

Abstract
BACKGROUND AND PURPOSE
Myocardial automatism and arrhythmias may ensue during strong sympathetic stimulation. We sought to investigate the relevant types of adrenoceptor, as well as the role of phosphodiesterase (PDE) activity, in the production of catecholaminergic automatism in atrial and ventricular rat myocardium.
EXPERIMENTAL APPROACH
The effects of adrenoceptor agonists on the rate of spontaneous contractions (automatic response) and the amplitude of electrically evoked contractions (inotropic response) were determined in left atria and ventricular myocytes isolated from Wistar rats.
KEY RESULTS
Catecholaminergic automatism was Ca2+-dependent, as it required a functional sarcoplasmic reticulum to be exhibited. Although both α- and β-adrenoceptor activation caused inotropic stimulation, only β1-adrenoceptors seemed to mediate the induction of spontaneous activity. Catecholaminergic automatism was enhanced and suppressed by β2-adrenoceptor blockade and stimulation respectively. Inhibition of either PDE3 or PDE4 (by milrinone and rolipram, respectively) potentiated the automatic response of myocytes to catecholamines. However, only rolipram abolished the attenuation of automatism produced by β2-adrenoceptor stimulation.
CONCLUSIONS AND IMPLICATIONS
α- and β2-adrenoceptors do not seem to be involved in the mediation of catecholaminergic stimulation of spontaneous activity in atrial and ventricular myocardium. However, a functional antagonism of β1- and β2-adrenoceptor activation was identified, the former mediating catecholaminergic myocardial automatism and the latter attenuating this effect. Results suggest that hydrolysis of cAMP by PDE4 is involved in the protective effect mediated by β2-adrenoceptor stimulation.
Keywords: α-adrenoceptor, β1-adrenoceptor, β2-adrenoceptor, spontaneous activity, sarcoplasmic reticulum, calcium, arrhythmia, catecholamines, phosphodiesterases
Introduction
Catecholaminergic signalling in the heart is one of the most physiologically relevant mechanisms for increase of cardiac output, which is achieved by chronotropic and inotropic stimulation. Both effects are mediated predominantly by β-adrenoceptors, whereas the contribution of post-junctional α-adrenoceptors to the positive inotropic effect is relatively smaller and variable among species (Brodde and Michel, 1999; Bers, 2001).
β-adrenoceptors are coupled to the Gαs subunit of GTP-binding proteins, which mediates stimulation of adenylate cyclase catalytic activity, leading to accumulation of cAMP and activation of the cAMP-dependent protein kinase (PKA). The latter phosphorylates several substrates in cardiac myocytes, including the sarcolemmal L-type Ca2+ channels, and phospholamban (PLB), a negative regulator of the sarco-endoplasmic reticulum Ca2+-ATPase (SERCA). The overall effect of PKA activation is enhancement of Ca2+ mobilization due to increase in both transmembrane Ca2+ influx and sarcoplasmic reticulum (SR) Ca2+ uptake. The latter contributes not only to accelerate relaxation, but also to increase SR Ca2+ content (Bers, 2001; ter Keurs and Boyden, 2007). Because the SR is the source of most of the Ca2+ that activates contraction in mammalian myocardium, and the fraction of the SR Ca2+ content released at systole is largely dependent on the amplitude of the L-type Ca2+ current (i.e. the trigger for release) and the SR Ca2+ content (Bassani et al., 1995), the systolic increase in cytosolic [Ca2+] is greatly amplified by β-adrenoceptor stimulation (Ginsburg and Bers, 2004), thus increasing contraction amplitude. However, augmented SR Ca2+ load may also result in enhanced diastolic Ca2+ release (Bassani et al., 1997; Shannon et al., 2002), which, by activation of sarcolemmal Ca2+-dependent inward currents, may give rise to delayed afterdepolarizations and spontaneous electrical activity in cardiac cells. While diastolic SR Ca2+ release has been implicated in the generation of pacemaker activity in nodal and subsidiary pacemaker cells from the right atrium, in the Ca2+-overloaded myocardium it may favour development of arrhythmia (Bassani et al., 1997; Bogdanov et al., 2001; Nam et al., 2005; ter Keurs and Boyden, 2007; Fujiwara et al., 2008; Venetucci et al., 2008).
Whereas α-adrenoceptor stimulation has been shown to favour Ca2+ influx/efflux balance due to action potential prolongation, and to produce modest increase in Ca2+ transient amplitude (Brodde and Michel, 1999; Bers, 2001), little is known on the role of this adrenoceptor type in the generation of Ca2+-dependent automatism. On the other hand, although both β1- and β2-adrenoceptor subtypes may exert positive inotropic action by coupling to the Gαs-adenylate cyclase-PKA signalling cascade, evidence shows that the latter subtype may be also coupled to other pathways,such as Gαi-containing G proteins, whose activation may antagonize some of the effects typically related to the cAMP cascade, and thus limit enhancement of cell Ca2+ cycling (Kuschel et al., 1999; Xiao et al., 2006; Siedlecka et al., 2008). Regarding β2-adrenoceptor stimulation, pro-arrhythmic effects have been described in the myocardium from humans (Kaumann and Sanders, 1993), rabbits (DeSantiago et al., 2008) and dogs (Billman et al., 1997; Altschuld and Billman, 2000), whereas Sosunov et al. (2000) reported arrhythmia facilitation by β2-adrenoceptor blockade in canine ventricle. Thus, it is not clear yet whether this subtype is involved in the stimulation of myocardial automatism by catecholamines.
The main goal of the present study was the identification of the adrenoceptor types involved in the mediation of catecholamine-induced spontaneous activity in left atrial and ventricular myocardium. In addition, because of recent evidence suggesting a link between cAMP hydrolysis by phosphodiesterases (PDEs) and arrhythmogenesis (Lehnart et al., 2005; Galindo-Tovar and Kaumann, 2008; Penna and Bassani, 2010), we also investigated whether catecholaminergic automatism was affected by PDE activity.
Methods
Myocardial preparations
Animal care and experimental protocols were in compliance with international and institutional guidelines, and were approved by the Committee on Ethics in Animal Research of the University of Campinas (CEEA/IB/UNICAMP, protocols No. P636-1 and P776-1). Left atria (LA) and left ventricular myocytes (VM) were isolated from adult (4–6-month-old) male Wistar rats (128 animals). Rats were maintained at 23 ± 2°C, under a 12 h : 12 h light–dark cycle, and had free access to food and water. Animals were killed by exsanguination following cerebral concussion.
Left atria were mounted in an organ bath containing Krebs–Henseleit solution at 36.5°C gassed with 95% O2/5% CO2. The preparation was attached to a force transducer (F-60, Narco Biosystems, Houston, TX, USA), and electrically stimulated with voltage pulses (1.2× threshold amplitude, 3 ms duration, 0.5 Hz) through a pair of piercing platinum electrodes. After application of 5 mN preload, the muscle was allowed to equilibrate for 45 min.
Ventricular myocytes were enzymatically isolated via retrograde perfusion at 37°C (Penna and Bassani, 2010). Briefly, after aorta cannulation, the heart was perfused with Ca2+-free Krebs–Henseleit solution for 5 min, and then with the same solution containing collagenase type I (0.5 mg·mL−1, Worthington Biochem, Lakewood, NJ, USA) for 15–20 min. After perfusion with enzyme-free solution for 5 min, the left ventricle was removed, and myocytes were mechanically dissociated, washed and stored at 4°C in glutamate solution. Cells were plated on a collagen-coated perfusion chamber placed on the stage of a videomicroscopy system, and perfused with modified Tyrode's solution at 23 or 36°C. Field electric stimulation (same parameters as for LA, except for the rate at 36°C, which was 1 Hz) was delivered through a pair of platinum electrodes. Cell shortening was measured with a video-edge detector (Centro de Engenharia Biomédica, UNICAMP, pat# MPI-300.834-7) and converted to percentage of the resting cell length (RL).
