCI21625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Germain M, Gagnon V, Parent S, Asselin E. Regulation of COX-2 protein expression by Akt in endometrial cancer cells is mediated through NF-κB/IκB pathway. Mol Cancer. 2004;3:17–26. doi: 10.1186/1476-4598-3-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorgoni B, Caivano M, Arizmendi C, Poli V. The transcription factor C/EBPβ is essential for inducible expression of the COX-2 gene in macrophages but not in fibroblasts. J Biol Chem. 2001;276:40769–40777. doi: 10.1074/jbc.M106865200. [DOI] [PubMed] [Google Scholar]
- Haque ME, Shekhar HU, Mohamad AU, Rahman H, Islam AMI, Hossain MS. Triterpenoids from the stem bark of Avicennia officinalis. Dhaka Univ J Pharm Sci. 2006;5:53–57. [Google Scholar]
- Huch HY, Pearce SF, Tersner JM, Schindler IL, Silverstein RL. Regulated expression of CD36 in foam cell formation. Blood. 1996;87:2025. [PubMed] [Google Scholar]
- Janssen-Heininger YM, Poynter ME, Baeuerle PA. Recent advances towards understanding redox mechanisms in the activation of nuclear factor-κB. Free Radic Biol Med. 2000;28:1317–1327. doi: 10.1016/s0891-5849(00)00218-5. [DOI] [PubMed] [Google Scholar]
- Jones PF, Jakubowicz T, Pitossi FJ, Maurer F, Hemmings BA. Molecular cloning and identification of a serine/threonine protein kinase of the second-messenger subfamily. Proc Natl Acad Sci USA. 1991;88:4171–4175. doi: 10.1073/pnas.88.10.4171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kane LP, Shapiro VS, Stokoe D, Weiss A. Induction of NF-κB by the Akt/PKB kinase. Curr Biol. 1999;9:601–604. doi: 10.1016/s0960-9822(99)80265-6. [DOI] [PubMed] [Google Scholar]
- Karin M, Ben-Neriah Y. Phosphorylation meets ubiquitination: the control of NF-κB activity. Annu Rev Immunol. 2001;18:621–663. doi: 10.1146/annurev.immunol.18.1.621. [DOI] [PubMed] [Google Scholar]
- Lee SJ, Lim KT. Inhibitory effect of 30 kDa phytoglycoprotein on expression of TNF-α and COX-2 via activation of PKC-α and ERK 1/2 in LPS-stimulated RAW 264.7 cells. Mol Cell Biochem. 2008;317:151–159. doi: 10.1007/s11010-008-9843-0. [DOI] [PubMed] [Google Scholar]
- Lee YM, Seon MR, Cho HJ, Kin J, Park HJY. Benzyl isothiocyanate exhibits anti-inflammatory effects in murine macrophages and in mouse skin. J Mol Med. 2009;87:1251–1261. doi: 10.1007/s00109-009-0532-6. [DOI] [PubMed] [Google Scholar]
- Li Q, Verma IM. NF-κB regulation in the immune system. Nat Rev Immunol. 2001;2:725–734. doi: 10.1038/nri910. [DOI] [PubMed] [Google Scholar]
- Maus UA, Waelsch K, Kuziel WA, Delbeck T, Mack M, Blackwell TS, et al. Monocytes are potent facilitators of alveolar neutrophil emigration during lung inflammation: role of the CCL2-CCR2 axis. J Immunol. 2003;170:3273–3278. doi: 10.4049/jimmunol.170.6.3273. [DOI] [PubMed] [Google Scholar]
- Meng F, Liu L, Chin PC, D'Mello SR. Akt is a downstream target of NF-κB. J Biol Chem. 2002;277:29674–29680. doi: 10.1074/jbc.M112464200. [DOI] [PubMed] [Google Scholar]
- Meng F, Liu L, Chin PC, D'Mello SR. Akt is a downstream target of NF-κB. J Biol Chem. 2010;277:29674–29680. doi: 10.1074/jbc.M112464200. [DOI] [PubMed] [Google Scholar]
