

Abstract
BACKGROUND AND PURPOSE
Tobacco and alcohol are often co-abused producing interactive effects in the brain. Although nicotine enhances memory while ethanol impairs it, variable cognitive changes have been reported from concomitant use. This study was designed to determine how nicotine and alcohol interact at synaptic sites to modulate neuronal processes.
EXPERIMENTAL APPROACH
Acute effects of nicotine, ethanol, and both drugs on synaptic excitatory glutamatergic and inhibitory GABAergic transmission were measured using whole-cell recording in hippocampal CA1 pyramidal neurons from brain slices of mice on control or nicotine-containing diets.
KEY RESULTS
Acute nicotine (50 nM) enhanced both GABAergic and glutamatergic synaptic transmission; potentiated GABAA receptor currents via activation of α7* and α4β2* nAChRs, and increased N-methyl-d-aspartate (NMDA) and α-amino-3-hydroxy-5-methylisoxazole-4-propionic acid (AMPA) receptor currents through α7* receptors. While ethanol (80 mM) also increased GABAA currents, it inhibited NMDA currents. Although ethanol had no effect on AMPA currents, it blocked nicotine-induced increases in NMDA and AMPA currents. Following chronic nicotine treatment, acute nicotine or ethanol did not affect NMDA currents, while the effects of GABAergic responses were not altered.
CONCLUSIONS AND IMPLICATIONS
Acute ethanol ingestion selectively attenuated nicotine enhancement of excitatory glutamatergic NMDA and AMPA receptor function, suggesting an overall reduction in excitatory output from the hippocampus. It also indicated that ethanol could decrease the beneficial effects of nicotine on memory performance. In addition, chronic nicotine treatment produced tolerance to the effects of nicotine and cross-tolerance to the effects of ethanol on glutamatergic activity, leading to a potential increase in the use of these drugs.
Keywords: ethanol, alcohol, GABAA receptor, NMDA receptor, AMPA receptor, nicotinic receptor
Introduction
Previous studies showed that acute ethanol consumption causes cognitive deficits in humans (Koelega, 1995) and in rats (Givens and McMahon, 1997). Nicotine, however, was found to improve some forms of cognitive performance in mammals (Stolerman et al., 1995). Also, when these two drugs were used together, nicotine was usually found to ameliorate (Gould et al., 2001), but in some cases, to potentiate (Rezvani and Levin, 2002) ethanol-induced deleterious effects on cognitive performance. Acute alcohol consumption can increase cigarette smoking in both alcohol-dependent and non-alcohol-dependent subjects (Mitchell et al., 1995). Other evidence also shows that nicotine can increase ethanol consumption in rats (Clark et al., 2001; Lêet al., 2003) and in mice (Marks et al., 1994). As a part of an extended amygdala circuitry, the hippocampus plays an important role in regulating motivated behaviours, including drug self-administration (Risold and Swanson, 1996; Kelley, 2004), and each of these two drugs has been shown to affect GABAergic and glutamatergic transmission in the hippocampus (Harris and Mihic, 2004; Dani and Harris, 2005). Therefore, functional interactions between nicotine and ethanol in the hippocampus could affect ethanol consumption and smoking in humans.
Nicotine activates brain nicotinic acetylcholine receptors (nAChRs), which are pentameric cation conductance channels composed of various combinations of α (α2–α7) and β (β2–β4) subunits (Leonard and Bertrand, 2001; receptor nomenclature follows Alexander et al., 2009). The two most common nAChRs in the hippocampus are α7* and α4β2* (Role and Berg, 1996), the asterisks denote possible inclusion of other subunits (Lukas et al., 1999). These nAChRs are found in high density on inhibitory hippocampal interneurons (Freedman et al., 1993; Sudweeks and Yakel, 2000) and astrocytes (Gahring et al., 2004). Nicotine enhances inhibitory postsynaptic currents (IPSCs) through GABAA receptors (Dani, 2001) and N-methyl-d-aspartate (NMDA) receptor excitatory postsynaptic currents (EPSCs; Ge and Dani, 2005). Ethanol is also known to enhance GABAA receptor IPSCs and to inhibit glutamatergic NMDA receptor EPSCs (Harris and Mihic, 2004; Proctor et al., 2006), suggesting these transmitter systems are possible sites for acute and chronic functional interaction between these two drugs. The acute interactions between nicotine and ethanol have been demonstrated previously in cognitive (memory/learning) tasks (Rezvani and Levin, 2002; Gulick and Gould, 2008; Robles and Sabria, 2008), but there is a lack of interactive effects on hypothermia (Rezvani and Levin, 2002). Additional support that ethanol and nicotine have common sites of action include reports showing that chronic ethanol treatment resulted in the development of tolerance to ethanol-induced hypothermia and cross-tolerance to nicotine-induced hypothermia (Collins et al., 1996) and fear conditioning (Gulick and Gould, 2008; Lallemand et al., 2009). In the present study, we examined the interactive effects of acute ethanol and nicotine, and also of chronic nicotine treatment on the modulation of functional glutamatergic and GABAergic synaptic activities by nAChRs, in hippocampal CA1 pyramidal neurons of mice.
Methods
Animals
All animal care and experimental procedures complied fully with all NIH, VA and University of Colorado Denver IACUC guidelines. Adult male (6–10 weeks old) C57BL/6 mice were group-housed with a 12 h light–dark cycle with lights on at 0600. The animals had free access to water and standard rodent chow, except during the nicotine liquid diet (described below).
Chronic nicotine diet procedure
In some experiments, mice consumed control liquid diet (Bio-Serv, Frenchtown, NJ, USA) or the same liquid diet containing nicotine. The animals started with control liquid diet for 3 days, which was followed by control liquid diet (control group) or liquid diet containing 0.022 mM (for 2 days), 0.087 mM (3 days) and 0.15 mM (6 days) of nicotine. The doses of nicotine used were those reported by Pekonen et al. (1993) with specific modification to take into account the amount of nicotine taken by liquid diet instead of nicotine in the drinking water. Fresh liquid diet was provided at 0700 and 1700 each day, at which times the night and day consumption was measured for each animal. Body weight changes were measured throughout the entire experiment to monitor for nicotine taste aversion, which was not found in this study. Brain slices were prepared 3 h following the termination of the nicotine liquid diet, which was replaced with control liquid diet. An average of 13.2 ± 0.7 g·day−1 of the liquid diet (for either control or nicotine-containing diet) was consumed throughout the 14 day treatment. A group of mice was also tested for blood, hippocampus and total brain nicotine levels using LC-MS/MS (iC42 Laboratory, Aurora, CO, USA). At the time of brain slice preparation, no measurable nicotine remained in the blood, hippocampus or total brain tissue, while cotinine (nicotine metabolite) levels were 8.27 ± 1.24 ng·mL−1, 3.53 ± 0.83 ng·g−1 and 2.19 ± 0.53 ng·g−1 respectively.
Brain slice preparation and storage
Mice were killed by quick cervical dislocation and the brains were rapidly removed and immersed in an ice-cold, sucrose-containing modified artificial cerebrospinal fluid (aCSF) for 60 s to chill the interior of the brain. The sucrose-containing aCSF consisted of (in mM): 87 NaCl, 2.5 KCl, 7.0 MgCl2, 0.5 CaCl2, 1.2 NaH2PO4, 25 d-glucose, 25.9 NaHCO3 and 75 sucrose, and was continuously oxygenated with 95% O2 and 5% CO2. Coronal slices (300 µm thick) were made using a Leica vibratome (model VT-1000S, Heerbrugg, Switzerland). The slices were temporarily submerged in the ice-cold sucrose-containing aCSF until all slices were collected, and then were transferred to individual compartments in a submersion storage system (Proctor and Dunwiddie, 1999) containing a standard aCSF buffer: (in mM) 126 NaCl, 3.0 KCl, 1.5 MgCl2, 2.4 CaCl2, 1.2 NaH2PO4, 11 d-glucose and 25.9 NaHCO3. The storage container was maintained at a constant temperature of 32.5°C and was continuously gassed with 95% O2 and 5% CO2 providing an oxygenated and pH 7.25 buffered environment. For electrophysiological whole-cell recordings, individual slices were transferred to a submersion recording chamber (Warner Instruments, Hamden, CT, USA) also maintained at a constant temperature of 32.5°C and superfused with gassed aCSF at a flow rate of 2 mL·min−1.
Electrophysiological recording
Whole-cell recording microelectrodes were constructed from borosilicate glass capillary tubing (1.5 mm o.d., 0.86 mm i.d.; Sutter Instrument Co., Novato, CA, USA) and pulled inside a heated platinum/iridium box filament with a model P-87 micro-pipette puller (Sutter Instrument Co) to a tip size of approximately 1.0–1.5 µm in diameter. These microelectrodes had electrical resistances of 6 to 9 MΩ when filled with a K+-gluconate internal solution containing (in mM): 130 K+-gluconate, 0.8 KCl, 0.1 CaCl2, 2.0 MgCl2, 1.0 EGTA, 10.0 HEPES, 2.0 Mg-ATP and 0.3 Na-GTP, adjusted to pH 7.25 and 290 mOsm. CA1 pyramidal neurons were recorded in the whole–cell configuration. GABAA receptor-mediated IPSC responses were evoked via an electrical pulse (200 µs, 4–8 V) with a bipolar tungsten stimulating electrode at 60 s intervals. The stimulating electrode was positioned in the stratum pyramidale within 300 µm of the whole-cell recording electrode. The cells were voltage-clamped to −55 mV (corrected for the liquid-junction potential) from the control resting membrane potential (−71.3 ± 0.65 mV; mean ± SEM) to enhance the recorded GABAA receptor currents. Similar conditions were also used for recording NMDA receptor EPSCs and α-amino-3-hydroxy-5-methylisoxazole-4-propionic acid (AMPA) receptor EPSCs. This stimulation-recording paradigm evoked synaptic responses predominantly from proximal inputs (i.e. GABAA receptor responses from interneuron axon terminals that synapse on or near the soma of the recorded pyramidal cell, and glutamatergic responses from proximal pyramidal cell excitatory terminals). We have used this paradigm to study the effects of ethanol in several rat and mouse strains (Wu et al., 2005; Proctor et al., 2006). The recording of miniature IPSCs (mIPSCs) or miniature EPSCs (mEPSCs) was carried out using microelectrodes filled with K+-gluconate (for mEPSC recordings) or CsCl (for mIPSC determinations; Sanna et al., 2004). The CsCl solution consisted of (in mM): 140 CsCl, 2 MgCl2, 1 CaCl2, 10 EGTA, 10 HEPES, 2 Na-ATP, adjusted to pH 7.25 with CsOH and 290 mOsm.
