

Abstract
BACKGROUND AND PURPOSE
Lithium chloride (LiCl) inhibits inositol monophosphatase (IMPase) at therapeutic concentrations. Given that LiCl induces death in cultured macrophages and that macrophages play an active role in atherosclerotic plaque destabilization, we investigated whether LiCl would induce selective macrophage death to stabilize the structure of the plaque.
EXPERIMENTAL APPROACH
The effect of LiCl was assessed on macrophages and smooth muscle cells (SMCs) in culture, in isolated atherosclerotic carotid arteries from rabbits and after local in vivo treatment via osmotic minipumps to rabbits with collared atherosclerotic carotid arteries. In addition, in vitro experiments were performed to elucidate the mechanism of LiCl-induced macrophage death.
KEY RESULTS
In vitro, whereas SMCs were highly resistant, LiCl induced macrophage death characterized by externalization of phosphatidylserine, caspase-3 cleavage and DNA fragmentation, all indicative of apoptosis. LiCl reduced inositol-1,4,5-trisphosphate levels in macrophages. Moreover, the IMPase inhibitor L-690 330 as well as IMPase gene silencing induced macrophage apoptosis. Both in vitro treatment of rabbit atherosclerotic carotid arteries with LiCl and local in vivo administration of LiCl to the plaques decreased plaque macrophages through apoptosis, as shown by terminal deoxynucleotidyl transferase deoxyuridine triphosphate (dUTP) nick-end labelling (TUNEL), without affecting SMCs. Vasomotor studies in vitro showed that LiCl did not affect the functionality of SMCs and endothelial cells.
CONCLUSIONS AND IMPLICATIONS
LiCl selectively decreased the macrophage load in rabbit atherosclerotic plaques via IMPase inhibition without affecting the viability or functionality of SMCs and endothelial cells. These data provide evidence for local administration of an IMPase inhibitor to stabilize atherosclerotic plaques.
Keywords: apoptosis, atherosclerosis, inositol monophosphatase, lithium, macrophage, plaque stability
Introduction
Lithium is a mood-stabilizing drug that has been used for decades as the primary therapy for bipolar disorder. Although its mode of action is not fully understood, a series of experiments have shown that lithium reduces the cellular concentration of inositol and increases that of inositol-1-phosphate in the brain (Allison et al., 1976). These findings suggest that inhibition of the enzyme inositol monophosphatase (IMPase) is involved. Inositol is the substrate for the synthesis of membrane-bound phosphatidylinositol diphosphate (PIP2) that is hydrolysed by the enzyme phospholipase C to release diacylglycerol (DAG) and soluble inositol-1,4,5-trisphosphate (IP3). The latter is a second messenger produced in response to the stimulation of G-protein-coupled receptors. It controls the release of Ca2+ from intracellular stores (Mikoshiba et al., 1993) and acts as a substrate for the inositol kinases that generate further phosphorylated inositolphosphate forms (Shears et al., 2004). Interfering with phosphatidylinositol cell signalling through inhibition of IMPase leads to decreased free inositol and consequently to decreased IP3 levels (Berridge et al., 1989). Lithium-evoked depletion of the second messenger IP3 can therefore have profound effects on signal transduction in cells. Lithium-sensitive IMPase (IMPA1) is the only gene known to encode IMPase activity that can be inhibited by therapeutic concentrations of lithium (∼1 mM) (Ohnishi et al., 2007). Recently, also IMPA2 has been shown to exhibit IMPase activity, although to a lower extent than IMPA1. IMPA2 can be inhibited by lithium chloride (LiCl) but only at higher concentrations (Ohnishi et al., 2007). Lithium also inhibits glycogen synthase kinase-3β (GSK-3β), a serine-threonine kinase with pro-apoptotic properties (Lin et al., 2007) that is constitutively active (Stambolic et al., 1996; Phiel & Klein, 2001). Mechanisms regulating GSK-3β such as the wingless-int (Wnt) pathway (Cook et al., 1996; Chen et al. 2000; Sarkar et al., 2005) can play a role in the control of cell survival.
In a recent study, lithium was a potent inducer of apoptosis in bone marrow-derived macrophages from C57BL/6 mice (Zhang et al., 2009). Macrophages play a key role in atherosclerotic plaque destabilization and rupture (Kockx and Herman, 2000; Lafont, 2003). Rupture-prone atherosclerotic plaques have a large necrotic core, numerous macrophages and a thin fibrous cap that consists of collagen-synthesizing smooth muscle cells (SMCs) and extracellular matrix (Virmani et al., 2002). Plaques tend to rupture as a consequence of a weakened fibrous cap, particularly in the shoulder regions where most macrophages reside (van der Wal et al., 1994). Lesional macrophages produce matrix metalloproteinases that degrade the extracellular matrix (Newby et al., 2009; Back et al. 2010), and induce SMC apoptosis, which leads to a decrease in collagen-producing cells (Boyle et al., 2002). Removal of macrophages from plaques by inducing macrophage-specific cell death could represent a novel approach in cardiovascular research to stabilize the structure of atherosclerotic plaques (Martinet et al., 2007). In the present study, we show that LiCl decreases the macrophage content in collar-induced atherosclerotic plaques by selective induction of macrophage death via inhibition of IMPase without affecting the viability and functionality of SMCs and endothelial cells.
Methods
Animals
All animal care and experimental procedures were approved by the Ethical Committee of the University of Antwerp.
Cell culture
The murine macrophage cell line J774A.1 was grown in RPMI 1640 medium supplemented with 100 U·mL−1 penicillin, 100 µg·mL−1 streptomycin, 50 µg·mL−1 gentamycin, 20 U·mL−1 polymyxin B and 10% heat-inactivated fetal bovine serum. Bone marrow-derived macrophages were obtained from C57BL/6 mice by flushing femur and tibia with RPMI 1640 medium containing 20 mg·mL−1 heparin. These bone marrow cells were cultured in RPMI 1640 medium supplemented with 20% L929-cell conditioned medium, antibiotics and 10% heat-inactivated fetal bovine serum. SMCs were isolated from rabbit aorta by collagenase type 2 and elastase digestion (60 to 90 min at 37°C) at final concentrations of 300 U·mL−1 and 5 U·mL−1 respectively. Mouse SMCs were isolated as reported previously (Ray et al., 2001). Mouse and rabbit SMCs were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 1% glutamine and F-10 Ham's medium respectively. Cells were cultured in a humidified 5% CO2 incubator at 37°C in the presence or absence of LiCl, NaCl, L-690 330, SB216763 or cycloheximide at various concentrations for several hours.
Osmolality of solutions was determined by a Fiske 110 osmometer (Fiske Associates, Boston, MA, USA) using freeze point depression. Evaluation of cell viability was based on the incorporation of the supravital dye neutral red by viable cells (Lowik et al., 1993). Levels of IP3 were measured using the [3H] Biotrak assay system (GE Healthcare, Buckinghamshire, UK) according to the manufacturer's instructions.
Detection of apoptotic cells
Apoptosis was detected with an Annexin V-FITC staining kit (BD Biosciences, San Jose, CA, USA) in combination with 7-amino-actinomycin D (7-AAD) as previously described (Lecoeur et al., 1997). Chromatin condensation and necrotic cells were studied by fluorescence microscopy after staining with Hoechst 33342 (Invitrogen, Carlsbad, CA, USA) and propidium iodide (PI; BD Biosciences) respectively. Cells were incubated with PI for 5 min, fixed in 4% formaldehyde for 10 min, washed with phosphate-buffered saline (PBS) and permeabilized with 0.2% Triton X-100 in PBS for 5 min. After washing with PBS, cells were labelled with 10 µM Hoechst 33342 for 5 min. For DNA fragmentation assays, the Vindelov method was used to evaluate the subG1 DNA content (Vindelov and Christensen, 1994). Cell samples were analysed by flow cytometry (FACsCantoII, BD Biosciences). Forward and side scatter gates were set to include only single cells.
siRNA-mediated gene silencing
J774A.1 macrophages and SMCs were transfected with 10–100 nmol·L−1 ON-TARGETplus SMARTpool siRNA (Dharmacon, Chicago, IL, USA) specific to IMPase, 10 nmol·L−1 ON-TARGETplus SMARTpool siRNA specific to GSK-3β or 10 nmol·L−1 non-targeting siControl RISC-free siRNA (negative control) using HiPerfect transfection reagent (Qiagen, Valencia, CA, USA). Down-regulation of mRNA was examined by real-time RT-PCR. Briefly, cDNA was prepared using a Fastlane Cell cDNA kit (Qiagen) at 42°C for 30 min, followed by inactivation of the reverse transcriptase at 95°C for 3 min. Relative abundance of IMPA1, IMPA2 and GSK-3β cDNA was then examined using a TaqMan gene expression assay (assay ID: Mm00497770_m1, Mm00475141_m1 and Mm00444911_m1, respectively (Applied Biosystems, Foster City, CA, USA) on a ABIPrism 7300 sequence detector system in 25 µL reaction volumes containing Universal PCR Master Mix. The parameters for PCR amplification were 50°C for 2 min, 95°C for 10 min, followed by 40 cycles of 95°C for 15 s and 60°C for 1 min. Relative expression of cDNA species was calculated using the comparative threshold cycle method. All data were controlled for quantity of cDNA by performing measurements on the endogenous reference gene, β-actin (assay ID: Mm00607939_s1).
