

Abstract
The stalk proteins P1 and P2, which are fundamental for ribosome activity, are the only ribosomal components for which there is a cytoplasmic pool. Accumulation of these two proteins is differentially regulated in Saccharomyces cerevisiae by degradation. In the absence of P2, the amount of P1 is drastically reduced; in contrast, P2 proteins are not affected by a deficiency in P1. However, association with P2 protects P1 proteins. The half-life of P1 is a few minutes, while that of P2 is several hours. The proteasome is not involved in the degradation of P1 proteins. The different sensitivity to degradation of these two proteins is associated with two structural features: phosphorylation and N-terminus structure. A phosphorylation site at the C-terminus is required for P1 proteolysis. P2 proteins, despite being phosphorylated, are protected by their N-terminal peptide. An exchange of the first five amino acids between the two types of protein makes P1 resistant and P2 sensitive to degradation.
Keywords: degradation signals/N-terminus/phosphorylation/ribosomal proteins/yeast
Introduction
The expression of all ribosomal proteins is tightly controlled in yeast, as well as in all organisms studied so far, in such a way that they are not found free in the cell cytoplasm (for reviews see Raué and Planta, 1991; Woolford and Warner, 1991). An excess of free ribosomal proteins seems to be damaging to the cell, probably because of the tendency of these proteins to bind RNA, interfering with the translation machinery. All ribosomal components are simultaneously down-regulated when the assembly of the particles is blocked (Gorenstein and Warner, 1976, 1977). In yeast, regulation at the level of transcription seems to be the most common mechanism, but feedback control of intron splicing and mRNA degradation have been reported in some instances (Presutti et al., 1991; Vilardell and Warner, 1997; Fewell and Woolford, 1999). Any ribosomal protein that eludes these controls is eliminated. Thus, it has been shown that an increase in gene dosage of some ribosomal proteins does not result in a proportional increase in the corresponding proteins in the cell, since the overproduced polypeptides are quickly degraded (El Baradi et al., 1986; Tsay et al., 1988). Nothing is known about the mechanisms of degradation responsible for the removal of free ribosomal proteins except that vacuolar proteases seem not to be involved in the process (Tsay et al., 1988). Similarly, nothing is known about whether degradation signals are present in ribosomal proteins.
The only ribosomal components that are found free in the cytoplasm are the ribosomal stalk acidic proteins. A cytoplasmic pool of these proteins has been reported in yeast (Zinker, 1980; Sanchez-Madrid et al., 1981; Mitsui et al., 1988), other eukaryotes (van Agthoven et al., 1978; Tsurugi and Ogata, 1985; Elkon et al., 1986) and bacteria (Ramagopal, 1976). The reported size of this pool varies from <1% (Mitsui et al., 1988) up to 75% (van Agthoven et al., 1978; Zinker, 1980) of the acidic stalk proteins in the cell. These discrepancies probably result from differences between organisms and in metabolic conditions of the cell (Saenz-Robles et al., 1990).
The ribosomal stalk proteins L7 and L12 were initially characterized in bacteria (Möller et al., 1972), and correspond to N-terminally blocked and unblocked forms of the same polypeptide. Two proteins with similar characteristics were later reported in Artemia salina (Möller et al., 1975); they were called phosphoproteins 1 and 2 (P1 and P2) because they were phosphorylated in the ribosome (Tsurugi et al., 1978). In some organisms, such as yeast and protozoa, several subtypes of P1 and P2 have been reported. Thus, two proteins of each type, P1α/P1β and P2α/P2β, are found in Saccharomyces cerevisiae. A third type of stalk protein, P3, has recently been found in plants (Szick et al., 1998).
The acidic stalk proteins, the only components that are present in multiple copies in the ribosome, are involved in the interaction and function of the translation factors during initiation (Heimark et al., 1976; Schwartz et al., 1983), elongation (Möller and Maassen, 1986) and termination (Tate et al., 1990) in bacteria. Less is known about their functions in eukaryotes, although they seem to be similar to those in bacteria (Sanchez-Madrid et al., 1979; MacConnell and Kaplan, 1982; Uchiumi et al., 1999).
The significance of the acidic protein pool in prokaryotes is unclear, but in eukaryotes the free proteins have been shown to participate in an exchange process with the ribosomes in yeast (Zinker and Warner, 1976), plants (Scharf and Nover, 1987) and mammals (Tsurugi and Ogata, 1985). This exchange seems to be connected with a ribosome-modulating mechanism involving the ribosomal stalk (Ballesta and Remacha, 1996). There is experimental evidence that the conformation of the yeast stalk, which is determined by its acidic protein content (Remacha et al., 1995) and/or the phosphorylation state of its components (Zambrano et al., 1997; Rodriguez-Gabriel et al., 1998), can affect the expression of specific proteins and the manifestation of particular phenotypes.
The existence of a cytoplasmic pool and the presence of multiple copies of the acidic proteins in each ribosome indicate that expression of these ribosomal components is probably controlled by a mechanism that must be, at least in part, different from that controlling other ribosomal proteins.
Since the size of the cytoplasmic pool of stalk proteins will have a direct effect on the exchange process, it is important to understand the mechanisms that allow this pool to form and control its size. These mechanisms are at present totally unknown.
Results
Expression of the 12 kDa acidic proteins in the double-disrupted S.cerevisiae strains D45 and D67
Ribosomes in S.cerevisiae strains D45 and D67 completely lack the P2 or P1 type of acidic proteins, respectively, because of gene disruption (Table I; Remacha et al., 1992). Western blotting of cell extracts showed that the amount of P2-type proteins in strain D67 was similar to that in the control, W303-1b, while P1 proteins were practically absent from strain D45 (Figure 1). Similar results were obtained when proteins were assayed by ELISA (data not shown). Thus, while the amount of P2 proteins in the cell is not affected by the absence of P1 proteins, expression of the latter proteins seems to be dependent on simultaneous expression of P2 proteins. These results suggest that some regulatory mechanism controls the accumulation of P1 proteins in the cytoplasm. Given that both P1 proteins behave similarly, P1β was chosen to study the mechanism of regulation in detail.
Table I. Saccharomyces cerevisiae strains used in this study.