Drugs and drug administration
All drugs were purchased from Sigma (St. Louis, MO, USA) except as noted. The drugs were prepared as 100-fold concentrates in 12 mL syringes (Monojet, polypropylene; Sherwood-Davis & Geck, St. Louis, MO, USA) and were added to the flowing aCSF superfusate (2 mL·min−1) via calibrated syringe pumps (Razel Scientific Instruments, Stamford, CT, USA). Ethanol was diluted in deionized water from a 95% stock solution and stored cold in a sealed glass bottle before loading into a 12 mL syringe.
Measurement of NMDA and AMPA receptor EPSCs, GABAA receptor IPSCs, mEPSCs and mIPSCs
N-methyl-d-aspartate receptor EPSCs were pharmacologically isolated by bath application of 6-cyano-7-nitroquinoxaline-2,3-dione disodium salt (CNQX, 20 µM), 3[[(3,4-dichlorophenyl)methyl]amino]propyl](diethoxymethyl) phosphinic acid (CGP 52432) (0.5 µM) and bicuculline methiodide (BMI, 20 µM) to block AMPA receptor EPSCs, GABAB and GABAA receptor IPSCs respectively. Similarly, isolated AMPA receptor EPSCs were measured after superfusion with d-2-amino-5-phosphonovaleric acid (D-APV, 50 µM) to block NMDA receptor responses, BMI (20 µM) and CGP 52432 (0.5 µM). To measure GABAA IPSCs, CNQX, D-APV and CGP 52432 were superfused to pharmacologically block the other current responses. mIPSCs and mEPSCs were pharmacologically isolated as above for IPSCs and EPSCs, as well as recorded in the presence of tetrodotoxin (0.5 µM) added to the perfusate to block action potential-generated spontaneous events. pClamp 9.0 software (Molecular Devices, Silicon Valley, CA, USA) was used to measure the mini events. Clampfit (Molecular Devices) was used to initially identify the events, and then each of these events was visually inspected and obvious spurious events were rejected (less than 5%) before further data analysis. Events were then averaged for each period of control, drug and washout (for non-nicotinic drugs) with Clampfit, and amplitudes, areas (charge transfer) and statistics were determined. The mIPSCs were completely blocked by bath application of the GABAA receptor antagonist, BMI (20 µM). The mEPSCs were recorded in the constant presence of picrotoxin (10 µM), a GABA receptor antagonist, and were completely blocked by the bath application of glutamate receptor antagonists, CNQX (20 µM) and D-APV (50 µM).
Acute application of nicotine and ethanol
Nicotine and the nAChR antagonists, methyllycaconitine (MLA), dihydro-β-erythroidine (DHβE) and mecamylamine were prepared fresh daily in deionized water. When needed, these drugs were added to the superfusate to provide final recording chamber concentrations as follows: nicotine (50 nM), MLA (10 nM), DHβE (10 µM) and mecamylamine (10 µM). A 50 nM acute nicotine concentration was chosen because: (i) this is the approximate blood-brain concentration in humans after smoking one or two cigarettes (Benowitz et al., 1989); (ii) desensitization of nAChRs occurs slowly over several minutes at low nicotine concentrations (Marks et al., 1994); and (iii) significant effects were observed on functional neurotransmitter systems (Figure 1A). Due to the effects of nicotine-induced long-lasting desensitization of the nAChRs (Fenster et al., 1997), nicotine was always the last drug to be added to the brain slices during multiple drug experiments. To determine the effects of nicotine, the synaptic responses were averaged over the peak nicotine effect, usually between 1 and 5 min following the nicotine application.
Figure 1.
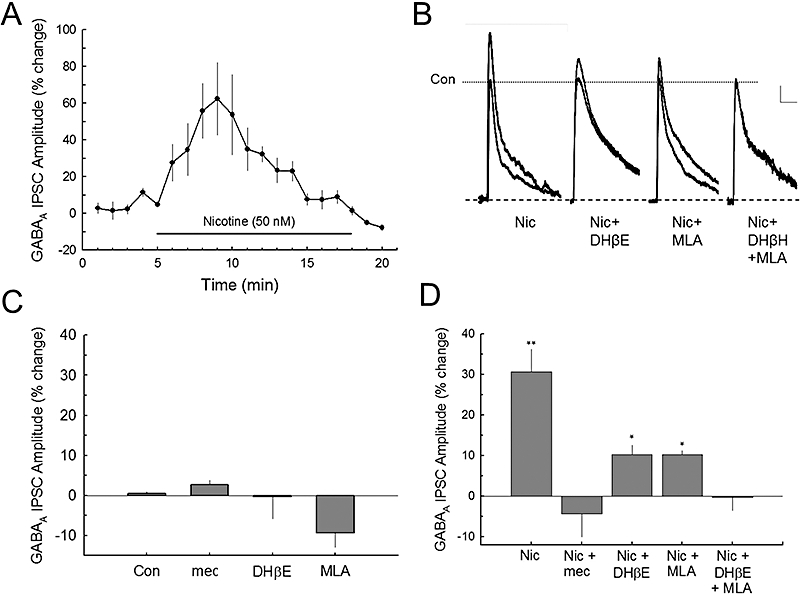
Nicotine enhances GABAA receptor inhibitory postsynaptic currents (IPSCs) via two nAChR subtypes. A. Bath perfusion of nicotine (50 nM) produces an increase in evoked GABAA receptor IPSCs recorded from CA1 pyramidal neurons. B. Traces show the effects of nicotine and the addition of nicotine antagonists on the GABAA receptor current responses. Scale: 50 pA, 100 ms. (Note: In these cells, the GABAB receptor was not blocked with 3[[(3,4-dichlorophenyl)methyl]amino]propyl](diethoxymethyl) phosphinic acid (CGP 52432) so that the late GABAB component can be seen in some cell traces.) Nicotine enhanced responses of both GABAA and GABAB receptors. C. Bath superfusion of the nicotinic antagonists [mecamylamine (mec, 10 µM), DHβE (10 µM) and MLA (10 nM)] did not significantly affect control synaptic GABAA IPSCs. The number of cells (n) recorded for each condition: Con (18), mec (7), DHβE (6), MLA (5). D. The composite data show that the effects of nicotine on GABAA IPSCs are partially blocked by either DHβE (10 µM) or MLA (10 nM), and are completely blocked by mec or the combination of DHβE and MLA, indicating the involvement of both α4β2* and α7* nACh receptors. Number of recorded cells: Nic (7), Nic + mec (6), Nic + DHβE (6), Nic + MLA (5), Nic + DHβE + MLA (5). *P < 0.05; **P < 0.01.
Results from our previous studies show that ethanol produces incremental neurotransmitter effects between 20 and 120 mM in both mice and rats (Proctor et al., 2004; 2006; Wu et al., 2005). Therefore, except as noted, a final recording chamber concentration of 80 mM ethanol was used to provide sufficiently robust effects (about 30% enhancement of GABAA receptor IPSCs and 30% inhibition of NMDA receptor EPSCs). Furthermore, this concentration (80 mM) approximates the average blood and brain level (360 mg·100 mL−1) measured when mice regain their righting reflex following an i.p. injection of a hypnotic/sedative dose of ethanol (4.1 g·kg−1; (Draski et al., 1992). This ethanol concentration was also shown to produce robust enhancement of hippocampal GABAA IPSCs in Sprague-Dawley rats (Weiner et al., 1997; Wu et al., 2005).
Statistics
Drug effects were quantified as the percent change in the amplitude of GABAA, NMDA and AMPA receptor current responses following drug application relative to the mean of control and recovery values. Statistical analyses were carried out using Student's t-test or anova with post hoc pairwise comparisons (Holm-Sidak). The probability distributions for miniature responses were all analysed by the Kolmogorov–Smirnov test (K–S-test), and the minimal significance level was set at P < 0.05.