Western blot analysis
Cultured cells were lysed in an appropriate volume of Laemmli sample buffer (Bio-Rad Laboratories, Hercules, CA, USA). Cell lysates were then heat-denatured for 4 min in boiling water and loaded on a NuPAGE 4–12% Bis-Tris gel (Invitrogen). After gel electrophoresis, proteins were transferred to an Immobilon-P Transfer Membrane (Millipore, Billerica, MA, USA) according to standard procedures. Membranes were blocked in Tris-buffered saline containing 0.05% Tween 20 and 5% nonfat dry milk (Bio-Rad Laboratories) for 1 h. After blocking, membranes were probed overnight at 4°C with primary antibodies in antibody dilution buffer (Tris-buffered saline containing 0.05% Tween 20 and 1% non-fat dry milk), followed by 1 h incubation with secondary antibody in antibody dilution buffer at room temperature. Antibody detection was accomplished with SuperSignal West Pico or SuperSignal West Femto Maximum Sensitivity Substrate (Pierce, Rockford, IL, USA) using a Lumi-Imager (Roche Diagnostics, Mannheim, Germany). The following primary antibodies were used: mouse anti-β-actin (clone AC-15, Sigma-Aldrich, St. Louis, MO, USA), rabbit anti-caspase-3 (Cell Signaling Technology, Danvers, MA, USA), goat anti-IMPA1 (Santa Cruz Biotechnology, Santa Cruz, CA, USA), rabbit anti-GSK-3β (Cell Signaling Technology) and rabbit anti-β-catenin (Cell Signaling Technology). Peroxidase-conjugated secondary antibodies were purchased from Dako (Glostrup, Denmark).
Animal studies
In vitro treatment of rabbit atherosclerotic carotid arteries
Male New Zealand White rabbits (2.5 to 4.0 kg, n= 9) were fed a diet supplemented with 1.5% cholesterol for 14 days. After anesthesia with sodium pentobarbital (30 mg·kg−1 i.v.) (CEVA Santé animale, Brussels, Belgium), a nonocclusive, flexible, silicone collar was placed around both carotid arteries and closed with silicone glue to induce atheroma-like lesions (i.e. intimal thickness consisting of SMCs and macrophages) (Kockx et al., 1992; De Meyer et al. 1997; Croons et al., 2009). After another 14 days, while continuing the cholesterol diet, the animals were killed by an overdose of sodium pentobarbital. Carotid arteries were dissected and released from the collars. Four rings were cut from each collared segment and were incubated in serum-containing F-10 Ham's medium for 3 or 7 days in the presence or absence of 30 mM LiCl. After treatment, the rings were formalin-fixed for 24 h.
In vivo treatment of rabbit atherosclerotic carotid arteries
Atheroma-like lesions were induced in the carotid artery of male New Zealand White rabbits (n= 10) as described earlier. Fourteen days after collar placement, an osmotic minipump (model 2ML1, Alzet, Cupertino, CA, USA) was connected to each collar and placed subdermally in the thoracic region. The pumps delivered 10 µL of solution (1 M LiCl) per hour locally to the carotid artery for 3 days. Because high concentrations of NaCl in the osmotic minipumps (up to 6 M NaCl) did not affect the cellular composition of atheroma-like lesions (data not shown), saline (0.9% NaCl) was used in all experiments as negative control. Serum samples were collected via the ear vein to determine lithium levels using an Advia Chemistry Analyzer (Siemens, Holliston, MA, USA). The rabbits were heparinized (150 U·kg−1) and killed by an overdose of sodium pentobarbital. Carotid arteries were dissected and released from the collars. Rings were cut from each collared segment and were either formalin-fixed for 24 h, snap-frozen in liquid nitrogen or used for vascular reactivity studies.
Histological examination
Formalin-fixed carotid artery rings were embedded in paraffin and stained with haematoxylin/eosin (H/E). Macrophages, SMCs and endothelium were immunohistochemically detected by an indirect peroxidase antibody conjugate technique (Kockx et al., 1992; De Meyer et al. 2000). The following primary antibodies were used: anti-rabbit macrophage (clone RAM 11, Dako), anti-α-SMC actin (clone 1A4, Sigma-Aldrich), anti-CD31 (Dako), anti-CD43 (Serotec, Oxford, UK), anti-rabbit neutrophil (clone RPN3/57, Serotec) and anti-S100 (dendritic cells) (Thermo Fisher Scientific, Fremont, CA, USA). The following secondary antibodies were used: Vectastain (Vector Laboratories, Burlingame, CA, USA) and rabbit anti-mouse horseradish peroxidase-conjugated antibody (Dako). For the detection of oligonucleosomal DNA cleavage, a terminal deoxynucleotidyl transferase dUTP nick-end labelling (TUNEL) technique was used (Kockx et al., 1998b; Schrijvers et al. 2004). RAM 11-positivity, α-SMC actin-positivity, CD31-positivity and TUNEL-positivity in six adjacent areas (600 × 450 µm each) covering one section were analysed, without knowledge of the treatments, using a colour image analysis system (Image Pro Plus 4.1, Media Cybernetics Inc., Silver Spring, MD, USA). Fragmented nuclei were counted in six random areas (160 × 120 µm each) on H/E-stained sections and expressed as the number of fragmented nuclei per 10−2 mm2.
The presence of lithium in the atheroma-like lesions and in cultured macrophages and SMCs was analysed using time-of-flight (TOF) static secondary ion mass spectrometry (S-SIMS) (De Mondt et al., 2009). Positive ion mass spectra were recorded with an Ion TOF V (IonTOF, Muenster, Germany) instrument by the application of 25 keV Bi3+ primary ions over a surface of 100 × 100 µm. The ion dose density was 4.5 1011 ions cm−2. Electron flooding was applied to compensate sample charging.
Vascular reactivity studies
Segments (2 mm long) from each collared carotid were mounted in organ baths (10 mL) filled with Krebs-Ringer solution (37°C, continuously aerated with 95% O2/5% CO2, pH 7.4) for force measurements at 59 mN preload (De Meyer et al., 1994). Tension was measured isometrically with a Statham UC2 force transducer (Gould, Cleveland, OH, USA) connected to a data acquisition system (Powerlab 8/3, ADInstruments, Spechbach, Germany). Contractility of the segments was determined by performing concentration-response curves for 5-HT and KCl. The organ baths were washed three times with physiological Krebs solution between consecutive concentration-response curves. In relaxation studies, segments were precontracted with 5-HT or KCl, followed by cumulative concentration-response curves for ACh or diethylamine NONOate (DEANO) respectively. The pD2 is the negative logarithm of the molar concentration producing half of the maximal response (Emax) and reflects the sensitivity to an agonist.
Statistical analysis
All data are presented as mean ± SEM and were processed with SPSS 16.0 software (SPSS Inc., Chicago, IL, USA). Differences were considered significant at P < 0.05. The applied statistical tests are reported in the figure legends and table.
Materials
The J774A.1 macrophages were obtained from American Type Culture Collection (Manassas, VA); RPMI 1640 medium, DMEM, F-10 Ham's medium and antibiotics from Invitrogen; foetal bovine serum and neutral red from Sigma-Aldrich; heparin from Leo Pharmaceutical Products (Ballerup, Denmark). Collagenase type 2 was from Worthington (Lakewood, NJ, USA) and elastase from Sigma-Aldrich. LiCl, NaCl, SB216763 and cycloheximide were from Sigma-Aldrich and L-690 330 from Tocris (Ellisville, MO, USA).
Results
Lithium chloride induces apoptosis in macrophages in vitro
Cell death was initiated by LiCl in a concentration-dependent (3–30 mM) manner in the macrophage cell line J774A.1 as well as in primary bone marrow-derived macrophages, but not in SMCs (Figure 1A). Because LiCl can exert osmotic effects at high concentrations, cells were also treated with NaCl in equimolar amounts and the osmolality of solutions was determined. Similar to LiCl, NaCl induced cell death in both macrophages and SMCs, but this effect was only minor and seen from 100 mM onward (Figure 1A), presumably due to the hyperosmolality of solutions (427 mOsm·kg−1 H2O and 426 mOsm·kg−1 H2O for 100 mM LiCl and 100 mM NaCl, respectively, vs. 291 mOsm·kg−1 H2O for control medium). Because the viability of both J774A.1 cells and bone marrow-derived macrophages decreased by more than 70% after 24 h exposure to 30 mM LiCl, these conditions were chosen for further experiments with J774A.1 macrophages. Even long-term (up to 16 days) in vitro treatment of SMCs with LiCl did not induce a significant reduction in cell viability (94 ± 3% vs. medium-treated SMCs). However, SMCs could not withstand this long-term treatment with cycloheximide (viability: 1 ± 1% vs. medium-treated SMCs), a protein synthesis inhibitor known to be a selective inducer of macrophage apoptosis during short term treatment (<8 h in vitro) (Croons et al., 2007). TOF-S-SIMS analysis showed that lithium was equally taken up by macrophages and SMCs (0.30% and 0.31% vs. total ion count). Annexin V and 7-AAD labelling of LiCl-treated macrophages showed a significant increase in apoptotic cells (Annexin V-positive and 7-AAD-negative) at 8 h and (secondary) necrotic cells (Annexin V-positive and 7-AAD-positive) at 16 h (Figure 2A). Moreover, macrophage death induced by LiCl was characterized by caspase-3 cleavage (Figure 2B), chromatin condensation (Figure 3B and C) and DNA fragmentation (Figure 3A), which are all markers of apoptosis. Necrotic cells were visualized using PI (Figure 3D). These characteristics were not seen after treatment with 30 mM NaCl (Figures 2–3, right-hand panels).
