

Abstract
The crystal structure of the cyclic phosphodiesterase (CPDase) from Arabidopsis thaliana, an enzyme involved in the tRNA splicing pathway, was determined at 2.5 Å resolution. CPDase hydrolyzes ADP-ribose 1″,2″-cyclic phosphate (Appr>p), a product of the tRNA splicing reaction, to the monoester ADP-ribose 1″-phosphate (Appr-1″p). The 181 amino acid protein shows a novel, bilobal arrangement of two αβ modules. Each lobe consists of two α-helices on the outer side of the molecule, framing a three- or four-stranded antiparallel β-sheet in the core of the protein. The active site is formed at the interface of the two β-sheets in a water-filled cavity involving residues from two H-X-T/S-X motifs. This previously noticed motif participates in coordination of a sulfate ion. A solvent-exposed surface loop (residues 100–115) is very likely to play a flap-like role, opening and closing the active site. Based on the crystal structure and on recent mutagenesis studies of a homologous CPDase from Saccharomyces cerevisiae, we propose an enzymatic mechanism that employs the nucleophilic attack of a water molecule activated by one of the active site histidines.
Keywords: ADP-ribose 1″,2″-cyclic phosphate/Arabidopsis/2′,3′-cyclic nucleotide phosphodiesterase/tRNA splicing/X-ray crystallography
Introduction
Splicing of pre-tRNA in the nuclei of eukaryotic cells is initiated by endonucleolytic cleavages that result in removal of the intron and formation of two tRNA half-molecules. In yeast and plants, the two tRNA halves are ligated together via an unusual 3′,5′-phosphodiester-2′-phosphomonoester linkage (reviewed by Phizicky and Greer, 1993; Westaway and Abelson, 1995). In eukaryotes, fractions of pre-tRNAs are processed by this pathway as well (Zillmann et al., 1991; Phizicky and Greer, 1993; Westaway and Abelson, 1995). Generation of functional tRNAs requires removal of the 2′-phosphate, which is present at the splice junction close to the anticodon (Culver et al., 1997). The dephosphorylation is catalyzed by a specific phosphotransferase (Zillmann et al., 1992; Culver et al., 1993). In this reaction, the 2′-phosphate is transferred to NAD+; the resulting products are nicotinamide and ADP-ribose 1″,2″-cyclic phosphate (Appr>p) (Culver et al., 1993). Interestingly, Appr>p is not the final product of this complex series of reactions. In the following step, catalyzed by a specific cyclic phosphodiesterase (CPDase), Appr>p is converted to ADP-ribose 1″-phosphate (Appr-1″p) through hydrolysis of the 2″-phosphoester bond (Culver et al., 1994; Genschik et al., 1997a).
Most of the tRNA splicing in vertebrates appears to occur by a pathway different from that described above. The ligation of tRNA halves in the latter pathway results in the conventional 3′,5′-phosphodiester linkage, with no 2′-phosphorylated tRNAs formed (Filipowicz and Shatkin, 1983; Laski et al., 1983; Westaway and Abelson, 1995). However, as indicated above, the tRNA splicing pathway leading to the generation of Appr>p is also present in vertebrates (Zillmann et al., 1991, 1992; Culver et al., 1993; Spinelli et al., 1998). This redundancy has fueled speculations about the importance of Appr>p and/or its hydrolysis product, Appr-1″p, for some as yet unspecified regulatory processes in the cell (Culver et al., 1993; Westaway and Abelson, 1995; Genschik et al., 1997a). In this context, it is important to note that in addition to tRNAs, other 2′-phosphorylated RNAs, which are likely to act as substrates for the Appr>p-forming phosphotransferase, are produced in eukaryotes. The 2′-phosphate-forming tRNA ligase functions in some unusual mRNA splicing events during the unfolded protein response in yeast (Gonzalez et al., 1999 and references therein), and probably mammals (Niwa et al., 1999), and may also be involved in ligation of virusoid and viroid RNAs in plants (for references see Genschik et al., 1997b).
The CPDase mentioned earlier was purified originally from wheat germ as an enzyme catalyzing hydrolysis of nucleoside 2′,3′-cyclic phosphates (N>p) to nucleoside 2′-phosphates (N-2′p) (Tyc et al., 1987). Further characterization of CPDases from wheat and Arabidopsis thaliana revealed that the enzyme also hydrolyzes Appr>p to Appr-1″p (Culver et al., 1993; Genschik et al., 1997a). The partially purified enzyme from yeast was found to use specifically Appr>p but not N>p as a substrate (Culver et al., 1993). The CPDase gene in Saccharomyces cerevisiae was identified recently (Martzen et al., 1999; Nasr and Filipowicz, 2000), and a bacterially expressed recombinant yeast protein was shown to hydrolyze both Appr>p and N>p (Nasr and Filipowicz, 2000); it is likely that the specificity of this enzyme is controlled by some additional proteins or other factors. Deletion analysis indicated that, under laboratory conditions, the CPDase gene is not essential for yeast viability (Nasr and Filipowicz, 2000). On the other hand, this gene was identified recently in a screen designed to isolate genes whose expression elicits the SOS response in Escherichia coli (Perkins et al., 1999). It is important to note that E.coli and some other prokaryotes contain the phosphotransferase that is capable of synthesizing Appr>p, using the 2′-phosphorylated RNA and NAD+ as substrates (Spinelli et al., 1998). The physiological role of this protein in E.coli is unknown.