| Strain | Genotype | Stalk proteins in cells |
Reference | |||
|---|---|---|---|---|---|---|
| P1α | P1β | P2α | P2β | |||
| W303-1b | MATα his3-11,15 leu2-3,112 ura3-1 trp1-1 ade2-1 can1-10 | + | + | + | + | Thomas and Rothstein (1989) |
| D67 | MATα his3-11,15 leu2-3,112 ura3-1 trp1-1 ade2-1 can1-10 RPP1A::LEU2 RPP1B::TRP1 | – | – | + | + | Remacha et al. (1992) |
| D45 | MATα his3-11,15 leu2-3,112 ura3-1 trp1-1 ade2-1 can1-10 RPP2A::URA3 RPP2B::HIS3 | + | + | – | – | Remacha et al. (1992) |
| D56 | MATα his3-11,15 leu2-3,112 ura3-1 trp1-1 ade2-1 can1-10 RPP2B::HIS3 RPP1B::TRP1 | + | – | + | – | Remacha et al. (1992) |
| D456 | MATα his3-11,15 leu2-3,112 ura3-1 trp1-1 RPP2A::URA3 RPP2B::HIS3 RPP1B::TRP1 | + | – | – | – | Remacha et al. (1995) |
| D567 | MATα his3-11,15 leu2-3,112 ura3-1 trp1-1 ade2-1 can1-10 RPP1A::LEU2 RPP1B::TRP1 RPP2B::HIS3 | – | – | + | – | Remacha et al. (1995) |
| NAT1 | MATα his3-11,15 leu2-3,112 ura3-1 trp1-1 ade2-1 can1-10 NAT1::LEU2 | + | + | + | + | Takakura et al. (1992) |
| FY1679 | MATa ura3-1 his3-11 leu2-3 | + | + | + | + | Thierry et al. (1990) |
| FS 11-1a | MATa leu2-3 rpn6 pFl38/RPN6 GAL1pr | + | + | + | + | González-Santamaría (unpublished data) |
| WCG4a | MATa his3-11 leu2-3 ura3-1 | + | + | + | + | Heinemeyer et al. (1993) |
| WCG4a-11/21a | MATa his3-11 leu2-3 ura3-1 pre1-1 pre2-1 | + | + | + | + | Heinemeyer et al. (1993) |
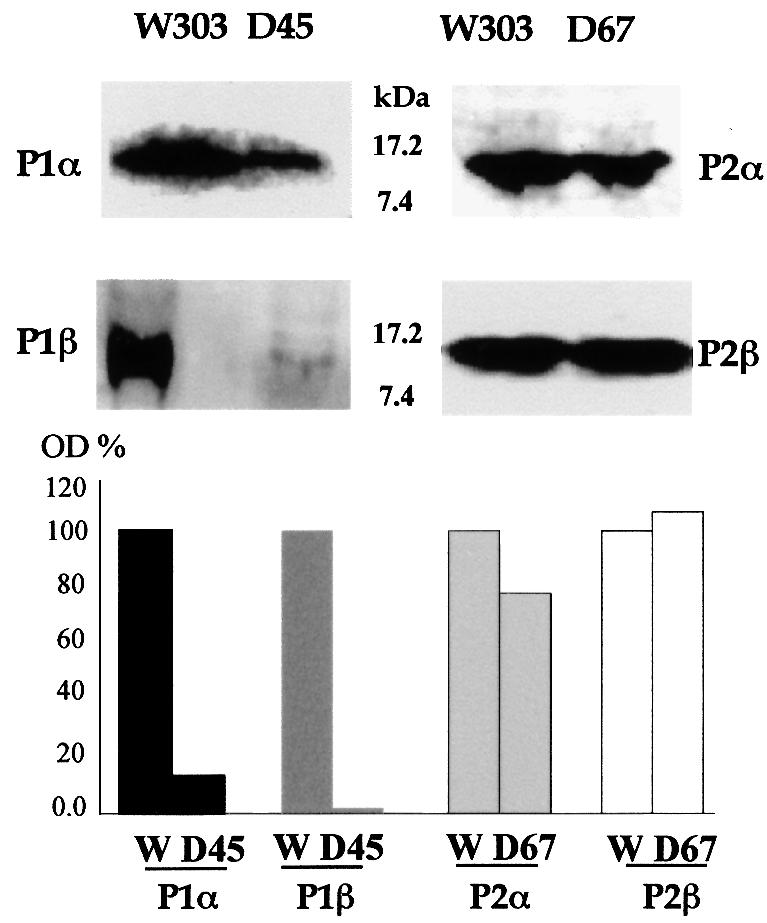
Fig. 1. Immunoblot analysis of P1/P2 levels in S.cerevisiae W303-1b, D45 and D67 strains. Total extracts (100 µg) from cells grown to mid-logarithmic phase in rich medium were separated by 15% SDS–PAGE followed by immunoblot analysis using monoclonal antibodies to P1β (1CE1), P2α (1BE3) and P2β (1AA9) and a rabbit polyclonal antibody to P1α. Densitometric estimation of the intensity of the bands was performed; results are shown as a percentage of the S.cerevisiae W303-1b band intensity.
Regulation does not take place at the level of mRNA accumulation or splicing
The amount of P1β mRNA was detected by northern blotting on total RNA from the D45 and W303-1b strains using a specific probe for the RPP1B gene, which encodes protein P1β, and the density of bands was measured. Actin mRNA was used as an internal control. As shown in Figure 2, the amount of RPP1B mRNA is the same in both strains, ruling out regulation of P1 protein expression at the level of mRNA accumulation.

Fig. 2. Estimation of P1β mRNA in S.cerevisiae W303-1b and D45 by a northern assay. Total RNA was resolved by electrophoresis, blotted onto a membrane and detected by hybridization using a 32P-labelled 1.3 kb PstI–HindIII DNA fragment containing the RPP1B coding sequence derived from pMRH46 (Remacha et al., 1988). A 1.0 kb 32P-labelled AvaI–HindIII DNA fragment from pYactII containing actin gene ACT1 was used as a standard. The results of the densitometric estimation are plotted as an RPP1B/ACT1 ratio.
Since the RPP1B gene has an intron, processing of the pre-mRNA could not be excluded as a potential regulatory mechanism. However, accumulation of RPP1B pre-mRNA was not observed by northern blotting and removal of the intron did not affect expression of P1β (results not shown), indicating that splicing is not involved in the regulatory process.
Identification of the mRNA region involved in P1/P2 regulation
The results described above indicate that accumulation of P1β is controlled post-transcriptionally. To identify the part of the mRNA that is responsible for the control, chimeric genes were constructed by swapping the promoter plus 5′-untranslated region (UTR) and/or 3′-UTR regions of the RPP1B and RPP2B genes, which encode the P1β and P2β proteins, respectively, and the resulting genes were cloned in the centromeric plasmids pFL36 and pFL38. Constructs, including the open reading frame (ORF) encoding P1β, were used to transform S.cerevisiae D456, which lacks the genes encoding proteins P1β, P2α and P2β, while constructs including the ORF encoding the P2β protein were introduced into the D567 strain, in which genes encoding proteins P1α, P1β and P2α were disrupted (Table I), yielding the strains indicated in Table II. The P1β and P2β proteins present in these strains were quantified either by inhibition ELISA (Table II) or by western blotting (data not shown); the two methods gave similar results. Neither the 5′-UTR nor the 3′-UTR from RPP1B affected the P2β accumulation in D567/122 and D567/121, which was as high as in the control D567/222 strain. Similarly, replacement of RPP1B 5′-flanking sequences by those from RPP2B did not increase the amount of P1β protein in D456/211, the level being similar to that of D456/111. These data indicate that the amount of acidic proteins in the cells extracts is not affected by the flanking regions and is exclusively determined by the gene coding sequence.
Table II. Expression of proteins from gene chimerasa.
| Strain | Promoter | 5′-UTR | ORF | 3′-UTR | Expressed protein (ng/mg S100) |
|
|---|---|---|---|---|---|---|
| P1β | P2β | |||||
| D456/111 | P1β | P1β | P1β | P1β | 13 | 0 |
| D567/222 | P2β | P2β | P2β | P2β | 0 | 333 |
| D456/211 | P2β | P2β | P1β | P1β | 9 | 0 |
| D567/122 | P1β | P1β | P2β | P2β | 0 | 345 |
| D567/121 | P1β | P1β | P2β | P1β | 0 | 327 |
aThe indicated strains, transformed with plasmids containing the described constructs, were grown to exponential phase. Extracts deprived of ribosomes (S100 fraction) were prepared and the amount of acidic proteins was estimated by inhibition ELISA using monoclonal antibodies 1CE1 (specific to P1β) and 1AA9 (specific to P2β).
There are two mechanisms that could explain a difference in accumulation of proteins in the cell, considering only the coding sequence of a gene: either a difference in the translation rate or a difference in the protein’s half-life. The high sequence homology and the same CAI (codon adaptation index) value (0.7) for both proteins makes the first possibility unlikely, so the second alternative was explored in more detail.
Estimation of the half-lives of P1β and P2β
The half-lives of proteins P1β and P2β were estimated by pulse–chase experiments. Saccharomyces cerevisiae D45/RPP1B, which overexpresses P1β protein, was obtained by transforming S.cerevisiae D45 with a multicopy plasmid carrying the RPP1B gene (YEp13/RPP1B); it was used to favour the detection of the protein. D45/RPP1B and D67 cells were pulse-labelled with [35S]Met–Cys as indicated in Materials and methods. Protein P1β was detected at time zero, but the amount was halved <15 min after the chase and there was practically no P1β after 2 h (Figure 3). For P2β, although a similar decay was observed in the first minutes, 5 h after the chase >50% of the original amount of P2β was still present in S.cerevisiae D67. These results indicate that P1β is highly unstable and is rapidly degraded when it is not bound to the ribosome. P2β is more stable and can accumulate free in the cytoplasm for a long period of time.