Figure 1.
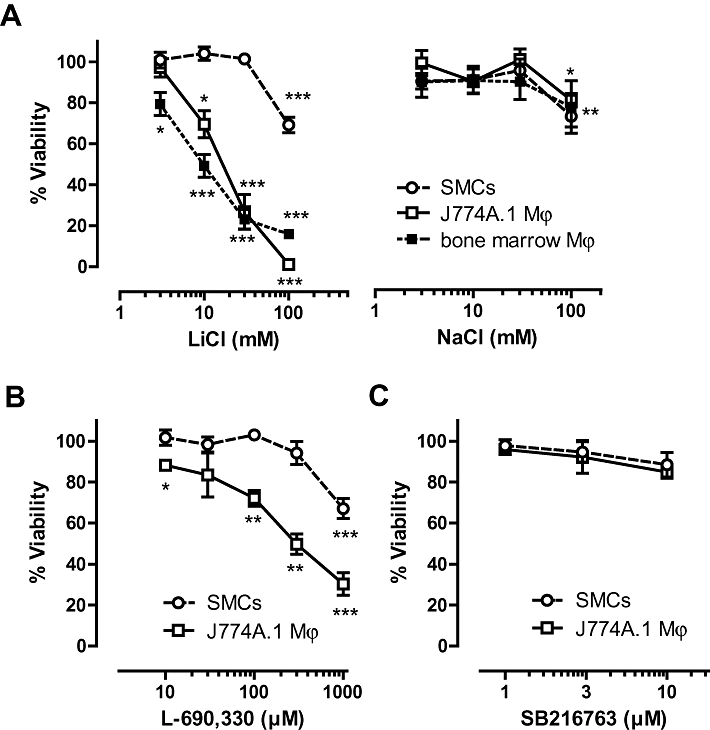
Effect of LiCl, NaCl, inositol monophosphatase (IMPase) inhibitor L-690 330 and glycogen synthase kinase-3β (GSK-3β) inhibitor SB216763 on the viability of macrophages and smooth muscle cells (SMCs). Cell viability was examined by neutral red assays. (A) J774A.1 macrophages (Mϕ), bone marrow-derived macrophages and rabbit aortic SMCs were treated with LiCl or NaCl (3–100 mM) for 24 h. Results represent mean ± SEM of four independent experiments, each performed in duplicate. J774A.1 macrophages (Mϕ) and rabbit aortic SMCs were also treated with IMPase inhibitor L-690 330 (10–1000 µM) (B) and GSK-3β inhibitor SB216763 (1–10 µM) (C) for 24 h. Results represent mean ± SEM of three independent experiments, each performed in duplicate. *P < 0.05; **P < 0.01 and ***P < 0.001 versus control (0 h) (one-sample t-test).
Figure 2.
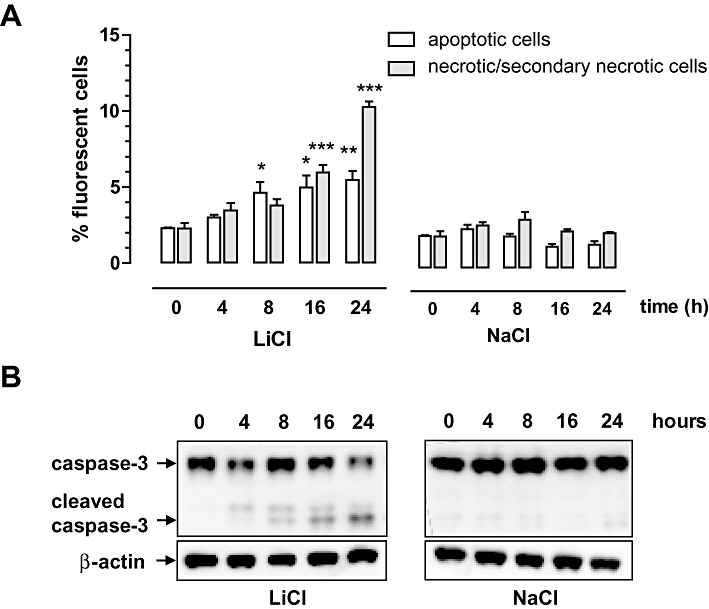
Characterization of LiCl-induced macrophage death. J774A.1 macrophages were treated with 30 mM LiCl or 30 mM NaCl for 0–24 h. (A) Quantification of apoptotic cells (Annexin V-positive and 7-AAD-negative cells) and (secondary) necrotic cells (Annexin V- and 7-AAD-positive cells) via flow cytometry. Data represent mean ± SEM of three independent experiments. *P < 0.05; **P < 0.01 and ***P < 0.001 versus control (anova, followed by Dunnett test). (B) Western blot analysis of caspase-3 cleavage.
Figure 3.
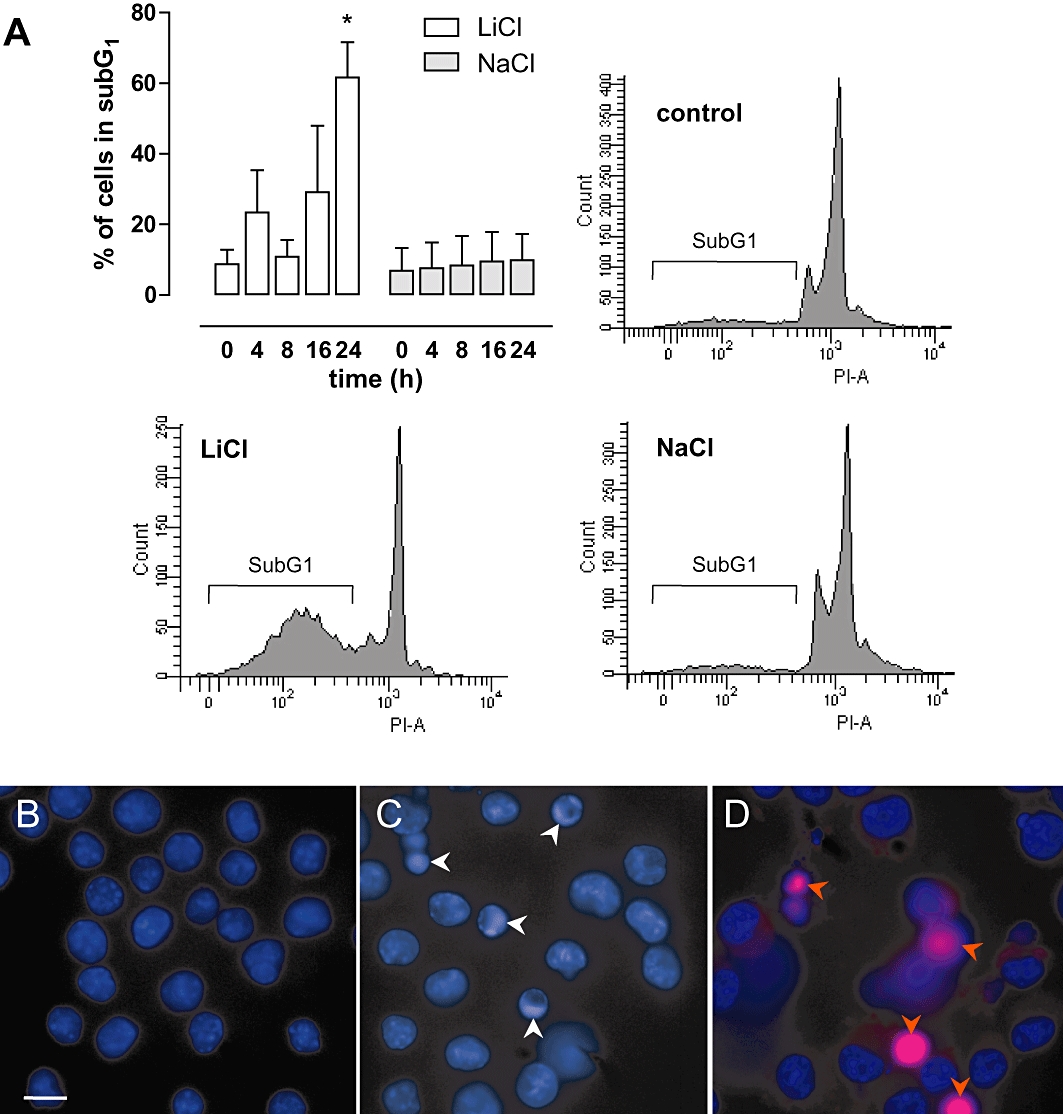
DNA fragmentation and chromatin condensation in LiCl-treated macrophages. (A) Quantification of apoptotic cells according to the Vindelov method. J774A.1 macrophages were treated with 30 mM LiCl or 30 mM NaCl for 0–24 h. DNA histograms of LiCl- and NaCl-treated macrophages, as well as untreated cells, are shown. Fragmented DNA appears in a subG1-peak on DNA histograms (PI-A, area of PI-positive cells). Data represent mean ± SEM of three independent experiments. *P < 0.05 versus control (anova, followed by Dunnett test). Evaluation of chromatin condensation (white arrowheads) and necrotic cells (red arrowheads) after staining with Hoechst 33342 and PI in J774A.1 macrophages treated with 30 mM LiCl for 0 h (B), 4 h (C) and 24 h (D).
