

Abstract
We show here that Vav-2 is tyrosine phosphorylated following antigen receptor engagement in both B- and T-cells, but potentiates nuclear factor of activated T cells (NFAT)-dependent transcription only in B cells. Vav-2 function requires the N-terminus, as well as functional Dbl homology and SH2 domains. More over, the enhancement of NFAT-dependent transcription by Vav-2 can be inhibited by a number of dominant-negative GTPases. The ability of Vav-2 to potentiate NFAT-dependent transcription correlates with its ability to promote a sustained calcium flux. Thus, Vav-2 augments the calcium signal in B cells but not T cells, and a truncated form of Vav-2 can neither activate NFAT nor augment calcium signaling. The CD19 co-receptor physically interacts with Vav-2 and synergistically enhances Vav-2 phosphorylation induced by the B-cell receptor (BCR). In addition, we found that Vav-2 augments CD19-stimulated NFAT- dependent transcription, as well as transcription from the CD5 enhancer. These data suggest a role for Vav-2 in transducing BCR signals to the transcription factor NFAT and implicate Vav-2 in the integration of BCR and CD19 signaling.
Keywords: CD19/lymphocyte/RhoG/signaling/Vav
Introduction
Signaling cascades emanating from the antigen receptors of B- and T-lymphocytes are required for the development and survival of mature lymphocytes, the deletion of autoreactive B- and T-cells, and lymphocyte activation in response to foreign antigen (Benschop and Cambier, 1999; van Leeuwen and Samelson, 1999). Upon antigen receptor stimulation, the activation of tyrosine kinases is one of the earliest events measurable and increases in tyrosine phosphorylation of a substantial number of proteins can be visualized by immunoblotting. The identification and characterization of these proteins have contributed significantly to our understanding of the mechanism by which antigen receptors function.
Among the molecules phosphorylated on tyrosine following antigen receptor engagement is the Vav-1 proto-oncogene (Bustelo and Barbacid, 1992; Bustelo et al., 1992; Margolis et al., 1992). Vav-1 consists of an N-terminal calponin homology (CH) domain, followed by an acidic region, central Dbl homology (DH), pleckstrin homology (PH) and zinc finger domains, and a SH3-SH2-SH3 region. Vav-1 has been shown to function as a guanine nucleotide exchange factor (GEF) for the Rho/Rac/Cdc42 family of low molecular weight Ras-like GTPases (Crespo et al., 1997; Han et al., 1998). These GTPases act as molecular switches for multiple effectors (Van Aelst and D’Souza-Schorey, 1997) and their activation by antigen receptors may permit coupling to a great number of signaling pathways. The exchange activity of Vav-1 is regulated by phosphorylation mediated by antigen receptor-activated tyrosine kinases and by inositol lipids that interact with the Vav-1 PH domain (Crespo et al., 1997; Han et al., 1997, 1998; Abe et al., 1999; Billadeau et al., 2000b; Kuhne et al., 2000; Lopez-Lago et al., 2000). The characterization of mice genetically altered to lack Vav-1 expression has emphasized the importance of Vav-1 at multiple stages of T-lymphocyte development and function. Significantly, it has been shown that overexpression of Vav-1 in T cells enhances basal and T-cell receptor (TCR)-activated transcription of the interleukin-2 (IL-2) promoter as well as nuclear factor of activated T cells (NFAT)-dependent transcription (Holsinger et al., 1995; Wu et al., 1995). Curiously, removal of the CH domain converts Vav-1 into its oncogenic state, but also causes a loss of this NFAT enhancing activity. Moreover, recent evidence suggests that Vav-1 is necessary for calcium mobilization and activation-induced assembly of the actin cytoskeleton, both of which are required for NFAT-dependent gene transcription (Turner et al., 1997; Fischer et al., 1998a; Holsinger et al., 1998; O’Rourke et al., 1998). However, the exact mechanism by which Vav-1 participates in antigen receptor signaling is not fully understood (Bustelo, 2000).
B-cell development and function in Vav-1-deficient mice are relatively normal, suggesting the presence of a GEF that can compensate for the loss of Vav-1 (Gulbranson-Judge et al., 1999). Recently, Vav-2, a new family member, was identified that is structurally related to Vav-1 and displays the characteristic domain structure that distinguishes Vav-1 from other GEFs (Schuebel et al., 1996). While Vav-1 is predominantly expressed in tissues and cells of hematopoietic origin, Vav-2 is more widely expressed (Schuebel et al., 1996). Both Vav-1 and Vav-2 are capable of transforming fibroblasts, but each induces a distinct morphology, suggesting non-redundant functions in vivo (Schuebel et al., 1998). It has been suggested that Vav-1 and Vav-2 may activate distinct GTPases (Schuebel et al., 1998). The role of Vav-2 in transducing signals downstream of lymphocyte antigen receptors is not clear.
We demonstrate that Vav-2 lies downstream of the antigen receptor in both B- and T-lymphocytes. Our findings suggest that Vav-1 and Vav-2 are not equivalent, as both Vav-1 and Vav-2 promote B-cell receptor (BCR)-stimulated calcium signals and NFAT-dependent transcription in B cells, but Vav-2 cannot perform this function in T cells. We also show that Vav-2 participates in CD19 co-receptor signaling in B lymphocytes. Interestingly, CD19 co-engagement with BCR lowers the threshold for Vav-2 phosphorylation and Vav-2 is capable of interacting with phosphorylated tyrosine 391 found in the CD19 cytoplasmic tail. Both BCR and CD19 stimulation result in the activation of NFAT-containing CD5 regulatory elements, which is further enhanced by the overexpression of Vav-2. These data implicate Vav-2 in the pathway of antigen receptor-elicited changes in gene expression in B lymphocytes.
Results
Vav-2 is a substrate for antigen receptor-activated tyrosine kinases
Vav-2-specific antibodies were used to establish whether Vav-2 was tyrosine phosphorylated following antigen receptor engagement. We stimulated freshly isolated murine B cells with BCR-specific antibody, precipitated Vav-2 and reacted immunoblots with antibody to phosphotyrosine. Increased Vav-2 phosphorylation was apparent following 15 s of stimulation, with peak levels detected 1–2 min following BCR crosslinking (Figure 1A). Vav-2 tyrosine phosphorylation remained elevated throughout the time course, up to 20 min. In contrast, and as previously described (Bustelo and Barbacid, 1992), Vav-1 was constitutively phosphorylated on tyrosine residues in freshly isolated B cells, but could be induced to become phosphorylated further upon BCR crosslinking (Figure 1A). To measure the stimulus dependence of Vav-2 tyrosine phosphorylation, we activated primary mouse B cells with titrated doses of antibody to BCR. Little Vav-2 phosphorylation was induced with 0.2 or 1 µg/ml of F(ab′)2 anti-IgM, while a 5-fold higher dose resulted in a considerable increase in detectable phosphorylation (Figure 1B). Similar to Vav-2, only the highest dose of anti-IgM induced a notable increase in Vav-1 tyrosine phosphorylation above background levels. Comparable results to those obtained with primary splenic B cells were observed when kinetic and anti-BCR dose–response analysis of Vav-2 tyrosine phosphorylation was performed using the IgM+ mouse B-cell lymphoma Bal-17, a widely used model for studying BCR signaling (Venkataraman et al., 1994) (data not shown). These data demonstrate that Vav-2 is a substrate for activated protein tyrosine kinases downstream of the BCR.

Fig. 1. Vav-2 is tyrosine phosphorylated after antigen–receptor ligation in primary B lymphocytes. (A) Vav-2 displays similar kinetics of tyrosine phosphorylation to Vav-1 in response to BCR ligation. Resting B cells were purified from mouse spleen and incubated with 10 µg/ml polyclonal F(ab′)2 anti-IgM and aliquots of cells were removed at timed intervals to make lysates. Lysates were divided in half and immunoprecipitates were prepared with irrelevant non-immune (NI) and polyclonal Vav-1- or Vav-2-specific antibodies. These were immunoblotted with antibody to phosphotyrosine (top panels). The blots were stripped and reprobed with monoclonal anti-Vav-1 or pooled mAbs specific for Vav-2 (bottom panels). (B) Vav-2 is tyrosine phosphorylated in a dose-dependent manner. B cells were isolated from mouse spleen and incubated with incremental doses of polyclonal F(ab′)2 anti-IgM. After 2 min, cells were lysed and divided in half; immunoprecipitates were prepared with non-immune (NI) and Vav-1- or Vav-2-specific polyclonal antibodies, and immunoblotted with antibody to phosphotyrosine (top panels). The blots were stripped and reprobed as above (bottom panels).
Jurkat T cells are a useful model for studies of TCR-mediated signal transduction; therefore, we compared the kinetics of Vav-1 and Vav-2 phosphorylation following activation of Jurkat cells by crosslinking the CD3 component of the TCR complex. Basal levels of tyrosine phosphorylation of both Vav-1 and Vav-2 were detectable, and activation resulted in a rapid increase in both Vav-1 and Vav-2 phosphorylation (Figure 2A). However, the tyrosine phosphorylation of Vav-2 was less sustained than that of Vav-1, with phosphorylation of Vav-2 becoming undetectable 10 min post-stimulation, while Vav-1 was still clearly phosphorylated (Figure 2A). When Jurkat cells were incubated with incremental amounts of anti-CD3, ligation of CD3 at all doses led to increases in phosphorylation of Vav-1, while in contrast only the highest dose of stimulating antibody resulted in a marked increase in Vav-2 tyrosine phosphorylation (Figure 2B). These data identify Vav-2 as an intracellular target downstream of TCR signaling. However, compared with Vav-1 tyrosine phosphorylation, the duration of Vav-2 tyrosine phosphorylation is much less extensive.