Although the sequence similarity (Figure 1) between plant and yeast CPDases is low (18% identity and 36% similarity), the proteins contain two prominent tetrapeptide motifs, H-X-T/S-X (X generally represents a hydrophobic amino acid) (Nasr and Filipowicz, 2000). Mutagenesis of the yeast protein has indicated that the two conserved histidines are essential for CPDase activity, whereas mutations of the threonine/serine residues inhibit the enzyme only partially, with the extent of inhibition being dependent on the nature of the substrate utilized (Nasr and Filipowicz, 2000). The importance of the H-X-T/S-X motifs is emphasized further by the fact that three other families of proteins with 2′,3′-cyclic nucleotide phosphodiesterase or related activities also share two copies of this tetrapeptide signature sequence (Nasr and Filipowicz, 2000). The first family comprises fungal RNA ligases involved in tRNA splicing. The ∼200 amino acid C-terminal domains of these enzymes have phosphodiesterase activity that opens the terminal cyclic phosphate in RNA to the 2′-phosphate (Xu et al., 1990; Westaway and Abelson, 1995); for the related RNA ligase from wheat germ, it has been shown that it also hydrolyzes 2′,3′-cyclic mononucleotides inefficiently (Tyc et al., 1987). To the second family belong brain CNPases, which hydrolyze 2′,3′-cyclic phosphates to 2′-phosphates in mono- or oligonucleotides (Sprinkle, 1989; Ballestero et al., 1999 and references therein). These proteins are abundant in the central nervous system of vertebrates. In goldfish and zebrafish, the protein expression is induced during optic nerve regeneration, but the precise function of the protein remains unknown (Ballestero et al., 1999). Importantly, a limited mutagenesis of the zebrafish CNPase identified His334 and Thr336, falling into one of the tetrapeptide signatures, as important for catalytic activity (Ballestero et al., 1999). A third family of proteins is constituted by bacterial and archaeal RNA ligases (Arn and Abelson, 1998), which are able to ligate tRNA half-molecules containing 2′,3′-cyclic phosphate and 5′-hydroxyl termini to products containing the 2′,5′- phosphodiester linkage (Arn and Abelson, 1996, 1998). Physiological substrates of these enzymes are not known. These enzymes have not been demonstrated to have a cyclic phosphodiesterase activity. However, the reaction they catalyze must involve hydrolysis of the 2′,3′-cyclic phosphate into 2′-phosphate (Arn and Abelson, 1998). Altogether, it appears that the four classes of enzymes discussed above belong to one protein superfamily; they all share two similarly spaced histidine-containing tetrapeptides (Nasr and Filipowicz, 2000), the proteins (Arn and Abelson, 1998; Nasr and Filipowicz, 2000) or their catalytic domains (Sprinkle, 1989; Xu et al., 1990) are ∼200 amino acids long, and all proteins catalyze hydrolysis of 2′,3′-cyclic phosphates to 2′-phosphates, or take part in mechanistically highly related reactions.

Fig. 1. Sequence alignment of CPDases from A.thaliana (SwissProt Y11650; first row) and S.cerevisiae (P53314; second row) and the phosphodiesterase domain of the yeast tRNA splicing ligase (S0003623, residues 657–827; third row). The numbering corresponds to the sequence of the Arabidopsis CPDase. Active site residues are red, and strictly conserved residues are blue. The assignment of secondary structure elements refers to the Arabidopsis CPDase. Because of an insertion in the yeast CPDase, β-strand β4 is depicted as two arrows while being one continuous strand. The figure was prepared with the program ALSCRIPT (Barton, 1993).
Results and discussion
We have solved the structure of the CPDase from A.thaliana, the first for any member of a family of cyclic nucleotide phosphodiesterases with a recently identified common sequence motif. The initial difficulties with phasing were overcome by employing the multiwavelength anomalous diffraction (MAD) methods, using a mercury derivative that yielded an electron density suitable for backbone tracing of the protein. Detailed analysis of the heavy atom positions showed trigonal arrangements for six of the eight mercury atoms that form two congruent equilateral triangles (a ∼25 Å) arranged in a parallel fashion. This geometry enabled positioning of a 3-fold non-crystallographic symmetry (NCS) axis, which was used to average the initial electron density with a correlation coefficient of 0.71. The averaged density allowed building of all three molecules in the asymmetric unit, with the exception of a surface loop constituted by residues 100–115. In the course of the refinement, this loop could be traced in molecule 1, yet the connection with the protein core (residues 112–115) is missing. In molecules 2 and 3, a total of 11 and seven residues, respectively, could not be located within this region.
Overall structure and topology
The CPDase from A.thaliana shows a previously unknown three-dimensional fold comprising a bilobal arrangement of two ααβ modules (Figure 2A) with the β-strands of each lobe interlaced with the other lobe (Figure 2B). Thus, this intrinsically symmetrical fold does not consist of separate domains, but rather represents a compact fold with two similar lobes. The primary topological structure is a tandem repeat of [α–β–α–β–turn–β] (see Figure 1 for assignment). Two antiparallel helices are located on the outer side of each lobe framing an antiparallel β-sheet that is wrapped around an accessible cleft of ∼12 Å diameter (Figure 2C). The lobe harboring the N- and C-terminal regions (‘terminal lobe’) consists of β1, β2, α2, α3, β6 and β7, and spans a four-stranded β-sheet, whereas the other lobe (‘transit lobe’, α1, β3, β4, β5 and α4) possesses a three-stranded β-sheet facing the central cavity, with an extra extended chain emulating one of the β-strands. However, this chain is not involved in proper hydrogen bonds to make it a true part of the sheet. The arrangement of the two β-sheets is reminiscent of an open barrel.



Fig. 2. Fold of the CPDase from A.thaliana and the active site. The ‘terminal lobe’ is green and the ‘transit lobe’ is blue. The exposed surface loop is red. (A) Schematic diagram of the secondary structure topology in CPDase. (B) View of the CPDase molecule. The active site is in the center of the protein and delimited by the β-sheets of both lobes and the helical turns at the bottom. The active site residues His42, Thr44, His119 and Ser121 are shown explicitly. (C) Stereo drawing of the Cα plot of molecule 1.
The lower side of the central water-filled cavity opposing the protein core is limited by α-turns from each of the two lobes connecting β3/β4 (transit lobe) and β6/β7 (terminal lobe), respectively. As observed with molecule 1, helix α3 leads into a surface-exposed loop pointing away from the protein core. The protein is ∼45 Å in width, 22 Å in depth and 30 Å in height. Four out of the six cysteine residues form disulfide bridges. Cys64 and Cys177 connect helix α3 and β-strand β7 in the terminal lobe, whereas Cys104 and Cys110 form a covalent link of the upstream and downstream parts of the exposed surface loop between α3 and β5. The two free cysteines were not involved in binding of the mercury compound used for heavy atom phasing. While Cys86 is shielded in a hydrophobic cavity at the boundaries of α1, α4 and β5, Cys159 is deeply buried in the active site cavity and thus not directly accessible as well.