Lithium chloride-induced cell death is mediated by IMPase inhibition
Because previous studies have demonstrated that lithium is an IMPase inhibitor (Berridge et al., 1989; Sarkar et al. 2005), we examined IP3 as downstream target of IMPase in macrophages and SMCs at various time points. Within 8 h, IP3 levels decreased in LiCl-treated macrophages, but not in SMCs (Figure 4A). IMPA1 and IMPA2 expression was examined in macrophages and SMCs. IMPA1 mRNA was equally present in both cell types (ΔCT of 9.5 ± 0.2 in macrophages and 9.3 ± 0.3 in SMCs vs. β-actin, P= 0.530, independent t-test), whereas IMPA2 expression was only detected in macrophages (ΔCT of 10.8 ± 0.2 vs. β-actin).
Figure 4.
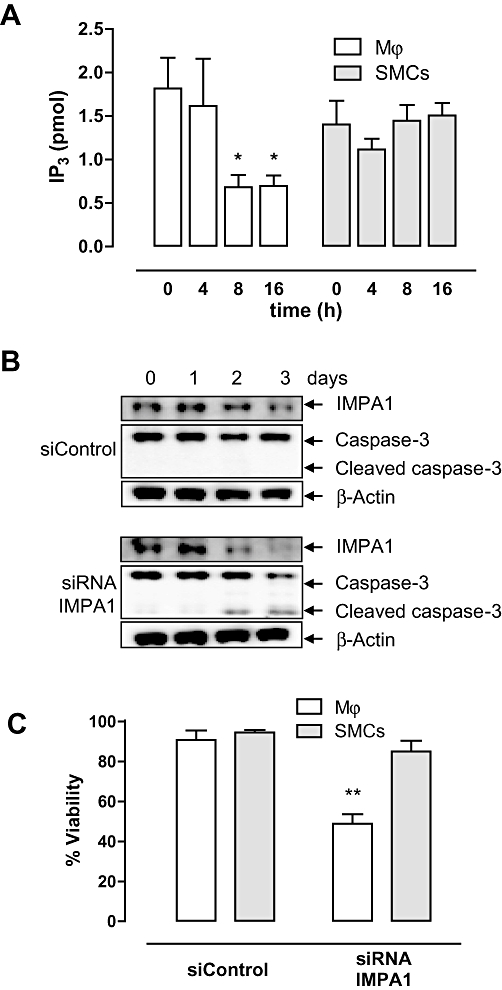
Lithium-sensitive inositol monophosphatase (IMPase) (IMPA1) is inhibited by LiCl in macrophages. (A) Levels of IP3 in J774A.1 macrophages and smooth muscle cells (SMCs) treated with 30 mM LiCl for 0–16 h. Results represent mean ± SEM of three independent experiments. *P < 0.05 versus 0 h (control) (anova, followed by Dunnett test). (B) Western blot analysis of IMPA1 and caspase-3 cleavage in J774A.1 macrophages transfected with siControl or siRNA IMPA1 for 0–3 days. (C) Viability of J774A.1 macrophages and mouse SMCs transfected with siControl or siRNA specific for IMPA1 at day 3. Bar graphs represent mean ± SEM of four independent experiments. **P < 0.01 versus siControl (paired-samples t-test).
Transfection of J774A.1 macrophages and SMCs with siRNA specific for IMPA1 decreased IMPA1 mRNA expression in both cell types after 72 h (relative expression 31 ± 11% in macrophages vs. siControl, P= 0.007, and 12 ± 11% in SMCs vs. siControl, P < 0.001, independent t-test) as well as protein expression (Figure 4B). However, only macrophages showed a 40% reduction in cell viability (Figure 4C), which was characterized by caspase-3 cleavage (Figure 4B). Moreover, the IMPase inhibitor L-690 330 induced selective macrophage death (Figure 1B).
Lithium chloride-induced cell death is not mediated by GSK-3β inhibition
Besides IMPase, GSK-3β is an enzyme well known to be sensitive to lithium (Klein and Melton, 1996) and GSK-3β controls the degradation of β-catenin. Macrophages treated with the GSK-3β inhibitor SB216763 showed accumulation of β-catenin in the cytosol, whereas LiCl-treated macrophages did not (Figure 5A). Transfection of J774A.1 macrophages with specific GSK-3β siRNA decreased GSK-3β mRNA expression (relative expression 20 ± 7% vs. siControl, P= 0.001, independent t-test) as well as protein expression (Figure 5B). Down-regulation of GSK-3β expression in macrophages did not induce a reduction in cell viability (Figure 5C) nor caspase-3 cleavage (Figure 5B). Moreover, cell death was not initiated in macrophages or SMCs after treatment with SB216763 (Figure 1C).
Figure 5.
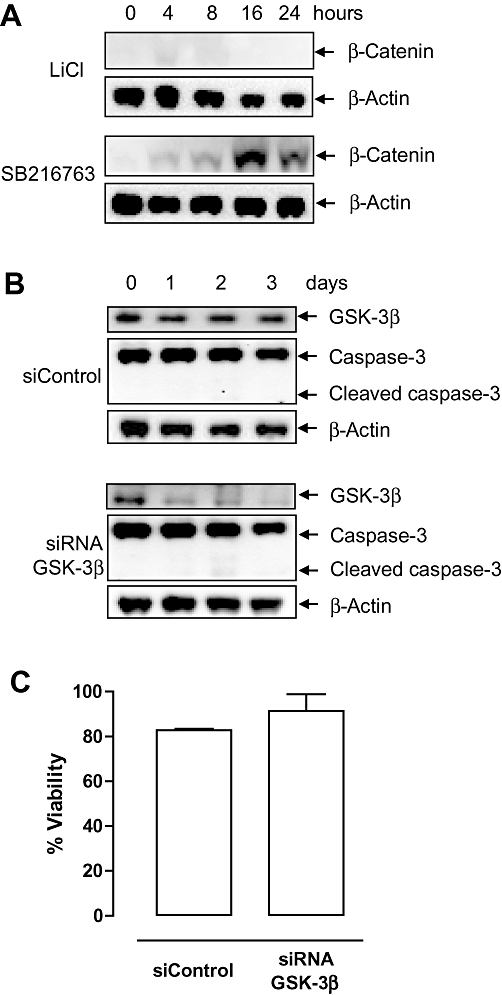
Glycogen synthase kinase-3β (GSK-3β) is not inhibited by LiCl in macrophages. (A) Western blot analysis of β-catenin in J774A.1 macrophages treated with 30 mM LiCl or 10 µM SB216763 for 0–24 h. (B) Western blot analysis of IMPase expression and caspase-3 cleavage in J774A.1 macrophages transfected with siControl or siRNA GSK-3β for 0–3 days. (C) Viability of macrophages transfected with siControl or siRNA specific for GSK-3β at day 3. Bar graphs represent mean ± SEM of three independent experiments (paired-samples t-test).
Lithium chloride decreases macrophage content in rabbit atheroma-like lesions in vitro
To investigate whether LiCl was able to clear macrophages from atherosclerotic plaques, collared carotid artery segments from hypercholesterolemic rabbits were exposed in vitro to 30 mM LiCl for 3 or 7 days. The RAM 11-positive area (macrophages) in the intima was not affected after 3 days (data not shown), but decreased significantly after 7 days of in vitro treatment (Figure 6A and C). In contrast, the α-SMC actin-positive area in the intima and media was not altered by LiCl (Figure 6B and C).
Figure 6.