Results
Acute nicotine increases GABAergic neurotransmission via α7* and α4β2* nAChR subtypes
After blocking glutamatergic currents with CNQX (20 µM) and D-APV (50 µM), and GABAB receptor currents with CGP 52432 (0.5 µM), electrical stimulation near the soma of CA1 pyramidal neurons (proximal terminal activation; Wu et al., 2005) produced synaptic GABA receptor-mediated responses (GABAA IPSCs). Bath superfusion of nicotine (50 nM) produced robust increases in synaptic GABAA IPSCs in hippocampal CA1 pyramidal neurons (Figure 1A). This effect of nicotine on synaptic GABAA IPSCs usually peaked within 3–5 min during nicotine application and decayed to baseline with a half-decay time of 4.9 ± 0.28 min during nicotine perfusion. Thus, acute nicotine application modulated GABAA IPSCs by an activation that was followed by desensitization or deactivation of nAChRs in the continuing presence of nicotine. To identify the nAChR subtypes that mediated this nicotine effect, we used (i) a selective α7 nAChR antagonist, MLA (10 nM); (ii) a selective α4β2* antagonist, DHβE (10 µM); or (iii) a non-selective antagonist, mecamylamine (10 µM), to characterize the subtypes of nAChR that may mediate this nicotine potentiation of GABAA IPSCs (Figure 1B). When applied alone, these nAChR antagonists each did not significantly alter the control GABAA receptor IPSC responses [F(3,29) = 2.794, P > 0.058, one-way anova] (Figure 1C). Pretreatment with the antagonists for at least 4 min was required to block the effects of 50 nM nicotine. Nicotine enhancement of GABAA IPSCs was partially blocked [F(4,25) = 8.763, P < 0.001, one-way anova] by either MLA (P < 0.05) or DHβE (P < 0.05); but was completely blocked by the co-application of MLA plus DHβE (P < 0.01), or by mecamylamine (P < 0.001; Figure 1D). These results suggest that the nicotine potentiation of the GABAA receptor IPSCs is likely to be mediated by activation of both α7* and α4β2* nAChRs.
Acute ethanol attenuates nicotinic effects on GABAA receptor IPSCs
Like nicotine, ethanol alone (80 mM) also enhanced GABAA receptor IPSCs in C57BL/6 mice (Figure 2A). This result is consistent with our previous data in Sprague-Dawley rats (Wu et al., 2005) and inbred long-sleep mice (Proctor et al., 2006). However, following pretreatment with ethanol, nicotine did not provide a significantly greater increase of the ethanol enhancement of GABAA IPSCs. One-way anova analysis of the data revealed that treatment with nicotine or ethanol each produced significant enhancement of GABAA receptor IPSCs [F(6,70) = 43.047, P < 0.001, one-way anova]. The post hoc Holm-Sidak pairwise analysis showed that separately, nicotine (50 nM) enhanced GABAA IPSCs by 32.0 ± 6.2% (mean ± SEM; n= 7; P < 0.01), and ethanol (80 mM) enhanced GABAA IPSCs by 26.9 ± 3.7% (n= 8; P < 0.01) compared WITH control responses (Figure 2B). However, the combined ethanol and nicotine application increased GABAA IPSCs by 40.1 ± 6.39% (n= 7; P < 0.01), which differed from that of ethanol alone (P < 0.01), but did not differ from that of nicotine alone (P > 0.05). Pretreatment with mecamylamine (10 µM), a non-selective nAChR antagonist, did not affect the ethanol-enhanced GABAA IPSCs (P > 0.7), but it effectively blocked the increases in GABAA responses by nicotine application (P < 0.01, Figure 2B). In the presence of mecamylamine, acute ethanol followed by acute nicotine treatment produced a GABAA IPSC enhancement that was similar to ethanol alone, but significantly different to that of nicotine alone (P < 0.01). The data show that there was a larger, but not significant additive or synergistic effect of nicotine and ethanol together on GABAA IPSCs. This finding that acute ethanol application did not significantly further enhance the nicotine potentiation of the GABAA IPSCs, may indicate that GABAA receptor responses had reached their maximum stimulation by either drug. To test whether a ceiling effect for either drug had been achieved, we measured the effects of 120 mM ethanol on synaptic GABAA IPSCs in a subgroup of animals. As in previous studies (Weiner et al., 1997; Proctor et al., 2006), 120 mM ethanol significantly further increased the GABAA receptor response above the 80 mM concentration by an additional 22.2 ± 3.3% (P < 0.05, Student's t-test). Thus, a ceiling effect for 80 mM ethanol is an unlikely explanation for the lack of augmentation between ethanol and nicotine during the co-application of these two drugs.
Figure 2.
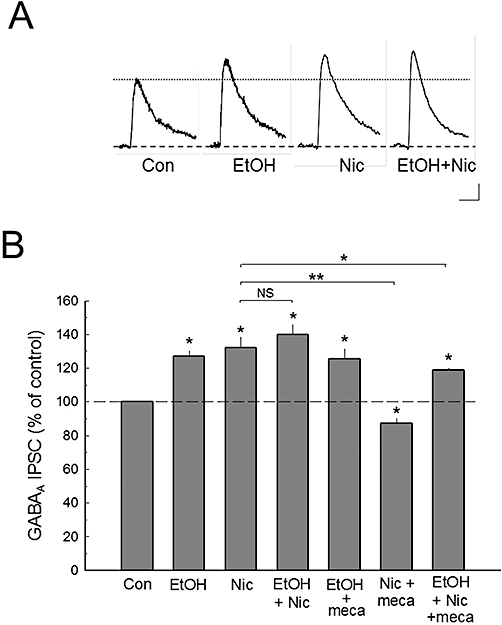
Ethanol modulates the effects of nicotine on GABAA receptor inhibitory postsynaptic currents (IPSCs). A. The evoked and pharmacologically isolated GABAA receptor IPSCs show that ethanol (EtOH) and nicotine (Nic) each enhances GABAA IPSCs, but they do not have a significant additive or synergistic effect. B. The composite data show that the enhancement effect of nicotine on GABAA IPSCs can be completely blocked by pretreatment of the slices with mecamylamine (mec; 10 µM), while the effect of ethanol on GABAA IPSCs is not affected by the presence of mecamylamine. Although individual nicotine or ethanol application increases GABAA IPSCs to a similar extent, the combined ethanol plus nicotine treatment does not significantly increase GABAA IPSCs further than that produced by each drug alone. Number of cell recorded: Con (30), EtOH (8), Nic (7), EtOH + Nic (7), EtOH + mec (7), Nic + mec (7), EtOH + Nic + mec (5). Scale bars in A: 10 pA, 25 ms; *P < 0.05; **P < 0.01.
Acute ethanol modulates nicotine-induced GABA release
To determine whether GABAergic interneurons (the major presynaptic inhibitory inputs onto pyramidal cells) are sites for functional interactions between nicotine and ethanol, we measured the effects of nicotine and/or ethanol on mIPSCs in CA1 pyramidal neurons. Ethanol application significantly enhanced mIPSC amplitudes (P < 0.001) and frequency (P < 0.001), and nicotine also significantly increased mIPSC amplitude (P < 0.001), as well as frequency (P < 0.001) as shown in Figure 3. The combined ethanol and nicotine application also resulted in significant increases of mIPSC amplitude (P < 0.001) and frequency (P < 0.001) when compared WITH control baseline conditions. The potentiation of mIPSC amplitudes by the combined ethanol and nicotine application was different from that of nicotine alone (P < 0.01), but was not different from that of ethanol alone (P > 0.05). Also, the combined drugs potentiated mIPSC frequency compared WITH nicotine (P < 0.001), but was not different from that of ethanol application alone (P > 0.05). One-way anova analysis showed that both nicotine and ethanol had overall effects on mIPSC amplitude [F(3,21) = 65.614, P < 0.001] and frequency [F(3,21) = 224.104, P < 0.001]. These results suggest that individual application of either ethanol or nicotine enhances GABA release, and that ethanol blocks further increases in GABA release by nicotine (Figure 3D). These data also suggest that both ethanol and nicotine have a postsynaptic effect due to the increase in mIPSC amplitudes. At this time, these measurements are difficult to reconcile with other reports indicating that nAChRs are localized primarily on the presynaptic (interneuronal) processes that synapse on CA1 hippocampal pyramidal cells in rodents.
Figure 3.
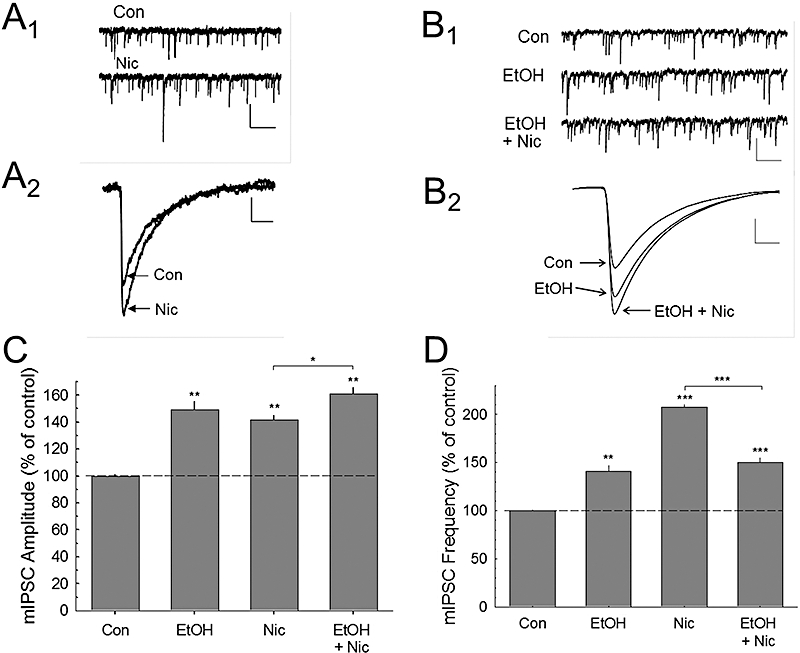
Nicotine and ethanol modulation of miniature inhibitory postsynaptic current (mIPSC) activity. The upper traces (A1) show raw control (Con) and during nicotine application (Nic) on mIPSC responses after abolishing glutamatergic transmission with 6-cyano-7-nitroquinoxaline-2,3-dione disodium salt and d-2-amino-5-phosphonovaleric acid, and tetrodotoxin (0.5 µM) to block action potential induced spontaneous IPSCs. (A2) Traces of the averaged mIPSCs before (Con) and during nicotine (Nic) treatment. (B1) These traces show the effects of ethanol (EtOH) or ethanol plus nicotine application on spontaneous mIPSCs, and (B2) the averaged waveforms for each condition. The summary data for nicotine and ethanol effects on mIPSC amplitude (C) and frequency (D) are shown. These data indicate that ethanol selectively inhibits nicotine-enhanced mIPSC frequency. Number of cells recorded in C and D: Con (10), EtOH (5), Nic (5), EtOH + Nic (5). Scale bars – in A1 and B1: 15 pA, 1.0 s; in A2: 5 pA, 15 ms; B2: 10 pA, 10 ms. **P < 0.01; ***P < 0.001.