Crystallographic arrangement
The NCS trimer (Figure 3) shows only a few specific intermolecular interactions, mainly of polar nature (e.g. ArgA155–GluB131 and GluA131–ArgC155). The most prominent location is certainly the path of the 3-fold NCS axis. Two arginine residues of each molecule (Arg31 and Arg38) gather in this region and coordinate a sulfate ion. The conical channel formed at the interface of the three NCS monomers is 7 and 4 Å in diameter and delimited by polar residues (Arg31/Arg38 on the wide side and Glu169 on the narrow side).

Fig. 3. The NCS trimer of CPDase (A.thaliana). Molecule 1 is green, molecule 2 is blue and molecule 3 is red. The view is along the 3-fold symmetry axis.
The presence of only three molecules in the asymmetric unit within a hexagonal cell of such large dimensions gives rise to a packing where the protein molecules form long and broad solvent channels parallel to the crystallographic 6-fold axis. As indicated by the Matthews parameter (Matthews, 1968), the solvent content of 67% is rather high and certainly accounts for the comparatively poor diffraction power of these crystals. This type of packing also reduces the contact sites between symmetry-related molecules; furthermore, these contacts are mostly non-specific. It is noteworthy that the exposed loop (residues 100–115) is one of the prominent contact sites between two symmetry partners and that aromatic stacking is observed with residues Phe108 and His103. Apart from this loop (see below) and minor variations in the N-terminal regions, the three NCS monomers agree quite well in their overall conformation (Table I).
Table I. Global geometrical analysis.
| Molecules 1/2 | Molecules 1/3 | Molecules 2/3 | |
|---|---|---|---|
| Agreement between NCS molecules | |||
| r.m.s.a distance (Cα) | 0.22 Å | 0.18 Å | 0.23 Å |
| r.m.s.a ΔB | 3.6 Å2 | 3.8 Å2 | 3.3 Å2 |
| Buried surfaceb | 862 Å2 | 701 Å2 | 786 Å2 |
Disordered residues (100–115) were excluded from the calculations.
aCalculated with LSQMAN (Kleywegt and Jones, 1997).
bCalculated with CNS (Brünger et al., 1998).
Active site
The repeated signature motif H-X-T/S-X and subsequent mutagenesis studies with S.cerevisiae CPDase (Nasr and Filipowicz, 2000) suggested that the active site should include His42 and His119, and thus these amino acids were predicted to be close to each other. Indeed, the crystal structure shows that both residues are located in a water-filled cavity and that the distance between them is ∼7 Å. The cavity constitutes the center of the protein and is accessible from one side only. In the structure presented here, a sulfate ion found in this region is coordinated by His42, His119 and Tyr124 (Figure 4A), and is surrounded by several water molecules. The arrangement is slightly reminiscent of that found in RNase A (Wlodawer and Sjolin, 1983; Wlodawer, 1985), where both catalytically active histidine residues are positioned at an acute angle, ready to clamp the substrate from two adjacent sides (Figure 4B). Unlike RNase A, however, the CPDase active site possesses different supporting residues, Ser121 and Thr44, which are located close to His119 and His42. The role of these residues during catalysis does not seem to be histidine activation but rather substrate coordination (see below). Because Tyr124 is involved in coordination of the active site sulfate as well, one might speculate about a possible role for this residue in the hydrolysis reaction. However, there are no biochemical data available yet to verify the importance of this residue. The putative active site cavity is lined with several aromatic residues (Trp12, Phe39, Phe84 and Trp171), where Phe39 is in a stacking conformation with Tyr124. The close spatial arrangement of both tryptophan residues of CPDase leads to considerable intrinsic fluorescence quenching, which was observed experimentally (data not shown). It is noteworthy that the unique triplet of phenyl groups in CPDase is located at the entry to the cavity.


Fig. 4. The active site of CPDase. (A) Stereo drawing of the active site showing the important residues (His42, Thr44, His119, Ser121 and Tyr124; see text) as well as the coordinated sulfate ion. The electron density (2Fo – Fc) was contoured at 1.0σ. (B) Comparison of the active sites of CPDase (A.thaliana) (left) and RNase A (Bos taurus) (right) (Wlodawer and Sjolin, 1983).
Proposed catalytic mechanism
A mechanism of enzymatic action for this family of CPDases can be proposed (Figure 5) based on the wild-type crystal structure and biochemical information about related proteins. His119 in its unprotonated form is probably activated by the backbone carbonyl of Met117 and interacts with a water molecule by removing a proton from it. The remaining hydroxide ion attacks the cyclic phosphate as a nucleophile. It cannot be decided at this stage whether this occurs as a concerted or a two-step reaction. The complex most likely passes through trigonal-bipyramidal geometry as in similar reactions, including RNase A (Wlodawer and Sjolin, 1983; Wlodawer, 1985) and ‘classical’ phosphorus chemistry (March, 1985). The leaving oxygen will then be in the apical position and is helped to leave by protonation from His42. Thr44, Ser121 and Tyr124 probably act as stabilizing groups by interacting with the phosphate oxygens. Tyr124 is not part of the previously identified motif but is also conserved in other proteins belonging to this subfamily of CPDases. Whereas in RNase A, Asp121 acts as an activator (Lewis base) for the initial base, in the case of CPDase the additional residues of the active site motif (Thr44, Ser121 and Tyr124) do not seem to fulfill the same role; rather, they keep the substrate locally fixed by providing the appropriate coordination. In the last step, the proton from the iminium group of His42 is transferred to the free 2′-oxygen of the phospho-ribosyl moiety. Because this transfer renders His42 in the unprotonated form and His119 in the protonated form, the system must be restored after the product leaves the cavity by transferring the iminium proton from His119 to His42, most likely via fast proton transfer using two ordered water molecules (TIP59 and TIP112) present in the cavity bridging both histidine residues with distances between 3.6 and 4.9 Å. From the coordination geometry found in the present structure, it seems very likely that His119 acts as the base. An ordered water molecule (TIP65) is found at the same position in all three molecules of the present structure with distances of 2.7 Å to His119 (atom Nε) and 3.3 Å to the active site sulfate (atom S). Based on these findings, we favor His119 as the initiating residue of catalysis, although the reverse mechanism cannot be excluded without more detailed studies determining the individual pKa values for the two catalytic histidine residues, as well as without additional structural data for complexes of CPDases with inhibitors.