Fig. 2. Vav-2 is tyrosine phosphorylated in Jurkat T cells. (A) Vav-2 undergoes rapid but transient tyrosine phosphorylation following CD3 ligation on Jurkat T cells. Jurkat cells were incubated with 10 µg/ml OKT3 anti-CD3 and aliquots of cells were removed at timed intervals to make lysates. Immunoprecipitates were prepared with irrelevant non-immune (NI) and polyclonal Vav-1- or Vav-2-specific antibodies and immunoblotted with antibody to phosphotyrosine (top panels). The blots were stripped and reprobed with polyclonal antibodies to either Vav-1 or Vav-2 (bottom panels). (B) Vav-2 is inducibly tyrosine phosphorylated in a dose-dependent manner following CD3 ligation on Jurkat T cells. Jurkat cells were incubated with incremental doses of OKT3 monoclonal anti-CD3. After 2 min, cells were lysed and immunoprecipitates prepared with non-immune (NI) and anti-Vav-1 or anti-Vav-2, and immunoblotted with anti-phosphotyrosine (top panels). The blots were stripped and reprobed as described in Figure 1 (bottom panels).
Vav-2 potentiates NFAT in B cells but not T cells
As Vav-1 has been shown to regulate NFAT-dependent transcription in T cells, we assessed whether Vav-1 and Vav-2 could control NFAT in B cells. We co-expressed Vav-1 or Vav-2 together with a luciferase reporter under the control of NFAT regulatory elements derived from the IL-2 promoter. These are known to respond to antigen receptor stimulation in B lymphocytes (Choi et al., 1994; Venkataraman et al., 1994). NFAT-dependent transcription in unstimulated Bal-17 was not enhanced by co-transfection of either Vav-1 or Vav-2 (Figure 3A). Co-transfection of either Vav-1 or Vav-2 did enhance the anti-IgM-stimulated increase in NFAT activity in a dose-dependent manner (Figure 3A). In these experiments, Vav-1 and Vav-2 appeared equal in their ability to enhance NFAT-dependent transcription. However, we cannot show that the levels of protein expressed in the transfected cells are equivalent because the threshold for detection of expressed protein was 1 µg of transfected DNA and no expression could be detected when Bal-17 was transfected with 0.5 or 0.1 µg of DNA. These results indicate that both Vav-1 and Vav-2 can participate in the pathway that links the BCR to increases in NFAT-dependent transcription.

Fig. 3. Vav-2 augments NFAT activation upon antigen–receptor ligation in B cells but not T cells. (A) Overexpression of Vav-2 enhances anti-BCR-induced NFAT activation in Bal-17. Bal-17 B cells were co-transfected with p(NFAT)3IL-2-luc together with empty vector or incremental doses (in micrograms) of plasmids encoding Vav-1 or Vav-2. Cells were cultured in medium alone (unstimulated) or were incubated with b7.6 anti-IgM and assayed for luciferase activity. (B) Vav-2 fails to augment CD3-induced NFAT activation. T Ag Jurkat cells were co-transfected with p(NFAT)3IL-2-luc together with empty vector or incremental doses (in micrograms) of plasmids encoding Vav-1 or Vav-2. Cells were cultured in medium alone (unstimulated) or were incubated with OKT3 anti-CD3 and assayed for luciferase activity. The bottom panel shows the relative expression of the transfected Vav-1 and Vav-2 proteins in this experiment.
We next sought to establish a role for Vav-2 in NFAT-dependent transcription in T lymphocytes. Jurkat T lymphocytes were co-transfected with the NFAT- containing reporter and varying amounts of the Vav-2 expression plasmid or Vav-1 as a control. Consistent with previous results, overexpression of Vav-1 resulted in enhanced basal and CD3-induced NFAT-mediated gene transcription (Figure 3B). However, no positive effect of Vav-2 was observed on basal NFAT activity or CD3-stimulated NFAT activation (Figure 3B). The inability of Vav-2 to augment NFAT-mediated gene transcription could not be explained by low level expression of transfected Vav-2, as transfected Vav-2 was detected with all doses of transfected plasmid. Furthermore, there was no evidence of cell death induced by overexpression of Vav-2 (data not shown). Thus, Vav-2 potentiation of NFAT-dependent transcription is B-cell specific.
Mechanism of action of Vav-2 in B cells
We wished to determine whether the Vav-2-mediated effects on NFAT-dependent transcription in B cells required GEF activity. Several structural models of DH domains have been reported (Aghazadeh et al., 1998; Soisson et al., 1998) and several critical regions in Vav-1 required for exchange activity identified. A mutation of leucine at position 213 within conserved region 1 (CR1) of the Vav-1 DH domain abolishes Rac exchange activity (Crespo et al., 1997), as does mutation of a conserved leucine-rich sequence in the CR3 of the DH domain (Kuhne et al., 2000). We prepared two separate Vav-2 mutants at homologous residues (L212R and LLL336– 338QIF) and tested their ability to augment NFAT- dependent transcription in Bal-17. Although both the L212R and the QIF Vav-2 mutants were expressed at high levels, neither was able to augment BCR-stimulated NFAT-dependent transcription (Figure 4). This suggests that exchange activity is essential for Vav-2 to link the BCR to NFAT-dependent transcription. We next mutated a critical arginine residue in the SH2 domain of Vav-2 (R688A Vav-2) so that it would no longer be able to interact with phosphotyrosine. This mutation also resulted in the loss of NFAT potentiation, suggesting a requirement for Vav-2 to interact with a tyrosine phosphoprotein to mediate its effects on NFAT. Taken together, these results suggest that both GEF activity and a functional SH2 domain are required for the ability of Vav-2 to enhance BCR-mediated NFAT-dependent gene transcription.
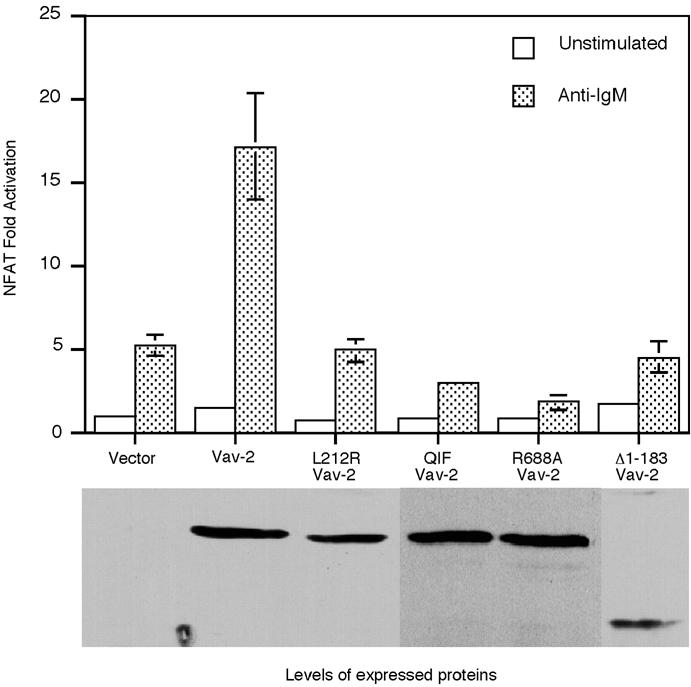
Fig. 4. Vav-2 requires intact CH, DH and SH2 domains to augment anti-IgM-induced NFAT activation in Bal-17. Bal-17 B cells were co-transfected with p(NFAT)3IL-2-luc together with 1 µg of empty vector or plasmid DNA encoding wild-type Vav-2, L212R Vav-2, Δ1–183 Vav-2, QIF Vav-2 or R688A Vav-2. Cells were incubated with medium alone (unstimulated) or stimulated by addition of b7.6 anti-IgM and then assayed for luciferase activity. The bottom panel shows expression of the Vav-2 proteins in Bal-17 cells in a single representative experiment.
Oncogenic Vav-1 contains a deletion of the CH domain and is mildly transforming towards 3T3 cells but unable to potentiate NFAT-dependent transcription (Katzav et al., 1991; Wu et al., 1995). Deletion of the N-terminal 187 amino acids of Vav-1 substantially enhances transforming potential; however, this mutant is still unable to potentiate NFAT-dependent transcription in Jurkat T cells (Lopez-Lago et al., 2000). We prepared an analogous truncation mutation of Vav-2 (Δ1–183) and assayed its ability to enhance NFAT-dependent transcription in Bal-17 B cells. Truncation of Vav-2 resulted in a loss of enhancement of anti-IgM-stimulated NFAT activity (Figure 4). This effect was not due to lack of expression of truncated Vav-2 as this protein was expressed at high levels in Bal-17 (Figure 4). Our results suggest that Vav-2 requires an intact N-terminus to potentiate NFAT-dependent transcription in B cells.
To evaluate potential effectors of Vav-2 in B cells, we co-transfected Bal-17 B cells with wild-type Vav-2 in combination with a panel of dominant-negative mutants or inhibitors of GTPase function. Blockade of Ras and Cdc42 activation led to substantial inhibition of NFAT potentiation by Vav-2 (Figure 5). In contrast, dominant-negative Rac-1 did not inhibit the Vav-2 effect on NFAT. Vav-2 has been shown to promote guanine nucleotide exchange on Rho family proteins RhoA, RhoB and RhoG. To evaluate the contribution of RhoA and RhoB to the effects of Vav-2 on NFAT-dependent transcription, we co-transfected C3 toxin from Clostridium botulinum. C3 toxin modifies a subset of Rho family proteins by ADP-ribosylation of the effector domain at position Asn41, leading to their inactivation (Hill et al., 1995). Overexpression of C3 toxin did not inhibit the effect of Vav-2 on transcription from NFAT-dependent reporter plasmids. C3 toxin was functional in Bal-17 cells, as transfection with a serum response element (SRE) luciferase reporter plasmid resulted in a V14 RhoA-inducible luciferase activity that was inhibited by co-transfection with C3 toxin (data not shown). However, overexpression of N17 RhoG exerted an inhibitory effect on Vav-2 potentiation of NFAT activity (Figure 5). The inhibition of Vav-2 potentiation of NFAT activity by dominant-negative G proteins was not due to non-specific toxicity, since co-transfection of these constructs with a control luciferase reporter driven by an SV40 promoter did not alter the relative luciferase activity (data not shown). These experiments indicate that Vav-2 potentiation of NFAT requires the function of Ras, Cdc42 and RhoG, but not Rac or Rho proteins.