Acute nicotine enhances glutamatergic transmission via α7* nAChRs
In the presence of the GABA receptor antagonists, electrical stimulation of the proximal excitatory hippocampal inputs to CA1 pyramidal cells evoked glutamatergic synaptic responses consisting of both AMPA and NMDA receptor currents. The pharmacologically isolated AMPA or NMDA receptor EPSCs were enhanced by bath perfusion of nicotine (50 nM; Figure 4A). This enhancement of AMPA and NMDA EPSCs in the CA1 pyramidal neurons occurred within 3–4 min of bath nicotine application, and returned to baseline levels during the next 8 min for AMPA EPSCs and 5 min for NMDA EPSCs with an average half-decay time of 4.7 ± 0.25 and 2.4 ± 0.42 min for AMPA and NMDA respectively (Figure 4A). Because nicotine induced similar peak enhancement of AMPA and NMDA receptor responses, we tested the effects of nicotine and nAChR antagonists on the glutamate EPSCs (the combined AMPA and NMDA receptor currents) (Figure 4B–D). None of the nAChR antagonists tested (mecamylamine, DHβE or MLA) significantly altered the control glutamate receptor EPSCs [F(3,29) = 1.463, P > 0.245, one-way anova] (Figure 4C). Moreover, the nicotine potentiation of the glutamate EPSC was not significantly attenuated (P > 0.05) by 10 µM DHβE, a concentration that was effective in blocking α4β2 nAChR subtypes. To determine whether other subtypes of αβ nAChRs were involved, we treated neurons with 50 µM DHβE, a concentration that was shown to inhibit αβ nAChRs (Harvey et al., 1996). This concentration of DHβE was also ineffective in blocking the nicotine enhancement of glutamate EPSCs (there was only 3.9 ± 10.2% reduction) [t= 0.177; P > 0.868, Student's t-test], which was not significantly different from the nicotine potentiation of glutamate responses. However, the nicotine potentiation of glutamate receptor EPSCs was completely blocked by 10 nM MLA, a selective α7 nAChR antagonist (P < 0.001), or by the non-selective nAChR antagonist, mecamylamine (10 µM; P < 0.001; Figure 4D).
Figure 4.
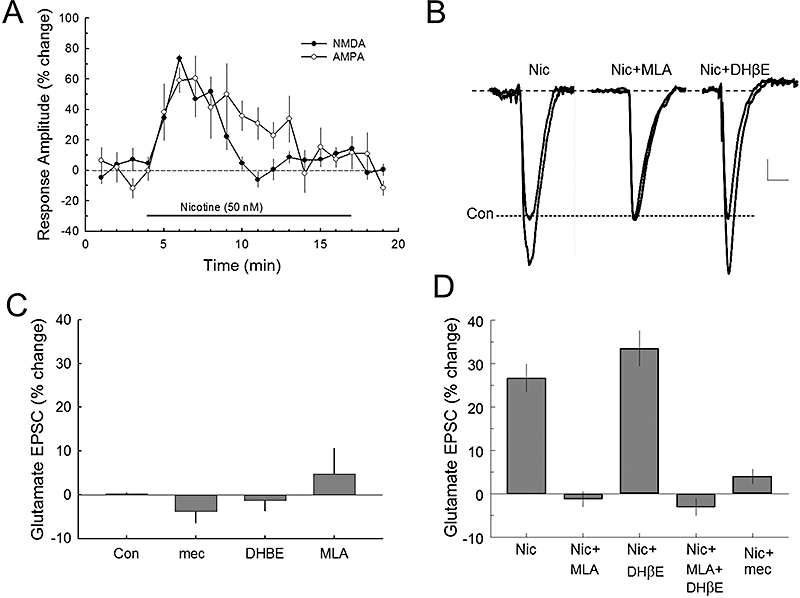
Nicotine increases glutamate release via the α7* nAChR subtype. A. Nicotine increases glutamatergic α-amino-3-hydroxy-5-methylisoxazole-4-propionic acid (AMPA) and N-methyl-d-aspartate (NMDA) excitatory postsynaptic currents (EPSCs) in CA1 pyramidal neurons. These responses were recorded in bicuculline methiodide (BMI) and 3[[(3,4-dichlorophenyl)methyl]amino]propyl](diethoxymethyl) phosphinic acid (CGP 52432) to block GABA activity. B. Averaged traces of glutamate receptor (combining AMPA and NMDA subtypes) responses to nicotine and nAChR antagonists. Scale: 50 pA, 30 ms. C. Bath superfusion of the nAChR antagonists, mecamylamine (mec; 10 µM), DHβE (10 µM) or MLA (10 nM), did not significantly affect synaptic glutamate receptor EPSCs. Number of cells recorded (n): Con (13), mec (6), DHβE (6), MLA (6). D. The composite data show that the 50 nM nicotine effect on glutamate (AMPA plus NMDA) receptor EPSCs can be completely blocked by pretreatment of brain slices with MLA, but not with DHβE. This result suggests that these effects of nicotine on glutamate receptors is mediated by the α7*, but not by the α4β2* nAChR subtype. Number of cells recorded: Nic (8), Nic + MLA (6), Nic + DHβE (8), Nic + MLA + DHβE (6), Nic + mec (5). *P < 0.05.
Acute ethanol blocks nicotinic effects on AMPA and NMDA receptor-mediated EPSCs
Nicotine significantly increased AMPA receptor-mediated EPSCs [F(4,27) = 17.468, P < 0.001; one-way anova, Figure 5A and B]. Holm-Sidak pairwise analysis revealed that ethanol alone had no significant effect on AMPA EPSCs, while nicotine enhanced AMPA EPSCs by 26.4 ± 3.5% (P < 0.01). However, pretreatment with ethanol effectively blocked the nicotine enhancement of AMPA EPSCs (P < 0.001; Figure 5B). This increase in AMPA EPSCs by nicotine was also blocked by mecamylamine (P < 0.001). These results show that while ethanol alone did not affect the synaptic AMPA EPSCs directly, ethanol blocked the nicotine enhancement of AMPA EPSCs.
Figure 5.
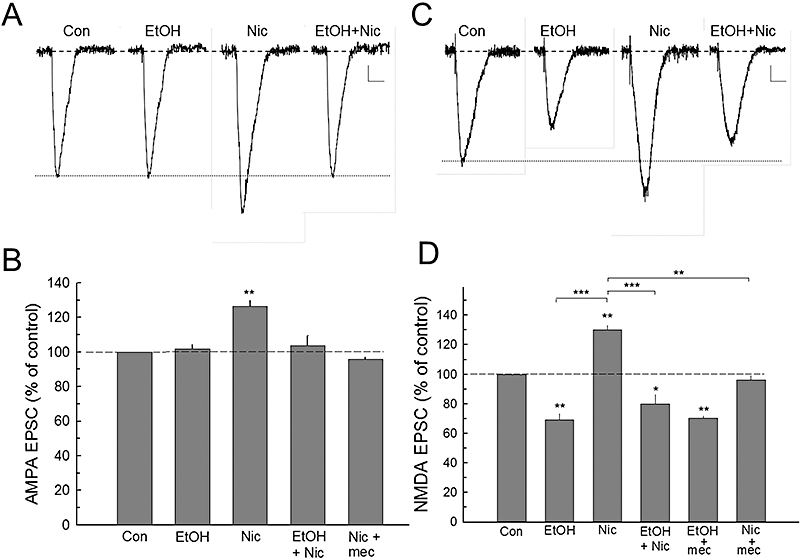
Ethanol attenuates the effects of nicotine on α-amino-3-hydroxy-5-methylisoxazole-4-propionic acid (AMPA) and N-methyl-d-aspartate (NMDA) receptor excitatory postsynaptic currents (EPSCs). A. Evoked response waveforms show that pharmacologically isolated AMPA receptor EPSCs are increased by nicotine (Nic), whereas ethanol (EtOH) has no significant effect on these responses. However, the enhancement of nicotine on AMPA EPSCs is prevented by ethanol pretreatment. B. The composite data show that the effects of nicotine on AMPA EPSCs are blocked by either ethanol (80 mM) or by mecamylamine (mec; 10 µM). Number of cells recorded (n): Con (8), EtOH (7), Nic (6), EtOH + Nic (6), Nic + mec (5). C. Ethanol depresses NMDA responses, while nicotine enhances this response. Pretreatment with ethanol essentially blocks the effects of subsequent nicotine application. D. Summary data of ethanol and nicotine action on the NMDA EPSC responses. Note that the general nAChR antagonist, mecamylamine, shows no alteration of the ethanol effect, but it blocks nicotine modulation. Cells recorded (n): Con (8), EtOH (7), Nic (6), EtOH + Nic (6), EtOH + mec (5), Nic + mec (4). Scale bars – A: 4 pA, 50 ms; C: 4 pA, 100 ms. *P < 0.05; **P < 0.01.