- Mukherjee DG, Dey CD. Clinical trial on Brahmi. J Exp Med Sci. 1996;10:5–9. [PubMed] [Google Scholar]
- Murakami A, Ohigashi H. Targeting NOx, iNOS and COX-2 in inflammatory cells: chemoprevention using food phytochemicals. Int J Cancer. 2007;121:2357–2363. doi: 10.1002/ijc.23161. [DOI] [PubMed] [Google Scholar]
- Piwien-Pilipuk G, Van Mater D, Ross SE, MacDougald OA, Schwartz J. Growth hormone regulates phosphorylation and function of CCAAT/enhancer-binding protein-β by modulating Akt and glycogen synthase kinase-3. J Biol Chem. 2001;276:19664–19671. doi: 10.1074/jbc.M010193200. [DOI] [PubMed] [Google Scholar]
- Plummer SM, Holloway KA, Manson MM, Munks RJ, Kaptein A, Farrow S, et al. Inhibition of cyclooxygenase 2 expression in colon cells by the chemopreventive agent curcumin involves inhibition of NF-κB activation via the NIK/IKK signaling complex. Oncogene. 1999;18:6013–6020. doi: 10.1038/sj.onc.1202980. [DOI] [PubMed] [Google Scholar]
- Reddy ST, Wadleigh DJ, Herschman HR. Transcriptional regulation of the cyclooxygenase-2 gene in activated mast cells. J Biol Chem. 2000;275:3107–3113. doi: 10.1074/jbc.275.5.3107. [DOI] [PubMed] [Google Scholar]
- Ren W, Qin L, Xu V, Cheng N. Inhibition of betulinic acid to growth and angiogenesis of human colorectal cancer cell in nude mice. Chinese-German J Clin Oncol. 2010;9:153–157. [Google Scholar]
- Rohini G, Devi CS. Bacopa monniera extract induces apoptosis in murine sarcoma cells (S-180) Phytother Res. 2008;22:1595–1598. doi: 10.1002/ptr.2515. [DOI] [PubMed] [Google Scholar]
- Sairam K, Rao CV, Babu MD, Goel RK. Prophylactic and curative effects of Bacopa monniera in gastric ulcer models. Phytomedicine. 2001;8:423–430. doi: 10.1078/S0944-7113(04)70060-4. [DOI] [PubMed] [Google Scholar]
- Santos M, Costa JF, Krettli AU, Zalis MG, Maia GL, Sette IM, et al. Anti-malarial activity of betulinic acid and derivatives in vitro against Plasmodium falciparum and in vivo in P. berghei-infected mice. Parasitol Res. 2009;105:275–279. doi: 10.1007/s00436-009-1394-0. [DOI] [PubMed] [Google Scholar]
- Schachter M. COX-2 inhibitors and cardiovascular risk. Br J Cardiol. 2003;10:288–292. [Google Scholar]
- Sezler E, Thallinger C, Hoeller C, Oberkleiner P, Wachek V, Pehamberger H, et al. Betulinic acid induced Mcl-1 expression in human melanoma-mode of action and functional significance. Mol Med. 2002;8:887–884. [PMC free article] [PubMed] [Google Scholar]
- Shacter E, Weitzman SA. Chronic inflammation and cancer. Oncology. 2002;16:217–226. [PubMed] [Google Scholar]
- Singh A, Singh SK. Reversible suppression of spermatogenesis and fertility without toxic effects. Contraception. 2009;79:71–79. [Google Scholar]
- Stephens L, Anderson K, Stokoe D, Erdjument-Bromage H, Painter GF, Holmes AB, et al. Protein kinase B kinases that mediate phosphatidylinositol 3,4,5-trisphosphate-dependent activation of protein kinase B. Science. 1998;279:710–714. doi: 10.1126/science.279.5351.710. [DOI] [PubMed] [Google Scholar]