Concentration–effect curves
In this study, the automatic response to adrenoceptor agonists was considered as the development of spontaneous contractions (SC) in the absence of electrical stimulation (Boer and Bassani, 2004; Carvalho et al., 2006; Penna and Bassani, 2010). For determination of the concentration–effect relationship in LA, we used an experimental protocol developed for multicellular myocardial preparations (Boer and Bassani, 2004), in which the high variability of response among preparations precludes determination of individual curves. Briefly, the number of SCs was computed during three successive 1 min rest periods intercalated by stimulation at 5 Hz for 30 s. This stimulation/rest protocol was applied in the absence of drugs, as well as after 2 min exposure to the agonist. The average SC rate at each agonist concentration was used for determination of the parameters of the concentration–effect curve. For VM experiments, a similar protocol was used, except that the stimulation rate was not increased before the rest periods. In this case, it was possible to obtain concentration–effect curves for individual cells. The SC rate was computed by visual inspection and from force/cell length traces recorded during the rest periods. We have previously reported that, with catecholamine exposure, the SC rate in the absence of electric stimulation is strongly correlated to the rate of extrasystolic contractions during electric stimulation (Penna and Bassani, 2010).
All experiments with LA were performed in the presence of 0.1 µM atropine to avoid interference from endogenously released acetylcholine. Preparations were incubated (LA) or perfused (VM) with receptor antagonists for 50 or 30 min, respectively, before agonist addition and throughout the experiment. Twitch amplitude, measured immediately before application of the stimulation/rest protocol, was used for assessment of the agonist inotropic effect. Preliminary experiments showed that sensitivity, but not the maximum inotropic response to agonists, was affected by the stimulation/rest protocol in LA. Thus, only the maximum response (Rmax) was used for comparisons.
To investigate the role of the SR in the generation of spontaneous activity, VM were treated with 5 µM thapsigargin for 5 min, which inhibits SERCA completely and irreversibly. The effectiveness of the treatment was confirmed by abolition of SR reloading upon electrical stimulation, tested by challenge with 10 mM caffeine (Bassani et al., 1995). In LA, SERCA was inhibited by incubation with 50 µM 2,5-di-(tert-butyl)-1,4-benzohydroquinone (Nakamura et al., 1992) for at least 20 min.
Solutions
The composition of the Krebs–Henseleit solution was (mM): 115 NaCl, 4.5 KCl, 25 NaHCO3, 2.5 CaCl2, 1.2 KH2PO4, 1.2 MgSO4, 11 glucose; pH 7.4. For experiments with catecholamines, 1 mM ascorbic acid was added. The composition of the Tyrode's solution was (mM): 140 NaCl, 6 KCl, 1 CaCl2, 1 MgCl, 5 HEPES, 11 glucose; pH adjusted to 7.4 with NaOH. The composition of the glutamate solution was (mM): 30 KCl, 70 glutamic acid, 1 MgCl2, 10 KH2PO4, 20 taurine, 10 HEPES, 11 glucose; pH adjusted to 7.4 with KOH.
All solutions were prepared with salts of analytical grade and type I water. Stock solutions of the drugs were prepared with water (except for milrinone, rolipram and thapsigargin, which were dissolved in dimethylsuphoxide), stored at −20°C, and diluted immediately before use. Except for thapsigargin (Calbiochem, La Jolla, CA, USA), all drugs were from Sigma Chem Co (St Louis, MO, USA).
Data analysis
A sigmoidal function was adjusted to the concentration–effect relationship for estimation of the Rmax and the negative logarithm of the agonist molar concentration that evoked half-maximal response (pD2). For LA, the standard error (SE) of each parameter was obtained from the curve fitting to the average points. For VM, mean and SE were calculated from values determined in individual cells. These values were similar to the mean parameters of the average curve determined in the same way as for LA, which indicates that both approaches produce equivalent results. For concentration–effect curves determined in VM, n refers to the number of cells studied, which were isolated from at least four hearts for a given experimental protocol.
Data were compared by either Student's t-test for unpaired or paired samples, or by one- or two-way analysis of variance followed by Bonferroni t-test for multiple comparisons, when appropriate. P < 0.05 was considered to show statistical significance.
Results
Under basal conditions, contraction amplitude was 2.98 ± 0.37 mN (n= 81) in LA, and 9.31 ± 0.24% and 6.47 ± 0.41 of RL in VM at 23 (n= 185) and 36°C (n= 59) respectively. In both preparations, the SC rate during stimulatory rest in the absence of agonists was low, especially in LA, in which it was not statistically different from zero (0.2 ± 0.1 SC·min−1 in LA; 0.6 ± 0.1 and 1.1 ± 0.2 SC·min−1 in VM at 23 and 36°C respectively). Such low rates indicate that the stimulation/rest protocol per se is not effective at promoting automatism. None of the adrenoceptor antagonists alone changed significantly the contraction amplitude or SC rate in either LA or VM.
Spontaneous activity was greatly enhanced by exposure to catecholamines (Figure 1A,B). In LA, SCs commonly developed at a quite regular rate (∼3–5 Hz), as described by Boer and Bassani (2004). In VM, spontaneous activity was manifested mainly as propagated contractile waves, or less often as contractions nearly as rapid as electrically evoked twitches. However, after treatment with thapsigargin, not only did twitch amplitude markedly decrease, but also spontaneous activity was totally abolished, even in the presence of high agonist concentrations (Figure 1C) in all of the six studied cells. In LA, di-(tert-butyl)-1,4-benzohydroquinone decreased the developed force by ∼60% both in the absence and presence of 10 µM noradrenaline, whereas the increase in SC rate in response to the agonist was reduced by 90% (not shown). These results confirm that spontaneous activity in both preparations depends strongly, if not completely, on the SR function.
Figure 1.
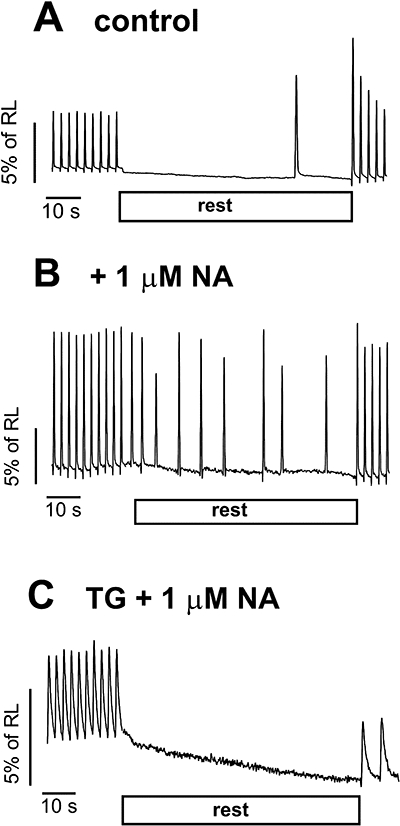
Records of ventricular myocyte shortening showing development of spontaneous contractions during interruption of electric stimulation (rest), in the absence (A) and presence (B and C) of noradrenaline (NA). Disabling sarcoplasmic reticulum function by thapsigargin (TG) treatment abolished spontaneous activity, even in the presence of noradrenaline (C). RL, resting cell length.
Adrenoceptor types involved in the mediation of the automatic effect of noradrenaline
The potency of catecholamines was greater for the production of positive inotropic response than for automatism stimulation during rest (Figure 2), as previously observed (Boer and Bassani, 2004; Carvalho et al., 2006; Penna and Bassani, 2010). Extrasystolic contractions, on the other hand, developed in some cells, at agonist concentrations that evoked inotropic responses close to the maximum (not shown). Figure 3 shows the automatic response to noradrenaline, which stimulated spontaneous activity in both LA and VM, although with greater potency in the latter (Table 1). This figure also shows concentration–effect curves to noradrenaline obtained in the presence of the competitive α- and β-adrenoceptor antagonists phentolamine (1 µM) and propranolol (1 µM), respectively, which are not selective for receptor subtypes. At these concentrations, phentolamine and propranolol did not depress LA (3.26 ± 0.30 and 3.35 ± 0.29 mN before and after phentolamine, respectively; 3.25 ± 0.58 and 2.88 ± 0.50 mN before and after propranolol, respectively; P > 0.65, paired t-test) or VM basal contraction amplitude (8.82 ± 1.20 and 8.61 ± 0.48% of RL before and after phentolamine, respectively; 8.22 ± 1.53 and 7.95 ± 1.38% of RL before and after propranolol, respectively; P > 0.46). These phentolamine and propranolol concentrations were previously shown to largely suppress the development of contraction and relaxation in response to 1 µM noradrenaline and isoprenaline, respectively, in endothelium-free rat aorta (Lai and Bassani, unpublished results).
Figure 2.