Acute ethanol application decreased NMDA receptor EPSC responses (Figure 5C), while application of nicotine increased this response, which was blocked by mecamylamine (Figure 5D). Pretreatment with ethanol attenuated the effects of nicotine (Figure 5C and D). Analysis of the data indicated that nicotine and ethanol produced significant overall effects [F(5,70) = 67.061, P < 0.001, one-way anova], and that the post hoc pairwise comparisons showed that ethanol produced a 31.7 ± 4.4% (P < 0.01) inhibition of NMDA EPSCs, and that this effect of ethanol was not altered by mecamylamine treatment (P > 0.05; Figure 5D). In contrast, nicotine produced a 30.4 ± 3.4% (P < 0.01) increase in NMDA EPSCs, and this increase was blocked by mecamylamine (P < 0.01) and by ethanol (−20.3 ± 6.7%, n= 7; P < 0.001). We then determined if nicotine increased glutamate release, and whether the modulatory effect of nicotine by ethanol was mediated by blocking this release. To investigate the possible presynaptic sites of interaction between ethanol and nicotine on glutamatergic EPSCs, we measured mEPSCs before and during nicotine and/or ethanol application (Figure 6A and B). Nicotine significantly increased the amplitude [F(3,32) = 1667.556, P < 0.001, one-way anova] and frequency [F(3,32) = 739.329, P < 0.001, one-way anova], and ethanol blocked these nicotine enhancements (Figure 6C and D).
Figure 6.
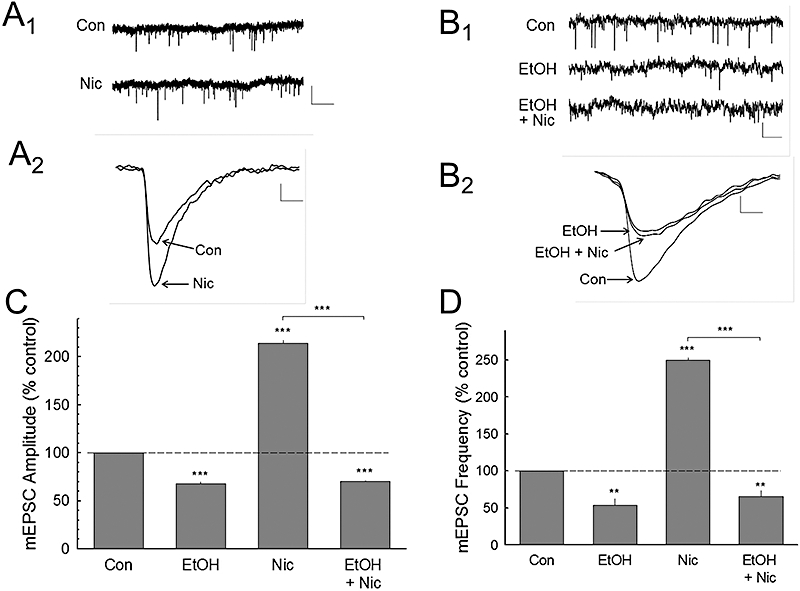
Nicotine and ethanol modulate miniature excitatory postsynaptic current (mEPSC) activity. A. The upper traces (A1) show raw control (Con) and during nicotine application (Nic) on mEPSC responses after abolishing GABAergic transmission with bicuculline methiodide (BMI) and CGP 52432, and tetrodotoxin (0.5 µM) to block action potential induced spontaneous EPSCs. (A2) Traces of the averaged mEPSCs before (Con) and during nicotine (Nic) treatment. (B1) These traces show the effects of ethanol (EtOH) or ethanol plus nicotine application on mEPSCs, and the averaged waveforms for each condition (B2). The summary data for nicotine and ethanol effects on mEPSC amplitude (C) and frequency (D) are shown. These data indicate that ethanol inhibits nicotine-enhanced both amplitude and frequency of mEPSCs. Number of cells recorded in C and D: Con (12), EtOH (7), Nic (9), EtOH + Nic (6). Scale bars – in A1 and B1: 10 pA, 2.0 s; in A2: 2 pA, 4 ms; B2: 4 pA, 4 ms. *P < 0.05; **P < 0.01; ***P < 0.001.
Chronic nicotine consumption attenuated nicotine and ethanol effects on NMDA receptor-mediated currents
Animals were maintained on control or nicotine-containing liquid diet for 2 weeks. There were no differences in body weight changes between the group on control liquid diet (starting weight: 19.0 ± 1.44 g; ending 20.0 ± 0.73 g) and the mice on the nicotine-containing liquid diet (starting weight 18.3 ± 0.62 g; ending 20.0 ± 0.89 g). Due to the unlimited access to the liquid diet (88% was consumed during the dark cycle), it is difficult to determine the actual nicotine levels in the blood and brain of each animal. The average consumption was 13.2 mL during the last 6 days of the 0.15 mM nicotine concentration in the nicotine liquid diet (see Methods section). Thus, each animal consumed approximately 320 µg of nicotine per day (average animal body weight was 20 g). At the time of brain slice preparation at 0900, no detectable levels of nicotine were measured in blood, hippocampus or total brain tissue. However, the relatively rapid conversion of nicotine to cotinine (half-life of 9.2 min in C57BL/6 mice; Siu and Tyndale, 2007) resulted in detectable levels of the more slowly metabolized cotinine (clearance half-life: 37.5 min) of 8.27 ± 1.24 ng·mL−1 in the blood, and 3.53 ± 0.83 and 2.19 ± 0.53 ng·g−1 in hippocampal and total brain tissue, respectively, at the time of slice preparation. To estimate the peak nicotine concentration for this liquid diet paradigm, we back-calculated from these average cotinine concentrations measured in plasma and brain tissue. This rough estimation suggests that the tissue nicotine concentration could have been as high as 22 µM, providing that all the nicotine-containing diet was consumed 3 h into the dark phase (Rhodes et al., 2005). Because consumption of the liquid diet likely occurred at various times throughout the dark cycle, as well as more than 10% during the light phase, we would predict a peak blood/tissue concentration closer to 1–10 µM. For comparison, this range is about 3- to 10-fold higher than that found in humans after smoking a single cigarette as reported in a recent study using PET scanning and cigarettes containing 11C-nicotine cigarettes (Rose et al., 2010).
Although there was no measurable amount of nicotine in plasma or brain tissue at the time of brain slice recording, acute ethanol (80 mM) application enhanced synaptic GABAA receptor IPSCs by 30.7 ± 5.4% and 43.1 ± 8% from mice on control diet and nicotine-containing diet respectively (Figure 7A). The enhancing effect of nicotine (50 nM) application on synaptic GABAA IPSCs, also was not significantly altered from those in control animals (Figure 7A). Although acute ethanol (80 mM) application inhibited synaptic NMDA receptor EPSCs by 33.8 ± 5.6% in mice on the control diet, this concentration of ethanol failed to inhibit NMDA EPSCs from animals on the nicotine-containing diet (3.3 ± 3.1%; Figure 7B). When the effects of acute nicotine (50 nM) on synaptic NMDA EPSCs were determined, the results showed that while nicotine application produced a 28.2 ± 2.1% enhancement of NMDA EPSCs from the animals on the control diet, nicotine failed to produce a significant enhancement from the animals on the nicotine-containing diet (1.45 ± 4.3%; Figure 7B). Thus, this 2 week chronic nicotine liquid diet treatment resulted in a differential ethanol effect on GABAA and NMDA receptor synaptic activity.
Figure 7.
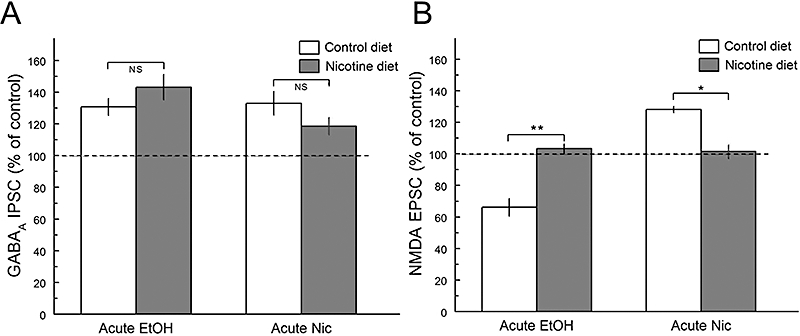
Chronic nicotine treatment alters acute ethanol and nicotine effects on N-methyl-d-aspartate (NMDA) receptor excitatory postsynaptic currents (EPSCs), but not on GABAA receptor inhibitory postsynaptic currents (IPSCs). A. After a 2 week chronic nicotine treatment, the acute effects by ethanol (EtOH) or nicotine (Nic) on GABAA receptor responses are not significantly different from those in control animals. B. However, after chronic nicotine treatment, animals showed a lack of ethanol inhibition of NMDA receptor EPSCs by an acute ethanol challenge application, as well as blocking the effects of acute nicotine. Therefore, besides the differential effects between inhibitory GABAA receptor IPSCs and excitatory NMDA receptor EPSCs by acute nicotine application, the chronic 2 week nicotine liquid diet produced tolerance to the acute effects of nicotine and cross-tolerance to the acute effects of ethanol on the NMDA receptor-ion channel complex. The number of cells for each condition: NMDA EPSCs: Con (8), EtOH (5), Nic (4). GABAA IPSCs: Con (5), EtOH (5), Nic (5).*P < 0.05; **P < 0.01.
Discussion and conclusions
The hippocampus receives rich intrinsic (interneurons) and extrinsic (septal) cholinergic innervation, and lesions of the extrinsic cholinergic pathways have been shown to impair memory and learning (McKinney and Jacksonville, 2005), suggesting the involvement of cholinergic transmission in these cognitive measures. In addition, septal cholinergic innervation can regulate the function of α7* nAChRs in CA1 hippocampal interneurons (Thinschmidt et al., 2005), where the majority of α-bungarotoxin binding occurs (Freedman et al., 1993). In the hippocampal CA1 neuronal circuit, the activation of nAChRs triggers the release of GABA from interneurons and glutamate from several excitatory terminal sources (Wonnacott et al., 2002). Thus, nAChRs are important modulators of GABAergic and glutamatergic synaptic transmission in the hippocampus.