Fig. 5. Vav-2 utilizes selective downstream effectors to potentiate NFAT activation. Bal-17 B cells were co-transfected withp(NFAT)3IL-2-luc together with 1 µg of Vav-2 expression vector and 1 µg of dominant-negative Ras, Rac, RhoG or Cdc42 or C3 transferase. Cells were cultured in medium alone or were incubated with b7.6 anti-IgM and assayed for luciferase activity. Results are shown as the percentage inhibition of Vav-2 potentiation of NFAT activity after IgM ligation (100%).
The ability of Vav-1 to regulate antigen receptor-triggered calcium flux in T cells has been clearly established from studies by a number of groups, including our own (Turner et al., 1997; Fischer et al., 1998b; Holsinger et al., 1998; Costello et al., 1999; Billadeau et al., 2000b). A sustained calcium signal is critical for NFAT-dependent transcription, therefore we sought to compare the ability of Vav-1 and Vav-2 to regulate calcium levels following overexpression in B- and T-cell lines. Overexpression of Vav-1 in Jurkat cells increased the peak levels of intracellular calcium and potentiated the sustained influx of calcium following TCR crosslinking (Figure 6). In contrast, while overexpression of Vav-2 in Jurkat cells increased the initial peak of calcium release, Vav-2 was less able than Vav-1 to potentiate the sustained phase of the calcium signal. We next compared the ability of Vav-1 and Vav-2 to potentiate BCR-triggered calcium signals in Bal-17. Vav-2 was consistently more effective than Vav-1 at potentiating both the initial peak and the sustained influx of calcium in Bal-17 (Figure 6). Truncation of the N-terminus of Vav-1 abrogates the ability of Vav-1 to enhance TCR-triggered calcium flux in T cells (Billadeau et al., 2000b). The N-terminus of Vav-2 is similarly required for potentiation of the BCR-elicited calcium signal, as its removal blocks the ability of Vav-2 to activate calcium flux following BCR crosslinking (Figure 6). These data indicate that one mechanism by which Vav-2 can enhance NFAT-mediated gene transcription downstream of the BCR is to lead to a sustained increase in intracellular calcium. In addition, like Vav-1, removal of the N-terminal CH domain uncouples Vav-2 from its ability to regulate increased calcium levels downstream of the BCR.
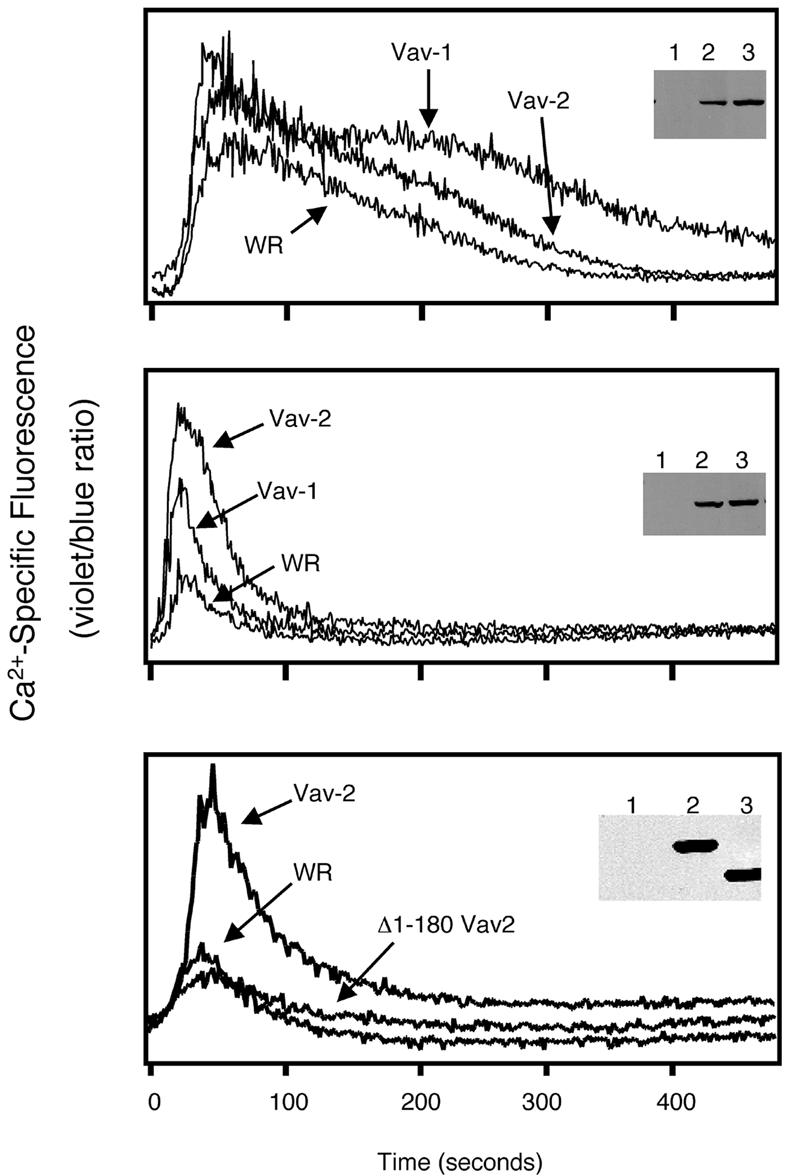
Fig. 6. Vav-2 potentiates antigen receptor-induced Ca2+ mobilization in B cells but not T cells. Jurkat T cells or Bal-17 B cells were infected with non-recombinant vaccinia (WR) or recombinant vaccinia expressing FLAG-tagged versions of Vav-1, Vav-2 or Δ1–180 Vav-2, as indicated. After infection, the cells were loaded with Indo-1. The Jurkat T cells (upper panel) were stimulated with a combination of anti-CD3 mAb (2.5 µg/ml) and goat anti-mouse IgG F(ab′)2. The Bal-17 B cells (middle and lower panels) were stimulated with the anti-murine IgM mAb b7.6 (5 µg/ml). The samples were immediately analyzed by flow cytometry over the indicated time course. Panel insets: protein expression in the infected cells was verified by immunoblotting of whole-cell lysates with the anti-FLAG mAb (lanes 1, 2 and 3 represent WR, Vav-1 and Vav-2 in the upper and middle panels, and WR, Vav-2 and Δ1–180 Vav-2 in the lower panel, respectively). The data shown are representative of at least three independent experiments.
Vav-2 potentiates BCR and CD19 signals
Previous studies have shown that ligation of CD19 with specific antibodies activates tyrosine kinases, leading to the phosphorylation of a number of intracellular substrates and increases in the concentration of intracellular free calcium ions (Weng et al., 1994; Sato et al., 1997a,b; O’Rourke et al., 1998; Fujimoto et al., 1999b). Importantly, co-crosslinking of the CD19 signaling complex and the BCR lowers the threshold for antigen-specific B-cell activation in vivo (Dempsey et al., 1996). To determine whether Vav-2 is activated downstream of CD19, we stimulated Bal-17 B cells using incremental doses of biotinylated anti-CD19 antibody Fab fragments crosslinked by avidin. We found little or no tyrosine phosphorylation of Vav-2 at 0.2 µg/ml, but higher doses of anti-CD19 Fab induced detectable tyrosine phosphorylation of Vav-2 (Figure 7A), although the magnitude of the response is less than that induced by antigen receptor ligation (Figure 7C and data not shown). This observation suggests that Vav-2 is a substrate for CD19-activated tyrosine kinases.

Fig. 7. Ligation of CD19 results in tyrosine phosphorylation of Vav-2 and the activation of NFAT. (A) CD19 ligation induces tyrosine phosphorylation of Vav-2. Bal-17 B cells were incubated with the indicated doses of biotinylated Fab fragment of antibody to CD19 and then stimulated by the addition of soluble avidin for 2 min. Cells were lysed and immunoprecipitates prepared with non-immune (NI) or polyclonal Vav-2 antibodies, and immunoblotted with antibody to phosphotyrosine (top panel). The blot was stripped and reprobed with pooled mAbs to Vav-2. (B) Tyrosine phosphorylation of Vav-2 is enhanced by co-ligation of CD19 to BCR. Bal-17 B cells were incubated with the indicated doses of either control Fab, biotinylated Fab anti-CD19, or biotinylated Fab anti-κ alone or combined with 0.2 µg of biotinylated Fab anti-CD19, followed by crosslinking with soluble avidin for 2 min. Cells were lysed and immunoprecipitates prepared with non-immune (NI) or polyclonal Vav-2 and immuno blotted with antibody to phosphotyrosine (top panel). The blot was stripped and reprobed as above. (C) This experiment was performed as in (B) except that the stimulating dose of Fab anti-CD19 was 5 µg/ml. (D) Vav-2 potentiates CD19 induction of NFAT. Bal-17 B cells were co-transfected with p(NFAT)3IL-2-luc together with 1 µg of empty vector or plasmids encoding Vav-2, L212R Vav-2 or R688A Vav-2. Cells were cultured in medium alone (unstimulated) or were incubated with 1D3 anti-CD19, which was crosslinked with F(ab′)2 goat anti-rat IgG before cell lysates were prepared and assayed for luciferase activity.