Fig. 3. Half-life of P1β and P2β proteins estimated by pulse–chase labelling. Saccharomyces cerevisiae D45/RPP1B and D67 were labelled with [35S]Met–Cys for 10 min and then chased with an excess of cold methionine and cysteine. At the time points indicated, cell aliquots were withdrawn, and the amount of P1β (closed circles) and P2β (open circles) proteins was estimated in D45/RPP1B and D67, respectively, by immunoprecipitation and SDS–PAGE as indicated in Materials and methods.
Similar results were obtained by inhibiting de novo protein synthesis with cycloheximide. The analysis was extended to the four acidic proteins in this case. In addition to D67 and D45/RPP1B, a P1α-overexpressing strain, D45/RPP1A, was used. This strain was also derived from D45 by transformation with an RPP1A-containing multicopy plasmid (YEp365 Trp P1α) in order to overexpress P1α. Exponentially growing cells were treated with the drug for different times and the amount of acidic proteins in the total cell extracts was estimated by western blotting using specific antibodies (Figure 4). Neither P1 protein was detected 15 min after cycloheximide addition, while P2β remained at similar levels after 2 h of treatment. Protein P2α was more stable than the P1 proteins, but was partially degraded after 120 min of cycloheximide treatment.

Fig. 4. Stability of acidic proteins determined by cycloheximide inhibition. Saccharomyces cerevisiae strains D45/RPP1B, D45/RPP1A and D67 growing exponentially in SC medium were treated with cycloheximide to inhibit protein synthesis. Aliquots were taken at the indicated times after the addition of cycloheximide and the amount of proteins in the cell extracts was estimated by western blotting using the corresponding specific antibodies (see Figure 1).
Protein P2α protects protein P1β from degradation
The high susceptibility of P1 proteins to proteolysis suggests that they must be protected from degradation in the cytoplasmic pool of wild-type cells, where they are found in similar amounts to the P2 proteins (Table III). Interaction between the two protein types has been detected in vitro (Zurdo et al., 2000a) and it is possible that P2 proteins protect P1 proteins from degradation in this way. To test this hypothesis, P1β was overexpressed in yeast either alone or together with P2α or P2β and the amount of each protein was estimated by ELISA in the cell supernatant (fraction S100) (Table III). As shown above, transformation of S.cerevisiae W303-1b with the P1β-encoding multicopy plasmid alone causes an increase in expression of this protein. This increase was notably smaller than the increase detected when P2 protein was overexpressed, due to the higher sensitivity of P1 proteins to degradation. However, simultaneous transformation with the P1β- and P2α-encoding multicopy plasmids dramatically increased the cytoplasmic pool of P1β, which became similar to the size of the pool in cells overexpressing P2α. In contrast, co-transformation with the P1β- and P2β-encoding plasmids does not stimulate accumulation of the P1 protein.
Table III. Estimation of acidic protein in the cytoplasmic pool of S.cerevisiae by ELISAa.
| Plasmid transforming S.cerevisiae W303 | Amount of protein in S100 fraction (ng/mg S100) |
||
|---|---|---|---|
| P1β | P2α | P2β | |
| Experiment I | |||
| none | 67 | 70 | n.t. |
| YEp13/RPP1B | 240 | 90 | n.t. |
| YEp356/RPP2A | 93 | 1200 | n.t. |
| YEp13/RPP1B and YEp356/RPP2A | 885 | 1320 | n.t. |
| Experiment II | |||
| none | 72 | n.t | 68 |
| YEp13/RPP1B | 240 | n.t. | 72 |
| YEp356/RPP2B | 68 | n.t. | 1230 |
| YEp13/RPP1B and YEp356/RPP2B | 110 | n.t. | 930 |
n.t., not tested.
aCells were grown to late exponential phase in SC medium and broken using glass beads; the S100 fraction was obtained by centrifugation to remove ribosomes. Proteins were assayed by inhibition ELISA using specific monoclonal antibodies.
Identification of the degradation signals in protein P1β
It has already been shown that protein P1β can be found in a processed form lacking the first eight amino acids (Santos et al., 1993). The presence of this protein form is prevented by the addition of protease inhibitors to the preparation, indicating that it must be an artefact produced during cell fractionation. However, the existence of this cleavage site suggested that the N-terminus could be important for P1β degradation in normal conditions. To test this possibility, the N-terminal peptides of P1β and P2β were swapped. Two pairs of chimeras of each protein were prepared, P1β(5N2β) and P1β(10N2β), and P2β(5N1β) and P2β(10N1β), carrying the first five and 10 amino acids of the opposite protein, respectively.
We also investigated whether phosphorylation could be a degradation signal for the ribosomal acidic proteins, as it is known to be for some other proteins (Hershko and Ciechanover, 1998). The last serine in the amino acid sequence (Ser96 in P1β and Ser100 in P2β), which has been shown to be the site of phosphorylation in vivo (Zambrano et al., 1997), was mutated in the native polypeptide as well as in the protein chimeras. All the P1β constructs were expressed in S.cerevisiae D456, while the P2β constructs were used to transform strain D567. The results are summarized in Figure 5.

Fig. 5. Effect of N-terminus and phosphorylation on accumulation of acidic proteins. (A) Effects on P1β. P1β chimeric genes carrying different modifications as listed below, cloned in pFL36, were expressed in S.cerevisiae D456. Strains W303-1b (1) and D456/111 (2) were included as controls. The amount of P1β protein in the extracts was estimated by western blotting using monoclonal antibody 1CE1. P1β modifications: (3) P1β(10N2β), first 10 amino acids from P2β; (4) P1β(5N2β), first five amino acids from P2β; (5) P1β(S96C), Ser96 mutated to cysteine; (6) P1β(S96F), Ser96 mutated to phenylalanine; (7) P1β(5N2β/S96C), first five amino acids from P2β and Ser96 mutated to cysteine. A representative western blot experiment is shown below the histogram. Error bars show the standard deviation of three independent experiments. (B) Effects on P2β. Modified P2β genes cloned in pFL38 were expressed in S.cerevisiae D567. Strains W303-1b (1) and D567/222 (8) were used as controls. Samples were processed as in (A) and protein P2β was assayed using monoclonal antibody 1AA9. P2β modifications: (9) P2β(10N1β), first 10 amino acids from P1β; (10) P2β(10N1β/S100F), first 10 amino acids from P1β and Ser100 mutated to phenylalanine; (11) P2β(5N1β), first five amino acids from P1β; (12) P2β(5N1β/S100F), first five amino acids from P1β and Ser100 mutated to phenylalanine. A representative western blot experiment is shown below the histogram. Error bars show the standard deviation of three independent experiments.
Replacing the first five or 10 amino acids of P1β with the equivalent sequence from P2β resulted in a stable protein that accumulated in the transformed D456 strain at a level similar to that in the parental W303-1b strain (Figure 5A, samples 3 and 4). The stability of each of the chimeric proteins was similar. A similar effect was produced by mutating Ser96 to cysteine or phenylalanine (Figure 5A, samples 5 and 6). The presence of both modifications together did not substantially affect accumulation of the protein compared with the effect produced by each modification independently (Figure 5A, sample 7).
In contrast, P2β accumulation was drastically reduced by the presence of five or 10 amino acids of the P1β N-terminal peptide (Figure 5B, samples 9 and 11). The reduction in the amount of modified P2β was completely reversed by mutation of Ser100 to phenylalanine (Figure 5B, samples 10 and 12).