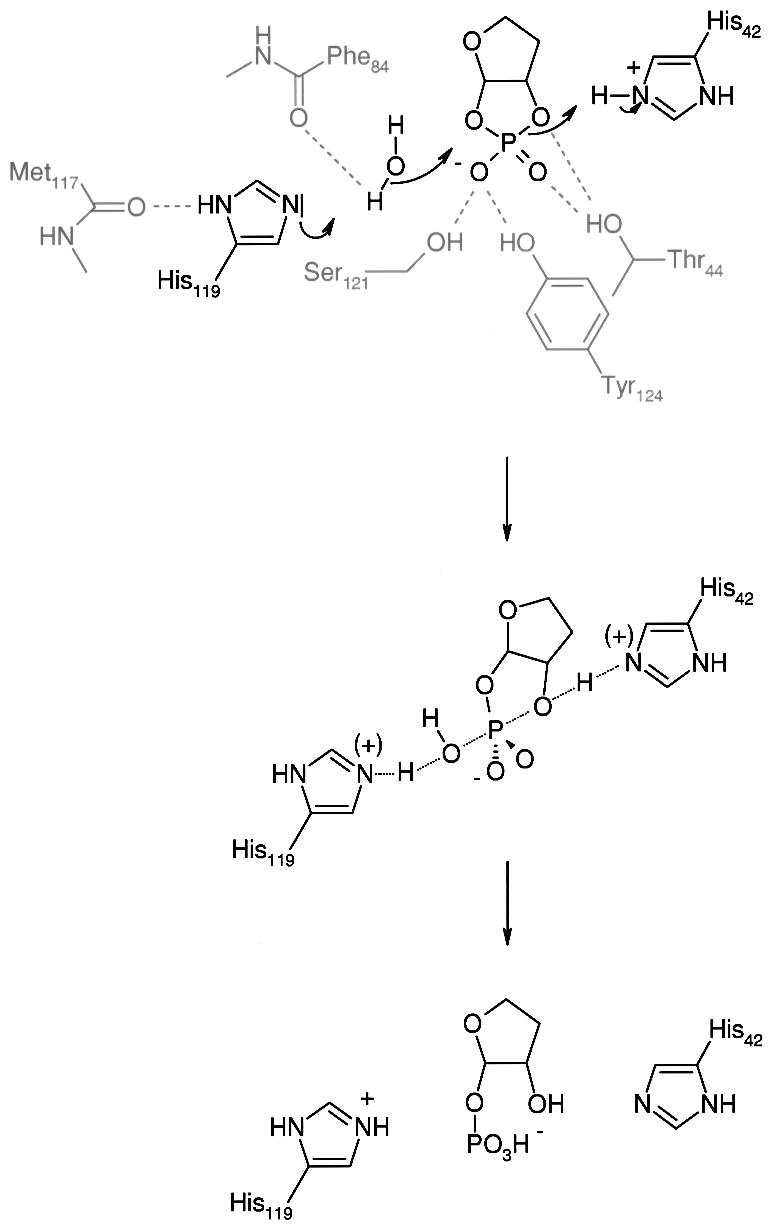
Fig. 5. Proposed enzymatic mechanism for the cleavage of cyclic phosphates by CPDase. The substrate drawn shows the ribosyl moiety of Appr>p. The 4″-extension has been omitted for clarity. The first row shows the starting situation as seen in the current crystal structure. A water molecule attacks the phosphate as nucleophile, leading to a trigonal-bipyramidal geometry for the phosphorus species (second row). After protonation of the 2″-oxygen, a 1″-phospho-ribosyl species remains (third row). Restoration of the active site histidines probably occurs via fast proton transfer involving conserved water molecules of the hydrophilic cavity (see text).
In a recent study (Nasr and Filipowicz, 2000) with the CPDase from S.cerevisiae, site-directed mutagenesis was performed within the two conserved tetrapeptide signature motifs. This work showed that both of the conserved histidine residues (His39 and His150; numbers refer to S.cerevisiae CPDase) are essential for CPDase activity. Furthermore, whereas mutations of hydrophobic residues of the motif had no effect on the enzymatic activity, mutation of Thr41 and Ser152 (S.cerevisiae CPDase numbering) to alanine reduced CPDase activity to 2 and 30%, respectively, with A>p as substrate. With Appr>p, in contrast, the reduction of activity was significantly lower: the T41A mutant displayed 49% of the wild-type activity and the S152A mutant showed no change in activity at all. These results support the proposed mechanism as outlined above and also emphasize the close relationship between CPDases from different organisms. Several attempts to obtain a crystal structure of the CPDase in complex with a substrate-mimicking agent (A>p and uridine vanadate), which were performed in order to validate the structural assumption needed for the elucidation of the mechanism, have not been successful so far (data not shown).
It is noteworthy that an exposed surface loop connecting α3 and β5 (residues 101–115) is positioned in the immediate vicinity of the active site. The loop is poorly defined in the present structure, most likely due to the inherent flexibility of this region. It is highly disordered and could not be traced at all in molecules 2 and 3. However, in molecule 1, it was possible to build a model lacking only residues 112–115, which connect one end of the loop with the protein core. Remarkably, one of the two disulfide bridges is located in this region, apparently connecting the upstream and downstream parts of the loop. From the considerable disorder within this region, one can conclude that the conformation of the loop is most likely not the same for the three molecules present in this crystal structure. Furthermore, we speculate that its functional role involves covering/uncovering of the active site cavity. The conformation as seen in molecule 1 is an obtuse angle between the protein core and the surface loop (∼120°). One can easily envisage a closed conformation of the loop shutting the active site cavity by reducing the angle between the loop and the protein core in a swing-like manner (see Figure 6).

Fig. 6. The exposed surface loop probably acts as a flap. GRASP (Nicholls, 1992) representation of molecule 1 (acidic residues are red, basic residues are blue). The exposed surface loop (on the right) is likely to perform a loop motion in a swing-like manner. Lys105 and His107/Phe108 might play a role as locking residues in the ‘closed’ conformation.