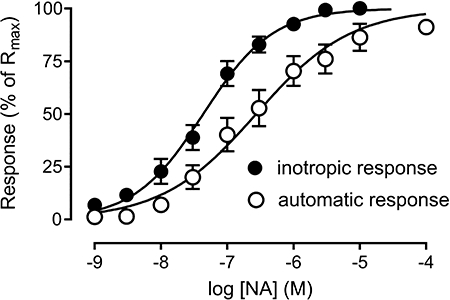
Inotropic (increase in systolic cell shortening) and automatic (rate of spontaneous contractions in the absence of electric stimulation) responses to noradrenaline (NA) determined in the same set of ventricular myocytes (n= 18). The responses were normalized to the respective maximum responses (Rmax). The mean pD2 value was significantly greater for the inotropic (7.36 ± 0.14) than for the automatic response (6.76 ± 0.16; P < 0.05).
Figure 3.
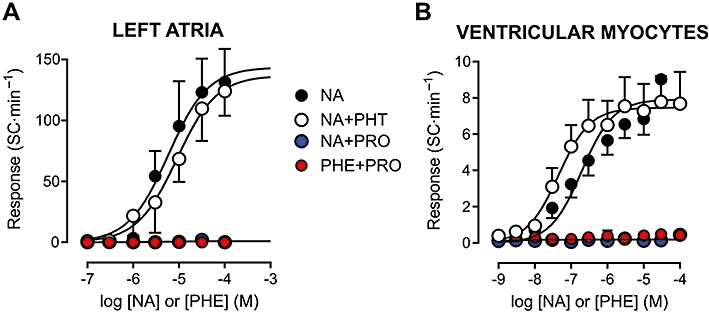
Concentration–effect curves for adrenoceptor agonists in isolated rat left atria (A) and ventricular myocytes (B). The automatic response is expressed as the rate of spontaneous contractions (SC) during rest. Curves to noradrenaline (NA) were obtained in the absence of antagonists, as well as in the presence of the α- and β-adrenoceptor antagonists phentolamine (PHT, 1 µM) and propranolol (PRO, 1 µM) respectively. The response to the α-adrenoceptor agonist phenylephrine (PHE) was determined in the presence of PRO. Atropine (0.1 µM) was present throughout in all experiments with left atria. Values shown are means and SE. Curve parameters are shown in Table 1.
Table 1.
Automatic response to adrenoceptor agonists, in the presence and absence of other compounds. Data are expressed as mean ± SE. Experiments with left atria were performed in the presence of 0.1 µM atropine
| Left atria | Ventricular myocytes | |||||
|---|---|---|---|---|---|---|
| Agonist | Basal (SC·s−1) | Rmax (SC·s−1) | pD2 | Basal (SC·s−1) | Rmax (SC·s−1) | pD2 |
| NA | 0.6 ± 0.4 (n= 13) | 161 ± 12 | 5.34 ± 0.15 | 1.3 ± 0.6 (n= 18) | 8.6 ± 1.0 | 6.76 ± 0.16 |
| NA + PHT | 0.4 ± 0.2 (n= 10) | 152 ± 13 | 4.99 ± 0.14 | 2.5 ± 1.3 (n= 10) | 7.5 ± 0.9 | 7.13 ± 0.11 |
| NA + PRO | 0.2 ± 0.2 (n= 6) | 0.6 ± 0.5a,b | – | 1.3 ± 0.7 (n= 11) | 0.5 ± 0.2a,b | – |
| PHE + PRO | 0.2 ± 0.1 (n= 10) | 0.2 ± 0.1a,b | – | 0.4 ± 0.2 (n= 13) | 0.5 ± 0.2a,b | – |
| ISO | 0.5 ± 0.4 (n= 12) | 83 ± 5b,d | 7.12 ± 0.18d | 0.5 ± 0.4 (n= 13) | 4.4 ± 0.4b,d | 7.64 ± 0.09b,d |
| ISO + BUT | 0.9 ± 0.5 (n= 6) | 122 ± 15c | 6.99 ± 0.23 | 0.1 ± 0.1 (n= 11) | 9.5 ± 1.3c | 7.16 ± 0.07c |
| ISO + ICI | – | – | – | 0.4 ± 0.2 (n= 15) | 11.9 ± 1.1c | 7.43 ± 0.10 |
| ISO + SAL | 0.4 ± 0.2 (n= 6) | 38 ± 4d | 6.85 ± 0.23 | – | – | – |
SC rate recorded at the agonist concentration of 100 µM.
P < 0.01 versus NA.
P < 0.01 versus ISO.
P < 0.01 versus NA + PHT.
BUT, butoxamine (0.3 µM); ICI, ICI118551 (0.1 µM); ISO, isoprenaline; NA, noradrenaline; PHE, phenylephrine; PHT, phentolamine (1 µM); PRO, propranolol (1 µM); Rmax, maximum responses; SAL, salbutamol (10 µM); SC, spontaneous contractions; SE, standard error.
In both preparations, the automatic Rmax to noradrenaline was significantly influenced by the antagonist used (P < 0.001, one-way analysis of variance). Whereas α-adrenoceptor blockade with phentolamine did not significantly change Rmax or pD2 values (P > 0.11), β-adrenoceptor antagonism by propranolol abolished noradrenaline-induced spontaneous activity (non significant regression, R2 < 0.17; Table 1 and Figure 3). Additionally, α1-adrenoceptor blockade by prazosin (0.3–1 µM) in VM produced results similar to those with phentolamine (not shown). Selective α-adrenoceptor stimulation with phenylephrine in the presence or absence of propranolol also failed to evoke automatism in both LA and VM (Table 1 and Figure 3).
Adrenoceptor type-dependent differences were observed also in the inotropic Rmax to noradrenaline (P < 0.001). In LA, Rmax was comparable in the absence and presence of phentolamine (2.92 ± 0.34 and 2.76 ± 0.59 mN respectively). β-Adrenoceptor blockade by propranolol did not abolish inotropic responsiveness to noradrenaline, but decreased the response at 100 µM noradrenaline by ∼45% (1.66 ± 0.14 mN, P < 0.01). phenylephrine at this concentration evoked a response similar to that to noradrenaline in the presence of propranolol (1.78 ± 0.12 mN). Likewise, the Rmax to noradrenaline in VM was not significantly affected by phentolamine (11.2 ± 1.1 and 10.8 ± 1.2% of RL in the absence and presence of phentolamine, respectively), but the response to 100 µM noradrenaline was reduced to 4.4 ± 0.4% of RL by propranolol a value comparable to the inotropic response to phenylephrine in the presence of propranolol (4.0 ± 0.4% of RL).
Functional antagonism of β1- and β2-adrenoceptor s in the mediation of automatic effects of catecholamines
As the results above indicate that stimulation of atrial and ventricular spontaneous activity by noradrenaline is mediated by β-adrenoceptors, it might be expected that isoprenaline, a full β-adrenoceptor agonist, would exert a comparable effect. However, the Rmax to isoprenaline was only 50% of that to noradrenaline under α-adrenoceptor blockade, although isoprenaline potency was considerably higher (P < 0.01; Table 1, Figure 4). It is interesting to notice that the absolute increase in SC rate in VM was similar for noradrenaline and isoprenaline up to the concentration of 30 nM, above which noradrenaline was more effective in stimulating spontaneous activity (Figure 4B). The inotropic Rmax values to isoprenaline and to (noradrenaline + phentolamine) were similar (LA: 2.73 ± 0.73 and 2.76 ± 0.59 mN, respectively; VM: 10.2 ± 1.4 and 10.8 ± 1.2% of RL, respectively; P > 0.75). Thus, the discrepant automatic responses to noradrenaline and isoprenaline were not paralleled by differences in the inotropic responses recorded in the same preparations. The apparent efficacy of isoprenaline in LA could be increased by manipulations aimed at enhancing diastolic SR Ca2+ release, such as addition of 0.5 mM caffeine (Rmax= 121 ± 13 SC·min−1, n= 11, P < 0.05) or diastolic membrane depolarization by raising extracellular K+ concentration from 5.7 to 8.2 mM (Rmax= 142 ± 16 SC·min−1, n= 5, P < 0.05).
Figure 4.