Effect of acetylation on degradation of protein P1β
One of the most significant differences in the structure of the N-terminus of eukaryotic acidic proteins is acetylation, which takes place at Ser2 after removal of the initial methionine, but only in P1 proteins (Santos et al., 1993). Since acetylation is thought to be a degradation signal in some proteins (Mayer et al., 1989), the effect of this modification in the case of P1β was studied using S.cerevisiae NAT1, a mutant strain having an inactive N-terminal acetyltransferase (Nat1p), which is responsible for the acetylation of P1 proteins (Takakura et al., 1992; Santos et al., 1993). In W303-1b and NAT1 strains, most P1β is ribosome bound and, therefore, inaccessible to the degradation machinery. For that reason, gene dosage experiments were performed in order to overexpress P1β in the cytoplasmic pool from the multicopy plasmid YEp356/RPP1B in the mutant and parental strains. As expected, a change in the mobility of P1 proteins was observed in NAT1 and NAT1/RPP1B due to the presence of a free amino group (P1α* and P1β* in Figure 6A). However, despite the absence of the acetyl group, an increase in the P1β cytoplasmic pool could not be detected in the mutant as compared with the parental strain (Figure 6B). This result confirms that inactivation of the N-acetyltransferase does not affect the stability of P1β.

Fig. 6. Effect of N-acetyltransferase (NAT1) inactivation on P1β protein accumulation. (A) Ribosomes from untransformed S.cerevisiae W303-1b and NAT1, and from the same two strains transformed with plasmid YEp356/RPP1B, were resolved by isoelectrofocusing in the pH range 2.0–5.0. Proteins were silver stained. P1α* and P1β* mark the positions of the non-acetylated forms. (B) The amount of free P1β protein in these strains was estimated by inhibition ELISA in cell extracts deprived of ribosomes (S100 fractions). Error bars show the standard deviation of three independent experiments.
The proteasome is not involved in degradation of P1β
The proteasome participates in the degradation of many short-lived proteins (Hershko and Ciechanover, 1998). Different proteasomal mutant strains have been used to check whether this pathway is involved in the degradation of P1β. The protein was overexpressed from the previously described YEp356/RPP1B multicopy plasmid in S.cerevisiae FS11-1b and WCG4a-11/21a and in the corresponding parental strains (Table I). In FS11-1b, the essential gene RPN6, which encodes a regulatory protein located in the lid of the proteasome cap, is under the control of the GAL1 promoter (P.González-Santamaría, J.P.G.Ballesta and M.Remacha, unpublished results). As a consequence, cells transferred from medium containing galactose to one containing glucose accumulate ubiquitylated proteins after 6–9 h (Figure 7A). Nevertheless, the amount of P1β in the cytoplasmic pool (S100 fraction) did not increase in these conditions; in fact, it was smaller than that in the control after 16 h in glucose (Figure 7B). Similar results were obtained using S.cerevisiae WCG4a-11/21a, a strain in which the PRE1 and PRE2 genes encoding the proteasome core β subunits are mutated, inactivating its protease activity (Heinemeyer et al., 1993). Polyubiquitylated proteins accumulate after 5 h growth at 38°C in mineral medium (Figure 7C); nevertheless, a reduction in the amount of P1β is also detected in this case (Figure 7D).

Fig. 7. P1β degradation is proteasome independent. Gene dosage experiments were carried out by overexpressing P1β under restrictive conditions in proteasomal mutant strains FS11-1b (M1) and WCG4a-11/21 (M2), and in parental strains FY1679 (C1) and WCG4a (C2). In the case of FS11-1b, S100 extracts were prepared 10 or 16 h after the change of medium (A and B). For WCG4a-11/21, S100 fractions were prepared after 5 h at 38°C in mineral medium (C and D). Western blot analysis using anti-ubiquitin (A and C) and anti-P1β (B and D) antibodies was performed.
Discussion
The cellular requirement for stalk acidic proteins, namely bacterial proteins L7/12 and eukaryotic proteins P1/P2, is higher than for the other ribosomal components since not only are they present in multiple copies in the ribosome, but also there is a cytoplasmic pool of free proteins not detected in other instances. However, in S.cerevisiae, the only eukaryote in which this question has been studied, the overall regulation of the P1/P2 genes seems to conform to that of genes for other ribosomal proteins, which takes place at the level of transcription using RAP1 (repressor-activator protein) or ABF1 (autonomously replicating sequence binding factor) as activators (Raué and Planta, 1991). Nevertheless, the previous finding that acidic proteins of one type respond differently to elimination of proteins of the opposite type (Remacha et al., 1992) suggested the existence of additional regulatory mechanisms specific for these ribosomal components. Taking proteins P1β and P2β as models, a search for these possible mechanisms was carried out.
An assay of acidic proteins in the protein P2-deficient S.cerevisiae D45 has confirmed that the amount of P1β is drastically reduced, while disruption of the P1 genes in the D67 strain does not affect expression of the P2β protein. The decrease in P1β in D45 cannot be due to an effect at the level of transcription, splicing or transcript stability, since no reduction in the amount of the corresponding mRNA was detected.
The data indicate that the reduction in P1β is closely related to its sensitivity to degradation. The half-life of both P1 proteins is very short when they are not bound to the ribosome. Consequently, newly synthesized P1 proteins are degraded in strain D45 when the absence of P2 proteins prevents their binding to the ribosomes. In contrast, P2 proteins are much more stable: the half-life of P2β is >5 h, so this protein accumulates in the cytoplasmic pool in the absence of P1 proteins. P2α has a half-life shorter than that of P2β, but also accumulates in the cytoplasm. P1 proteins seem to be similar to other ribosomal proteins with respect to their sensitivity to degradation and, certainly, they behave like the latter in gene dosage experiments. Thus, transformation of a wild-type strain with a multicopy plasmid carrying the P1β gene does not result in the expected increase of protein. On the other hand, the amount of P2 proteins increases dramatically in similar gene dosage tests, consistent with their higher resistance to degradation.
Interestingly, P2-type proteins seem able to protect the P1 type from degradation since the amount of P1β is similar to that of P2α when both are overexpressed simultaneously. These results indicate that an interaction between P1 and P2 proteins takes place outside the ribosome in the cytoplasmic pool and that this interaction is strong enough to protect the sensitive proteins from degradation. The interaction is quite specific and only seems to take place between P1β and P2α. Very probably, P1α and P2β also interact in the same way; although this association has not been tested in vivo, experimental data using purified proteins confirm that it takes place in vitro (Zurdo et al., 2000a). Similar associations of acidic protein in the ribosome, P1α/P2β and P1β/P2α, have already been reported (Ballesta et al., 2000), but the results in this report indicate that they also exist in the cytoplasmic pool, suggesting that the P proteins probably assemble into the ribosome as heterodimers. Exchange between acidic proteins in the ribosome and those in the cytoplasmic pool may also involve these protein couples rather than individual proteins.
This information is relevant to our understanding of the stalk assembly process since it has previously been shown that only P1 proteins, and not P2 proteins, are able to interact directly with protein P0 in the ribosome to form the stalk (Zurdo et al., 2000b). The different sensitivity of P1 and P2 proteins to degradation, together with the fact that free P2 proteins cannot bind independently to the ribosome, would allow the cell to control the assembly of a correct ribosomal stalk simply by degrading the excess of P1 proteins.
One of the most interesting results of this report is the first identification of degradation signals in ribosomal proteins. The high sensitivity of free ribosomal components to proteolysis was found in the initial studies on eukaryotic ribosome synthesis. However, no explanations for this high sensitivity have been reported so far. The striking difference in sensitivity to proteolysis of P1 and P2 proteins, despite their close structural similarity, prompted us to approach this question by looking for structural peculiarities that could be responsible for this difference in behaviour. One of the most obvious structural differences between P1 and P2 proteins is the state of the N-terminus. In P1 proteins, the first methionine is removed and the following serine residue is acetylated, while in P2 proteins the initial methionine is unblocked (Santos et al., 1993). Acetylation may be a signal for degradation for some proteins (Mayer et al., 1989). However, we found that the cytoplasmic pool of P1β was not affected in S.cerevisae NAT1, a strain with an inactive NAT1 acetylase, which accumulates unblocked P1 proteins, so N-terminal acetylation seems not to be the signal for degradation in these polypeptides.