To establish a role for Vav-2 in BCR/CD19 signaling we used an activation system that mimics the effects of C3d-bound antigen and co-crosslinks surface Ig with the CD19–CD21 complex (Tooze et al., 1997). In this system, biotinylated Fab fragments prepared from monoclonal antibodies (mAbs) to κ light chain and CD19 are crosslinked by the addition of avidin. When Bal-17 B cells were stimulated in this way, anti-CD19 Fab at 0.2 µg/ml and anti-κ Fab at 0.2 µg/ml were suboptimal stimuli for Vav-2 tyrosine phoshorylation (Figure 7B). Combination of these stimuli, however, led to synergistic increases in the levels of Vav-2 tyrosine phosphorylation (Figure 7B). This synergy was also evident when the amount of anti-CD19 Fab was increased to 5 µg/ml. As shown in Figure 7B and C, stimulation with anti-κ Fab alone at 0.2 µg/ml elicited no detectable Vav-2 tyrosine phosphorylation, but at concentrations of 1 and 5 µg/ml led to more tyrosine phosphorylation of Vav-2 than did anti-CD19 stimulation alone. Combination of the agonists resulted in synergistic levels of Vav-2 tyrosine phosphorylation at all three doses of anti-κ Fab (Figure 7C). Stimulation of freshly isolated splenic B cells from either wild-type or Vav-1–/– mice by this method gave similar results (data not shown). Taken together, these data suggest that co-crosslinking of the BCR and CD19 can result in a synergistic increase in the levels of Vav-2 tyrosine phosphorylation.
Although CD19 crosslinking has been shown to promote activation of multiple signaling cascades in B cells, it has not previously been shown to activate transcription factors directly. Therefore we sought to establish whether CD19 ligation influenced NFAT transcriptional activity and whether this was potentiated by Vav-2. CD19 crosslinking induced a 2- to 3-fold induction of NFAT activity in Bal-17 cells, which was further augmented by overexpression of Vav-2 (Figure 7D). This enhanced NFAT activation is dependent upon both an intact exchange domain and a functional SH2 domain, since inactivating mutations within these domains blocked the ability of Vav-2 to promote CD19-stimulated NFAT activation (Figure 7D). Thus, signaling through the CD19 receptor regulates NFAT-dependent gene transcription and this activation can be enhanced by overexpression of Vav-2.
Vav-2 associates with phosphorylated Tyr391 of CD19
Co-crosslinking of the BCR–CD19 complex with all concentrations of anti-κ Fab in combination with 5 µg/ml anti-CD19 Fab resulted in co-precipitation of a 100– 110 kDa protein with Vav-2, which was reactive with antibody to phosphotyrosine (Figure 7C). Based on the observed size, we reasoned that this protein was likely to be CD19; however, the 1D3 anti-CD19 mAb does not react with CD19 on immunoblots and a rabbit polyclonal anti-CD19 serum was not sufficiently sensitive to confirm the identity of the protein (data not shown). To determine whether Vav-2 was capable of interacting with CD19, COS7 cells were transfected with expression plasmids encoding full-length Vav-2, and lysates were incubated with specific antibodies or synthetic peptides corresponding to the sequences surrounding the nine potential tyrosine phosphorylation sites of CD19. We found that Vav-2 interacts specifically with phosphorylated peptide corresponding to the region surrounding Tyr391 of CD19 and does not bind to a non-phosphorylated version of the same peptide (Figure 8A). Using the same approach we determined that Vav-2 does not bind to any other of the eight remaining tyrosine motifs located within the cytoplasmic tail of CD19 (data not shown). We next used far-western blotting to demonstrate that the interaction between Vav-1, Vav-2 and the phosphorylated CD19 peptide was direct and not mediated by a bridging protein. The SH2 domains of Vav-1 and Vav-2 were expressed as fusion proteins in Escherichia coli together with glutathione S-transferase (GST). These were purified, resolved by SDS–PAGE and transferred to nitrocellulose. Filters were then incubated with phosphorylated or non-phosphorylated versions of the biotinylated Tyr391 peptide, the binding of which was visualized using streptavidin–horseradish peroxidase (HRP) and enhanced chemiluminescence (ECL). The SH2 domains of both Vav-1 and Vav-2 specifically interacted with the phosphorylated, but not the unphosphorylated version of the peptide (Figure 8B). The phosphorylated peptide failed to interact with GST, indicating the specificity of the interaction. Taken together, these data indicate that Vav-2 can interact with Y319 of CD19 in a phospho-specific manner.
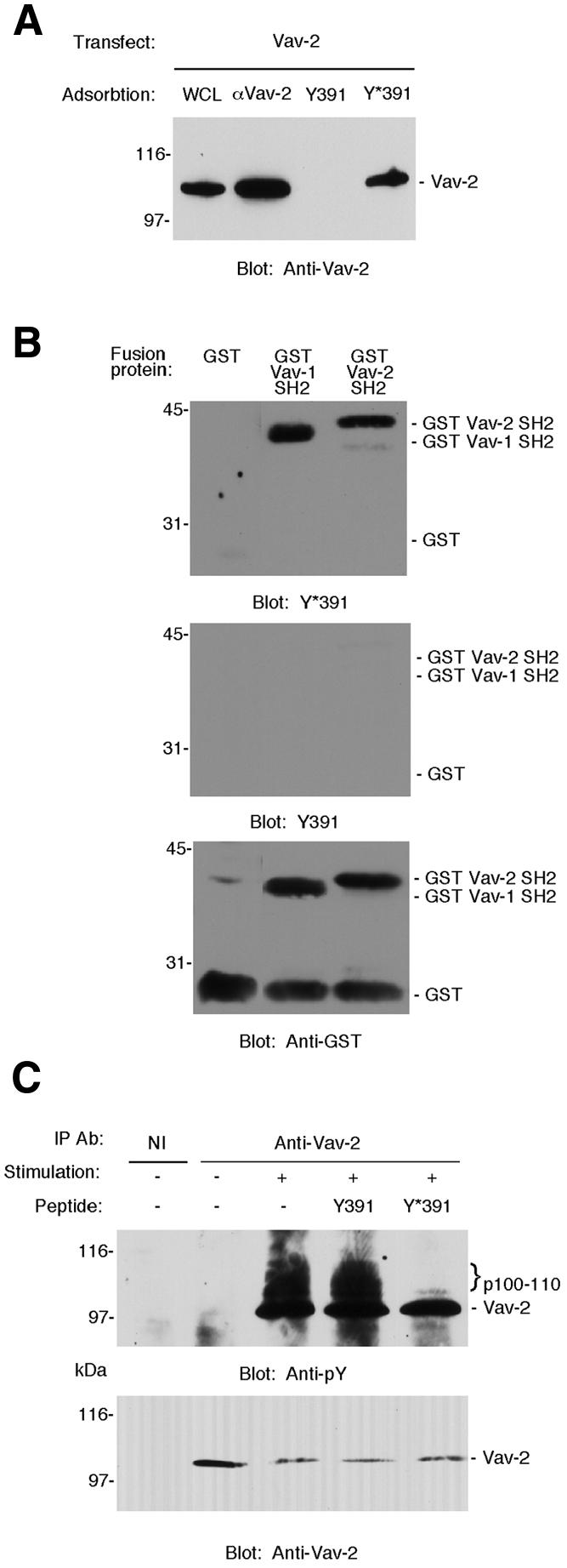
Fig. 8. Vav-2 interacts with tyrosine phosphorylated CD19. (A) Interaction of Vav-2 with Tyr391 (Y391) of CD19. Biotinylated peptides representing sequence surrounding Tyr391 of human CD19 in phosphorylated (Y*391) or non-phosphorylated (Y391) versions (O’Rourke et al., 1998) or antibody to Vav-2 (αVav-2) were incubated with lysates prepared from COS7 cells that had been transfected with Vav-2. The resulting complexes were probed with pooled mAbs to Vav-2. A portion of the transfected COS7 cell lysate (WCL) was loaded onto the gel as a control. (B) Y*391 binds directly to the SH2 domain of Vav-2. Equal amounts of fusion proteins representing GST alone or the SH2 domain of either Vav-1 or Vav-2 immobilized on nitrocellulose were incubated with biotinylated peptide Y*391 (top panel), biotinylated peptide Y391 (middle panel) or anti-GST (bottom panel). (C) Y*391 specifically inhibits co-precipitation of a 100–110 kDa tyrosine phosphoprotein with Vav-2. Bal-17 B cells were incubated with either 10 µg of control Fab (–) or 5 µg of biotinylated Fab anti-κ combined with 5 µg of biotinylated Fab anti-CD19 (+), which were crosslinked by the addition of soluble avidin for 2 min. Following cell lysis, immunoprecipitates were prepared with non-immune or polyclonal Vav-2 antibodies in the absence of peptide or in the presence of 100 µM Y391 or Y*391 and immunoblotted with antibody to phosphotyrosine (top panel). The positions of the co-precipitating 100–110 kDa proteins are marked with curly brackets. The blot was stripped and reprobed with pooled mAbs to Vav-2 (bottom panel).
To establish whether such a CD19–Vav-2 complex exists in vivo, we stimulated Bal-17 cells by CD19–BCR co-ligation and prepared Vav-2 immunoprecipitates in the absence or presence of CD19 peptides. An anti-phosphotyrosine immunoblot revealed co-immunoprecipitation of a tyrosine phosphorylated protein of 100–110 kDa (the molecular weight of CD19) with Vav-2. Co-immunoprecipitation of this protein was abolished in the presence of phosphorylated Y391 peptide, but not its unphosphorylated counterpart (Figure 8C).