Comparison with other proteins
Although the current structure of CPDase from A.thaliana displays a previously unknown fold, there are nevertheless some structural similarities with other proteins, especially some kinases and proteins involved in RNA processing. The topological arrangement of one half of the CPDase molecule resembles a structural motif known from the ααβ module in glycerol kinase (Feese et al., 1998), which is responsible for the catalytic activity of that protein. However, the catalytic site in this case is formed at the tetrameric interface of four glycerol kinase monomers and therefore is not applicable to the interpretation of the mechanism employed by the CPDase.
Many proteins involved in RNA processing or modification contain a highly modular structure where the RNA-binding module is constituted by a 60–90 residue domain. Three commonly identified RNA-binding modules include the ribonucleoprotein domain (RNP, also known as the RBD or RRM domain), the K-homology domain (KH) and the double-stranded RNA-binding domain (dsRBD) (for a review see Varani, 1998). Numerous structures for the representatives of each of the three types of RNA-binding modules are known already, and they all share the common topology of two (anti-)parallel α-helices on one side of the protein and a three- or four-stranded β-sheet on the other side, where the latter is involved in RNA binding. These topologies show a striking similarity to either of the two lobes of the CPDase from A.thaliana, although the connectivity between the chains is different in each CPDase lobe from that in the three RNA-binding domains. This comparison shows that although a different fold was adopted for this representative of a new class of cyclic nucleotide phosphodiesterases, related structural elements are employed for similar tasks. This particular example might be interesting in terms of structural evolution of proteins involved in RNA and nucleotide metabolism.
Correlation between structural and biochemical data
The physiologically active species of CPDase is most likely to be monomeric, because no experimental data have been reported that would support the requirement for a CPDase multimer for activity. The structure reported here also supports the assumption of a monomeric active species. When calculating the buried surface areas between pairs of each of the three monomers in the present structure, values between 700 and 860 Å2 are found (see Table I). These numbers are slightly higher than the ‘threshold’ given in Janin (1997) for non-specific protein–protein interactions that are due simply to crystal packing effects. However, we still suggest that the trimeric NCS arrangement is elicited by the crystal packing since the intermolecular contacts are of non-specific nature and a trimeric aggregate in solution can be excluded based on our ultracentrifugation results (see below). However, SDS–PAGE under denaturing/reducing conditions indicated that the protein could apparently exist as a dimer. To elucidate this behavior in more detail, we subjected samples of CPDase from A.thaliana to analytical ultracentrifugation. Global fitting of sedimentation equilibrium data to a self-association model works best with the assumption of a monomer–dimer equilibrium and an apparent association constant of Ka = 1 × 104 M–1, corresponding to ∼15% dimer content in the concentration range tested (data not shown). This weak association constant does not support a physiological role for the dimer, nor does it explain the presence of the dimer band in the SDS–PAGE experiments sufficiently, since a weak dimeric species should not survive under these conditions. We assume that the second species observed in electrophoresis experiments was a non-specific assembly of two CPDase monomers that was formed during denaturation of the native sample. During denaturation, the protein populates this assembled state, probably due to kinetic reasons. A possible mechanism would be the unfolding of the dimeric species where the two molecules cannot separate their chains efficiently because of special conformations in the native (dimeric) state.
With respect to substrate specificity, the plant CPDase of the current study is clearly different from other proteins with cyclic phosphodiesterase activity. The tRNA splicing ligase acts on 2′,3′-cyclic phosphate-terminated RNA (Phizicky et al., 1986); it accepts 2′,3′-cyclic mononucleotides as substrates only very inefficiently (Tyc et al., 1987). CNPase from vertebrate brain hydrolyzes 2′,3′-cyclic phosphates in mono- and oligoribonucleosides (Sprinkle, 1989); however, neither one of these two families of enzymes is able to hydrolyze the 1″,2″-cyclic phosphate of Appr>p (Culver et al., 1994; our unpublished results). In this context, structural elucidation of protein– substrate complexes of CPDase will help to clarify the specificity mechanism of this enzyme.
Arabidopsis thaliana CPDase as a model enzyme for a family of proteins
Appr>p is a product generated during tRNA splicing in plants, yeast and also vertebrates. It is processed further into Appr-1″p in a reaction catalyzed by a CPDase, which cleaves the 2″-phosphoester. CPDases have, to date, been characterized in wheat, A.thaliana and yeast. These CPDases constitute one group within a large family of proteins, which includes at least four different classes of enzymes having cyclic phosphodiesterase or related activities. Members of this family, or enzymatically competent domains thereof, are of comparable lengths (∼200 residues) and share two similarly spaced tetrapeptide signature motifs (see Introduction).
The structure of CPDase, as described in the present study, is the first for a member of the cyclic phosphodiesterase family discussed above and therefore serves as a model for characterization of this group of enzymes. Topologically, the structural elements, as determined in the current crystal structure, show similarities to proteins involved in RNA binding and to kinases. However, the arrangement of structural elements within A.thaliana CPDase results in a new protein fold with unique features. A surface loop shows high disorder in the crystal structure but could be traced in one of the three molecules in the asymmetric unit. The loop is in an ‘open’ conformation, granting unlimited access to a water-filled cavity that contains the active site, harboring the residues of the tandem signature motif. These residues, as well as Tyr124, participate in coordination of a sulfate ion in the apo enzyme. We propose an enzymatic mechanism, which employs the nucleophilic attack of a water molecule that is activated by His119. Further catalysis follows similar reactions, as, for instance, observed with RNase A. However, additional residues of the signature motif (Thr44 and Ser121) and also Tyr124 seem not to take part in the reaction itself, but serve rather as coordinating residues, helping to fix the substrate geometrically. Based on the current structure, a loop motion seems possible, which leads to a ‘closed’ conformation where the active site is covered. Further studies will have to be undertaken to test this hypothesis, as well as to determine the mechanism of substrate specificity.