Nevertheless, the structure of the N-terminus is essential for determining the sensitivity of these proteins to proteolysis. Replacement of fragments of different sizes at the N-terminus of P1β and P2β by the equivalent fragment of the other protein clearly indicated that the first five amino acids of P1β are sufficient to induce degradation of P2β, while the P2β N-terminus stabilizes P1β. Since the methionine is removed, the actual signal in fact corresponds to only four residues: SDSI. The equivalent sequence in P1α, STES, is closely related. These sequences do not correspond to any known degradation signal. Moreover, the well known N-end rule pathway does not seem to work in these proteins, since serine is considered a stabilizing residue in that degradation system (Varshavsky, 1997). In agreement with this conclusion, experiments carried out with different proteasomal mutant strains seem to exclude the involvement of this structure in the P1 degradation process. Indeed, lower amounts of P1β were detected in these mutants after prolonged incubation under restrictive conditions. These results were unexpected, since most short-lived proteins seem to be degraded by the proteasome pathway. Since ribosomal proteins are not degraded by vacuolar proteases either (Tsay et al., 1988), as we have confirmed for protein P1 using vacuolar protease mutants (results not shown), some unknown degradation pathway must be responsible for P1 protein proteolysis.
On the other hand, P2β protein is resistant to degradation due to the protective effect of its highly conserved N-terminal peptide, which is also able to protect the otherwise sensitive P1β. Interestingly, protein P2α, which carries the same N-terminal sequence, seems to be less stable than P2β. This protein probably has some additional destabilizing structural elements, which we are trying to identify.
The N-terminal structure is not the only degradation signal: phosphorylation is also required for proteolysis of the acidic proteins. Thus, mutation of Ser96 in the C-terminus of P1β, which has been shown to be the site of phosphorylation in vivo (Zambrano et al., 1997), results in complete resistance of the protein to degradation. Similarly, the sensitive chimeric P2β carrying the P1β N-terminus becomes resistant upon mutation of its phosphorylation site at Ser100, proving that phosphorylation is absolutely required.
Phosphorylation is a degradation signal in other cellular proteins, which are degraded either through the vacuolar or the proteasome pathway (Hershko and Ciechanover, 1998). In these cases, a ubiquitin ligase would recognize only the phosphorylated form of the protein and ubiquitylate it, making it susceptible to the corresponding degradation pathway. Since, as commented on earlier, the acidic proteins seem to be degraded by neither the vacuolar nor the proteasome pathway, these data indicate that phosphorylation may be a signal for other uncharacterized degradation processes, which may or may not require ubiquitylation. Ubiquitylated forms of P1β were not detected, but since ubiquitin can be removed from the proteins by isopeptidases (Hershko and Ciechanover, 1998), the possibility that this modification is not involved in the P1 protein degradation pathway cannot be ruled out completely.
Collectively, our results show the existence of two types of signal involved in yeast P1β degradation: phosphorylation at the last serine residue and N-terminal structure. The cell can exploit these two signals in order to control (i) the cytoplasmic pool of the two types of acidic protein in different ways and (ii) the stalk composition through the exchange process. P2 proteins are more resistant to degradation and so can accumulate free in the cytoplasm. However, they cannot bind to the ribosome alone and require the presence of P1 proteins, which are protected from degradation by association with P2. Thus, the cell in normal conditions will control the existence of ribosomes carrying a stalk with both types of acidic proteins. However, it might be possible to increase the amount of P1 over that of P2 in the pool by dephosphorylating these proteins and, consequently, to have a ribosome population carrying a higher proportion of P1.
The dependence on phosphorylation makes the degradation of acidic proteins clearly different from the process whereby excesses of other ribosomal proteins are mopped up. Except for the stalk components and proteins S10 (Kruse et al., 1985) and L3 (Campos et al., 1990), yeast ribosomal proteins are not phosphorylated, so their degradation cannot depend on this modification. It is possible, however, that the difference affects mainly the initial control mechanism, while the basic degradation machinery is the same. Additional experimental evidence will be required to identify both processes and to clarify this question.
Materials and methods
Strains and media
Saccharomyces cerevisiae strains used in this study are listed in Table I. Cells were grown to mid-log phase in rich (YPD and YPG), synthetic (SD) or mineral media (Sherman et al., 1986). Transformation of S.cerevisiae was carried out as described elsewhere (Gietz et al., 1995). Escherichia coli DH5α was used for propagating cloning vectors and was grown in LB medium. Bacteria were transformed according to standard procedures (Hanahan, 1983).
Cell fractionation
Yeasts were fractionated as previously described (Rodriguez-Gabriel et al., 2000). In summary, cells were broken using glass beads in the presence of protease inhibitors. The extract was centrifuged in a Sorval SS-34 rotor at 12 000 r.p.m. for 15 min, yielding the S30 supernatant fraction. The S100 supernatant fraction and ribosome pellet were obtained by high-speed centrifugation of S30 at 90 000 r.p.m. for 30 min in a Beckman TL100.3 rotor. The crude ribosome pellet was washed by centrifugation through a discontinuous sucrose gradient.
Plasmids
Standard DNA cloning and manipulation were carried out as described (Sambrook et al., 1989). All constructions were checked by automatic sequencing.
BS/RPP1B, pFL36/RPP1B, YEp356/RPP1B and YEp13/RPP1B. These were obtained by inserting a 1.98 kb HindIII fragment containing the protein P1β-encoding RPP1B gene from pMRH46 (Remacha et al., 1988) in the multiple cloning site (MCS) of Bluescript KS (Stratagene, La Jolla, USA), pFL36 (Bonneaud et al., 1991), YEp356 (Myers et al., 1986) and YEp13 (Broach et al., 1979), respectively.
YEp356 Trp P1α and YEp356/RPP2A. A 2.3 kb EcoRI fragment including the RPP2A gene from pMRE44 (Remacha et al., 1988) was inserted in the corresponding sites of plasmid YEp356. In addition, YEp356 Trp P1α carries TRP1 as a marker, inserted as previously described (Zurdo et al., 2000b).
BS/RPP2B, pFL38/RPP2B and YEp356/RPP2B. These were generated by cloning a 1.0 kb HindIII–PstI fragment containing the P2β-encoding RPP2B gene from pRVE45 (Remacha et al., 1988) in the MCS of Bluescript KS, pFL38 (Bonneaud et al., 1991) and YEp356, respectively.
Plasmids with chimeric genes. All chimeric constructs carrying different parts of the RPP1B and RPP2B genes were made following a similar strategy. The fragments of the two genes to be fused were obtained by PCR from the plasmids BS/RPP1B and BS/RPP2B. In each case, one of the primers was complementary to a region outside the gene in the MCS of the plasmid, while the second primer was complementary to the end of the internal region of the gene to be fused. The two PCR products, previously phosphorylated at the 5′ end using T4 polynucleotide kinase, were ligated to the MCS of Bluescript by the specific restriction sites present in one of the ends and the plasmid was circularized by blunt-end ligation at the other end. The oligonucleotides used as primers for each construct are listed in Table IV. The P1β and P2β chimeric constructs were then subcloned in pFL36 and pFL38, respectively. Site-directed mutagenesis of serine residues was performed by PCR (Dieffenbach and Dveksler, 1995) as previously reported (Zambrano et al., 1997).