In rodent brain slices, brief application of nAChR agonists directly onto hippocampal interneurons, but not onto CA1 pyramidal neurons, produces inward current responses (Frazier et al., 1998; Sudweeks and Yakel, 2000; Alkondon and Albuquerque, 2004). This indicates that nAChRs may be localized mainly on the presynaptic terminals of interneurons that synapse on CA1 pyramidal neurons. Therefore, the enhanced synaptic neurotransmission via GABAA, NMDA and AMPA receptor-ion channel complexes on CA1 neurons by nicotine is most likely due to increased release of transmitters from inhibitory GABAergic interneurons and excitatory glutamatergic terminals. This nicotine modulation is further confirmed by experiments in which the effects of nicotine and/or ethanol on mIPSCs or mEPSCs are measured. Our results show that both nicotine and ethanol enhance the release of GABA (Figure 3), but the nicotine potentiation of the mIPSC frequency is partially inhibited by co-application of ethanol, suggesting that ethanol can attenuate the nicotine-induced increases in GABA release. This is consistent with earlier reports showing that ethanol (100 mM or higher concentrations) inhibited the function of α7*, and enhanced the activity of α4β2* nAChRs (Covernton and Connolly, 1997; Narahashi et al., 2001).
Acute application of nicotine and ethanol
Nicotine rapidly activates and desensitizes nAChRs in a concentration- and nAChR subtype-dependent manner (Marks et al., 1994). Here, we used a 50 nM nicotine concentration in the bath superfusion because: (i) this concentration is within the range of nicotine levels found in the blood of smokers 5–10 min following 1–2 cigarettes (Benowitz et al., 1989); and (ii) studies have shown that in neurons exposed to more than 100 nM nicotine, greater than 90% of their nAChRs were desensitized within 10 min (Marks et al., 1994). We delivered nicotine by bath superfusion because this mode of agonist application more closely mimics the condition that occurs in a smoker in which nicotine-rich blood delivers nicotine to neurons in the brain.
We used an acute application of 80 mM ethanol concentration throughout this study because, in rodent brain slices, previous studies of ethanol inhibition of long-term potentiation reported effective concentrations ranging between 50–100 mM (Sinclair and Lo, 1986; Schummers et al., 1997; Wayner et al., 2004; Zhang et al., 2005; Tokuda et al., 2007), although Blitzer et al. (1990) found effects at lower concentrations. In addition, Pyapali et al. (1999) showed that while 10–30 mM ethanol can block long-term potentiation in adolescent rat brain slices, these concentrations of ethanol had no effect on adult rat brain slices. The ethanol concentration used in our study is consistent with most of the reports in the literature. However, this is a high concentration of ethanol by human standards and would be lethal for most individuals.
Subtypes of nAChRs involved in GABAergic and glutamatergic transmission
In rodent hippocampus, α4β2* and α7* subtypes of nAChRs predominate (Jones et al., 1999; Sudweeks and Yakel, 2000). Nicotine modulates both inhibitory and excitatory synaptic transmission (Dani, 2001). We found that nicotine enhancement of GABAA receptor IPSCs was completely blocked by the combination of MLA and DHβE, but it was only partly attenuated by each drug alone. These results suggest that nicotine potentiation of GABAA IPSCs is mediated by both α7* and α4β2* nAChR subtypes. The effects of nicotine on glutamate AMPA and NMDA receptor EPSCs was completely blocked by MLA and not by 10 or 50 nM DHβE, providing strong evidence that glutamatergic transmission was mediated primarily by α7* nAChRs. Furthermore, Le Magueresse et al. (2006) showed that during early post-natal periods, α7* and β2* nAChRs regulate GABAergic transmission, but only α7* receptors modulate glutamatergic transmission in the hippocampus. α7 nAChRs are low-affinity receptors therefore one would anticipate that higher concentrations of nicotine would further increase NMDA receptor EPSCs. Indeed, a 500 nM nicotine application was found to produce repetitive, depolarizing currents (action potentials), which were not reversed by ethanol or mecamylamine application (data not shown). This result is consistent with behavioural data that a high dose of nicotine can produce convulsions (Dobelis et al., 2003).
Ethanol modulates the effects of nicotine on glutamatergic synapses
Ethanol has been shown to inhibit NMDA receptor EPSCs, but has very little effect on AMPA EPSCs (Allgaier, 2002; current results). In the present study, ethanol (80 mM) inhibited NMDA EPSCs by about 30%, while it had no effect on AMPA EPSCs. These results are similar to what we observed in other rodent hippocampal CA1 neurons (Proctor et al., 2006). In contrast, nicotine significantly increased both NMDA and AMPA receptor EPSCs, results that are consistent with nicotinic regulation of presynaptic glutamate release. Moreover, these increases in EPSCs were completely blocked by MLA, but not altered by 10 or 50 µM DHβE, suggesting that the effect of nicotine on glutamatergic transmission is mediated mainly by α7* nAChRs. As ethanol does not directly inhibit AMPA EPSCs, and therefore is an ineffective inhibitor of postsynaptic AMPA receptors, the ethanol blockade of the nicotine potentiation of both NMDA and AMPA receptor EPSCs is likely to be due to the inhibition of presynaptic α7* nAChRs. This mechanism of ethanol modulation of nicotinic effects is further supported by the mEPSC studies, in which ethanol was able to attenuate the effect of nicotine alone on NMDA and AMPA mEPSCs. Ethanol inhibition of mEPSC amplitude in this experiment seems to suggest that ethanol has direct effects on the postsynaptic glutamate receptors. This result may be consistent with the notion of a weak inhibition of ethanol on presynaptic glutamatergic transmission (Proctor et al., 2006). However, ethanol could still block nicotine potentiation of glutamate receptor EPSCs by antagonizing presynaptic α7 nAChRs. Taken together, the current results suggest that ethanol inhibits the α7* nAChR subtype, which mediates the nicotine potentiation of the presynaptic release of glutamate. Thus, the overall effect of nicotine on enhancing CA1 excitatory glutamatergic transmission is significantly attenuated by ethanol when these two drugs are used together.
Cross-tolerance between nicotine and ethanol
Previous studies (Collins et al., 1993; de Fiebre and Collins, 1993) have shown that high ethanol-sensitive inbred long sleep mice are also more sensitive to acute nicotine effects than low ethanol-sensitive, short sleep mice on various behavioural measures, including Y-maze crossing, rearing activities, heart rate and body temperature. Following chronic nicotine administration, the long sleep mice developed nicotine tolerance to three of these four nicotine-induced behavioural measures, and cross-tolerance to ethanol on all four of these measures, suggesting common sites of action. Recently, Gulick and Gould (2008) showed that following chronic nicotine treatment, tolerance to nicotine and cross-tolerance to ethanol occurred in fear conditioned learning in C57BL/6 mice. In the current report, we showed that following chronic treatment with dietary nicotine, these mice developed functional tolerance to the acute effects of nicotine on synaptic NMDA receptor EPSCs and functional cross-tolerance to the effects of ethanol on this neurotransmitter system. However, neither tolerance to nicotine's effects, nor cross-tolerance to ethanol's effects on synaptic GABAA receptor IPSCs occurred in this chronic 2 week nicotine treatment paradigm.
Significance of nicotine and ethanol interactions in the brain
This study provides evidence that α7* nAChRs modulate functional glutamatergic neurotransmission, and that α7* and α4β2* nAChRs mediate the enhancement of GABAergic transmission in the hippocampal CA1 neuronal circuitry. As ethanol inhibits the α7* nAChRs, it neutralizes the nicotine potentiation of glutamatergic neurotransmission. In contrast, ethanol potentiates the effects of nicotine on GABAergic synaptic transmission. Such differences in the underlying neuronal mechanisms are likely to modulate the pharmacological effects on individuals during concurrent use (Fertig, 2002; Rose et al., 2004). nAChRs are important in many clinical psychiatric disorders such as schizophrenia (Freedman et al., 1994; Leonard and Bertrand, 2001), anxiety (Picciotto et al., 2002) and depression (Bertrand, 2005), as well as post-traumatic stress disorder (Hapke et al., 2005). Thus, concurrent use or ‘self-medication’ of nicotine and ethanol could alter the symptoms of these disorders, and may lead to an increased drive for the use of both substances.
Acknowledgments
This study was supported by a VA Merit Review Grant (to WRP), the National Institute on Alcohol Abuse and Alcoholism (AA016548 to WRP) and the Colorado Tobacco Research Program Grant (4I-007 to WRP).
Glossary
Abbreviations
- aCSF
artificial cerebrospinal fluid
- AMPA
α-amino-3-hydroxy-5-methylisoxazole-4-propionic acid
- BMI
bicuculline methiodide
- CA1
CA1 hippocampal region
- CGP 52432
3[[(3,4-dichlorophenyl)methyl]amino]propyl](diethoxymethyl) phosphinic acid
- CNQX
6-cyano-7-nitroquinoxaline-2,3-dione disodium salt
- D-APV
d-2-amino-5-phosphonovaleric acid
- DHβE
dihydro-beta-erythroidine
- EPSCs
excitatory postsynaptic currents
- GABAA
γ-amino butyric acid type A receptor
- GABAB
γ-amino butyric acid type B receptor
- IPSCs
inhibitory postsynaptic currents
- mEPSC
miniature excitatory postsynaptic current
- mIPSC
miniature inhibitory postsynaptic current
- MLA
methyllycaconitine
- nAChR
nicotinic acetylcholine receptor
- NMDA
N-methyl-d-aspartate
- α4β2
α4β2 nicotinic ACh receptor
- α7
α7 nicotinic ACh receptor
Conflict of interest
The authors declare that, except for income received from primary employers, no financial support or compensation has been received from any individual or corporate entity over the past 3 years for research or professional service and there are no personal financial holdings that could be perceived as constituting a potential conflict of interest.