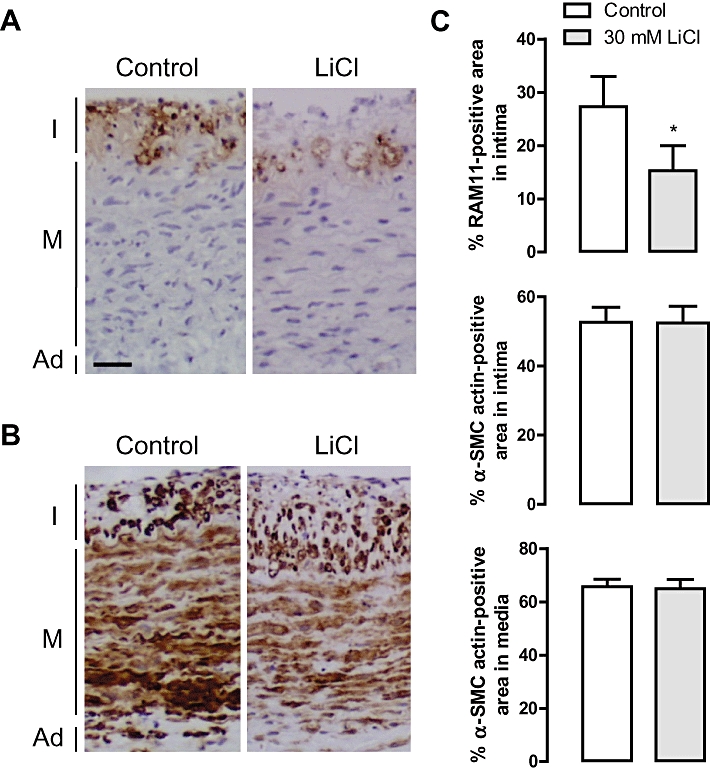
Effects of 7 days of treatment in vitro with 30 mM LiCl on rabbit carotid artery rings with collar-induced atheroma-like lesions. (A) Immunoreactivity of RAM 11 (macrophages, brown). (B) Immunoreactivity of α-smooth muscle cell (SMC) actin (SMCs, brown). I, intima; M, media; Ad, adventitia; scale bar = 50 µm. (C) Percentage RAM 11-positive area in the intima, and percentage α-SMC actin-positive area in the intima or in the media after treatment for 7 days with serum-containing F-10 Ham's medium in the presence or absence of 30 mM LiCl. Results are shown as mean ± SEM. *P < 0.05 versus control (n= 9, Wilcoxon paired samples test).
Lithium chloride induces selective macrophage apoptosis in rabbit atheroma-like lesions in vivo
LiCl was locally administered in vivo by connecting an osmotic minipump to a collar around the rabbit carotid artery. The presence of lithium in the LiCl-treated atheroma-like lesions was confirmed by TOF-S-SIMS analysis (Figure 7A). However, lithium levels in serum samples were below the detection limit (0.10 mM). LiCl decreased the RAM 11-positive area in the atheroma-like lesions (Figure 7B and E) without affecting the α-SMC actin positivity in intima or media (Figure 7C and E). CD31-staining showed that the integrity of the endothelial layer of lithium-treated segments was not different from saline-treated arteries (Figure 7D and E). In addition, CD43-staining showed the presence of few T-lymphocytes in the intimal layer of the atheroma-like lesions but these cells were not affected by LiCl treatment (data not shown). Neutrophils and dendritic cells could not be detected in the atheroma-like lesions. The intima of lithium-treated segments were characterized by TUNEL-positivity (Figure 8B) that co-localized with macrophages (Figure 8A) but not with SMCs (Figure 8C) or T-lymphocytes (data not shown). The media was TUNEL negative (Figure 8A and C). In addition, the number of intimal cells with fragmented nuclei was significantly increased in LiCl-treated segments, whereas nuclear fragmentation was not detected in the media (Figure 9A and B). All these findings indicate that LiCl-induced apoptosis occurred only in macrophages.
Figure 7.
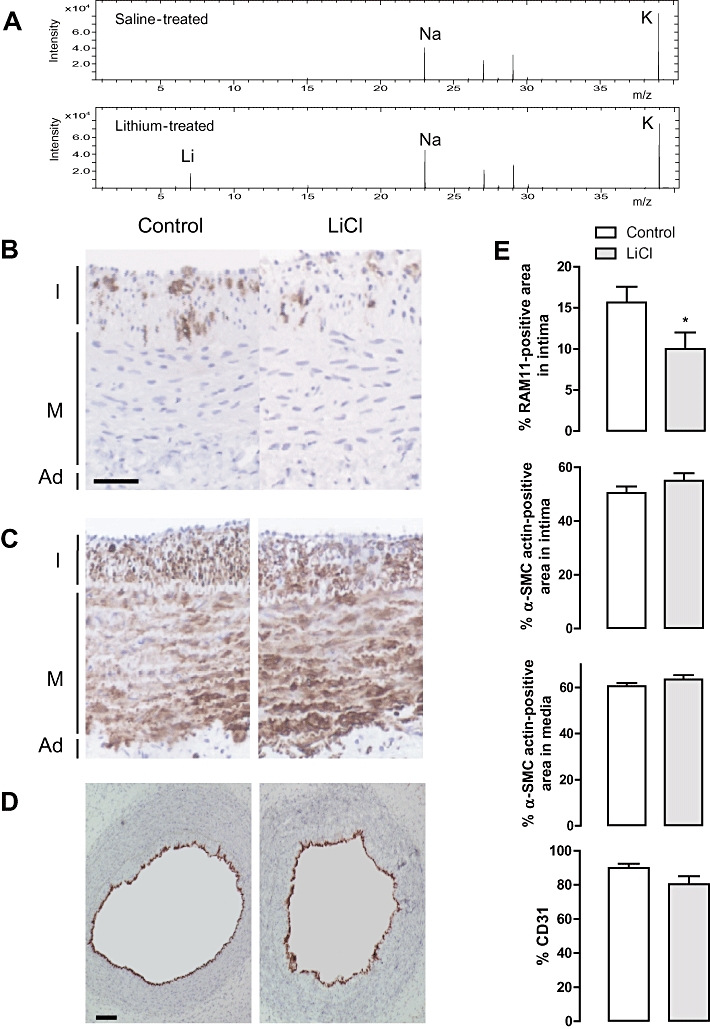
Effects of LiCl locally administered in vivo, via an osmotic minipump that was connected to a collar around atherosclerotic carotid arteries of hypercholesterolemic rabbits. (A) Low mass over charge ratio (m/z) range of positive ion mass spectra recorded with time-of-flight static secondary ion mass spectrometry (TOF-S-SIMS) of saline-treated (upper panel) or LiCl-treated (lower panel) carotid arteries. Only the positive ion mass spectrum of LiCl-treated segments showed a signal at m/z 7 due to the 7Li+ ions (lower panel). Immunoreactivity of RAM 11 (macrophages, brown; scale bar = 50 µm) (B), α-SMC actin (SMCs, brown) (C) and CD31 (endothelial cells, red; scale bar = 100 µm) (D). I, intima; M, media; Ad, adventitia. (E) Percentage RAM 11-positive area in the intima, α-SMC actin-positive area in the intima or in the media and CD31-positivity after treatment for 3 days with saline (control) or 1 M LiCl in the osmotic minipump. Results are shown as mean ± SEM. *P < 0.05 versus control (n= 10, Mann-Whitney U-test).
Figure 8.
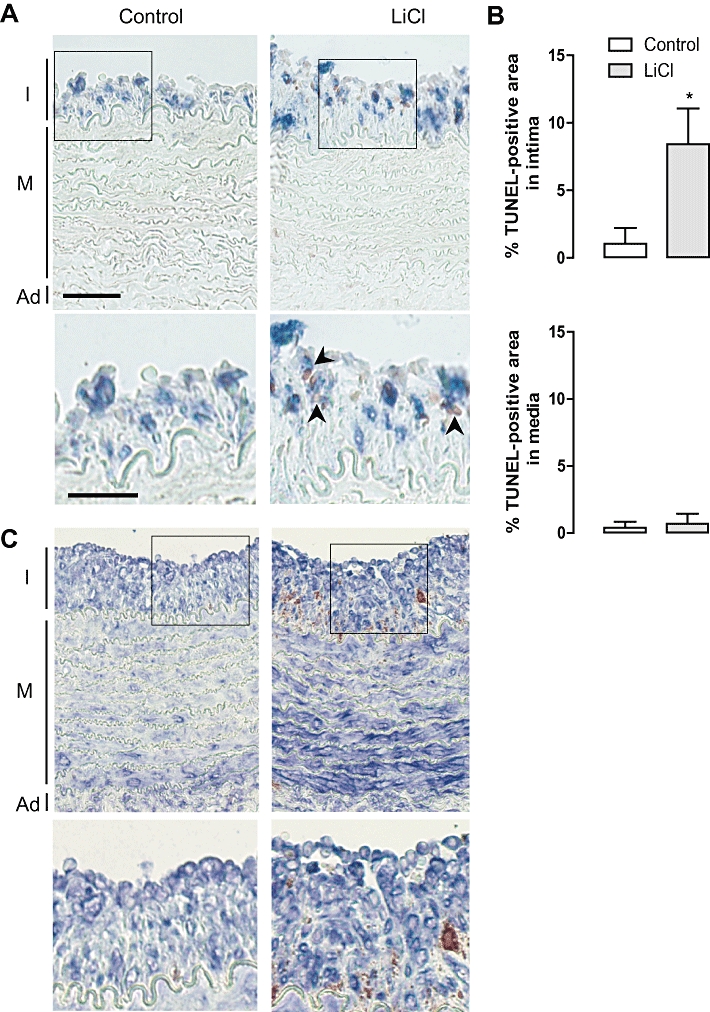
Terminal deoxynucleotidyl transferase dUTP nick-end labelling (TUNEL) labelling as a measure for apoptosis after local in vivo treatment of rabbit atherosclerotic carotid arteries with saline (control) or LiCl for 3 days. (A) Double staining of RAM 11 (macrophages, blue) with TUNEL (DNA fragmentation, red) I, intima; M, media; Ad, adventitia; scale bar = 50 µm. Magnification of boxed areas shows co-localization of RAM 11 with TUNEL (arrowheads), only in the intimal area of LiCl-treated atherosclerotic carotid arteries. Scale bar = 30 µm. (B) Percentage TUNEL-positive area in the intima or in the media. Results are shown as mean± SEM. *P < 0.05 versus control (n= 10, Mann–Whitney U-test). (C) Double staining of α-SMC actin (SMCs, blue) with TUNEL (DNA fragmentation, red) I, intima; M, media; Ad, adventitia. Magnification of boxed areas shows no co-localization of α-SMC actin with TUNEL.