Vav-2 links BCR and CD19 to the control of CD5 transcription
To establish that the effects we were measuring were more generally applicable to NFAT-dependent genes in B cells, we determined the effects of Vav-2 on BCR- and CD19-induced CD5 expression. CD5 transcription is a consequence of BCR ligation that is contingent upon occupation of NFAT regulatory sites in the CD5 enhancer (Teutsch et al., 1995; Berland and Wortis, 1998). Ligation of either BCR or CD19 with specific antibodies led to induction of CD5 enhancer-driven luciferase activity in Bal-17 cells. Overexpression of Vav-2 heightened the BCR- and CD19-mediated transcriptional activation of a CD5 reporter (Figure 9A). Mutations affecting either the GEF domain or the SH2 domain of Vav-2 abrogated the ability of Vav-2 to potentiate transcription from the CD5 reporter.

Fig. 9. Vav-2 potentiates BCR- and CD19-driven NFAT-dependent CD5 transcription. (A) Overexpression of Vav-2 augments BCR- and CD19-stimulated CD5 enhancer activity. Bal-17 B cells were co-transfected with pCD5-luc122R together with 5 µg of empty vector or plasmid encoding Vav-2 or L212R Vav-2. Cells were cultured in medium alone (unstimulated) or were incubated with either b7.6 anti-IgM or 1D3 anti-CD19 with F(ab′)2 goat anti-rat IgG and assayed for luciferase activity. (B) Induction of CD5 by Vav-2 requires intact NFAT regulatory sites. Bal-17 B cells were co-transfected with 10 µg of enhancerless CD5 reporter construct, the wild-type enhancer pCD5-luc122R construct or a derivative with point mutations in both the proximal and distal NFAT sites together with 5 µg of empty vector or 5 µg of plasmid encoding Vav-2. Stimulations are as in (A).
The 122 bp enhancer element that is required for BCR induction of the CD5 promoter is comprised of sites that interact with multiple transcription factors. Mutational analysis demonstrated that two NFAT sites contribute to 80% of CD5 enhancer activity and identified three additional transcription factor binding sites, which also contribute to CD5 induction (Berland and Wortis, 1998). Given the complex nature of the enhancer element, we wished to determine whether the effect of Vav-2 was specific for NFAT regulation. Bal-17 B cells were transfected with CD5 reporter constructs lacking the enhancer, with wild-type enhancer or with both NFAT sites altered by a point mutation, and the effects of overexpression of Vav-2 on BCR- and CD19-mediated activation were assessed. Significantly, the presence of intact NFAT binding sites was required for Vav-2 enhancement of both anti-BCR- and anti-CD19-stimulated CD5 enhancer activity (Figure 9B).
Discussion
In this study we have demonstrated a previously unrecognized role for Vav-2 as a mediator of BCR and CD19 signaling in B lymphocytes. We have found that Vav-2 is rapidly and sustainably phosphorylated by tyrosine kinases activated through BCR ligation, and that the observed tyrosine phosphorylation is dependent on the dose of stimulus. These findings suggest that Vav-2 may play a role in the important first steps initiated following BCR engagement. Further evidence for this suggestion is provided by the finding that ligation of the CD19 co-receptor also stimulates Vav-2 tyrosine phosphorylation, and simultaneous engagement of CD19 and BCR synergistically lowers the threshold for Vav-2 tyrosine phosphorylation. Co-ligation of CD19 and BCR, which may be brought about in vivo by antigen–C3d complexes, may therefore quantitatively alter the signal produced by BCR ligation alone, in part by lowering the threshold for Vav-2 activation. We have shown that upon co-stimulation through the BCR–CD19 complex, Vav-2 is capable of binding to Tyr391 of CD19, the same tyrosine with which Vav-1 interacts (Figure 8) (O’Rourke et al., 1998). As both Vav-1 and Vav-2 bind to Tyr391 of CD19, it is unlikely that both exchange factors can simultaneously mediate the signal from the same CD19 molecule. Our peptide competition data suggest that a Vav-2–CD19 complex does exist inside the cell. Therefore, the activated B cell contains two pools of CD19: one associated with Vav-1 and one associated with Vav-2. Previous studies demonstrating that Tyr391 of CD19 was essential for Ca2+ mobilization and PIP5-K activation found that the requirement for Vav-1 was not absolute (O’Rourke et al., 1998), thus suggesting the existence of additional components of this signaling pathway that were recruited through Tyr391. Our data suggest that Vav-2 is one of these components that can mediate BCR and CD19 signals to potentiate NFAT-dependent transcription.
The phenotype of B cells from Vav-1-deficient mice is also consistent with the notion that exchange factors other than Vav-1 are important for BCR and CD19 signal transduction. Vav-1-deficient mice display profound deficiencies in the development and function of T lymphocytes, but display an altogether milder phenotype with respect to B-cell development and function. Vav-1 is not required for the normal development of the majority of B cells in the mouse. Thus, the receptor selection events required for development and survival of conventional (B-2) B cells appear to be Vav-1 independent. Vav-1 is also not required for the in vivo B-cell response to Type II thymus-independent antigens, which signal by extensively crosslinking the BCR. Furthermore, the response of Vav-1-deficient B cells to thymus-dependent antigens is normal when T-cell help is provided (Gulbranson-Judge et al., 1999). Taken together with our findings, these results suggest that Vav-2 is likely to be an important mediator of BCR and CD19 signaling.
Vav-2 was tyrosine phosphorylated following activation of Jurkat T cells with antibodies to the TCR. However, Vav-2 phosphorylation was not as prolonged as Vav-1 phosphorylation in these cells. As tyrosine phosphorylation of Vav proteins is necessary for their activation, this result may indicate that Vav-2 is more transiently activated. However, direct demonstration of this possibility will require measurements of Vav-1 and Vav-2 exchange activity at different times following TCR triggering. Our results show that Vav-2 cannot duplicate the effects of Vav-1 to augment NFAT-dependent transcription in T cells. Recent work complementing these data has shown that even though Vav-2 fails to activate NFAT, Vav-2 can function in T cells to promote cellular cytotoxicity (Billadeau et al., 2000a). Thus, although Vav-2 cannot duplicate all the functions of Vav-1 in T cells, it can play a functional role in TCR signaling. However, the observation that Vav-2 is expressed at high levels in the thymus (Fischer et al., 1998a), yet it can not compensate for the severe T-cell phenotype in Vav-1-deficient mice (Turner et al., 1997; Fischer et al., 1998a; Holsinger et al., 1998; Costello et al., 1999), implies differential roles for these two GEFs in TCR signaling. This finding further distinguishes Vav-1 and Vav-2, and suggests that despite their sequence similarity Vav-2 does not simply compensate for the lack of Vav-1.
We performed experiments to identify why the ability of Vav-2 to potentiate NFAT-dependent transcription was B-cell specific. We have shown previously (Billadeau et al., 2000a), and in this study, that Vav-2 cannot augment the sustained influx of calcium in Jurkat T cells. Since sustained influx of calcium is critical for the nuclear localization of components of the NFAT transcription factor complex (Dolmetsch et al., 1997, 1998; Crabtree, 1999), the failure of Vav-2 to potentiate the calcium signal is likely to be an important contribution to its failure to activate NFAT. In B lymphocytes, however, Vav-2 was a potent stimulator of the sustained calcium response and this is likely to be of importance for the potentiation of NFAT-dependent transcription by Vav-2. We showed that a mutant version of Vav-2 lacking the CH and acidic regions was unable to augment the BCR-triggered calcium signal and this loss of function correlated with a failure to activate NFAT. Previous studies have indicated a requirement for the N-terminal CH and acidic domains of Vav-1 for potentiation of NFAT-dependent transcription in T cells (Wu et al., 1995; Billadeau et al., 2000b; Lopez-Lago et al., 2000). Interestingly, the CH domain of Vav-1 was found to be necessary for the sustained calcium response in T cells (Billadeau et al., 2000b). A similar requirement for the homologous domains of Vav-2 was found in B cells. Elucidation of the mechanism of function of the CH and acidic regions of Vav-1 and Vav-2 should therefore provide insights into the regulation of Vav family proteins and NFAT activation.
We found that Vav-2 potentiation of NFAT-dependent transcription in B cells required a functional DH domain. This suggests that the observed effects on NFAT- dependent transcription are mediated through the activation of one or more small Ras-like GTPases. A recent report has shown that mutation of the Vav-1 GEF domain led to an increase in NFAT activity in unstimulated Jurkat cells (Kuhne et al., 2000). We did not observe similar findings with our two DH domain mutants in unstimulated B cells. This may reflect a difference between Vav-1 and Vav-2 or between T and B cells. In this regard, it is notable that while overexpression of wild-type Vav-1 potentiates NFAT in resting T cells, overexpression of Vav-1 or Vav-2 has little effect on NFAT activity in resting B cells (Wu et al., 1995) (Figure 3). Thus, our results indicate that Vav-2 must function as a GTPase exchange factor in order to potentiate NFAT-dependent transcription in B cells.
It is tempting to speculate that the differences between the roles of Vav-1 and Vav-2 in the two classes of lymphocytes may be partly explained by their previously reported distinct GTPase specificities (Schuebel et al., 1998). However, the identity of the GTPase targets downstream of Vav-2 is controversial. Schuebel et al. (1998) have shown that full-length Vav-2 has no exchange activity towards Cdc42 or Rac-1, while Abe et al. (1999) found that truncated proteins consisting of the Vav-2 DH domain or DH/PH/CR can activate Cdc42 and Rac-1. We found that Vav-2-mediated activation of NFAT activity in B cells was blocked by dominant-negative RhoG, Ras and Cdc42. Expression of C3 toxin, which inhibits RhoA and RhoB, and N17 Rac-1 failed to inhibit Vav-2 augmentation of IgM-stimulated NFAT activation. These data do not preclude an ability of Vav-2 to act as a GEF for these GTPases in B cells, but do suggest that these G proteins do not positively participate in NFAT regulation by Vav-2 downstream of the BCR. RhoG, despite its name, is more closely related in structure to Rac1 and Cdc42 than to Rho, and it is unlikely to be modified by ADP ribosylation (Vincent et al., 1992). N17 RhoG functions as a dominant negative and inhibited Vav-2 potentiation of NFAT in our cell studies; this observation is therefore consistent with the in vitro data (Schuebel et al., 1998). The Ras and Cdc42 pathways are required for NFAT-dependent transcription in T cells (Wu et al., 1995; Holsinger et al., 1998; Yablonski et al., 1998). Irrespective of whether these GTPases function in a parallel pathway necessary for NFAT-dependent transcription or are direct substrates of Vav-2, our findings indicate that the function of multiple GTPases is required for Vav-2 to promote NFAT- dependent transcription in B cells.