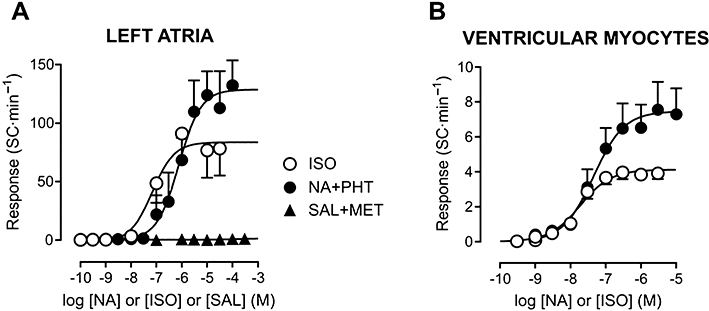
Concentration–effect curves for β-adrenoceptor agonists in isolated rat left atria (A) and ventricular myocytes (B). Curves are shown as in Figure 2. In both preparations, the maximum automatic response was greater for preferential activation of β1-adrenoceptors by noradrenaline (NA) under α-adrenoceptor blockade by phentolamine (PHT, 1 µM) than by non-selective β-adrenoceptor stimulation by isoprenaline (ISO). β2-adrenoceptor stimulation by salbutamol (SAL) in the presence of the β1-adrenoceptor antagonist metoprolol (MET, 0.5 µM) did not evoke automatism. All experiments with left atria were performed in the presence of 0.1 µM atropine. Values shown are means and SE. Curve parameters are shown in Table 1.
The major difference in the mode of action of isoprenaline and noradrenaline is that only the former acts on β2-adrenoceptors (Lands et al., 1967). Figure 4A shows that the β2-adrenoceptor agonist salbutamol was unable to induce spontaneous activity in LA exposed to the β1-adrenoceptor antagonist metoprolol (0.5 µM, 100-fold greater than the KB value in rat atrial tissue, Bassani and De Moraes, 1988). The SC rate was 0.2 ± 0.2 and 1.5 ± 1.2 SC·min−1 in the absence and presence of 300 µM salbutamol (n= 4; P > 0.32). Metoprolol-treated LA also failed to develop spontaneous activity when isoprenaline was used as the agonist (not shown).
Nevertheless, as shown in Figure 5, the automatic effect of isoprenaline was affected by the competitive β2-adrenoceptor antagonist butoxamine (0.3 µM, 100-fold greater than the KB value in rat atria, Bassani and De Moraes, 1988). In both LA (Figure 5A) and VM (Figure 5B), butoxamine significantly enhanced the automatic Rmax to isoprenaline (P < 0.01; Table 1). The pD2 value was not significantly changed by butoxamine in LA (P > 0.67), but was decreased in VM (P < 0.01; Table 1). However, it should be noted that, in the latter preparation, the absolute automatic response up to 30 nM isoprenaline was essentially the same in both conditions, whereas above this concentration isoprenaline produced higher SC rates in butoxamine-treated myocytes (Figure 5B), as observed for noradrenaline. The highly selective β2-adrenoceptor antagonist ICI118551 (0.1 µM; Brodde and Michel, 1999) also increased the automatic Rmax to isoprenaline in VM (P < 0.01), although isoprenaline pD2 value was unaffected (Table 1; Figure 5B). It is important to notice that, in the presence of butoxamine or ICI118551, the automatic Rmax to isoprenaline was not significantly different from that to noradrenaline in either LA (P > 0.07) or VM (P > 0.58). These results were reproduced in experiments with VM performed at 36°C (Table 2, Figure 5C), that is, potentiation of the automatic response to isoprenaline by ICI118551, resulting in a Rmax value similar to that to NA. At a supramaximal agonist concentration (3 µM), the percentage of cells that developed extrasystoles at 36°C was greater in the presence of noradrenaline (71%) than that of isoprenaline (30%). However, in the presence of ICI118551, 75% of the cells showed extrasystolic contractions in response to isoprenaline. Thus, in terms of apparent efficacy, the automatic response to isoprenaline under β2-adrenoceptor blockade resembles that to noradrenaline under α-adrenoceptor blockade; in both cases, the response is expected to be mediated preferentially by β1-adrenoceptors.
Figure 5.
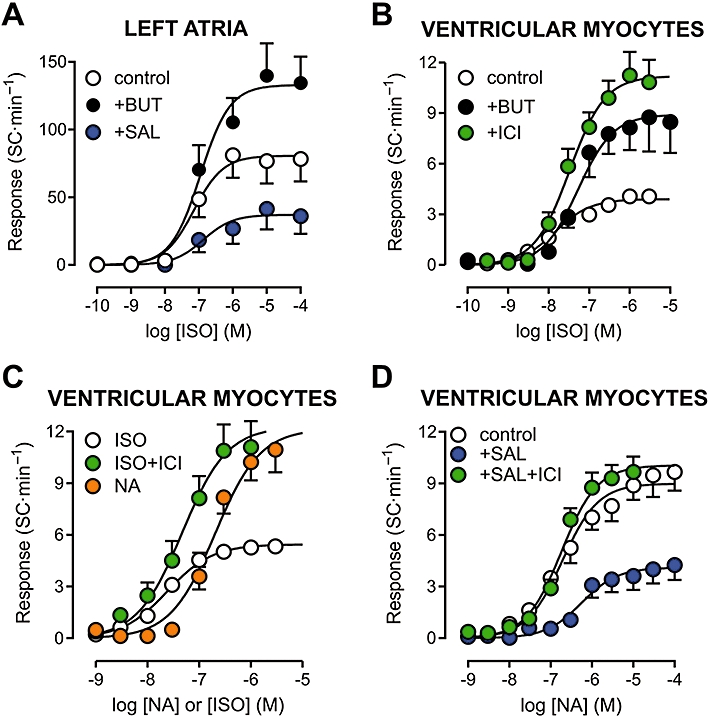
Concentration–effect curves for isoprenaline (ISO) and noradrenaline (NA) in isolated rat left atria (A) and ventricular myocytes (B–D). Curves were also determined during β2-adrenoceptor blockade with butoxamine (BUT, 0.3 µM) or ICI118551 (ICI, 0.1 µM), and in the presence of the β2-adrenoceptor agonist salbutamol (SAL, 10 µM). Experiments with left atria were performed in the presence of 0.1 µM atropine. Panels A and C show results obtained at 36°C, whereas the temperature was 23°C for experiments in panels B and D. Curve parameters are shown in Tables 1–3.
Table 2.
Automatic response to noradrenaline (NA) and isoprenaline (ISO) determined in rat ventricular myocytes at 36°C. The latter agonist was tested in the absence and presence of the β2-adrenoceptor antagonist ICI118551 (ICI, 0.1 µM). Data are presented as mean ± SE
| Agonist | Basal (SC·s−1) | Rmax (SC·s−1) | pD2 |
|---|---|---|---|
| ISO (n= 20) | 1.1 ± 0.3 | 5.7 ± 0.4 | 7.61 ± 0.08 |
| ISO + ICI (n= 20) | 0.9 ± 0.2 | 11.7 ± 1.4a | 7.47 ± 0.11 |
| NA (n= 19) | 1.0 ± 0.3 | 12.4 ± 1.4a | 6.70 ± 0.05 |
P < 0.01 versus ISO in the absence of ICI; one-way analysis of variance: P > 0.77 for basal values, and P < 0.001 for Rmax; t-test for ISO versus ISO + ICI: P > 0.31.
Rmax, maximum responses; SC, spontaneous contractions; SE, standard error.
In LA and VM, the inotropic Rmax to isoprenaline was 30–40% lower in the presence of butoxamine (1.81 ± 0.33 mN and 7.8 ± 1.8% of RL, respectively), but this decrease did not reach statistical significance (P > 0.14). ICI118551 did not affect significantly the inotropic Rmax to isoprenaline in VM (8.8 ± 2.2% of RL, P > 0.60). At 36°C, the inotropic Rmax in VM was comparable for noradrenaline and isoprenaline (12.6 ± 1.3 and 10.3 ± 1.0% of RL), and the latter was not significantly affected by ICI118551 (9.5 ± 1.1% of RL, P > 0.61).
The ability of β2-adrenoceptor antagonists to enhance isoprenaline-induced automatism suggests that some mechanism coupled to β2-adrenoceptors might limit the automatism stimulation mediated by β1-adrenoceptors. If so, one would expect that sustained β2-adrenoceptor activation could attenuate the development of spontaneous activity during β1-adrenoceptor stimulation. To test this possibility, the automatic response to catecholamines was determined in the presence of 10 µM salbutamol, which was applied 5 min before and during agonist exposure. Salbutamol alone did not produce significant increase in basal twitch force in LA (from 2.64 ± 0.27 to 2.94 ± 0.35 mN) or cell shortening in VM (from 7.8 ± 1.2 to 9.3 ± 1.3% of RL; P > 0.20, paired t-test). Continuous exposure to salbutamol did not significantly change the inotropic Rmax to isoprenaline (3.65 ± 0.63 mN) or noradrenaline (9.2 ± 1.6% of RL) in LA and VM respectively (P > 0.27).