Materials and methods
Preparation of proteins
The NcoI–BamHI fragment of Arabidopsis CPDase (SwissProt accession no. Y11650) as described by Genschik et al. (1997a) was inserted into the pET11d vector, yielding a construct with eight additional C-terminal residues (GSHHHHHH). Expression was carried out in E.coli BL21(DE3) cells. A 1 l culture of transformed BL21(DE3) cells was grown overnight at 37°C in Luria–Bertani (LB) medium containing 50 mg/l ampicillin. The overnight culture was diluted into 8 l of LB medium (50 mg/l ampicillin) and grown at 37°C until the absorbance at 600 nm reached values >1.0. Isopropyl-β-d-thiogalactopyranoside (IPTG) was then added to a final concentration of 0.5 mM and the concentration of ampicillin was increased 4-fold. Cell growth after induction was continued overnight.
After the cells were harvested, the pellet was resuspended in 100 ml of buffer D1 (1 mM EDTA, 100 mM NaCl, 5 mM benzamidinium chloride, 1 mM phenylmethylsulfonyl fluoride, 0.1% Triton X-100, 20 mM Tris–HCl pH 8.0) and sonicated. Insoluble components were removed by ultracentrifugation at 100 000 g (4 h, 8°C). The supernatant was diluted 5-fold and applied to Ni-nitrilotriacetic acid (Ni-NTA; Qiagen, Valencia, CA) resin, equilibrated with chromatography buffer D2 (100 mM NaCl, 50 mM Tris–HCl pH 8.0). Elution was done discontinuously with increasing imidazole concentrations (20, 50, 100 and 200 mM imidazole in buffer D2). CPDase was obtained from fractions eluting at 100–200 mM imidazole. Concentration and removal of imidazole were carried out via ultrafiltration with Centricon devices (Millipore, Bedford, MA).
Preparation of Se-methionine-labeled protein followed the protocols of Yang et al. (1990) and Van Duyne et al. (1993), which are based on methionine shutdown within the expression organism. Briefly, an overnight culture of transformed E.coli BL21(DE3) was grown in 1 l of LB medium and 50 mg/l ampicillin. The cells from the overnight culture were harvested and resuspended in 6 l of M9a medium (6 g/l Na2HPO4, 3 g/l K2HPO4, 0.5 g/l NaCl, 1 g/l NH4Cl); 20 ml of M9-add (20% glucose, 2.5 g/l bovine serum albumin, 0.5 M MgSO4, 5 g/l ampicillin) was added per 1 l of culture. When the absorbance at 600 nm reached A600 nm = 0.6, 10 ml of Met-shutdown mix (10 g/l lysine hydrochloride, 10 g/l threonine, 10 g/l phenylalanine, 5 g/l leucine, 5 g/l isoleucine, 5 g/l valine, 5 g/l seleno-l-methionine) were added per 1 l of culture. After additional incubation for 20 min, protein expression was induced by addition of IPTG to a final concentration of 0.5 mM. The culture was incubated overnight at 37°C. Further processing was as described for the preparation of wild-type protein.
Identification of proteins
The identity of recombinant wild-type CPDase was confirmed by N-terminal sequencing, which showed the presence of the initiating methionine. Mass spectra of diluted acidified samples were obtained by electrospray ionization on an HP1100 LCMS system (Agilent Technologies) and yielded a molecular mass of 21 477 g/mol compared with a calculated mass of 21 481 g/mol. The difference of 4 g/mol can be explained by the presence of two disulfide bridges. Mass spectra of crystals obtained from the Se-Met-labeled protein showed that only 5% of the protein had the methionine derivative incorporated, which explained our failure to obtain useful phases from crystals of this material.
Analytical ultracentrifugation
A Beckman Optima Model XL-A analytical ultracentrifuge equipped with a four-place An-Ti rotor was used for sedimentation equilibrium experiments. Three 12 mm cells equipped with carbon-filled, double channel centerpieces and plane quartz windows were used. Protein solutions with A280 nm ranging from 0.15 to 0.45 were loaded on the right (200 µl/channel), with the corresponding reference buffer on the left (220 µl/channel). The reference buffer was the dialysate buffer that contained 100 mM NaCl, 20 mM Tris–HCl pH 8.0 with a density of ρ = 1.0030 g/ml at 20°C, as determined with an Anton Paar Model DMA 58 densitometer. After equilibration at 3000 r.p.m. and 20°C, at which reference wavelength and radial scans were performed, the rotor was accelerated to the selected experimental speed where scans were collected at 4 h intervals for 48 h. The proteins were run at two speeds (17 000 and 20 000 r.p.m.). Radial scans were recorded at 280 nm in a step mode with 0.001 cm steps and 13 averages. Equilibrium was attained when two consecutive scans 4 h apart were indistinguishable. Analysis of ultracentrifugation data was performed with the software package from Beckman, Inc. and A.P.Minton (NIDDK, NIH). The partial specific volume of 0.710 ml/g was calculated from amino acid sequences and the values of Zamyatnin (1984).
Crystallization, data collection and derivatization
Crystals of CPDase were grown in acidic ammonium sulfate [1.0 M (NH4)2SO4, 0.1 M NaOAc pH 4.5] and ethanol/ammonium sulfate [0.9 M (NH4)2SO4, 10% EtOH] conditions using the vapor diffusion method (hanging drop); the drops consisted of 3 µl of reservoir solution and 3 µl of protein solution (15 mg/ml in 100 mM NaCl, 20 mM Tris–HCl pH 8.0). Both conditions yield obelisk-shaped crystals of 2–5 mm length and 0.1–0.5 mm width. In-house diffraction data, mostly limited to a resolution of 3.5–2.8 Å, were measured on an MAR image plate system (MAR Research, Hamburg, Germany) mounted on a Rigaku rotating anode generator (Cu-Kα radiation, λ = 1.5418 Å) under cryo conditions at 100 K using an Oxford Cryosystem (Oxford, UK). The crystals were prepared for cryo data collection by flash soaking (<1 min) in the cryo buffer [1.7 M (NH4)2SO4, 0.1 M NaOAc pH 5.0, 10% glycerol]. Heavy atom derivatives were prepared by flash-soaking of appropriate crystals in the cryo buffer containing varying amounts of the respective heavy atom compound. MAD data sets were collected at beamline X9B of the National Synchrotron Light Source, Brookhaven, NY, equipped with an ADSC Quantum-4 CCD detection system. Data analysis and reduction were performed with the HKL2000 software (Otwinowski and Minor, 1997); the statistics for different data sets are summarized in Table II.