Table IV. Oligonucleotides used to construct gene chimeras.
| Plasmid | Oligonucleotide |
Template | |
|---|---|---|---|
| Name | Sequence (5′ to 3′) | ||
| pFL36/211 | E | aatcaacaacagaaatgtctgactc | BS/RPP1B |
| BSDW | tgaattgtaatacgactcactat | ||
| FN | aatagatattactttctatatttg | BS/RPP2B | |
| BSDW | tgaattgtaatacgactcactat | ||
| pFL38/122 | B | ttcttagttgtttgatttctttg | BS/RPP1B |
| BSUP | tatgaccatgattacgccaag | ||
| A | gaagaaaatgaaatacttagc | BS/RPP2B | |
| BSUP | tatgaccatgattacgccaag | ||
| pFL38/121 | DN | gaaagtcattttttctgcaacg | BS/RPP1B |
| BSDW | tgaattgtaatacgactcactat | ||
| C | cttgtatgtttaatcgaataaacca | BS/122 | |
| BSUP | tatgaccatgattacgccaag | ||
| pFL36/P1β(10NP2β) | 3 | agcatcagctaggatgaacaataataagtaagcagc | BS/122 |
| BSUP | tatgaccatgattacgccaag | ||
| 4 | ggtaatcacctctgac | BS/RPP1B | |
| BSDW | tgaattgtaatacgactcactat | ||
| pFL36/ P1β(5NP2β) | R5aa2β | agctaagtatttcattttcttc | BS/122 |
| BSUP | tatgaccatgattacgccaag | ||
| aa61β | atttcctttgctgctttcatc | BS/RPP1B | |
| BSDW | tgaattgtaatacgactcactat | ||
| pFL38/P2β(10NP1β) | 1 | gcgttaccaccttgaacagcagcaaaggaaataat | BS/211 |
| BSUP | tatgaccatgattacgccaag | ||
| 2 | attatttcctttgctgctgttcaaggtggtaacgct | pFL38/RPP2B | |
| 383 | tgagcggataacaatttcacacag | ||
| pFL38/P2β(5NP1β) | 2B5N1B | atgtctgactctattgcttacttattattggttcaa | BS/RPP2B |
| BSUP | tatgaccatgattacgccaag | ||
| 5UTR2B | ttctgttgttgattaatagata | BS/RPP2B | |
| BSDW | tgaattgtaatacgactcactat | ||
| pFL36/P1β(S96C) | S1BS96C | gaatgtgacgacgacatgggtttcggt | BS/RPP1B |
| BSDW | tgaattgtaatacgactcactat | ||
| R1BS96C | ttcagcagcttcttcttctttttc | BS/RPP1B | |
| BSUP | tatgaccatgattacgccaag | ||
Determination of protein half-life
Pulse–chase labelling. Yeasts were grown in SC medium with methionine (minus leucine for D45 carrying YEp13/P1B) up to stationary phase, centrifuged and resuspended in 10 ml of SC without methionine. The cells were allowed to complete a cell cycle at 30°C and were supplemented with 700 µCi of [35S]methionine and cysteine (1.5 Ci/mmol) (Redimix; Amersham). After 10 min, the cells were centrifuged, washed twice with non-radioactive SC and resuspended in 10 ml of the same medium containing an excess of unlabelled methionine and cysteine. Aliquots (2 ml) were taken at time points between 0 and 120 min, washed in cold water and frozen at –80°C until protein extraction. Extracts were prepared by breaking cells with glass beads in a buffer containing 1% Triton X-100, 0.15 M NaCl, 5 mM disodium EDTA, 50 mM sodium HEPES pH 7.5 and protease inhibitors [2.5 µg/ml leupeptin, pepstatin, bestatin, aproptinin, chymostatin and antipain (Sigma), 1 mM phenylmethylsulfonyl fluoride (PMSF) and 10 mM N-ethylmaleimide]. The extracts were centrifuged at 12 000 g for 15 min. The amount of radiolabelled amino acid incorporated into the protein was estimated by trichloroacetic acid precipitation. Acidic proteins were immunoprecipitated from the extracts using specific monoclonal antibodies. The immunoprecipitated pellets were washed four times with 1 ml of lysis buffer, resuspended in SDS loading buffer and incubated at 100°C for 5 min. The samples were subjected to 15% SDS–PAGE and fluorography. Radioactive signals were visualized using a phosphoimager (Fujifilm BAS 1500, BAS reader program Tina2).
Inhibition by cycloheximide. Cells were grown in SD with the appropriate supplements until they reached mid-log phase (A600 = 0.4); 10 µg/ml cycloheximide was then added and aliquots were taken at time points between 0 and 120 min. Acidic proteins in the extracts were assayed by western blot analysis using specific antibodies.
Northern blot analysis. Total RNA, isolated from cells disrupted using glass beads (Bromley et al., 1982), was resolved by electrophoresis on formaldehyde–agarose gels and blotted onto nitrocellulose membranes. Hybridization was carried out as described (Sambrook et al., 1989) using probes corresponding to the coding region of the gene. Part of the actin gene was used as an internal standard. Probes were radioactively labelled using the Prime-a-Gene labelling system (Promega). Radioactive signals on the filters were detected using a phosphoimager.
Protein analysis. Proteins were resolved by 15% SDS–PAGE or isoelectrofocusing in 5% polyacrylamide gels using a 2.5–5.0 pH range (Rodriguez-Gabriel et al., 2000).
Western blotting. Proteins were separated by15% SDS–PAGE and blotted onto PVDF 0.45 µm membranes (Immobilon; Millipore) as previously described (Rodriguez-Gabriel et al., 2000). P1 and P2 proteins were detected using monoclonal antibodies 1CE1 (anti-P1β), 1AA9 (anti-P2β), 1BE3 (anti-P2α) (Vilella et al., 1991) and a rabbit antibody against P1α (Zurdo et al., 2000a). Antigen–antibody complexes was detected using horseradish peroxidase-conjugated rabbit anti-mouse or donkey anti-rabbit IgG diluted 1 in 2500 (Amersham) with an enhanced chemi luminescence system (Amersham) according to the manufacturer’s instructions.
Immunological techniques. Inhibition ELISA was performed using specific antibodies against the acidic proteins as previously detailed (Vilella et al., 1991). Immunoprecipitation of acidic proteins was carried out using monoclonal antibodies (1–5 µg/100 µl supernatant) specific for P1β (1CE1) and P2β (1AA9). Samples were incubated for 2 h on ice; 30 µl of 3.7 mg/ml protein G–agarose (Sigma) were added and the suspensions were incubated with rotation for 1 h at 4°C, followed by 15 s centrifugation at 12 000 g.
Acknowledgments
Acknowledgements
We thank M.C.Fernandez Moyano for expert technical assistance. This work was supported by grant PB94-0032 from the Dirección General de Política Científica (Spain) and by an institutional grant to the Centro de Biología Molecular ‘Severo Ochoa’ from the Fundación Ramón Areces (Madrid).