The basal SC rate was not significantly affected by salbutamol in LA (from 0.5 ± 0.3 to 0.4 ± 0.2 SC·min−1) or in VM (from 1.7 ± 0.7 to 1.1 ± 0.5 SC·min−1; P > 0.14, paired t-test). As shown in Figure 5A and Table 1, salbutamol decreased the automatic Rmax to isoprenaline by more than 50% in LA (P < 0.001), without affecting significantly isoprenaline pD2 (P > 0.38). In VM, salbutamol also suppressed the automatic Rmax to noradrenaline (P < 0.01; Table 3, Figure 5D), in addition to decreasing the sensitivity to the agonist (P < 0.05). The effects of salbutamol on the noradrenaline-stimulated automatism of VM were completely abolished by ICI118551 (Table 3; Figure 5D).
Table 3.
Automatic response to noradrenaline (NA) determined in rat ventricular myocytes in the presence of the compounds indicated in the left column. Data are presented as mean ± SE. The parameters of the response to NA in the absence of other drugs, shown in Table 1, are presented again in this table for clarity
| Pretreatment | Basal (SC·s−1) | Rmax (SC·s−1) | pD2 |
|---|---|---|---|
| None (n= 18) | 1.3 ± 0.6 | 8.6 ± 1.0 | 6.76 ± 0.16 |
| SAL (n= 17) | 1.1 ± 0.5 | 4.3 ± 1.0a | 6.30 ± 0.17a |
| ICI + SAL (n= 19) | 1.6 ± 0.3 | 10.7 ± 0.7b | 6.80 ± 0.06b |
| MLR (n= 19) | 0.4 ± 0.2 | 10.4 ± 0.5 | 7.44 ± 0.10a |
| MLR + SAL (n= 7) | 3.6 ± 0.8a,c | 7.6 ± 0.5 | 7.03 ± 0.20d |
| ROL (n= 16) | 0.1 ± 0.1a | 9.5 ± 0.5 | 7.36 ± 0.09a |
| ROL + SAL (n= 16) | 1.9 ± 0.6c | 9.1 ± 0.6 | 7.21 ± 0.1a,d |
P < 0.05 versus NA in the absence of other drugs.
P < 0.05 versus SAL alone.
P < 0.05 versus the same phosphodiesterase inhibitor in the absence of SAL.
P < 0.05 versus in the absence of the phosphodiesterase inhibitor.
ICI, ICI118551 (0.1 µM); MLR, milrinone (3 µM); Rmax, maximum responses; ROL, rolipram (3 µM); SAL, salbutamol (10 µM); SC, spontaneous contractions; SE, standard error.
Catecholaminergic automatism: effects of PDE inhibition
Because PDE activity seems able to prevent PKA phosphorylation of SR proteins in response to β2-adrenoceptor activation (Soto et al., 2009), we hypothesized that the anti-automatic effect of β2-adrenoceptor stimulation might involve PDEs. To test this hypothesis, the automatic response of VM to noradrenaline, both in the presence and absence of salbutamol, was determined under inhibition of PDE3 and PDE4 by milrinone and rolipram respectively (3 µM; 15 min pre-exposure and throughout the experiment).
Incubation with milrinone did not significantly affect contraction amplitude (9.9 ± 0.7 to 9.5 ± 0.7% of RL; n= 26; P > 0.17, paired t-test) or SC rate (1.0 ± 0.3 to 0.6 ± 0.2 SC·min−1; P > 0.10). The inotropic Rmax to noradrenaline was decreased by 25% by milrinone treatment (from 11.2 ± 1.1 to 8.6 ± 1.0% of RL), while the automatic Rmax was increased by 20% (Table 3), but these changes did not attain statistical significance (P > 0.05). However, the noradrenaline pD2 was significantly greater in the presence of milrinone (P < 0.001; Table 3).
In milrinone-treated cells, salbutamol addition increased both contraction amplitude (from 10.1 ± 1.1 to 11.9 ± 1.1% of RL; n= 7; P < 0.02; paired t-test) and SC rate (from 0.4 ± 0.4 to 3.6 ± 0.8 SC·min−1; P < 0.02). in contrast to the effects of salbutamol in the absence of milrinone, A two-way analysis of variance to investigate the combination of salbutamol and milrinone effects revealed that these compounds exerted significant (P < 0.01), but independent effects (P > 0.43 for milrinone–salbutamol interaction) on the automatic Rmax to noradrenaline, the former decreasing and the latter increasing it (Figure 6A). The post hoc Bonferroni test showed that milrinone attenuated the depression of the automatic Rmax by salbutamol (P < 0.01). A similar result was observed for noradrenaline potency: salbutamol decreased, whereas milrinone increased the pD2 value for noradrenaline (P < 0.02), and, again, their effects were independent (P > 0.88 for interaction). Treatment with milrinone plus salbutamol, but not with salbutamol alone, depressed the inotropic Rmax to noradrenaline (3.3 ± 0.7% of RL; P < 0.01).
Figure 6.
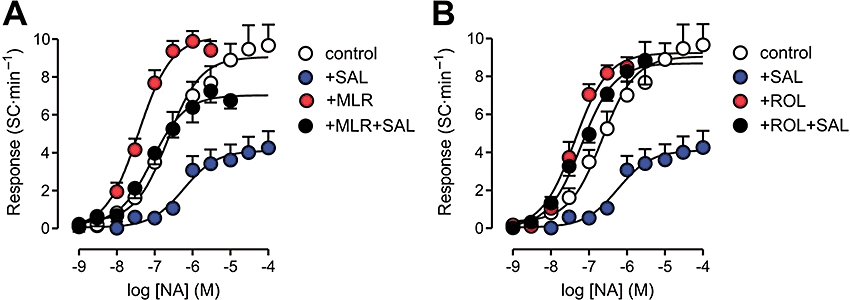
Concentration–effect curves for noradrenaline (NA) in isolated rat ventricular myocytes, determined in the presence and absence of the phosphodiesterase inhibitors milrinone (MLR, 3 µM, panel A) or rolipram (ROL, 3 µM, panel B). Some curves were determined during perfusion with the β2-adrenoceptor agonist salbutamol (SAL, 10 µM). Curve parameters are shown in Table 2.
As observed with milrinone, rolipram alone did not significantly affect baseline contraction amplitude (from 9.3 ± 0.6 to 8.8 ± 0.6% of RL; n= 32; P > 0.22, paired t-test), although the SC rate was decreased (from 0.8 ± 0.3 to 0.4 ± 0.2 SC·min−1; P < 0.04). Rolipram by itself increased the pD2, but did not affect the automatic (Table 3) or the inotropic (10.4 ± 1.6% of RL) Rmax to noradrenaline. In the presence of rolipram, salbutamol markedly enhanced basal contraction amplitude (from 9.2 ± 0.7 to 14.3 ± 1.4% of RL), and also increased and SC rate (from 0.7 ± 0.4 to 1.9 ± 0.6 SC·min−1; n= 16; P < 0.04; paired t-test). Nevertheless, in contrast with what happened with milrinone, a significant interaction (P < 0.03) was detected between the effects of rolipram and salbutamol on the automatic responsiveness to noradrenaline. In the presence of rolipram, salbutamol failed to produce the significant decrease in both pD2 and Rmax to noradrenaline that was observed in absence of rolipram (Table 3 and Figure 6B). In other words, exposure to rolipram abolished the effects of salbutamol on the automatic responsiveness to noradrenaline. The inotropic Rmax to noradrenaline was depressed after (rolipram + salbutamol) treatment (to 3.9 ± 0.7% of RL; P < 0.01), probably because before noradrenaline challenge, cells treated with salbutamol presented greater basal contraction amplitude. Accordingly, systolic shortening at maximal noradrenaline concentrations under rolipram exposure was not significantly different in the absence and presence of salbutamol (18.3 ± 1.5 and 17.6 ± 1.5% of RL, respectively; P > 0.75).