Figure 9.
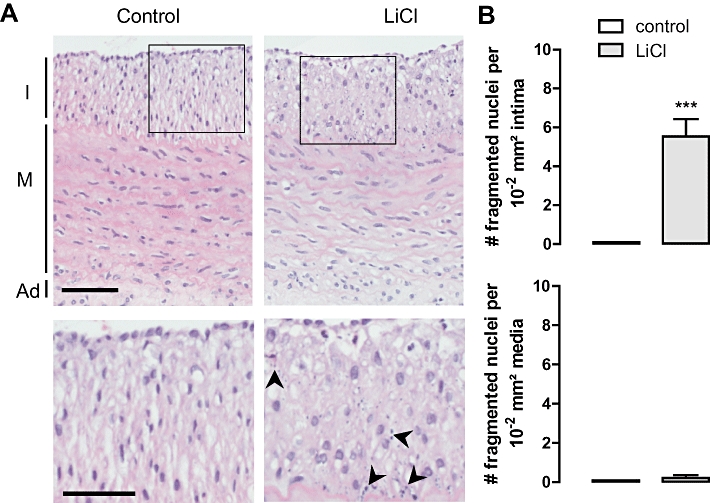
Fragmented nuclei as a measure of apoptosis after local in vivo treatment of rabbit atherosclerotic carotid arteries with saline (control) or LiCl for 3 days. (A) H/E staining I, intima; M, media; Ad, adventitia; scale bar = 50 µm. Magnification of boxed areas shows fragmented nuclei (arrowheads) only in the intimal area of LiCl-treated atherosclerotic carotid arteries. Scale bar = 30 µm. (B) Number of fragmented nuclei per 10−2 mm2 in the intima or in the media. No fragmented nuclei were detected in control, saline-treated atherosclerotic carotid arteries. Results are shown as mean ± SEM. ***P < 0.001 versus control (n= 10, Mann-Whitney U test).
To determine the functionality of the SMCs of the rabbit carotid arteries treated with saline or LiCl, receptor-dependent and -independent contractions were examined. The maximal contraction (Emax) of the segments to depolarizing KCl and to the receptor-dependent agonist 5-HT was unaltered (Table 1). Moreover, LiCl treatment did not affect the sensitivity (pD2) to contractile agonists. Endothelium-dependent and -independent relaxations to ACh and NO donor DEANO, respectively, did not show significant differences between saline- and LiCl-treated segments (Table 1).
Table 1.
Maximal response (Emax) and sensitivity (pD2) to contractile and relaxing agents of rabbit carotid arteries that were locally treated in vivo with saline or LiCl for 3 days
| Emax | pD2 (- log EC50) | |||
|---|---|---|---|---|
| Saline | LiCl | Saline | LiCl | |
| Contraction (mN) | ||||
| 5-HT | 41 ± 4 | 38 ± 4 | 6.96 ± 0.09 | 6.88 ± 0.08 |
| KCl | 32 ± 3 | 27 ± 3 | 1.38 ± 0.03 | 1.44 ± 0.02 |
| Relaxation (%) | ||||
| ACh | 20 ± 5 | 14 ± 3 | 7.05 ± 0.25 | 6.73 ± 0.27 |
| DEANO | 38 ± 2 | 35 ± 2 | 6.49 ± 0.09 | 6.61 ± 0.15 |
Values are shown as mean ± SEM of six saline-treated and seven LiCl-treated carotid arteries. There were no significant differences between arteries treated with LiCl or saline (unpaired Student's t-test).
DEANO, diethylamine NONOate.
Discussion and conclusions
In this study, we report that IMPase inhibition by LiCl reduces the macrophage content of atherosclerotic plaques through induction of apoptosis. Macrophages play a major role in atherosclerotic plaque rupture by weakening the fibrous cap (Kockx and Herman, 2000; Lafont, 2003). These lesional macrophages are capable of degrading the extracellular matrix by producing matrix metalloproteinases and inducing SMC apoptosis. Clinical studies have shown that the macrophage content is increased in unstable plaques (Moreno et al., 1994; Burke et al., 1997; Ward et al. 2000). Current therapies for stabilizing the vulnerable plaque are based on drugs that prevent acute myocardial infarction by reducing the lipid content, reducing plaque inflammation, improving endothelial function, and/or decreasing the risk of thrombosis (Ambrose and D'Agate, 2005). Despite their value in systemic therapy, these drugs are unsuitable for the acute management of rupture-prone plaques (Ambrose and Martinez, 2002; Cannon et al. 2004). Indeed, it has been reported that lipid-lowering in hypercholesterolemic rabbits reduces the number of plaque macrophages (Aikawa et al., 1998; Kockx et al. 1998a), but these beneficial effects were only seen after several months of discontinuation of the cholesterol-rich diet. Therefore, new (local) therapies as coadjutants to systemic therapy are needed.
Removal of macrophages from plaques via drug-induced macrophage death may be a promising alternative strategy to alter plaque composition in order to increase plaque stabilization (Martinet et al., 2007). The main challenges of this approach are to initiate macrophage death in a selective way, without affecting other cell types in the plaque, in particular SMCs and endothelial cells, and local application of the drug to avoid systemic loss of macrophages. Previous experiments have shown that local administration of protein synthesis inhibitors either via osmotic minipumps or drug-eluting stents leads to selective induction of macrophage death in atherosclerotic plaques (Croons et al., 2007; Verheye et al., 2007; Croons et al. 2009). It should be noted, however, that SMCs do not withstand long-term exposure (>8 h in vitro) to protein synthesis inhibitors (Croons et al., 2008). In this study, we demonstrated that LiCl induces apoptosis in cultured macrophages, but not in cultured SMCs, even after 16 days of treatment. Moreover, local in vivo administration of LiCl to collared atherosclerotic carotid arteries via osmotic minipumps reduced the macrophage content without changing the SMC and endothelial cell viability. TUNEL-positive staining and nuclear fragmentation were observed in the intima of LiCl-treated atheroma-like lesions, but not in the media, suggesting that selective macrophage apoptosis was induced. To confirm this finding, we also studied other cell types (neutrophils, dendritic cells and T-lymphocytes) in the atheroma-like lesions. Neutrophils and dendritic cells could not be detected in the intima of the atheroma-like lesions as demonstrated previously (Kockx et al., 1992), whereas CD43-staining showed that T-lymphocytes were scarcely present in the intima. The amount of T-lymphocytes did not change after LiCl treatment. Furthermore, TUNEL-positivity co-localized with macrophages, but not with SMCs and T-lymphocytes, indicating that only macrophages entered apoptosis after LiCl treatment. The functionality of LiCl-treated SMCs and endothelial cells remained unchanged. The contractile responses to KCl and 5-HT as well as endothelium-dependent and -independent relaxations were similar in saline- and LiCl-treated artery segments. In addition, no morphological or functional changes could be detected in SMCs of carotid artery rings distant from the local infusion site, most likely because lithium levels in serum samples were below the detection limit of 0.10 mM. These data suggest that macrophage reduction succeeded after local administration of LiCl, without triggering systemic effects.
Lithium inhibits several enzymes, including IMPase, inositol polyphosphatase, fructose 1,6-biphosphatase and bisphosphate nucleotidase. IMPase is the most direct biochemical lithium target at therapeutic concentrations (Berridge et al., 1989). This enzyme is involved in synthesis and recycling of inositol by catalysing the rate-limiting conversion of IP3 into inositol. Recycling of inositol is necessary to maintain PI-mediated cell signalling if the cells do not have an alternative source, for example, in absence of extracellular inositol. Depletion of inositol leads to a reduction of PIP2, the necessary precursor for the generation of DAG and IP3. In the present study, both IMPase gene silencing and treatment with the specific IMPase inhibitor L-690 330 induced apoptosis in macrophages, but not in SMCs. Moreover, LiCl led to decreased IP3 levels in macrophages, confirming the inositol depletion hypothesis (Berridge et al., 1989). These findings strongly indicate that LiCl-induced macrophage cell death was mediated by IMPase inhibition.