References
- Ballesta J.P.G. and Remacha,M. (1996) The large ribosomal subunit stalk as a regulatory element of the eukaryotic translational machinery. Prog. Nucleic Acid Res. Mol. Biol., 55, 157–193. [DOI] [PubMed] [Google Scholar]
- Ballesta J.P.G., Guarinos,E., Zurdo,J., Parada,P., Nusspaumer,G., Lalioti,V.S., Perez-Fernandez,J. and Remacha,M. (2000) Structure of the yeast ribosomal stalk. In Garrett,R., Douthwaite,S., Matheson,A., Liljas,A., Moore,P.B. and Noller,H.F. (eds), The Ribosome. Structure, Function, Antibiotics and Cellular Interactions. American Society for Microbiology, Washington, DC, pp. 115–125. [Google Scholar]
- Bonneaud N., Ozier-Kalogeropoulos,O., Li,G.Y., Labouesse,M., Minvielle-Sebastia,L. and Lacroute,F. (1991) A family of low and high copy replicative, integrative and single-stranded S. cerevisiae/E. coli shuttle vectors. Yeast, 7, 609–615. [DOI] [PubMed] [Google Scholar]
- Broach J., Strathern,J.N. and Hicks,J.B. (1979) Transformation in yeast: development of a hybrid cloning vector and isolation of the CAN1 gene. Gene, 8, 121–133. [DOI] [PubMed] [Google Scholar]
- Bromley S., Hereford,L. and Rosbash,M. (1982) Further evidence that the rna2 mutation of Saccharomyces cerevisiae affects mRNA processing. Mol. Cell. Biol., 2, 1205–1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campos F., Corona-Reyes,M. and Zinker,S. (1990) The yeast 5S rRNA binding ribosomal protein YL3 is phosphorylated in vivo. Biochim. Biophys. Acta, 1087, 142–146. [DOI] [PubMed] [Google Scholar]
- Dieffenbach C.W. and Dveksler,G.S. (1995) PCR Primer. A Laboratory Manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- el Baradi T.T., van der Sande,C., Mager,W.H., Raué,H.A. and Planta,R.J. (1986) The cellular level of yeast ribosomal protein L25 is controlled principally by rapid degradation of excess protein. Curr. Genet., 10, 733–739. [DOI] [PubMed] [Google Scholar]
- Elkon K.B., Skelly,S., Parnassa,A., Möller,W., Danho,W., Weissbach,H. and Brot,N. (1986) Identification and chemical synthesis of a ribosomal protein antigenic determinant in systemic lupus erythematosus. Proc. Natl Acad. Sci. USA, 83, 7419–7423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fewell S.W. and Woolford,J.L.,Jr (1999) Ribosomal protein S14 of Saccharomyces cerevisiae regulates its expression by binding to RPS14B pre-mRNA and to 18S rRNA. Mol. Cell. Biol., 19, 826–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gietz R.D., Schiestl,R.H., Willems,A.R. and Woods,R.A. (1995) Studies on the transformation of intact yeast cells by the LiAc/SS-DNA/PEG procedure. Yeast, 11, 355–360. [DOI] [PubMed] [Google Scholar]
- Gorenstein C. and Warner,J.R. (1976) Co-ordinate regulation of the synthesis of eukaryotic ribosomal proteins. Proc. Natl Acad. Sci. USA, 73, 1547–1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorenstein C. and Warner,J.R. (1977) Synthesis and turnover of ribosomal proteins in the absence of 60S subunit assembly in Saccharomyces cerevisiae. Mol. Gen. Genet., 157, 327–332. [DOI] [PubMed] [Google Scholar]
- Hanahan H.D. (1983) Studies on transformation of Escherichia coli with plasmids. J. Mol. Biol., 166, 557–580. [DOI] [PubMed] [Google Scholar]
- Heimark R.L., Hersehy,J.W. and Traut,R.R. (1976) Cross-linking of initiation factor IF2 to proteins L7/L12 in 70 S ribosomes of Escherichia coli. J. Biol. Chem., 251, 779–784. [PubMed] [Google Scholar]
- Heinemeyer W., Gruhler,A., Mohrle,V., Mahe,Y. and Wolf,D.H. (1993) PRE2, highly homologous to the human major histocompatibility complex-linked RING10 gene, codes for a yeast proteasome subunit necessary for chrymotryptic activity and degradation of ubiquitinated proteins. J. Biol. Chem., 268, 5115–5120. [PubMed] [Google Scholar]
- Hershko A. and Ciechanover,A. (1998) The ubiquitin system. Annu. Rev. Biochem., 67, 425–479. [DOI] [PubMed] [Google Scholar]
- Kruse C., Johnson,S.P. and Warner,J.R. (1985) Phosphorylation of the yeast equivalent of ribosomal protein S6 is not essential for growth. Proc. Natl Acad. Sci. USA, 82, 7515–7519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacConnell W.P. and Kaplan,N.O. (1982) The activity of the acidic phosphoproteins from the 80S rat liver ribosome. J. Biol. Chem., 257, 5359–5366. [PubMed] [Google Scholar]
- Mayer A., Siegel,N.R., Schwartz,A.L. and Ciechanover,A. (1989) Degradation of proteins with acetylated amino termini by the ubiquitin system. Science, 244, 1480–1483. [DOI] [PubMed] [Google Scholar]
- Mitsui K., Nakagawa,T. and Tsurugi,K. (1988). On the size and the role of a free cytosolic pool of acidic ribosomal proteins in yeast S. cerevisiae. J. Biochem., 104, 908–911. [DOI] [PubMed] [Google Scholar]
- Möller W. and Maassen,J.A. (1986) On the structure, function and dynamics of L7/12 from Escherichia coli ribosomes. In Hardesty,B. and Kramer,G. (eds), Structure, Function and Genetics of Ribosomes. Springer-Verlag, New York, NY, pp. 309–325. [Google Scholar]
- Möller W., Groene,A., Terhorst,C. and Amons,R. (1972) 50S ribosomal proteins. Purification and partial characterization of two acidic proteins, A1 and A2, isolated from 50S ribosomes from Escherichia coli. Eur. J. Biochem., 25, 5–12. [DOI] [PubMed] [Google Scholar]
- Möller W., Slobin,L.I., Amons,R. and Richter,D. (1975) Isolation and characterization of two acidic proteins of 60S ribosomes from Artemia salina cysts. Proc. Natl Acad. Sci. USA, 72, 4774–4778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers A.M., Tzagoloff,A., Kinney,D.M. and Lusty,C.J. (1986) Yeast shuttle and integrative vectors with multiple cloning sites suitable for constructions of LacZ fusions. Gene, 45, 299–310. [DOI] [PubMed] [Google Scholar]
- Presutti C., Ciafré,S.A. and Bozzoni,I. (1991) The ribosomal protein L2 in S.cerevisiae controls the level of accumulation of its own mRNA. EMBO J., 10, 2215–2221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramagopal S. (1976) Accumulation of free ribosomal proteins S1, L7 and L12 in E. coli.Eur. J. Biochem., 69, 289–297. [DOI] [PubMed] [Google Scholar]
- Raué H.A. and Planta,R.J. (1991) Ribosome biogenesis in yeast. Prog. Nucleic Acid Res. Mol. Biol., 41, 89–129. [DOI] [PubMed] [Google Scholar]
- Remacha M., Saenz-Robles,M.T., Vilella,M.D. and Ballesta,J.P.G. (1988) Independent genes coding for three acidic proteins of the large ribosomal subunit from Saccharomyces cerevisae. J. Biol. Chem., 263, 9094–9101. [PubMed] [Google Scholar]
- Remacha M., Santos,C., Bermejo,B., Naranda,T. and Ballesta,J.P.G. (1992) Stable binding of the eukaryotic acidic phosphoproteins to the ribosome is not an absolute requirement for in vivo protein synthesis. J. Biol. Chem., 267, 12061–12067. [PubMed] [Google Scholar]