A key to understanding how CD19 and Vav-2 regulate the magnitude of the immune response is to determine the identity of genes transcribed as a consequence of CD19- and Vav-2-dependent signaling. Of the biochemical responses initiated by CD19, to date no direct evidence for its control of gene transcription has been demonstrated. The ability of CD19 to modulate BCR-induced activation of MAP kinases (Tooze et al., 1997; O’Rourke et al., 1998) and Ca2+ mobilization (Dempsey et al., 1996; Sato et al., 1997b; Buhl and Cambier, 1999) is strongly suggestive of an involvement of NFAT in CD19-regulated responses. At present, only a few genes have been described that require NFAT site occupancy for expression in B lymphocytes; these include Igκ (Meyer and Ireland, 1998) and tumor necrosis factor-α (Tsai et al., 1996). BCR ligation has previously been shown to induce NFAT- dependent transcription of a CD5 reporter construct in primary B cells (Berland and Wortis, 1998). We have shown that ligation of CD19 on B cells resulted in the activation of NFAT. The overexpression of Vav-2 substantially enhanced the ability of CD19 to activate NFAT-dependent transcription and this required Vav-2 GEF activity. The availability of knockout mice should allow us to demonstrate the relative importance of each Vav family member in BCR- and CD19-mediated NFAT signaling. Our data demonstrate the potential of Vav-2 to participate in BCR and CD19 activation of CD5 transcription through an NFAT-dependent mechanism. It may be that the induction of CD5 in response to CD19–BCR ligation could function to mediate feedback inhibition of B-cell activation, as CD5 has been shown to be a negative regulator of signaling (Tarakhovsky et al., 1995; Bikah et al., 1996). Further work is required to verify this suggestion. The concept that CD19 may participate in limiting the ongoing B-cell response to antigen, in addition to its role in augmenting B-cell responses, is further strengthened by the recent finding that CD19 is necessary for inhibitory signaling through the CD22 co-receptor (Fujimoto et al., 1999a).
In conclusion, we have identified Vav-2 as a novel transducer of signals initiated by the BCR and CD19, and we have identified cell type-specific differences in function between Vav-1 and Vav-2. Vav-2 regulates NFAT- dependent transcription only in antigen receptor- stimulated B lymphocytes. Vav-2 may therefore control the expression of genes by B cells whose function is important in the initiation, maintenance and cessation of the immune response.
Materials and methods
Antibodies
Vav-2 mAbs were generated following immunization of female F344 rats with a GST fusion protein containing amino acids 552–868 of Vav-2 and fusion with the IR983F plasmacytoma (Bazin, 1982). Polyclonal antibodies to Vav-2 were obtained by immunization of rabbits with the same antigen. Polyclonal sera and four stable hybridomas secreting antibodies that immunoprecipitated Vav-2 and recognized Vav-2 on western blots were tested for reactivity with Vav-1, and neither precipitated Vav-1 nor reacted with Vav-1 on western blots (data not shown). Other polyclonal antibodies used in Figure 2 that recognize Vav-1 and Vav-2 have been described elsewhere (Billadeau et al., 2000a). Additional antibodies used include mAb 9E10 against the myc epitope tag, mAb M2 against the FLAG epitope tag (Sigma), 4G10 anti-phosphotyrosine, mAb to Vav-1 (UBI), 1D3 anti-murine CD19, b7.6 anti-murine IgM, LO-MK-1 rat IgG2a antibody to mouse κ light chain (Zymed), polyclonal antibody to GST generated by immunization of a rabbit with purified GST fusion protein, F(ab′)2 fragments of polyclonal goat anti-mouse IgM (Chemicon), OKT3 anti-CD3, F(ab′)2 polyclonal goat anti-rat IgG, and HRP-conjugated rabbit anti-mouse IgG, mouse anti-rabbit IgG and rabbit anti-rat IgG (Jackson ImmunoResearch). Fab fragments were obtained by papain digestion, purified by passing over an anti-rat IgG Fc fragment-specific column and biotinylated with ImmunoPure Sulfo-NHS-Biotin (Pierce and Warriner).
Plasmids and recombinant vaccinia
The luciferase reporter constructs used in this paper were as follows: p(NFAT)3IL2-Luc (a gift from David Williams), which contains three copies of the distal IL-2 promoter NFAT site upstream of the –80 to +35 region of the IL-2 promoter inserted into pGL2-Basic vector (Promega); and the CD5 promoter construct, pCD5Luc122R, which contains the murine CD5 promoter and 3′ enhancer cloned into pGL3-Basic (Promega) and its derivatives with mutations in both NFAT sites that have been described previously (Berland and Wortis, 1998). Wild-type Vav-1, Vav-2 and all its derivatives were in the expression plasmid pCMV3myc (Welch et al., 1998), which was provided by Heidi Welch. L212R Vav-2, QIF Vav-2 and R688A Vav-2 were generated by site-directed mutagenesis using a Stratagene Quickchange kit as per the manufacturer’s instructions. Expression plasmids for N17 Rac1, N17 Cdc42, V14 RhoA and C3 transferase were a gift of Doreen Cantrell. N17 Ras was provided by Simon Cook. A 517 bp fragment encoding RhoG was amplified by PCR from human genomic DNA and subcloned into pCMV3myc. N17 RhoG was prepared using the aforementioned DNA as a template for the Stratagene site-directed mutagenesis Quickchange protocol. The generation of recombinant vaccinia used to express wild-type Vav-1 and Vav-2 has been described previously (Billadeau et al., 2000a,b). The Δ1–180 Vav-2 recombinant vaccinia was generated using a site-directed mutagensis kit (Clonetech Laboratories, Inc.) to introduce a unique HindIII site used for digestion and removal of the first 179 amino acids of Vav-2. The resulting mutant was FLAG tagged and recombinant vaccinia virus was generated as described previously (Billadeau et al., 2000b). Constructs used to produce GST fusion proteins were generated by the directional insertion of DNA encoding amino acids 666–776 of Vav-1 and 663–762 of Vav-2 into pMEX-HMK vector. All constructs and derivatives were sequenced to verify their structure.
Mice
All mice were bred at the Babraham Institute Small Animal Barrier Unit (SABU) and housed according to UK Home Office guidelines.
Preparation of splenocytes
Spleens were disaggregated using a 70 µm nylon cell strainer in Dulbecco’s modified Eagle’s medium (DMEM) (Sigma) containing 2% heat-inactivated fetal calf serum (FCS). Single-cell suspensions were incubated with anti-Thy1.2 (Sigma) for 20 min on ice prior to the addition of ‘Low-Tox’ rabbit complement (Cedarlane) and incubation for 30 min at 37°C. The cell suspension was then layered onto Lympholyte-M (Cedarlane), centrifuged for 20 min at 1000 g, and the enriched B-cell fraction recovered. B cells purified in this manner were routinely >90% B220+ by flow cytometry.
Stimulations, immunoprecipitation and immunoblotting
Stimulations for analysis of tyrosine phosphorylation in primary B cells and Jurkat T cells were performed at 37°C. For stimulations of Bal-17 cells, as shown in Figure 7, cells were incubated for 10 min at room temperature with the indicated doses of biotinylated Fab fragments. Unbound Fabs were removed by washing and then cell-bound antibody fragments were crosslinked by the addition of 20 µg/ml soluble avidin at 37°C. After stimulation, cells were lysed in ice-cold buffer containing 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS, 100 mM Tris pH 7.4, 150 mM NaCl, 1 mM EDTA, 1 mM sodium orthovanadate, 10 mM sodium molybdate, 10 mM sodium fluoride, 5 mM iodoacetamide, 10 µM leupeptin, 2 µg/ml antipain, 1 µM pepstatin A, 6 µg/ml chymostatin, 1 µg/ml aprotinin, 1 µg/ml 4-(2-aminoethyl)-benzensulfonyl fluoride (Calbiochem). Clarified lysates were incubated with specific antibodies or non-immune control antibody followed by Protein G Fast Flow Sepharose (Pharmacia). Peptide adsorption was performed using peptides with the sequences described previously (O’Rourke et al., 1998) using lysates from COS7 cells transfected with plasmids encoding either Vav-1 or Vav-2. For the peptide competition experiment, 100 µM peptide (EGEGY*[391]EEPDSE or its non-phosphorylated equivalent) was included during the immunoprecipitation. All antibody or peptide complexes were washed four times with lysis buffer, boiled in reducing Laemmli buffer, separated by SDS–PAGE, transferred to nitrocellulose and immunoblotted with antibody to phosphotyrosine, Vav-2, Vav-1 or myc. Antibody binding was revealed using HRP-conjugated secondary antibodies followed by ECL (Pierce and Warriner). In some instances, membranes were stripped of blotting antibody in 62.5 mM Tris–HCl pH 6.7, 2% SDS, 100 µM 2-mercaptoethanol and subsequently reprobed.