Discussion
The general aim of this study was to investigate the mechanisms involved in the production of spontaneous activity in rat myocardium by catecholamines. The present results indicate that: (i) the SR is crucially involved in the production of this effect; (ii) enhancement of spontaneous activity is mediated by β1-adrenoceptors, whereas α- or β2-adrenoceptor stimulation is unable to evoke automatism in either atrial or ventricular myocardium; (iii) β2-adrenoceptor stimulation is able to antagonize the β1-mediated enhancement of spontaneous activity by catecholamines; and (iv) although both PDE3 and PDE4 seem to affect catecholaminergic automatism, only inhibition of PDE4 abolished the effect of β2-adrenoceptor stimulation.
The occurrence of spontaneous activity in myocardial cells exposed to high catecholamine concentrations is generally attributed to development of Ca2+ overload (Nam et al., 2005; ter Keurs and Boyden, 2007; DeSantiago et al., 2008; Venetucci et al., 2008). The primordial role of the SR in the generation of automatism by catecholamines in rat myocardium was confirmed by the marked suppression of SCs by inhibition of SR function, even at maximal agonist concentrations.
Ca2+-dependent triggered activity has been implicated in atrial fibrillation of sympathetic origin (Chen and Tan, 2007). Our findings in the isolated LA model of catecholaminergic automatism are in agreement with this main mechanism. For instance, spontaneous activity in this preparation seems to arise from focal electrical activity (Zafalon et al., 2009), requires previous rapid pacing (Boer and Bassani, 2004) and is sensitive to SERCA inhibition (present results). In addition, we observed that isoprenaline-induced automatism could be enhanced by maneuvers that increase diastolic SR Ca2+ release directly, such as exposure to low-caffeine concentration (Ferraz et al., 2001), or indirectly, such as increase in extracellular K+ concentration (which is expected to induce SR Ca2+ overload by impairment of Ca2+ extrusion via the Na+/Ca2+ exchanger due to sarcolemmal depolarization).
β1-adrenoceptors mediate catecholaminergic automatism in rat myocardium
While α-adrenoceptor blockade did not significantly affect the inotropic and automatic responses to noradrenaline, β-adrenoceptor blockade with propranolol reduced by 50% the inotropic response to 100 µM noradrenaline and phenylephrine, and totally abolished noradrenaline-induced automatism. These results indicate that α-adrenoceptor signalling, although mediating a modest inotropic stimulation, is not required for the production of catecholaminergic automatism in rat myocardium.
On the other hand, β2-adrenoceptor stimulation by either salbutamol or isoprenaline completely failed to induce spontaneous activity in LA treated with the β1-adrenoceptor antagonist metoprolol. Both β1- and β2-adrenoceptors are coupled to the Gs-PKA pathway. Nevertheless, selective β2-adrenoceptor stimulation typically produces smaller increase in Ca2+ transient and contraction amplitude than that of β1-adrenoceptors in the myocardium of several species (e.g. Xiao et al., 1994; 2006; Cui et al., 1996; Kuschel et al., 1999; DeSantiago et al., 2008), and may even depress contractility (Siedlecka et al., 2008). Although β2-adrenoceptors have been implicated in the generation of Ca2+-dependent arrhythmias, this was observed mostly under pathophysiological conditions, such as myocardial ischaemia or heart failure (Billman et al., 1997; Altschuld and Billman, 2000; DeSantiago et al., 2008), conditions in which β1-adrenoceptors are typically down-regulated, and β2-adrenoceptor involvement in Ca2+ cycling is abnormally enhanced (Brodde and Michel, 1999; Altschuld and Billman, 2000; DeSantiago et al., 2008). On the other hand, Steinberg et al. (2002) attributed to the β1-adrenoceptor subtype the induction of catecholaminergic spontaneous activity in canine ventricle, although β2-adrenoceptor activation was able to produce substantial inotropic stimulation. Accordingly, it has been shown that β1-, but not β2-adrenoceptor blockade, can suppress catecholaminergic automatism in ventricular myocardium (Galindo-Tovar and Kaumann, 2008; Penna and Bassani, 2010). These reports are in line with our present findings that β1-adrenoceptors play a major role in the mediation of myocardial automatism induction by catecholamines. On the other hand, Kaumann and Sanders, (1993) reported induction of β2-adrenoceptor-mediated automatism in right atrial tissue from non-failing human hearts. It remains to be determined whether this is also the case in human ventricular myocardium.
β2-adrenoceptors attenuate β1-adrenoceptor mediated catecholaminergic automatism in rat myocardium: a possible link with PDE4
Compared to β1-adrenoceptors, β2-adrenoceptor s are less effective in raising cAMP levels, as well as inducing positive inotropic and lusitropic effects, a difference largely attributed to the spatial restriction of cAMP accumulation (Nikolaev et al., 2006; Xiao et al., 2006). Compartmentalization of the β2-adrenoceptor signalling, associated with briefer PKA activation (Soto et al., 2009), may be the cause of the lack of significant PKA phosphorylation of internal proteins such as PLB, which is typically phosphorylated during β1-adrenoceptor activation (Xiao et al., 1994; 2006; Kuschel et al., 1999; DeSantiago et al., 2008; Soto et al., 2009). PLB phosphorylation by PKA is considered a requisite for increase in SR Ca2+ content and release during β-adrenergic stimulation (Li et al., 2002), and, as shown by us and others (Fujiwara et al., 2008), SR function is crucial to the generation of Ca2+-dependent spontaneous activity and/or arrhythmias. Here we found evidence of β2-adrenoceptor involvement in catecholaminergic automatism, but by antagonizing the β1-adrenoceptor-mediated enhancement in spontaneous activity, rather than contributing to it. This evidence is based on: (i) greater apparent efficacy of noradrenaline (preferential β1-adrenoceptor stimulation) over isoprenaline (similar potency on both subtypes) at inducing automatism; (ii) potentiation of isoprenaline-stimulated automatism by β2-adrenoceptor competitive antagonists; and (iii) attenuation of isoprenaline- and noradrenaline-evoked spontaneous activity by background β2-adrenoceptor stimulation with salbutamol, which was completely reversed by ICI118551.
Opposing influences of β1- and β2-adrenoceptor stimulation have been previously described. The former is associated with deleterious effects, such as worsening of doxorubicin and catecholamine cardiotoxicity and apoptosis, and the latter was shown to exert cardioprotective effects (see Communal et al., 1999; Fajardo et al., 2006; Xiao et al., 2006). Protection against arrhythmia by β2-adrenoceptors was previously reported by Sosunov et al., (2000) in a canine model of inherited arrhythmia, as well as in normal dogs. In the present study, we demonstrate that this functional antagonism of β1- and β2-adrenoceptor-mediated effects also extends to the potentially arrhythmogenic induction of myocardial automatism in apparently healthy, normal, wild type rodents. It remains to be determined if this type of β2-adrenoceptor-mediated cardioprotection is also observed in other species, including humans.
Increasing cAMP accumulation, independently of β-adrenoceptor stimulation, is sufficient to induce automatism in VM (Penna and Bassani, 2010). The PDE-catalysed degradation of cAMP limits the effects of activation of the β-adrenoceptor pathway, especially at high stimulation levels. Among the PDE families, only PDE3 and PDE4 are involved in the regulation of myocardial contractility (Osadchii, 2007). PDE families appear to be compartmentalized (Mongillo et al., 2004) and this would allow the creation of cAMP concentration gradients and/or microdomains that may favour or attenuate PKA activation in different regions of the cell. In this study, selective inhibition of PDE3 or PDE4 by non-saturating concentrations of milrinone and rolipram, respectively, did not produce significant inotropic or automatic effects, but markedly potentiated the automatic response to NA, increasing the pD2 to a value comparable to that observed for the inotropic response of VM in the absence of the inhibitors (Penna and Bassani, 2010). This result indicates that both isoenzymes seem to attenuate the production of spontaneous activity in rat ventricle, and is in agreement with the observation by Galindo-Tovar and Kaumann (2008) that rolipram can enhance the β1-adrenoceptor-mediated induction of spontaneous activity by catecholamines in mouse ventricle.