Although IMPase mRNA expression was similar in macrophages and SMCs, SMCs did not show a reduction in IP3 levels after LiCl treatment. SMCs might have a lower lithium uptake as compared with macrophages. However, we showed by TOF-S-SIMS that the lithium content was the same in both cell types. Another possibility is that SMCs can take up inositol from the extracellular environment. Because lithium also inhibits the transport of inositol into cells (Lubrich & van Calker, 1999), this possibility is unlikely. Furthermore, IMPase in SMCs might be insensitive to lithium. IMPA1 is inhibited by therapeutic concentrations of lithium (∼1 mM) and IMPA2, which has a lower IMPase activity, is inhibited at higher lithium concentrations (Ohnishi et al., 2007). IMPA1 and IMPA2 have a different expression pattern in tissues (Ohnishi et al., 2007) therefore it is conceivable that IMPA2 is more abundantly expressed in SMCs as compared with macrophages. However, our data showed that SMCs do not express IMPA2. Taken together, we do not have a clear-cut explanation why SMCs did not show a reduction in IP3 levels after LiCl treatment.
Besides IMPase inhibition, LiCl can also inhibit GSK-3β (Klein and Melton, 1996). This serine/threonine kinase has been associated with apoptosis of neurons (Pap and Cooper, 1998) and is inhibited by the Wnt pathway. GSK-3β phosphorylates β-catenin, which is degraded to prevent induction of gene expression (Wada, 2009). Activation of the Wnt pathway increases β-catenin levels, a response induced by inhibition of GSK-3β. Our data indicated that LiCl-induced macrophage apoptosis was not mediated by GSK-3β inhibition because β-catenin did not accumulate in the cytosol. Moreover, GSK-3β down-regulation by siRNA and a GSK-3β inhibitor did not induce cell death in macrophages.
In conclusion, LiCl selectively decreased the macrophage load in rabbit atherosclerotic plaques through apoptosis via inhibition of IMPase, without affecting the viability or functionality of SMCs and endothelial cells. Despite the fact that lithium has a narrow therapeutic range – limiting its clinical applications – our data provide evidence for the use of IMPase inhibitors as potential candidates to stabilize atherosclerotic plaques. Given that statin therapy reduces mortality in cardiovascular disease, but only after several weeks of treatment, additional local administration of an IMPase inhibitor applied via drug-eluting stents might help to reduce the cardiovascular risk and prevent plaque destabilization.
Acknowledgments
We are indebted to Rita Van Den Bossche, Anne-Elise Van Hoydonck, Peter Soete and Geert Frederix for excellent technical assistance. This work was supported by the Institute for the Promotion of Innovation through Science and Technology in Flanders (IWT-Vlaanderen), the Fund for Scientific Research (FWO)-Flanders (project G.0112.08) and the University of Antwerp (TOP-BOF). W. Martinet is a postdoctoral fellow of the FWO-Flanders.
Glossary
Abbreviations
- DAG
diacylglycerol
- Emax
maximal response
- GSK-3β
glycogen synthase kinase-3β
- IMPase
inositol monophosphatase
- IP3
inositol-1,4,5-trisphosphate
- pD2
negative logarithm of the molar concentration producing half of the maximal response
- PIP2
phosphatidylinositol diphosphate
- RT-PCR
reverse transcriptase-polymerase chain reaction
- SMC
smooth muscle cell
- TOF-S-SIMS
time-of-flight static secondary ion mass spectrometry
- TUNEL
terminal deoxynucleotidyl transferase dUTP nick-end labelling
- Wnt pathway
wingless-int pathway
Conflicts of interest
None.
Supplementary material
Supporting Information: Teaching Materials; Figs 1–9 as PowerPoint slide.
References
- Aikawa M, Rabkin E, Okada Y, Voglic SJ, Clinton SK, Brinckerhoff CE, et al. Lipid lowering by diet reduces matrix metalloproteinase activity and increases collagen content of rabbit atheroma: a potential mechanism of lesion stabilization. Circulation. 1998;97:2433–2444. doi: 10.1161/01.cir.97.24.2433. [DOI] [PubMed] [Google Scholar]
- Allison JH, Blisner ME, Holland WH, Hipps PP, Sherman WR. Increased brain myo-inositol 1-phosphate in lithium-treated rats. Biochem Biophys Res Commun. 1976;71:664–670. doi: 10.1016/0006-291x(76)90839-1. [DOI] [PubMed] [Google Scholar]
- Ambrose JA, D'Agate DJ. Classification of systemic therapies for potential stabilization of the vulnerable plaque to prevent acute myocardial infarction. Am J Cardiol. 2005;95:379–382. doi: 10.1016/j.amjcard.2004.09.037. [DOI] [PubMed] [Google Scholar]
- Ambrose JA, Martinez EE. A new paradigm for plaque stabilization. Circulation. 2002;105:2000–2004. doi: 10.1161/01.cir.0000012528.89469.8e. [DOI] [PubMed] [Google Scholar]
- Back M, Ketelhuth DF, Agewall S. Matrix metalloproteinases in atherothrombosis. Prog Cardiovasc Dis. 2010;52:410–428. doi: 10.1016/j.pcad.2009.12.002. [DOI] [PubMed] [Google Scholar]
- Berridge MJ, Downes CP, Hanley MR. Neural and developmental actions of lithium: a unifying hypothesis. Cell. 1989;59:411–419. doi: 10.1016/0092-8674(89)90026-3. [DOI] [PubMed] [Google Scholar]
- Boyle JJ, Weissberg PL, Bennett MR. Human macrophage-induced vascular smooth muscle cell apoptosis requires NO enhancement of Fas/Fas-L interactions. Arterioscler Thromb Vasc Biol. 2002;22:1624–1630. doi: 10.1161/01.atv.0000033517.48444.1a. [DOI] [PubMed] [Google Scholar]
- Burke AP, Farb A, Malcom GT, Liang YH, Smialek J, Virmani R. Coronary risk factors and plaque morphology in men with coronary disease who died suddenly. N Engl J Med. 1997;336:1276–1282. doi: 10.1056/NEJM199705013361802. [DOI] [PubMed] [Google Scholar]
- Cannon CP, Braunwald E, McCabe CH, Rader DJ, Rouleau JL, Belder R, et al. Intensive versus moderate lipid lowering with statins after acute coronary syndromes. N Engl J Med. 2004;350:1495–1504. doi: 10.1056/NEJMoa040583. [DOI] [PubMed] [Google Scholar]
- Chen RH, Ding WV, McCormick F. Wnt signaling to beta-catenin involves two interactive components. Glycogen synthase kinase-3beta inhibition and activation of protein kinase C. J Biol Chem. 2000;275:17894–17899. doi: 10.1074/jbc.M905336199. [DOI] [PubMed] [Google Scholar]
- Cook D, Fry MJ, Hughes K, Sumathipala R, Woodgett JR, Dale TC. Wingless inactivates glycogen synthase kinase-3 via an intracellular signalling pathway which involves a protein kinase C. EMBO J. 1996;15:4526–4536. [PMC free article] [PubMed] [Google Scholar]
- Croons V, Martinet W, Herman AG, Timmermans JP, De Meyer GRY. Selective clearance of macrophages in atherosclerotic plaques by the protein synthesis inhibitor cycloheximide. J Pharmacol Exp Ther. 2007;320:986–993. doi: 10.1124/jpet.106.113944. [DOI] [PubMed] [Google Scholar]
- Croons V, Martinet W, Herman AG, De Meyer GRY. Differential effect of the protein synthesis inhibitors puromycin and cycloheximide on vascular smooth muscle cell viability. J Pharmacol Exp Ther. 2008;325:824–832. doi: 10.1124/jpet.107.132944. [DOI] [PubMed] [Google Scholar]
- Croons V, Martinet W, Herman AG, Timmermans JP, De Meyer GRY. The protein synthesis inhibitor anisomycin induces macrophage apoptosis in rabbit atherosclerotic plaques through p38 mitogen-activated protein kinase. J Pharmacol Exp Ther. 2009;329:856–864. doi: 10.1124/jpet.108.149948. [DOI] [PubMed] [Google Scholar]
- De Meyer GRY, Bult H, Ustunes L, Kockx M, Jordaens FH, Zonnekeyn LL, et al. Vasoconstrictor responses after neo-intima formation and endothelial removal in the rabbit carotid artery. Br J Pharmacol. 1994;112:471–476. doi: 10.1111/j.1476-5381.1994.tb13097.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Meyer GRY, Van Put DJ, Kockx MM, Van Schil P, Bosmans R, Bult H, et al. Possible mechanisms of collar-induced intimal thickening. Arterioscler Thromb Vasc Biol. 1997;17:1924–1930. doi: 10.1161/01.atv.17.10.1924. [DOI] [PubMed] [Google Scholar]