Supplementary material
Supporting Information: Teaching Materials; Figs 1–7 as PowerPoint slide.
References
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 4th edition. Br J Pharmacol. 2009;158(Suppl 1):S1–S254. doi: 10.1111/j.1476-5381.2009.00499.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alkondon M, Albuquerque EX. The nicotinic acetylcholine receptor subtypes and their function in the hippocampus and cerebral cortex. Prog Brain Res. 2004;145:109–120. doi: 10.1016/S0079-6123(03)45007-3. [DOI] [PubMed] [Google Scholar]
- Allgaier C. Ethanol sensitivity of NMDA receptors. Neurochem Int. 2002;41:377–382. doi: 10.1016/s0197-0186(02)00046-3. [DOI] [PubMed] [Google Scholar]
- Benowitz NL, Porchet H, Jacob P., III Nicotine dependence and tolerance in man: pharmacokinetic and pharmacodynamic investigations. Prog Brain Res. 1989;79:279–287. doi: 10.1016/s0079-6123(08)62487-5. [DOI] [PubMed] [Google Scholar]
- Bertrand D. The possible contribution of neuronal nicotinic acetylcholine receptors in depression. Dialogues Clin Neurosci. 2005;7:207–216. doi: 10.31887/DCNS.2005.7.3/dbertrand. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blitzer RD, Gil O, Landau EM. Long-term potentiation in rat hippocampus is inhibited by low concentrations of ethanol. Brain Res. 1990;537:203–208. doi: 10.1016/0006-8993(90)90359-j. [DOI] [PubMed] [Google Scholar]
- Clark A, Lindgren S, Brooks SP, Watson WP, Little HJ. Chronic infusion of nicotine can increase operant self-administration of alcohol. Neuropharmacol. 2001;41:108–117. doi: 10.1016/s0028-3908(01)00037-5. [DOI] [PubMed] [Google Scholar]
- Collins AC, Romm E, Selvaag S, Turner S, Marks MJ. A comparison of the effects of chronic nicotine infusion on tolerance to nicotine and cross-tolerance to ethanol in long- and short-sleep mice. J Pharmacol Exp Ther. 1993;266:1390–1397. [PubMed] [Google Scholar]
- Collins AC, Wilkins LH, Slobe BS, Cao JZ, Bullock AE. Long-term ethanol and nicotine treatment elicit tolerance to ethanol. Alcohol Clin Exp Res. 1996;20:990–999. doi: 10.1111/j.1530-0277.1996.tb01936.x. [DOI] [PubMed] [Google Scholar]
- Covernton PJ, Connolly JG. Differential modulation of rat neuronal nicotinic receptor subtypes by acute application of ethanol. Br J Pharmacol. 1997;122:1661–1668. doi: 10.1038/sj.bjp.0701568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dani JA. Overview of nicotinic receptors and their roles in the central nervous system. Biol Psychiatry. 2001;49:166–174. doi: 10.1016/s0006-3223(00)01011-8. [DOI] [PubMed] [Google Scholar]
- Dani JA, Harris RA. Nicotine addiction and comorbidity with alcohol abuse and mental illness. Nat Neurosci. 2005;8:1465–1470. doi: 10.1038/nn1580. [DOI] [PubMed] [Google Scholar]
- Dobelis P, Hutton S, Lu Y, Collins AC. GABAergic systems modulate nicotinic receptor-mediated seizures in mice. J Pharmacol Exp Ther. 2003;306:1159–1166. doi: 10.1124/jpet.103.053066. [DOI] [PubMed] [Google Scholar]
- Draski LJ, Spuhler KP, Erwin VG, Baker RC, Deitrich RA. Selective breeding of rats differing in sensitivity to the effects of acute ethanol administration. Alcohol Clin Exp Res. 1992;16:48–54. doi: 10.1111/j.1530-0277.1992.tb00634.x. [DOI] [PubMed] [Google Scholar]
- Fenster CP, Rains MF, Noerager B, Quick MW, Lester RA. Influence of subunit composition on desensitization of neuronal acetylcholine receptors at low concentrations of nicotine. J Neurosci. 1997;17:5747–5759. doi: 10.1523/JNEUROSCI.17-15-05747.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fertig N. Overview – alcohol and tobacco: mechanisms and treatment. Proceedings of a workshop. Bethesda, Maryland, USA. May 6–7, 2002. Alcohol Clin Exp Res. 2002;26:1909–1910. doi: 10.1097/01.ALC.0000040845.55554.69. [DOI] [PubMed] [Google Scholar]
- de Fiebre CM, Collins AC. A comparison of the development of tolerance to ethanol and cross-tolerance to nicotine after chronic ethanol treatment in long- and short-sleep mice. J Pharmacol Exp Ther. 1993;266:1398–1406. [PubMed] [Google Scholar]
- Frazier CJ, Rollins YD, Breese CR, Leonard S, Freedman R, Dunwiddie TV. Acetylcholine activates an α-bungarotoxin-sensitive nicotinic current in rat hippocampal interneurons, but not pyramidal cells. J Neurosci. 1998;18:1187–1195. doi: 10.1523/JNEUROSCI.18-04-01187.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freedman R, Wetmore C, Stromberg I, Leonard S, Olson L. Alpha-bungarotoxin binding to hippocampal interneurons: immunocytochemical characterization and effects on growth factor expression. J Neurosci. 1993;13:1965–1975. doi: 10.1523/JNEUROSCI.13-05-01965.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freedman R, Adler LE, Bickford P, Byerley W, Coon H, Cullum CM, et al. Schizophrenia and nicotinic receptors. Harv Rev Psychiatry. 1994;2:179–192. doi: 10.3109/10673229409017136. [DOI] [PubMed] [Google Scholar]
- Gahring LC, Persiyanov K, Dunn D, Weiss R, Meyer EL, Rogers SW. Mouse strain-specific nicotinic acetylcholine receptor expression by inhibitory interneurons and astrocytes in the dorsal hippocampus. J Comp Neurol. 2004;468:334–346. doi: 10.1002/cne.10943. [DOI] [PubMed] [Google Scholar]
- Ge SY, Dani JA. Nicotinic acetylcholine receptors at glutamate synapses facilitate long-term depression or potentiation. J Neurosci. 2005;25:6084–6091. doi: 10.1523/JNEUROSCI.0542-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Givens B, McMahon K. Effects of ethanol on nonspatial working memory and attention in rats. Behav Neurosci. 1997;111:275–282. doi: 10.1037//0735-7044.111.2.275. [DOI] [PubMed] [Google Scholar]
- Gould TJ, Collins AC, Wehner JM. Nicotine enhances latent inhibition and ameliorates ethanol-induced deficits in latent inhibition. Nicotine Tob Res. 2001;3:17–24. doi: 10.1080/14622200020032060. [DOI] [PubMed] [Google Scholar]
- Gulick D, Gould TJ. Interactive effects of ethanol and nicotine on learning in C57BL/6J mice depend on both dose and duration of treatment. Psychopharmacology (Berl) 2008;196:483–495. doi: 10.1007/s00213-007-0982-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hapke U, Schumann A, Rumpf HJ, John U, Konerding U, Meyer C. Association of smoking and nicotine dependence with trauma and posttraumatic stress disorder in a general population sample. J Nerv Ment Dis. 2005;193:843–846. doi: 10.1097/01.nmd.0000188964.83476.e0. [DOI] [PubMed] [Google Scholar]
- Harris RA, Mihic SJ. Alcohol and inhibitory receptors: unexpected specificity from a nonspecific drug. Proc Natl Acad Sci USA. 2004;101:2–3. doi: 10.1073/pnas.0307281101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvey SC, Maddox FN, Luetje CW. Multiple determinants of dihydro-beta-erythroidine sensitivity on rat neuronal nicotinic receptor alpha subunits. J Neurochem. 1996;67:1953–1959. doi: 10.1046/j.1471-4159.1996.67051953.x. [DOI] [PubMed] [Google Scholar]
- Jones S, Sudweeks S, Yakel JL. Nicotinic receptors in the brain: correlating physiology with function. Trends Neurosci. 1999;22:555–561. doi: 10.1016/s0166-2236(99)01471-x. [DOI] [PubMed] [Google Scholar]
- Kelley AE. Memory and addiction: shared neural circuitry and molecular mechanisms. Neuron. 2004;44:161–179. doi: 10.1016/j.neuron.2004.09.016. [DOI] [PubMed] [Google Scholar]
- Koelega HS. Alcohol and vigilance performance: a review. Psychopharmacology (Berl) 1995;118:233–249. doi: 10.1007/BF02245951. [DOI] [PubMed] [Google Scholar]
- Lallemand F, Ward RJ, De WP. The influence of chronic nicotine administration on behavioural and neurochemical parameters in male and female rats after repeated binge drinking exposure. Alcohol. 2009;44:535–546. doi: 10.1093/alcalc/agp047. [DOI] [PubMed] [Google Scholar]
- Lê AD, Wang A, Harding S, Juzytsch W, Shaham Y. Nicotine increases alcohol self-administration and reinstates alcohol seeking in rats. Psychopharmacology (Berl) 2003;168:216–221. doi: 10.1007/s00213-002-1330-9. [DOI] [PubMed] [Google Scholar]