Several reports indicate that inhibition of PDE4 is more effective than that of PDE3 at potentiating catecholamine effects in rodent myocardium, particularly those mediated by β2-adrenoceptors (Mongillo et al., 2004; Rochais et al., 2006; Vargas et al., 2006), including PLB phosphorylation (Soto et al., 2009). Although the β2-adrenoceptor agonist salbutamol does not affect contraction amplitude in rat VM (Siedlecka et al., 2008; present results), we observed that inhibition of either PDE3 or PDE4 unmasked significant inotropic stimulation by this agonist. However, this effect was more prominent after PDE4 than PDE3 antagonism. Moreover, the attenuation of noradrenaline-induced automatism by salbutamol was abolished by rolipram, but not by milrinone. These observations indicate that cAMP degradation by PDEs seems to play a role in blunting the automatic response to β2-adrenoceptor stimulation in the rat ventricle. Additionally, they are suggestive that PDE4, but not PDE3, is closely involved in the functional antagonism between β1- and β2-adrenoceptor signalling pathways regarding the generation of catecholaminergic automatism.
The involvement of β2-adrenoceptors and PDE4 in the attenuation of catecholaminergic, Ca2+-dependent automatism in the myocardium may have pathophysiological implications. Deficiency of PDE4D results in cardiomyopathy and arrhythmias, and the amount of this isoform associated with the SR is decreased in human heart failure (Lehnart et al., 2005). Interestingly, in failing, but not in control hearts, β2-adrenoceptor stimulation is able to promote PLB phosphorylation by PKA, marked positive inotropic effect, SR Ca2+ overload and arrhythmias (DeSantiago et al., 2008). Decrease in ventricular PDE4 activity was observed in rats with compensated ventricular hypertrophy due to aortic banding (Abi-Gerges et al., 2009), a condition in which catecholaminergic automatism is enhanced (Carvalho et al., 2006). Finally, stress, recognized as a risk factor for arrhythmia in humans (see Lane et al., 2005), decreased cardiac PDE activity associated with the SR (Okruhlicováet al., 2004). We recently observed depressed inotropic responsiveness and greater apparent efficacy of catecholamines at increasing extrasystolic activity in VM from stressed rats, which was consistent with impaired cAMP degradation and loss of β2-adrenoceptor-mediated protection (Penna and Bassani, 2010). Although further investigation is required, the current information on the mechanisms involved in catecholaminergic arrhythmogenesis points out that β2-adrenoceptors and PDEs might be promising targets for the pharmacological treatment of arrhythmias.
Acknowledgments
We are indebted to Ms. Elizângela S. Oliveira and Ms. Ana Carolina Fantin for the technical support. This study was partially supported by CNPq (Proc N. 300632/2005-3 and 141175/2002-8).
Glossary
Abbreviations
- LA
left atria
- PDE
phosphodiesterase
- PKA
cAMP-dependent protein kinase
- PLB
phospholamban
- RL
resting cell length
- Rmax
maximum response
- SC
spontaneous contractions
- SERCA
sarco-endoplasmic reticulum Ca2+-ATPase
- SR
sarcoplasmic reticulum
- VM
ventricular myocytes
Conflicts of interest
None.
Supplementary material
Supporting Information: Teaching Materials; Figs 1–6 as PowerPoint slide.
References
- Abi-Gerges A, Richter W, Lefebvre F, Mateo P, Varin A, Heymes C, et al. Decreased expression and activity of cAMP phosphodiesterases in cardiac hypertrophy and its impact on β-adrenergic cAMP signals. Circ Res. 2009;105:784–792. doi: 10.1161/CIRCRESAHA.109.197947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschuld RA, Billman GE. β2-adrenoceptors and ventricular fibrillation. Pharmacol Ther. 2000;88:1–14. doi: 10.1016/s0163-7258(00)00075-9. [DOI] [PubMed] [Google Scholar]
- Bassani RA, De Moraes S. Effects of repeated footshock stress on the chronotropic responsiveness of the isolated pacemaker of the rat: role of beta-2 adrenoceptors. J Pharmacol Exp Ther. 1988;246:316–321. [PubMed] [Google Scholar]
- Bassani JWM, Yuan WL, Bers DM. Fractional SR Ca release is regulated by trigger Ca and SR Ca content. Am J Physiol. 1995;268:C1313–C1319. doi: 10.1152/ajpcell.1995.268.5.C1313. [DOI] [PubMed] [Google Scholar]
- Bassani RA, Bassani JWM, Lipsius SL, Bers DM. Diastolic Ca efflux in atrial pacemaker cells and Ca-overloaded myocytes. Am J Physiol. 1997;273:H886–H892. doi: 10.1152/ajpheart.1997.273.2.H886. [DOI] [PubMed] [Google Scholar]
- Bers DM. Excitation-Contraction Coupling and Cardiac Contractile Force. 2nd edn. Dordrecht: Kluwer Press; 2001. [Google Scholar]
- Billman GE, Castillo LC, Hensley J, Hohl CM, Altschuld RA. β2-adrenergic receptor antagonists protect against ventricular fibrillation. Circ Res. 1997;96:1914–1922. doi: 10.1161/01.cir.96.6.1914. [DOI] [PubMed] [Google Scholar]
- Boer DC, Bassani RA. Quantitation of the dose-response relationship for arrhythmogenic agents in isolated cardiac tissue. Braz J Biomed Engin. 2004;20:3–10. [Google Scholar]
- Bogdanov KY, Vinogradova TM, Lakatta EG. Sinoatrial nodal cell ryanodine receptor and Na-Ca exchanger: molecular partners in pacemaker regulation. Circ Res. 2001;88:1254–1258. doi: 10.1161/hh1201.092095. [DOI] [PubMed] [Google Scholar]
- Brodde OE, Michel MC. Adrenergic and muscarinic receptors in the human heart. Pharmacol Rev. 1999;51:651–681. [PubMed] [Google Scholar]
- Carvalho BMR, Bassani RA, Franchini KG, Bassani JWM. Enhanced calcium mobilization in rat ventricular myocytes during the onset of pressure overload-induced hypertrophy. Am J Physiol. 2006;291:H1803–H1813. doi: 10.1152/ajpheart.01345.2005. [DOI] [PubMed] [Google Scholar]
- Chen PS, Tan AY. Autonomic nerve activity and atrial fibrillation. Heart Rhythm. 2007;4(Suppl. 3):S61–S64. doi: 10.1016/j.hrthm.2006.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Communal C, Singh K, Sawyer DB, Colucci WS. Opposing effects of β1- and β2-adrenergic receptors on cardiac myocyte apoptosis. Role of a pertussis toxin-sensitive G protein. Circulation. 1999;100:2210–2212. doi: 10.1161/01.cir.100.22.2210. [DOI] [PubMed] [Google Scholar]
- Cui Y, Shen YT, Kalthof B, Iwasi M, Sato N, Uechi M, et al. Identification and functional role of beta-adrenergic receptor subtypes in primate and rodent: in vivo versus isolated myocytes. J Mol Cell Cardiol. 1996;28:1307–1317. doi: 10.1006/jmcc.1996.0121. [DOI] [PubMed] [Google Scholar]
- DeSantiago J, Ai X, Islam M, Acuna G, Ziolo MT, Bers DM, et al. Arrhythmogenic effects of β2-adrenergic stimulation in the failing heart are attributable to enhanced sarcoplasmic reticulum Ca load. Circ Res. 2008;102:1389–1397. doi: 10.1161/CIRCRESAHA.107.169011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fajardo G, Zhao M, Powers J, Bernstein D. Differential cardiotoxic/cardioprotective effects of β-adrenergic receptor subtypes in myocytes and fibroblasts in doxorubicin cardiomyopathy. J Mol Cell Cardiol. 2006;40:375–383. doi: 10.1016/j.yjmcc.2005.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferraz SA, Bassani JWM, Bassani RA. Rest dependence of twitch amplitude and sarcoplasmic reticulum calcium content in the developing rat myocardium. J Mol Cell Cardiol. 2001;33:711–722. doi: 10.1006/jmcc.2001.1337. [DOI] [PubMed] [Google Scholar]