- Takada V, Aggarwal BB. Betulinic acid suppresses carcinogen-induced NF-κB activation through inhibition of IκBα kinase and p65 phosphorylation: abrogation of cyclooxygenase-2 and matrix metalloprotease-9. J Immunol. 2003;171:3278–3286. doi: 10.4049/jimmunol.171.6.3278. [DOI] [PubMed] [Google Scholar]
- Tan Y, Yu R, Pezzuto JM. Betulinic acid induced programmed cell death in human melanoma cells involves mitogen activated protein kinase activation. Clin Cancer Res. 2003;9:2866–2875. [PubMed] [Google Scholar]
- Vijayan V, Helen A. Protective activity of Bacopa monniera Linn. on nicotine-induced toxicity in mice. Phytother Res. 2007;21:378–381. doi: 10.1002/ptr.2073. [DOI] [PubMed] [Google Scholar]
- Viji V, Helen A. Inhibition of lipoxygenases and cyclooxygenase-2 enzymes by extracts isolated from Bacopa monniera (L.) Wettst. J Ethnopharmacol. 2008;118:305–311. doi: 10.1016/j.jep.2008.04.017. [DOI] [PubMed] [Google Scholar]
- Viji V, Shobha B, Kavitha SK, Ratheesh M, Kripa K, Helen A. Betulinic acid isolated from Bacopa monniera (L.) Wettst suppresses lipopolysaccharide stimulated interleukin-6 production through modulation of nuclear factor-κB in peripheral blood mononuclear cells. Int Immunopharmacol. 2010;10:843–849. doi: 10.1016/j.intimp.2010.04.013. [DOI] [PubMed] [Google Scholar]
- Vohora D, Pal SN, Pillai KK. Protection from phenytoin-induced cognitive deficit by Bacopa monniera, a reputed Indian nootropic plant. J Ethnopharmacol. 2000;71:383–390. doi: 10.1016/s0378-8741(99)00213-5. [DOI] [PubMed] [Google Scholar]
- Wang L, Shao J, Muhlenkamp P, Liu S, Klepcyk P, Ren J, et al. Increased insulin receptor substrate-1 and enhanced skeletal muscle insulin sensitivity in mice lacking CCAAT/enhancerbinding protein-β. J Biol Chem. 2000;275:14173–14181. doi: 10.1074/jbc.m000764200. [DOI] [PubMed] [Google Scholar]
- Wang Y, Chang J, Li YC, Li YS, Shyy JY, Chien S. Shear stress and VEGF activate IKK via the Flk-1/Cbl/Akt signaling pathway. Am J Physiol Heart Circ Physiol. 2004;286:685–692. doi: 10.1152/ajpheart.00237.2003. [DOI] [PubMed] [Google Scholar]
- Watanabe K, Jinnouchi K, Yagi T. Immunoreactivity for myeloperoxidase in the vestibule after the injection of bacterial lipopolysaccharide into the middle ear. Auris Nasus Larynx. 2002;29:241–245. doi: 10.1016/s0385-8146(02)00002-0. [DOI] [PubMed] [Google Scholar]
- Yoon JJ, Lee YJ, Kim JS, Kang DG, Lee HS. Protective role of betulinic acid on TNF-α-induced cell adhesion molecules in vascular endothelial cells. Biochem Biophys Res Commun. 2010;391:96–101. doi: 10.1016/j.bbrc.2009.11.009. [DOI] [PubMed] [Google Scholar]
- Yun Y, Han S, Park E, Yim D, Lee S, Lee C, et al. Immunomodulatory activity of betulinic acid by producing pro-inflammatory cytokines and activation of macrophages. Archives Pharmacol Res. 2003;26:1087–1095. doi: 10.1007/BF02994763. [DOI] [PubMed] [Google Scholar]
- Zuchermann SH, Bendele AM. Regulation of serum tumor necrosis factor alpha in glucocorticoid sensitive and resistant rodent endotoxin shock models. Infect Immun. 1989;57:3009–3013. doi: 10.1128/iai.57.10.3009-3013.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.