Table II. Diffraction data statistics.
| Data set | CNP3 | CNP6 | CNP10 | CNP17 |
|---|---|---|---|---|
| X-ray source | In-house | In-house | In-house | In-house |
| Derivative | Native | (C2H5HgO)2HPO2 | GdCl3 | HoCl3 |
| Space group | P6122 | P6122 | P6122 | P6122 |
| Cell dimensions (Å) | 125.1, 125.1, 210.0 | 127.5, 127.5, 210.0 | 124.9, 124.9, 209.2 | 125.1, 125.1, 208.7 |
| Resolution (Å) | 2.85 | 3.8 | 3.0 | 3.5 |
| Total no. of measurements | 125 538 | 264 592 | 211 919 | 281 006 |
| No. of independent measurements | 21 901 | 10 561 | 18 028 | 12 841 |
| Completenessa (%) | 88.9 (87.0) | 88.0 (62.3) | 89.9 (75.7) | 80.3 (59.9) |
| Multiplicity | (8.5)b | (6)b | 4.1 | (6.5)b |
|
Rmergea (%) |
10.8 (23.2) |
12.8 (19.2) |
9.4 (11.6) |
14.7 (30.6) |
| Data set |
CNP19_IN |
CNP19_PK |
CNP19_RM |
CNP18_ALL |
| X-ray source | X9B | X9B | X9B | X9B |
| Derivative | (C2H5HgO)2HPO2 | (C2H5HgO)2HPO2 | (C2H5HgO)2HPO2 | Se-Met |
| Wavelength (Å) | 1.0094 | 1.0060 | 0.9537 | – |
| Space group | P6122 | P6122 | P6122 | P6122 |
| Cell dimensions (Å) | 126.7, 126.7, 210.3 | 124.6, 124.6, 206.8 | 126.7, 126.7, 210.2 | 125.9, 125.9, 209.9 |
| Resolution (Å) | 2.5 | 2.5 | 2.5 | 2.5 |
| Total no. of measurements | 337 838 | 333 441 | 366 016 | 1 085 650 |
| No. of independent measurements | 35 173 | 33 522 | 35 145 | 34 305 |
| Completenessa (%) | 92.1 (75.1) | 93.5 (80.7) | 94.9 (90.2) | 98.8 (97.8) |
| Multiplicity | (5)b | (5)b | (5)b | (18)b |
| Rmergea (%) | 8.2 (39.6) | 9.7 (34.2) | 10.1 (40.9) | 8.1 (14.4) |
aNumber for last shell is given in parentheses.
bEstimated from SCALEPACK output.
Rmerge = Σ|I – <I>|/ΣI, where I is the observed intensity and <I> is the average intensity obtained from multiple observations of symmetry-related reflections after rejections.
Indexing of diffraction patterns was successful, assuming hexagonal symmetry. Because the intensities along the [00l] direction showed significant values only for l = 6n, the presence of a screw operation (61 or 65) was indicated. Additionally, self-rotation functions were calculated with GLRF (Tong and Rossmann, 1990), starting with space group P6, revealing the presence of crystallographic 2-fold axes (κ = 180°) at Ψ = 0°, 30°, 60° and 90°. NCS could not be identified from self-rotation functions. Thus, the space group was determined to be P6122 or P6522, with an asymmetric unit containing three molecules, corresponding to the molar volume of 3.68 A3/Da [solvent content 67% (Matthews, 1968)].
Phase calculation and structure determination
Although a three-wavelength MAD data set was collected for the putative Se-Met form of the protein, the low level of Se-Met incorporation, determined subsequently, made any phasing by that approach impossible. Because the phases obtained from multiple isomorphous replacement (MIR) with CNP6 (mercury), CNP10 (gadolinium) and CNP17 (holmium) did not yield an interpretable electron density map, the structure of CNP was solved by a combination of MAD and single isomorphous replacement (SIR) methods, using the mercury derivative obtained from (C2H5HgO)2HPO2 as described above (data set CNP19). All derivative MIR data were analyzed with the native data set CNP3; CNP18_ALL was used as the native data set in the SIR/MAD approach because, as mentioned above, the Se-Met incorporation was <5%, making these data native for all practical purposes.
Heavy atom positions were found by inspection of isomorphous difference Patterson maps, as well as with direct methods using the programs SOLVE (Terwilliger and Berendzen, 1999) and SnB (Blessing and Smith, 1999; Weeks and Miller, 1999). The positions were confirmed by difference Fourier methods using PHASES (Furey and Swaminathan, 1995) and Patterson vector superposition methods as implemented in SHELXS-97 (Sheldrick, 1991). It became clear at this stage that the screw operation was 61 and not 65; thus, the space group was determined to be P6122. The refinement of heavy atom parameters and calculation of SIR/MAD phases were carried out with PHASES (see Table III for a summary of the phasing statistics). Even after phase improvement and solvent flattening using PHASES, DM (CCP4, 1994) or SHARP (de la Fortelle and Bricogne, 1997), the electron density maps were only interpretable to the extent of allowing us to trace the protein backbone. A more detailed analysis of the mercury and holmium positions revealed a trigonal arrangement within the unit cell. Assuming a 3-fold NCS using the axis as given by the heavy atom triangles and the constructed protein backbone model, NCS averaging was attempted.
Table III. MAD phasing statistics.
| |
Dispersive |
Anomalous |
||||
|---|---|---|---|---|---|---|
| Inflection | Peak | Remote | Inflection | Peak | Remote | |
| Data set | CNP19_IN | CNP19_PK | CNP19_RM | CNP19_IN | CNP19_PK | CNP19_RM |
| Wavelength (Å) | 1.0094 | 1.0060 | 0.9537 | 1.0094 | 1.0060 | 0.9537 |
| Figure of merit | 0.453 | 0.451 | 0.458 | – | – | – |
| Phasing powera | 3.66 | 3.81 | 3.88 | 1.35 | 1.59 | 1.42 |
| M.r.e.b | 0.50 | 0.50 | 0.49 | 0.51 | 0.50 | 0.51 |
| Mean bias | 100.6 | 100.8 | 100.2 | 89.9 | 89.9 | 89.7 |
| RCullisc | 0.495 | 0.483 | 0.476 | n.a.d | n.a.d | n.a.d |
| RKraute | 0.087 | 0.091 | 0.090 | – | – | – |
| No. of Hg sites | 8 | |||||
| Total no. of reflections | 21 679 | |||||
| Mean figure of merit | 0.769 | |||||
aPhasing power: 
The NCS operation was calculated and refined with PHASES using manually determined axis parameters as starting values. Briefly, the electron density map as calculated from initial phase refinement was averaged applying a proper 3-fold symmetry with the axis parameters φ = 103.84°, ψ = 111.95°, x0 = 8.04, y0 = 36.73, z0 = 103.83 (correlation coefficient: 0.71). The map was skewed and a mask was created, which was used for averaging of the initial electron density. The averaged map was vastly superior to the starting one, allowing identification of the amino acid sequence in the electron density map and model building.