- Fujiwara K, Tanaka H, Mani H, Nakagami T, Takamatsu T. Burst emergence of intracellular Ca2+ waves evokes arrhythmogenic oscillatory depolarization via the Na+/ Ca2+ exchanger: simultaneous confocal recording of membrane potential and intracellular Ca2+ in the heart. Circ Res. 2008;103:509–518. doi: 10.1161/CIRCRESAHA.108.176677. [DOI] [PubMed] [Google Scholar]
- Galindo-Tovar A, Kaumann AJ. Phosphodiesterase-4 blunts inotropism and arrhythmias but not sinoatrial tachycardia of (-)-adrenaline mediated through mouse cardiac β1-adrenoceptors. Br J Pharmacol. 2008;153:710–720. doi: 10.1038/sj.bjp.0707631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginsburg KS, Bers DM. Modulation of excitation-contraction coupling by isoproterenol in cardiomyocytes with controlled SR Ca2+ load and Ca2+ current trigger. J Physiol. 2004;556:463–480. doi: 10.1113/jphysiol.2003.055384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaumann AJ, Sanders L. Both beta 1- and beta2-adrenoceptors mediate catecholamine-evoked arrhythmias in isolated human right atrium. Naunyn-Schmiederberg's. Arch Pharmacol. 1993;348:536–540. doi: 10.1007/BF00173215. [DOI] [PubMed] [Google Scholar]
- ter Keurs HEDJ, Boyden PA. Calcium and arrhythmogenesis. Physiol Rev. 2007;87:457–506. doi: 10.1152/physrev.00011.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuschel M, Zhou YY, Spurgeon HA, Bartel S, Karczewski P, Zhang SJ, et al. 2-adrenergic cAMP signaling is uncoupled from phosphorylation of cytoplasmic proteins in canine heart. Circulation. 1999;99:2458–2465. doi: 10.1161/01.cir.99.18.2458. [DOI] [PubMed] [Google Scholar]
- Lands AM, Luduena FP, Buzzo HJ. Differentiation of receptor responsiveness to isoproterenol. Life Sci. 1967;6:2241–2249. doi: 10.1016/0024-3205(67)90031-8. [DOI] [PubMed] [Google Scholar]
- Lane RD, Laukes C, Marcus FI, Chesney MA, Sechrest L, Gear K, et al. Psychological stress preceding idiopathic ventricular fibrillation. Psychosom Med. 2005;67:359–365. doi: 10.1097/01.psy.0000160476.67536.41. [DOI] [PubMed] [Google Scholar]
- Lehnart SE, Wehrens XHT, Reiken S, Warrier S, Belevych AE, Harvey RD, et al. Phosphodiesterase 4D deficiency in the ryanodine-receptor complex promotes heart failure and arrhythmias. Cell. 2005;123:25–35. doi: 10.1016/j.cell.2005.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Kranias EG, Mignery GA, Bers DM. Protein kinase A phosphorylation of the ryanodine receptor does not affect calcium sparks in mouse ventricular myocytes. Circ Res. 2002;90:309–316. doi: 10.1161/hh0302.105660. [DOI] [PubMed] [Google Scholar]
- Mongillo M, McSorley T, Evellin S, Sood A, Lissandron V, Terrin A, et al. Fluorescence resonance energy transfer-based analysis of cAMP dynamics in live neonatal cardiac myocytes reveals distinct functions of compartmentalized phosphodiesterases. Circ Res. 2004;95:67–75. doi: 10.1161/01.RES.0000134629.84732.11. [DOI] [PubMed] [Google Scholar]
- Nakamura H, Nakasaki Y, Matsuda N, Shigekawa M. Inhibition of sarcoplasmic reticulum Ca2+-ATPase by 2,5-di(tert-butyl)-1,4-benzohydroquinone. J Biochem. 1992;112:750–755. doi: 10.1093/oxfordjournals.jbchem.a123970. [DOI] [PubMed] [Google Scholar]
- Nam GB, Burashnikov A, Antzelevich C. Cellular mechanisms underlying development of catecholaminergic ventricular tachycardia. Circulation. 2005;111:2727–2733. doi: 10.1161/CIRCULATIONAHA.104.479295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikolaev VO, Bünemann M, Schmitteckert E, Lohse MJ, Engelhardt D. Cyclic AMP imaging in adult cardiac myocytes reveals far-reaching β1-adrenergic but locally confined β2-adrenergic receptor-mediated signaling. Circ Res. 2006;99:1084–1091. doi: 10.1161/01.RES.0000250046.69918.d5. [DOI] [PubMed] [Google Scholar]
- Okruhlicová L, Klenerová V, Hynie S, Sida P. In situ detection of cyclic AMP-phosphodiesterase activity in the heart of Lewis and Sprague-Dawley rats: the effect of restraint stress or amphetamine application. Histol Histopathol. 2004;19:719–726. doi: 10.14670/HH-19.719. [DOI] [PubMed] [Google Scholar]
- Osadchii OE. Myocardial phosphodiesterases and regulation of cardiac contractility in health and cardiac disease. Cardiovasc Drugs Ther. 2007;21:171–194. doi: 10.1007/s10557-007-6014-6. [DOI] [PubMed] [Google Scholar]
- Penna LB, Bassani RA. Increased spontaneous activity and lower inotropic response to catecholamines in ventricular myocytes from footshock-stressed rats. Stress. 2010;13:73–82. doi: 10.3109/10253890902951778. [DOI] [PubMed] [Google Scholar]
- Rochais F, Abi-Gerges A, Horner K, Lefebvre F, Cooper DMF, Conti M, et al. A specific pattern of phosphodiesterase controls the cAMP signals generated by different Gs-coupled receptors in adult rat ventricular myocytes. Circ Res. 2006;98:1081–1088. doi: 10.1161/01.RES.0000218493.09370.8e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon TR, Ginsburg KS, Bers DM. Quantitative assessment of the SR Ca2+ leak-load relationship. Circ Res. 2002;91:594–600. doi: 10.1161/01.res.0000036914.12686.28. [DOI] [PubMed] [Google Scholar]
- Siedlecka U, Arora M, Kolettis T, Soppa GK, Lee J, Stagg MA, et al. Effects of clenbuterol on contractility and Ca2+ homeostasis in isolated rat ventricular myocytes. Am J Physiol. 2008;295:H1917–H1926. doi: 10.1152/ajpheart.00258.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sosunov EA, Gainullin RZ, Möise NS, Steinberg SF, Danilo JP, Rosen MR. β1- and β2-adrenergic receptor subtype effects in German shepherd dogs with inherited lethal ventricular arrhythmias. Cardiovasc Res. 2000;48:211–219. doi: 10.1016/s0008-6363(00)00171-1. [DOI] [PubMed] [Google Scholar]
- Soto D, Arcangelis V, Zhang J, Xiang Y. Dynamic protein kinase A activities induced by β-adrenoceptors dictate signaling propagation for substrate phosphorylation and myoycte contraction. Circ Res. 2009;104:770–779. doi: 10.1161/CIRCRESAHA.108.187880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinberg SF, Alcott S, Pak E, Hu D, Protas L, Möise NS, et al. β1-Receptors increase cAMP and induce abnormal Cai cycling in the German shepherd sudden death model. Am J Physiol. 2002;282:H1181–H1188. doi: 10.1152/ajpheart.00871.2001. [DOI] [PubMed] [Google Scholar]
- Vargas ML, Hernandez J, Kaumann AJ. Phosphodiesterase PDE3 blunts the positive inotropic and cyclic AMP enhancing effects of CGP12177 but not of noradrenaline in rat ventricle. Br J Pharmacol. 2006;147:158–163. doi: 10.1038/sj.bjp.0706498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venetucci LA, Trafford AW, O'Neill SC, Eisner DA. The sarcoplasmic calcium reticulum and arrhythmogenic calcium release. Cardiovasc Res. 2008;77:285–292. doi: 10.1093/cvr/cvm009. [DOI] [PubMed] [Google Scholar]
- Xiao RP, Hohl C, Altschuld R, Jones L, Livingston B, Ziman B, et al. β2-Adrenergic receptor-stimulated increase in cAMP in rat heart cells is not coupled to changes in Ca2+ dynamics, contractility, or phospholamban phosphorylation. J Biol Chem. 1994;269:19151–19156. [PubMed] [Google Scholar]
- Xiao RP, Zhu W, Zheng M, Cao C, Zhang Y, Lakatta EG, et al. Subtype-specific α1- and β-adrenoceptor signaling in the heart. Trends Pharmacol Sci. 2006;27:330–337. doi: 10.1016/j.tips.2006.04.009. [DOI] [PubMed] [Google Scholar]
- Zafalon JN, Bassani JWM, Bassani RA. Determination of the vectorelectrogram in isolated rat atria: application to the study of arrhythmias. Physiol Meas. 2009;30:1281–1291. doi: 10.1088/0967-3334/30/11/011. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.