Far-western blotting
These methods were adapted from those previously reported by Coggeshall and colleagues (Tridandapani et al., 1997). GST fusion proteins encoding amino acids 666–766 of Vav-1 and 663–762 of Vav-2 were purified from lysates of E.coli using standard protocols. Ten micrograms of each protein were resolved by SDS–PAGE and transferred to nitrocellulose. The filters were incubated overnight at 4°C with 1 µM phosphorylated or non-phosphorylated biotinylated peptide. After extensive washing in Tris-buffered saline (TBS)/0.05% Tween-20/0.4% NP-40, peptide binding was visualized using streptavidin-conjugated HRP (Southern Biotechnology Associates, Inc.) and ECL.
Transfection and luciferase assays
COS7 cells were cultured in DMEM containing 10% FCS, 2 mM glutamine, 100 U/ml penicillin, 100 mg/ml streptomycin. COS7 cells (2 × 105) from subconfluent cultures were transfected with 2 µg of plasmid DNA using LipofectAmine (Life Technologies) according to the manufacturer’s protocol. Bal-17 murine B-lymphoma cells were maintained in RPMI supplemented with 5% FCS, 2 mM glutamine, 100 U/ml penicillin, 100 mg/ml streptomycin. Bal-17 cells (2 × 107) growing in log phase were electroporated with 10 µg of reporter construct plus the indicated amount of co-transfected plasmid or empty expression vector at 300 mV and 960 µF discharged from a Bio-Rad gene pulser. After transfection, cells were cultured in supplemented RPMI and induced for 18–24 h with 2 µg/ml b7.6 monoclonal anti-mouse IgM or 1 µg/ml 1D3 anti-mouse CD19 plus 20 µg/ml F(ab′)2 goat anti-rat IgG or medium alone. T Ag Jurkat T cells, which stably express the SV40 virus large T-antigen, were grown in supplemented RPMI as described above. Log-phase Jurkat cells (1 × 107) were electroporated with 10 µg of reporter construct plus the indicated amount of co-transfected plasmid or empty expression vector at 250 mV and 960 µF using a Bio-Rad gene pulser. Eighteen hours post-transfection, triplicate samples of 2 × 105 T cells were incubated with medium alone or 5 µg/ml OKT3 anti-human CD3 for an additional 6 h. Cells were harvested, washed in phosphate-buffered saline (PBS), lysed in Cell Culture Lysis Reagent (Promega) and luciferase activity quantitated on a Top Count NXT Luminescence Counter (Packard) using the Luciferase Assay System (Promega) according to the manufacturer’s instructions. Lysates were corrected for protein concentration by a DC Protein Assay (Bio-Rad) determination. In all luciferase assay transfections, empty expression vector DNA was used to equalize the amount of input DNA so that the amounts of cytomegalovirus (CMV) promoter and other regulatory elements were kept constant. The results for all transfections are shown as the fold induction relative to activity in unstimulated cells co-transfected with empty expression vector and the reporter plasmid. The results are expressed as the means of values determined from 3–4 independent experiments (Bal-17 transfections) or from triplicate samples (Jurkat transfections) in one representative of three independent experiments. Error bars in all figures represent the standard errors of the means.
Calcium measurements
Measurements of intracellular Ca2+ mobilization were performed using Jurkat and Bal-17 cells infected with recombinant vaccinia viruses expressing FLAG-tagged Vav-1, Vav-2 or truncated Δ1–180Vav-2 as described previously (Billadeau et al., 2000b). In brief, cells were infected with control non-recombinant virus or recombinant viruses at a multiplicity of infection of 10:1 for 2 h. For the last 30 min of infection, the cells were loaded with 5 µM Indo-1AM (Calbiochem) and divided in half. Cells were washed with PBS containing 1% bovine serum albumin and stored in RPMI-10 until analyzed. The Jurkat T cells were treated with goat anti-mouse IgG F(ab′)2 alone or stimulated with a combination of anti-CD3 mAb (2.5 µg/ml) and goat anti-mouse IgG F(ab′)2. The Bal-17 B cells were left unstimulated or stimulated with the anti-murine IgM mAb b7.6 (5 µg/ml). The samples were immediately analyzed by flow cytometry for the relative blue:violet ratio for 500 s. Unstimulated cells did not demonstrate Ca2+ fluxes in the absence of the stimulatory antibody (data not shown).
Acknowledgments
Acknowledgements
We thank Geoff Butcher and Vikki Brew for mAb production, Babraham Institute Sequencing Support for DNA analysis and oligonucleotides, and SABU staff for animal husbandry. We thank Denis Alexander, Geoff Butcher, John Cambier, Doreen Cantrell, Simon Cook, Julian Downward, Doug Fearon, Alan Hall, Greg Huyer, Insong Lee, Michelle Morrow, Lill Martensson, Victor Tybulewicz, Elena Vigorito, Louise Webb, Art Weiss, Heidi Welch, David Williams and Henry Wortis for reagents and advice. D.D.B. is a Leukemia and Lymphoma Special Fellow. This work was funded in part by a Biotechnology and Biological Sciences Research Council Competitive Strategic Grant and by a grant from the Leukaemia Research Fund (to M.T.), and NIH grants AI15803 (to R.B.) and CA-47752 (to P.J.L.).
References
- Abe K., Whitehead,I.P., O’Bryan,J.P. and Der,C.J. (1999) Involvement of NH2-terminal sequences in the negative regulation of Vav signalling and transforming activity. J. Biol. Chem., 274, 30410–30418. [DOI] [PubMed] [Google Scholar]
- Aghazadeh B., Zhu,K., Kubiseski,T.J., Liu,G.A., Pawson,T., Zheng,Y. and Rosen,M.K. (1998) Structure and mutagenesis of the Dbl homology domain. Nature Struct. Biol., 5, 1098–1107. [DOI] [PubMed] [Google Scholar]
- Bazin H. (1982) Production of rat monoclonal antibodies with the LOU non-secreting IR983F myeloma cell line. In Peeters,H. (ed.), Protides of the Biological Fluids 29th Colloquium 1981. Pergamon Press, Oxford, UK. [Google Scholar]
- Benschop R.J. and Cambier,J.C. (1999) B cell development: signal transduction by antigen receptors and their surrogates. Curr. Opin. Immunol., 11, 143–151. [DOI] [PubMed] [Google Scholar]
- Berland R. and Wortis,H.H. (1998) An NFAT-dependent enhancer is necessary for anti-IgM-mediated induction of murine CD5 expression in primary splenic B cells. J. Immunol., 161, 277–285. [PubMed] [Google Scholar]
- Bikah G., Carey,J., Ciallella,J.R., Tarakhovsky,A. and Bondada,S. (1996) CD5-mediated negative regulation of antigen receptor-induced growth signals in B-1 B cells. Science, 274, 1906–1909. [DOI] [PubMed] [Google Scholar]
- Billadeau D.D., Mackie,S.M., Schoon,R.A. and Leibson,P.J. (2000a) The Rho family guanine nucleotide exchange factor Vav-2 regulates the development of cell-mediated cytotoxicity. J. Exp. Med., 192, 381–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Billadeau D.D., Mackie,S.M., Schoon,R.A. and Leibson,P.J. (2000b) Specific subdomains of Vav differentially affect T cell and NK cell activation. J. Immunol., 164, 3971–3981. [DOI] [PubMed] [Google Scholar]
- Buhl A.M. and Cambier,J.C. (1999) Phosphorylation of CD19 Y484 and Y515, and linked activation of phosphatidylinositol 3-kinase, are required for B cell antigen receptor-mediated activation of Bruton’s tyrosine kinase. J. Immunol., 162, 4438–4446. [PubMed] [Google Scholar]
- Bustelo X.R. (2000) Regulatory and signalling properties of the Vav family. Mol. Cell. Biol., 20, 1461–1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bustelo X.R. and Barbacid,M. (1992) Tyrosine phosphorylation of the vav proto-oncogene product in activated B cells. Science, 256, 1196–1199. [DOI] [PubMed] [Google Scholar]
- Bustelo X.R., Ledbetter,J.A. and Barbacid,M. (1992) Product of vav proto-oncogene defines a new class of tyrosine protein kinase substrates. Nature, 356, 68–71. [DOI] [PubMed] [Google Scholar]
- Choi M.S., Brines,R.D., Holman,M.J. and Klaus,G.G. (1994) Induction of NF-AT in normal B lymphocytes by anti-immunoglobulin or CD40 ligand in conjunction with IL-4. Immunity, 1, 179–187. [DOI] [PubMed] [Google Scholar]
- Costello P.S., Walters,A.E., Mee,P.J., Turner,M., Reynolds,L.F., Prisco,A., Sarner,N., Zamoyska,R. and Tybulewicz,V.L.J. (1999) The Rho-family GTP exchange factor Vav is a critical transducer of T cell receptor signals to the calcium, ERK and NF-κB pathways. Proc. Natl Acad. Sci. USA, 96, 3035–3040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crabtree G.R. (1999) Generic signals and specific outcomes: signalling through Ca2+, calcineurin and NFAT. Cell, 96, 611–614. [DOI] [PubMed] [Google Scholar]
- Crespo P., Schuebel,K.E., Ostrom,A.A., Gutkind,J.S. and Bustelo,X.R. (1997) Phosphotyrosine-dependent activation of Rac-1 GDP/GTP exchange by the Vav proto-oncogene product. Nature, 385, 169–172. [DOI] [PubMed] [Google Scholar]