- Remacha M., Jimenez-Diaz,A., Bermejo,B., Rodriguez-Gabriel,M.A., Guarinos,E. and Ballesta,J.P.G. (1995) Ribosomal acidic phospho proteins P1 and P2 are not required for cell viability but regulate the pattern of protein expression in Saccharomyces cerevisiae. Mol. Cell. Biol., 15, 4754–4762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Gabriel M.A., Remacha,M. and Ballesta,J.P.G. (1998) Phosphorylation of ribosomal protein P0 is not essential for ribosome function but affects translation. Biochemistry, 37, 16620–16626. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Gabriel M.A., Remacha,M. and Ballesta,J.P.G. (2000) The RNA interacting domain but not the protein interacting domain is highly conserved in ribosomal protein P0. J. Biol. Chem., 275, 2130–2136. [DOI] [PubMed] [Google Scholar]
- Saenz-Robles M.T., Remacha,M., Vilella,M.D., Zinker,S. and Ballesta,J.P.G. (1990) The acidic ribosomal proteins as regulators of the eukaryotic ribosomal activity. Biochim. Biophys. Acta, 1050, 51–55. [DOI] [PubMed] [Google Scholar]
- Sambrook J., Fritsch,E.F. and Maniatis,T. (1989) Molecular Cloning: A Laboratory Manual, 2nd edn. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- Sanchez-Madrid F., Reyes,R., Conde,P. and Ballesta,J.P.G. (1979) Acidic ribosomal proteins from eukaryotic cells. Effect on ribosomal functions. Eur. J. Biochem., 98, 409–416. [DOI] [PubMed] [Google Scholar]
- Sanchez-Madrid F., Juan-Vidales,F. and Ballesta,J.P.G. (1981) Effect of phosphorylation on affinity of acidic proteins from Saccharomyces cerevisiae for the ribosome. Eur. J. Biochem., 114, 609–613. [DOI] [PubMed] [Google Scholar]
- Santos C., Ortiz-Reyes,B.L., Naranda,T., Remacha,M. and Ballesta,J.P.G. (1993) The acidic phosphoproteins from Saccharomyces cerevisiae ribosomes. NH2-terminal acetylation is a conserved difference between P1 and P2 proteins. Biochemistry, 32, 4231–4236. [DOI] [PubMed] [Google Scholar]
- Scharf K.-D. and Nover,L. (1987) Control of ribosome biosynthesis in plant cell cultures under heat shock conditions. II. Ribosomal proteins. Biochim. Biophys. Acta, 909, 44–57. [DOI] [PubMed] [Google Scholar]
- Schwartz I., Vincent,M., Strycharz,W.A. and Kahan,L. (1983) Photo chemical cross-linking of translation initiation factor 3 to E. coli 50S ribosomal subunits. Biochemistry, 22, 1483–1489. [DOI] [PubMed] [Google Scholar]
- Sherman F., Fink,G.R. and Hicks,J.B. (1986) Methods in Yeast Genetics. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- Szick K., Springer,M. and Bailey-Serres,J. (1998) Evolutionary analyses of the 12-kDa acidic ribosomal P-proteins reveal a distinct protein of higher plant ribosomes. Proc. Natl Acad. Sci. USA, 95, 2378–2383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takakura H., Tsumasawa,S., Miyagi,M. and Warmer,J.R. (1992) NH2-terminal acetylation of ribosomal proteins of Saccharomyces cerevisiae. J. Biol. Chem., 267, 5442–5445. [PubMed] [Google Scholar]
- Tate W.P. et al. (1990) The ribosomal domain of the bacterial release factors. The carboxyl-terminal domain of the dimer of Escherichia coli ribosomal protein L7/L12 located in the body of the ribosome is important for release factor interaction. Eur. J. Biochem., 187, 543–548. [DOI] [PubMed] [Google Scholar]
- Thierry A., Fairhead,C. and Dujon,B. (1990) The complete sequence of the 8.2 kb segment left of MAT on chromosome III reveals five ORFs, including a gene for a yeast ribokinase. Yeast, 6, 521–534. [DOI] [PubMed] [Google Scholar]
- Thomas B.J. and Rothstein,R. (1989) Elevated recombination rates in transcriptionally active DNA. Cell, 56, 619–630. [DOI] [PubMed] [Google Scholar]
- Tsay Y.-F., Thompson,J.R., Rotenberg,M.O., Larkin,J.C. and Woolford,J.L.,Jr (1988) Ribosomal protein synthesis is not regulated at the translational level in Saccharomyces cerevisiae: balanced accumulation of ribosomal proteins L16 and rp59 is mediated by turnover of excess protein. Genes Dev., 2, 664–676. [DOI] [PubMed] [Google Scholar]
- Tsurugi K. and Ogata,K. (1985) Evidence for the exchangeability of acidic ribosomal proteins on cytoplasmic ribosomes in regenerating rat liver. J. Biochem., 98, 1427–1431. [DOI] [PubMed] [Google Scholar]
- Tsurugi K., Collatz,E., Todokoro,K., Ulbrich,N., Lightfoot,H. and Wool,I.G. (1978) Isolation of eukaryotic ribosomal proteins. Purification and characterisation of the 60S ribosomal subunit proteins La, Lb, Lf, P1, P2, L13′, L14, L18′, L20 and L38. J. Biol. Chem., 253, 946–955. [PubMed] [Google Scholar]
- Uchiumi T., Hori,K., Nomura,T. and Hachimori,A. (1999) Replacement of L7/L12⋅L10 protein complex in Escherichia coli ribosomes with the eukaryotic counterpart changes the specificity of elongation factor binding. J. Biol. Chem., 274, 27578–27582. [DOI] [PubMed] [Google Scholar]
- van Agthoven A., Kriek,J., Amons,R. and Möller,W. (1978) Isolation and characterisation of the acidic phosphoproteins of 60S ribosomes from Artemia salina and rat liver. Eur. J. Biochem., 91, 553–556. [DOI] [PubMed] [Google Scholar]
- Varshavsky A. (1997) The N-end rule pathway of protein degradation. Genes Cells, 2, 13–28. [DOI] [PubMed] [Google Scholar]
- Vilardell J. and Warner,J.R. (1997) Ribosomal protein L32 of Saccharomyces cerevisiae influences both the splicing of its own transcript and the processing of rRNA. Mol. Cell. Biol., 17, 1959–1965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilella M.D., Remacha,M., Ortiz,B.L., Mendez,E. and Ballesta,J.P.G. (1991) Characterization of the yeast acidic ribosomal phosphoproteins using monoclonal antibodies. Proteins L44/L45 and L44′ have different functional roles. Eur. J. Biochem., 196, 407–414. [DOI] [PubMed] [Google Scholar]
- Woolford J.L. and Warner,J.R. (1991) The ribosome and its synthesis. In Broach,J., Jones,E. and Pringle,J. (eds), The Molecular Biology of the Yeast Saccharomyces cerevisiae: Genome Dynamics, Protein Synthesis, and Energetics. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, pp. 587–626. [Google Scholar]
- Zambrano R., Briones,E., Remacha,M. and Ballesta,J.P.G. (1997) Phosphorylation of the acidic ribosomal P proteins in Saccharomyces cerevisiae. A reappraisal. Biochemistry, 36, 14439–14446. [DOI] [PubMed] [Google Scholar]
- Zinker S. (1980) P5/P5′, the acidic ribosomal phosphoprotein from S. cerevisiae. Biochim. Biophys. Acta, 606, 76–82. [DOI] [PubMed] [Google Scholar]
- Zinker S. and Warner,J.R. (1976) The ribosomal proteins of Saccharomyces cerevisiae. Phosphorylated and exchangeable proteins. J. Biol. Chem., 251, 1799–1807. [PubMed] [Google Scholar]
- Zurdo J., Sanz,J.M., Gonzalez,C., Rico,M. and Ballesta,J.P.G. (1997) The exchangeable yeast ribosomal acidic protein YP2β shows characteristics of a partly folded state under physiological conditions. Biochemistry, 36, 9625–9635. [DOI] [PubMed] [Google Scholar]
- Zurdo J., Gonzalez,C., Sanz,J.M., Rico,M., Remacha,M. and Ballesta,J.P.G. (2000a) Structural differences between Saccharomyces cerevisiae ribosomal stalk proteins P1 and P2 support their functional diversity. Biochemistry, 39, 8935–8943. [DOI] [PubMed] [Google Scholar]
- Zurdo J., van den Berg,A., Parada,P., Nusspaumer,G., Jimenez-Diaz,A., Remacha,M. and Ballesta,J.P.G. (2000b) Assembly of Saccharomyces cerevisiae ribosomal stalk: binding of P1 proteins is required for the interaction of P2 proteins. Biochemistry, 39, 8929–8934. [DOI] [PubMed] [Google Scholar]