- De Meyer GRY, Kockx MM, Cromheeke KM, Seye CI, Herman AG, Bult H. Periadventitial inducible nitric oxide synthase expression and intimal thickening. Arterioscler Thromb Vasc Biol. 2000;20:1896–1902. doi: 10.1161/01.atv.20.8.1896. [DOI] [PubMed] [Google Scholar]
- De Mondt R, Van Vaeck L, Heile A, Arlinghaus HF, Vangaever F, Lenaerts J. TOF-S-SIMS molecular depth profiling of organic bilayers using mechanical wear test methodology. Anal Bioanal Chem. 2009;393:1917–1921. doi: 10.1007/s00216-009-2657-4. [DOI] [PubMed] [Google Scholar]
- Klein PS, Melton DA. A molecular mechanism for the effect of lithium on development. Proc Natl Acad Sci U S A. 1996;93:8455–8459. doi: 10.1073/pnas.93.16.8455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kockx MM, Herman AG. Apoptosis in atherosclerosis: beneficial or detrimental? Cardiovasc Res. 2000;45:736–746. doi: 10.1016/s0008-6363(99)00235-7. [DOI] [PubMed] [Google Scholar]
- Kockx MM, De Meyer GRY, Jacob WA, Bult H, Herman AG. Triphasic sequence of neointimal formation in the cuffed carotid artery of the rabbit. Arterioscler Thromb. 1992;12:1447–1457. doi: 10.1161/01.atv.12.12.1447. [DOI] [PubMed] [Google Scholar]
- Kockx MM, De Meyer GRY, Buyssens N, Knaapen MWM, Bult H, Herman AG. Cell composition, replication, and apoptosis in atherosclerotic plaques after 6 months of cholesterol withdrawal. Circ Res. 1998a;83:378–387. doi: 10.1161/01.res.83.4.378. [DOI] [PubMed] [Google Scholar]
- Kockx MM, Muhring J, Knaapen MW, De Meyer GRY. RNA synthesis and splicing interferes with DNA in situ end labeling techniques used to detect apoptosis. Am J Pathol. 1998b;152:885–888. [PMC free article] [PubMed] [Google Scholar]
- Lafont A. Basic aspects of plaque vulnerability. Heart. 2003;89:1262–1267. doi: 10.1136/heart.89.10.1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lecoeur H, Ledru E, Prevost MC, Gougeon ML. Strategies for phenotyping apoptotic peripheral human lymphocytes comparing ISNT, annexin-V and 7-AAD cytofluorometric staining methods. J Immunol Methods. 1997;209:111–123. doi: 10.1016/s0022-1759(97)00138-5. [DOI] [PubMed] [Google Scholar]
- Lin CF, Chen CL, Chiang CW, Jan MS, Huang WC, Lin YS. GSK-3beta acts downstream of PP2A and the PI 3-kinase-Akt pathway, and upstream of caspase-2 in ceramide-induced mitochondrial apoptosis. J Cell Sci. 2007;120:2935–2943. doi: 10.1242/jcs.03473. [DOI] [PubMed] [Google Scholar]
- Lowik CW, Alblas MJ, van de Ruit M, Papapoulos SE, van der Pluijm G. Quantification of adherent and nonadherent cells cultured in 96-well plates using the supravital stain neutral red. Anal Biochem. 1993;213:426–433. doi: 10.1006/abio.1993.1442. [DOI] [PubMed] [Google Scholar]
- Lubrich B, van Calker D. Inhibition of the high affinity myo-inositol transport system: a common mechanism of action of antibipolar drugs? Neuropsychopharmacology. 1999;21:519–529. doi: 10.1016/S0893-133X(99)00037-8. [DOI] [PubMed] [Google Scholar]
- Martinet W, Verheye S, De Meyer GRY. Selective depletion of macrophages in atherosclerotic plaques via macrophage-specific initiation of cell death. Trends Cardiovasc Med. 2007;17:69–75. doi: 10.1016/j.tcm.2006.12.004. [DOI] [PubMed] [Google Scholar]
- Mikoshiba K, Furuichi T, Miyawaki A, Yoshikawa S, Nakade S, Michikawa T, et al. Structure and function of inositol 1,4,5-trisphosphate receptor. Ann N Y Acad Sci. 1993;707:178–197. doi: 10.1111/j.1749-6632.1993.tb38052.x. [DOI] [PubMed] [Google Scholar]
- Moreno PR, Falk E, Palacios IF, Newell JB, Fuster V, Fallon JT. Macrophage infiltration in acute coronary syndromes. Implications for plaque rupture. Circulation. 1994;90:775–778. doi: 10.1161/01.cir.90.2.775. [DOI] [PubMed] [Google Scholar]
- Newby AC, George SJ, Ismail Y, Johnson JL, Sala-Newby GB, Thomas AC. Vulnerable atherosclerotic plaque metalloproteinases and foam cell phenotypes. Thromb Haemost. 2009;101:1006–1011. [PMC free article] [PubMed] [Google Scholar]
- Ohnishi T, Ohba H, Seo KC, Im J, Sato Y, Iwayama Y, et al. Spatial expression patterns and biochemical properties distinguish a second myo-inositol monophosphatase IMPA2 from IMPA1. J Biol Chem. 2007;282:637–646. doi: 10.1074/jbc.M604474200. [DOI] [PubMed] [Google Scholar]
- Pap M, Cooper GM. Role of glycogen synthase kinase-3 in the phosphatidylinositol 3-Kinase/Akt cell survival pathway. J Biol Chem. 1998;273:19929–19932. doi: 10.1074/jbc.273.32.19929. [DOI] [PubMed] [Google Scholar]
- Phiel CJ, Klein PS. Molecular targets of lithium action. Annu Rev Pharmacol Toxicol. 2001;41:789–813. doi: 10.1146/annurev.pharmtox.41.1.789. [DOI] [PubMed] [Google Scholar]
- Ray JL, Leach R, Herbert JM, Benson M. Isolation of vascular smooth muscle cells from a single murine aorta. Methods Cell Sci. 2001;23:185–188. doi: 10.1023/a:1016357510143. [DOI] [PubMed] [Google Scholar]
- Sarkar S, Floto RA, Berger Z, Imarisio S, Cordenier A, Pasco M, et al. Lithium induces autophagy by inhibiting inositol monophosphatase. J Cell Biol. 2005;170:1101–1111. doi: 10.1083/jcb.200504035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrijvers DM, Martinet W, De Meyer GRY, Andries L, Herman AG, Kockx MM. Flow cytometric evaluation of a model for phagocytosis of cells undergoing apoptosis. J Immunol Methods. 2004;287:101–108. doi: 10.1016/j.jim.2004.01.013. [DOI] [PubMed] [Google Scholar]
- Shears SB, Yang L, Qian X. Cell signaling by a physiologically reversible inositol phosphate kinase/phosphatase. Adv Enzyme Regul. 2004;44:265–277. doi: 10.1016/j.advenzreg.2004.02.002. [DOI] [PubMed] [Google Scholar]
- Stambolic V, Ruel L, Woodgett JR. Lithium inhibits glycogen synthase kinase-3 activity and mimics wingless signalling in intact cells. Curr Biol. 1996;6:1664–1668. doi: 10.1016/s0960-9822(02)70790-2. [DOI] [PubMed] [Google Scholar]
- Verheye S, Martinet W, Kockx MM, Knaapen MWM, Salu K, Timmermans JP, et al. Selective clearance of macrophages in atherosclerotic plaques by autophagy. J Am Coll Cardiol. 2007;49:706–715. doi: 10.1016/j.jacc.2006.09.047. [DOI] [PubMed] [Google Scholar]
- Vindelov LL, Christensen IJ. Detergent and proteolytic enzyme-based techniques for nuclear isolation and DNA content analysis. Methods Cell Biol. 1994;41:219–229. doi: 10.1016/s0091-679x(08)61720-3. [DOI] [PubMed] [Google Scholar]
- Virmani R, Burke AP, Farb A, Kolodgie FD. Pathology of the unstable plaque. Prog Cardiovasc Dis. 2002;44:349–356. doi: 10.1053/pcad.2002.122475. [DOI] [PubMed] [Google Scholar]
- Wada A. Lithium and neuropsychiatric therapeutics: neuroplasticity via glycogen synthase kinase-3beta, beta-catenin, and neurotrophin cascades. J Pharmacol Sci. 2009;110:14–28. doi: 10.1254/jphs.09r02cr. [DOI] [PubMed] [Google Scholar]
- van der Wal AC, Becker AE, van der Loos CM, Das PK. Site of intimal rupture or erosion of thrombosed coronary atherosclerotic plaques is characterized by an inflammatory process irrespective of the dominant plaque morphology. Circulation. 1994;89:36–44. doi: 10.1161/01.cir.89.1.36. [DOI] [PubMed] [Google Scholar]
- Ward MR, Pasterkamp G, Yeung AC, Borst C. Arterial remodeling. Mechanisms and clinical implications. Circulation. 2000;102:1186–1191. doi: 10.1161/01.cir.102.10.1186. [DOI] [PubMed] [Google Scholar]
- Zhang M, Jin W, Zhou X, Yu J, Lee AJ, Sun SC. Deregulation of Tpl2 and NF-kappaB signaling and induction of macrophage apoptosis by the anti-depressant drug lithium. Cell Signal. 2009;21:559–566. doi: 10.1016/j.cellsig.2008.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.