- Dempsey P.W., Allison,M.E., Akkaraju,S., Goodnow,C.C. and Fearon,D.T. (1996) C3d of complement as a molecular adjuvant: bridging innate and acquired immunity. Science, 271, 348–350. [DOI] [PubMed] [Google Scholar]
- Dolmetsch R.E., Lewis,R.S., Goodnow,C.C. and Healy,J.I. (1997) Differential activation of transcription factors induced by Ca2+ response amplitude and duration [published erratum appears in Nature (1997), 388, 308)]. Nature, 386, 855–858. [DOI] [PubMed] [Google Scholar]
- Dolmetsch R.E., Xu,K. and Lewis,R.S. (1998) Calcium oscillations increase the efficiency and specificity of gene expression. Nature, 392, 933–936. [DOI] [PubMed] [Google Scholar]
- Fischer K.D. et al. (1998a) Vav is a regulator of cytoskeletal reorganisation mediated by the T-cell receptor. Curr. Biol., 8, 554–562. [DOI] [PubMed] [Google Scholar]
- Fischer K.D., Tedford,K. and Penninger,J.M. (1998b) Vav links antigen-receptor signaling to the actin cytoskeleton. Semin. Immunol., 10, 317–327. [DOI] [PubMed] [Google Scholar]
- Fujimoto M., Bradney,A.P., Poe,J.C., Steeber,D.A. and Tedder,T.F. (1999a) Modulation of B lymphocyte antigen receptor signal transduction by a CD19/CD22 regulatory loop. Immunity, 11, 191–200. [DOI] [PubMed] [Google Scholar]
- Fujimoto M., Poe,J.C., Jansen,P.J., Sato,S. and Tedder,T.F. (1999b) CD19 amplifies B lymphocyte signal transduction by regulating Src-family protein tyrosine kinase activation. J. Immunol., 162, 7088–7094. [PubMed] [Google Scholar]
- Gulbranson-Judge A., Tybulewicz,V.L., Walters,A.E., Toellner,K.M., MacLennan,I.C. and Turner,M. (1999) Defective immunoglobulin class switching in Vav-deficient mice is attributable to compromised T cell help. Eur. J. Immunol., 29, 477–487. [DOI] [PubMed] [Google Scholar]
- Han J., Das,B., Wei,W., Van Aelst,L., Mosteller,R.D., Khosravi Far,R., Westwick,J.K., Der,C.J. and Broek,D. (1997) Lck regulates Vav activation of members of the Rho family of GTPases. Mol. Cell. Biol., 17, 1346–1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han J. et al. (1998) Role of substrates and products of PI 3-kinase in regulating activation of Rac-related guanosine triphosphates by Vav. Science, 279, 558–560. [DOI] [PubMed] [Google Scholar]
- Hill C.S., Wynne,J. and Treisman,R. (1995) The Rho family GTPases RhoA, Rac1 and CDC42Hs regulate transcriptional activation by SRF. Cell, 81, 1159–1170. [DOI] [PubMed] [Google Scholar]
- Holsinger L.J., Spencer,D.M., Austin,D.J., Schreiber,S.L. and Crabtree,G.R. (1995) Signal transduction in T lymphocytes using a conditional allele of Sos. Proc. Natl Acad. Sci. USA, 92, 9810–9814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holsinger L.J., Graef,I.A., Swat,W., Chi,T., Bautista,D.M., Davidson,L., Lewis,R.S., Alt,F.W. and Crabtree,G.R. (1998) Defects in actin-cap formation in Vav-deficient mice implicate an actin requirement for lymphocyte signal transduction. Curr. Biol., 8, 563–572. [DOI] [PubMed] [Google Scholar]
- Katzav S., Cleveland,J.L., Heslop,H.E. and Pulido,D. (1991) Loss of the amino-terminal helix–loop–helix domain of the vav proto-oncogene activates its transforming potential. Mol. Cell. Biol., 11, 1912–1920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhne M.R., Ku,G. and Weiss,A. (2000) A guanine nucleotide exchange factor-independent function of Vav-1 in transcriptional activation. J. Biol. Chem., 275, 2185–2190. [DOI] [PubMed] [Google Scholar]
- Lopez-Lago M., Lee,H., Cruz,C., Movilla,N. and Bustelo,X.R. (2000) Tyrosine phosphorylation mediates both activation and down modulation of the biological activity of Vav. Mol. Cell. Biol., 20, 1678–1691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margolis B., Hu,P., Katzav,S., Li,W., Oliver,J.M., Ullrich,A., Weiss,A. and Schlessinger,J. (1992) Tyrosine phosphorylation of vav proto-oncogene product containing SH2 domain and transcription factor motifs. Nature, 356, 71–74. [DOI] [PubMed] [Google Scholar]
- Meyer K.B. and Ireland,J. (1998) PMA/ionomycin induces Ig κ 3′ enhancer activity which is in part mediated by a unique NFAT transcription complex. Eur. J. Immunol., 28, 1467–1480. [DOI] [PubMed] [Google Scholar]
- O’Rourke L., Tooze,R., Turner,M., Sandoval,D.M., Carter,R.H., Tybulewicz,V.L.J. and Fearon,D.T. (1998) CD19 as a membrane-anchored adaptor protein of B lymphocytes: costimulation of lipid and protein kinases by recruitment of Vav. Immunity, 8, 635–645. [DOI] [PubMed] [Google Scholar]
- Sato S., Jansen,P.J. and Tedder,T.F. (1997a) CD19 and CD22 expression reciprocally regulates tyrosine phosphorylation of Vav protein during B lymphocyte signalling. Proc. Natl Acad. Sci. USA, 94, 13158–13162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato S., Steeber,D.A., Jansen,P.J. and Tedder,T.F. (1997b) CD19 expression levels regulate B lymphocyte development: human CD19 restores normal function in mice lacking endogenous CD19. J. Immunol., 158, 4662–4669. [PubMed] [Google Scholar]
- Schuebel K.E., Bustelo,X.R., Nielsen,D.A., Song,B.J., Barbacid,M., Goldman,D. and Lee,I.J. (1996) Isolation and characterization of murine Vav-2, a member of the Vav family of proto-oncogenes. Oncogene, 13, 363–371. [PubMed] [Google Scholar]
- Schuebel K.E., Movilla,N., Rosa,J.L. and Bustelo,X.R. (1998) Phosphorylation-dependent and constitutive activation of Rho proteins by wild-type and oncogenic Vav-2. EMBO J., 17, 6608–6621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soisson S.M., Nimnual,A.S., Uy,M., Bar-Sagi,D. and Kuriyan,J. (1998) Crystal structure of the Dbl and pleckstrin homology domains from the human Son of Sevenless protein. Cell, 95, 259–268. [DOI] [PubMed] [Google Scholar]
- Tarakhovsky A., Kanner,S.B., Hombach,J., Ledbetter,J.A., Muller,W., Killeen,N. and Rajewsky,K. (1995) A role for CD5 in TCR-mediated signal transduction and thymocyte selection. Science, 269, 535–537. [DOI] [PubMed] [Google Scholar]
- Teutsch M., Higer,M., Wang,D. and Wortis,H.W. (1995) Induction of CD5 on B and T cells is suppressed by cyclosporin A, FK-520 and rapamycin. Int. Immunol., 7, 381–392. [DOI] [PubMed] [Google Scholar]
- Tooze R.M., Doody,G.M. and Fearon,D.T. (1997) Counterregulation by the coreceptors CD19 and CD22 of MAP kinase activation by membrane immunoglobulin. Immunity, 7, 59–67. [DOI] [PubMed] [Google Scholar]
- Tridandapani S., Kelley,T., Pradhan,M., Cooney,D., Justement,L.B. and Coggeshall,K.M. (1997) Recruitment and phosphorylation of SH2-containing inositol phosphatase and Shc to the B-cell Fc γ immunoreceptor tyrosine-based inhibition motif peptide motif. Mol. Cell. Biol., 17, 4305–4311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai E.Y., Yie,J., Thanos,D. and Goldfeld,A.E. (1996) Cell-type-specific regulation of the human tumor necrosis factor α gene in B cells and T cells by NFATp and ATF-2/JUN. Mol. Cell. Biol., 16, 5232–5244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner M., Mee,P.J., Walters,A.E., Quinn,M.E., Mellor,A.L., Zamoyska,R. and Tybulewicz,V.L.J. (1997) A requirement for the Rho-family GTP exchange factor Vav in positive and negative selection of thymocytes. Immunity, 7, 451–460. [DOI] [PubMed] [Google Scholar]
- Van Aelst L. and D’Souza-Schorey,C. (1997) Rho GTPases and signalling networks. Genes Dev., 11, 2295–2322. [DOI] [PubMed] [Google Scholar]
- van Leeuwen J.E.M. and Samelson,L.E. (1999) T cell antigen-receptor signal transduction. Curr. Opin. Immunol., 11, 242–248. [DOI] [PubMed] [Google Scholar]
- Venkataraman L., Francis,D.A., Wang,Z., Liu,J., Rothstein,T.L. and Sen,R. (1994) Cyclosporin-A sensitive induction of NF-AT in murine B cells. Immunity, 1, 186–196. [DOI] [PubMed] [Google Scholar]
- Vincent S., Jeanteur,P. and Fort,P. (1992) Growth-regulated expression of rhoG, a new member of the ras homolog gene family. Mol. Cell. Biol., 12, 3138–3148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welch H., Eguinoa,A., Stephens,L.R. and Hawkins,P.T. (1998) Protein kinase B and rac are activated in parallel within a phosphatidylinositide 3OH-kinase-controlled signaling pathway. J. Biol. Chem., 273, 11248–11256. [DOI] [PubMed] [Google Scholar]
- Weng W.K., Jarvis,L. and LeBien,T.W. (1994) Signaling through CD19 activates Vav/mitogen-activated protein kinase pathway and induces formation of a CD19/Vav/phosphatidylinositol 3-kinase complex in human B cell precursors. J. Biol. Chem., 269, 32514–32521. [PubMed] [Google Scholar]
- Wu J., Katzav,S. and Weiss,A. (1995) A functional T-cell receptor signaling pathway is required for p95vav activity. Mol. Cell. Biol., 15, 4337–4346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yablonski D., Kane,L.P., Qian,D. and Weiss,A. (1998) A Nck-Pak1 signaling module is required for T-cell receptor-mediated activation of NFAT, but not of JNK. EMBO J., 17, 5647–5657. [DOI] [PMC free article] [PubMed] [Google Scholar]