- Le Magueresse C, Safiulina V, Changeux JP, Cherubini E. Nicotinic modulation of network and synaptic transmission in the immature hippocampus investigated with genetically modified mice. J Physiol. 2006;576:533–546. doi: 10.1113/jphysiol.2006.117572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonard S, Bertrand D. Neuronal nicotinic receptors: from structure to function. Nicotine Tob Res. 2001;3:203–223. doi: 10.1080/14622200110050213. [DOI] [PubMed] [Google Scholar]
- Lukas RJ, Changeux JP, Le NN, Albuquerque EX, Balfour DJ, Berg DK, et al. International Union of Pharmacology. XX. Current status of the nomenclature for nicotinic acetylcholine receptors and their subunits. Pharmacol Rev. 1999;51:397–401. [PubMed] [Google Scholar]
- McKinney M, Jacksonville MC. Brain cholinergic vulnerability: relevance to behavior and disease. Biochem Pharmacol. 2005;70:1115–1124. doi: 10.1016/j.bcp.2005.05.019. [DOI] [PubMed] [Google Scholar]
- Marks MJ, Grady SR, Yang JM, Lippiello PM, Collins AC. Desensitization of nicotine-stimulated 86Rb+ efflux from mouse brain synaptosomes. J Neurochem. 1994;63:2125–2135. doi: 10.1046/j.1471-4159.1994.63062125.x. [DOI] [PubMed] [Google Scholar]
- Mitchell SH, De WH, Zacny JP. Effects of varying ethanol dose on cigarette consumption in healthy normal volunteers. Behav Pharmacol. 1995;6:359–365. [PubMed] [Google Scholar]
- Narahashi T, Soderpalm B, Ericson M, Olausson P, Engel JA, Zhang X, et al. Mechanisms of alcohol-nicotine interactions: alcoholics versus smokers. Alcohol Clin Exp Res. 2001;25:152S–156S. doi: 10.1097/00000374-200105051-00026. [DOI] [PubMed] [Google Scholar]
- Pekonen K, Karlsson K, Laakso I, Ahtee L. Plasma nicotine and cotinine concentrations in mice after chronic oral nicotine administration and challenge doses. Eur J Pharm Sci. 1993;1:13–18. [Google Scholar]
- Picciotto MR, Brunzell DH, Caldarone BJ. Effect of nicotine and nicotinic receptors on anxiety and depression. Neuroreport. 2002;13:1097–1106. doi: 10.1097/00001756-200207020-00006. [DOI] [PubMed] [Google Scholar]
- Proctor WR, Dunwiddie TV. Electrophysiological analysis of G protein-coupled receptors in mammalian neurons. In: Enna SJ, Williams M, editors. Current Protocols in Pharmacology. New York: John Wiley & Sons Inc.; 1999. pp. 11.2.1–11.2.22. [DOI] [PubMed] [Google Scholar]
- Proctor WR, Wu PH, Bennett B, Johnson TE. Differential effects of ethanol on gamma-aminobutyric acid-A receptor-mediated synaptic currents in congenic strains of inbred long and short-sleep mice. Alcohol Clin Exp Res. 2004;28:1277–1283. doi: 10.1097/01.alc.0000139816.32706.f1. [DOI] [PubMed] [Google Scholar]
- Proctor WR, Diao L, Freund RK, Browning MD, Wu PH. Synaptic GABAergic and glutamatergic mechanisms underlying alcohol sensitivitiy in mouse hippocampal neurons. J Physiol (Lond) 2006;575:145–159. doi: 10.1113/jphysiol.2006.112730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pyapali GK, Turner DA, Wilson WA, Swartzwelder HS. Age and dose-dependent effects of ethanol on the induction of hippocampal long-term potentiation. Alcohol. 1999;19:107–111. doi: 10.1016/s0741-8329(99)00021-x. [DOI] [PubMed] [Google Scholar]
- Rezvani AH, Levin ED. Nicotine-alcohol interactions and cognitive function in rats. Pharmacol Biochem Behav. 2002;72:865–872. doi: 10.1016/s0091-3057(02)00762-1. [DOI] [PubMed] [Google Scholar]
- Rhodes JS, Best K, Belknap JK, Finn DA, Crabbe JC. Evaluation of a simple model of ethanol drinking to intoxication in C57BL/6J mice. Physiol Behav. 2005;84:53–63. doi: 10.1016/j.physbeh.2004.10.007. [DOI] [PubMed] [Google Scholar]
- Risold PY, Swanson LW. Structural evidence for functional domains in the rat hippocampus. Science. 1996;272:1484–1486. doi: 10.1126/science.272.5267.1484. [DOI] [PubMed] [Google Scholar]
- Robles N, Sabria J. Effects of moderate chronic ethanol consumption on hippocampal nicotinic receptors and associative learning. Neurobiol Learn Mem. 2008;89:497–503. doi: 10.1016/j.nlm.2008.01.006. [DOI] [PubMed] [Google Scholar]
- Role LW, Berg DK. Nicotinic receptors in the development and modulation of CNS synapses. Neuron. 1996;16:1077–1085. doi: 10.1016/s0896-6273(00)80134-8. [DOI] [PubMed] [Google Scholar]
- Rose JE, Brauer LH, Behm FM, Cramblett M, Calkins K, Lawhon D. Psychopharmacological interactions between nicotine and ethanol. Nicotine Tob Res. 2004;6:133–144. doi: 10.1080/14622200310001656957. [DOI] [PubMed] [Google Scholar]
- Rose JE, Mukhin AG, Lokitz SJ, Turkington TG, Herskovic J, Behm FM, et al. Kinetics of brain nicotine accumulation in dependent and nondependent smokers assessed with PET and cigarettes containing 11C-nicotine. Proc Natl Acad Sci USA. 2010;107:5190–5195. doi: 10.1073/pnas.0909184107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanna E, Talani G, Busonero F, Pisu MG, Purdy RH, Serra M, et al. Brain steroidogenesis mediates ethanol modulation of GABAA receptor activity in rat hippocampus. J Neurosci. 2004;24:6521–6530. doi: 10.1523/JNEUROSCI.0075-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schummers J, Bentz S, Browning MD. Ethanol's inhibition of LTP may not be mediated solely via direct effects on the NMDA receptor. Alcohol Clin Exp Res. 1997;21:404–408. doi: 10.1111/j.1530-0277.1997.tb03783.x. [DOI] [PubMed] [Google Scholar]
- Sinclair JG, Lo GF. Ethanol blocks tetanic and calcium-induced long-term potentiation in the hippocampal slice. Gen Pharmacol. 1986;17:231–233. doi: 10.1016/0306-3623(86)90144-8. [DOI] [PubMed] [Google Scholar]
- Siu EC, Tyndale RF. Characterization and comparison of nicotine and cotinine metabolism in vitro and in vivo in DBA/2 and C57BL/6 mice. Mol Pharmacol. 2007;71:826–834. doi: 10.1124/mol.106.032086. [DOI] [PubMed] [Google Scholar]
- Stolerman IP, Mirza NR, Shoaib M. Nicotine psychopharmacology: addiction: cognition and neuroadaptation. Med Res Rev. 1995;15:47–72. doi: 10.1002/med.2610150105. [DOI] [PubMed] [Google Scholar]
- Sudweeks SN, Yakel JL. Functional and molecular characterization of neuronal nicotinic ACh receptors in rat CA1 hippocampal neurons. J Physiol (Lond) 2000;527(Pt 3):515–528. doi: 10.1111/j.1469-7793.2000.00515.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thinschmidt JS, Frazier CJ, King MA, Meyer EM, Papke RL. Medial septal/diagonal band cells express multiple functional nicotinic receptor subtypes that are correlated with firing frequency. Neurosci Lett. 2005;389:163–168. doi: 10.1016/j.neulet.2005.07.038. [DOI] [PubMed] [Google Scholar]
- Tokuda K, Zorumski CF, Izumi Y. Modulation of hippocampal long-term potentiation by slow increases in ethanol concentration. Neuroscience. 2007;146:340–349. doi: 10.1016/j.neuroscience.2007.01.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wayner MJ, Armstrong DL, Phelix CF. Ethanol effects on dentate granule cell LTP. Neurotox Res. 2004;5:599–604. doi: 10.1007/BF03033179. [DOI] [PubMed] [Google Scholar]
- Weiner JL, Gu C, Dunwiddie TV. Differential ethanol sensitivity of subpopulations of GABAA synapses onto rat hippocampal CA1 pyramidal neurons. J Neurophysiol. 1997;77:1306–1312. doi: 10.1152/jn.1997.77.3.1306. [DOI] [PubMed] [Google Scholar]
- Wonnacott S, Mogg A, Bradley A, Jones IW. Presynaptic nicotinic acetylcholine receptors: subtypes mediating neurotransmitter release. In: Levin ED, editor. Nicotinic Receptors in the Nervous System. Methods and New Frontiers in Neuroscience. Boca Raton, FL: CRC Press; 2002. pp. 29–49. [Google Scholar]
- Wu PH, Poelchen W, Proctor WR. Differential GABA-A receptor modulation of ethanol effects on GABA-A synaptic activity in hippocampal CA1 neurons. J Pharmacol Exp Ther. 2005;312:1082–1089. doi: 10.1124/jpet.104.075663. [DOI] [PubMed] [Google Scholar]
- Zhang TA, Hendricson AW, Wilkemeyer MF, Lippmann MJ, Charness ME, Morrisett RA. Synergistic effects of the peptide fragment D-NAPVSIPQ on ethanol inhibition of synaptic plasticity and NMDA receptors in rat hippocampus. Neuroscience. 2005;134:583–593. doi: 10.1016/j.neuroscience.2005.04.010. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.