Model building and refinement
Using the averaged electron density map, the protein model was built with the program O (Jones et al., 1991), starting with the backbone model. Initial refinement was performed by conjugate gradient protocols and NCS restraints with X-PLOR 3.1 (Brünger, 1992a). At a later stage, CNS 1.0 (Brünger et al., 1998) was used for refinement to include a bulk solvent model and overall anisotropic B-factor correction (B11 = –13.054, B22 = –13.054, B33 = 26.109, B12 = –13.356, B13 = 0, B23 = 0) with the conjugate gradient method and a maximum-likelihood residual target. Typical protocols consisted of two cycles of positional and individual isotropic B-factor refinement runs. The NCS restraints were released slowly and a manually built water model was included in the refinement. The initial R-factor of the unrefined model was 0.480 (Rfree = 0.494) and dropped to 0.255 (Rfree = 0.331) without the application of corrections. For the final model (including 229 water molecules and nine sulfate ions), the R-factor was 0.196 (Rfree = 0.265). Geometrical properties of the model were analyzed with the program PROCHECK (Laskowski et al., 1993); Table IV summarizes the refinement statistics.
Table IV. Refinement statistics.
| Molecule 1 or total | Molecule 2 | Molecule 3 | |
|---|---|---|---|
| Refinement | |||
| resolution | 20–2.5 Å | ||
| no. of reflections used for refinement (F >2σFo) | 28 263 | ||
| no. of non-H atoms | 1420 | 1346 | 1376 |
| visible residues | 179 | 170 | 174 |
| R-factora | |||
| no. of reflections in working set | 25 481 (90.2%) | ||
| no. of reflections in test set | 2782 (9.8%) | ||
| Rb | 0.196 (0.288) | ||
| Rfreeb | 0.265 (0.362) | ||
| Temperature factors | |||
| average B-factor (Å2) | 44.1 | 45.0 | 48.3 |
| r.m.s. deviation for bonded atoms (Å2) | 4.27 | 4.33 | 4.14 |
| Geometry | |||
| r.m.s. deviation of bond lengths | 0.018 | ||
| r.m.s. deviation of bond angles | 2.10° | ||
| r.m.s. deviation of dihedral angles | 25.1° | ||
| r.m.s. deviation of improper angles | 1.2° | ||
| Ramachandran plot | |||
| residues in most favored regions | 86.4% | ||
| residues in additionally allowed regions | 13.6% | ||
| Solvent statistics | |||
| no. of water molecules | 81 | 85 | 63 |
| no. of sulfate ions | 2 + 1 | 3 | 3 |
aR-factor = Σ||Fo| – |Fc||/Σ|Fo|, where Fo and Fc are the observed and calculated structure factors, respectively; Rfree defined in Brünger (1992b).
bNumber for last shell is given in parentheses.
Graphical representation
Ribbon drawings and graphical representations of electron densities and protein models in this work were generated by MOLSCRIPT (Kraulis, 1991) or BOBSCRIPT (Esnouf, 1999) and were rendered with POVRay (http://www.povray.org).
Accession numbers
Coordinates and structure factors have been deposited in the Protein Data Bank with accession number 1fsi.
Acknowledgments
Acknowledgements
This paper is dedicated to Professor Hans-Jürgen Bestmann on the occasion of his 75th birthday. We thank Hal Dixon (King’s College, Cambridge, UK) and Hans-Georg Beisel (MPI für Biochemie, Martinsried) for helpful discussions, Lewis Pannell (Structural Mass Spectrometry Facility, NIDDK, NIH) for mass spectrometry, Young Kim (Protein Chemistry Laboratory, SAIC, Frederick) for N-terminal sequencing, Zbigniew Dauter (beamline X9B, NSLS, Brookhaven National Laboratory) for advice on data collection and analysis, and Anne Arthur for editorial comments. A.H. wishes to thank Robert Huber for access to the resources of MPI für Biochemie, Martinsried. The Friedrich Miescher-Institut is part of the Novartis Research Foundation.
References
- Arn E.A. and Abelson,J.N. (1996) The 2′–5′ RNA ligase of Escherichia coli. Purification, cloning, and genomic disruption. J. Biol. Chem., 271, 31145–31153. [DOI] [PubMed] [Google Scholar]
- Arn E. and Abelson,J. (1998) RNA ligases: function, mechanism and sequence conservation. In Simons,R. and Grunberg-Manago,M. (eds), RNA Structure and Function. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, pp. 695–726. [Google Scholar]
- Ballestero R., Dybowski,J., Levy,G., Agranoff,B. and Uhler,M. (1999) Cloning and characterization of zRICH, a 2′,3′-cyclic-nucleotide 3′-phosphodiesterase induced during zebrafish optic nerve regeneration. J. Neurochem., 72, 1362–1371. [DOI] [PubMed] [Google Scholar]
- Barton G.J. (1993) ALSCRIPT: a tool to format multiple sequence alignments. Protein Eng., 6, 37–40. [DOI] [PubMed] [Google Scholar]
- Blessing R. and Smith,G. (1999) Difference structure factor normalisation for heavy-atom or anomalous-scattering substructure determinations. J. Appl. Crystallogr., 32, 664–670. [Google Scholar]
- Brünger A.T. (1992a) X-PLOR Version 3.1: System for X-ray Crystallography and NMR. Yale University Press, New Haven, CT. [Google Scholar]
- Brünger A.T. (1992b) The free R value: a novel statistical quantity for assessing the accuracy of crystal structures. Nature, 355, 472–474. [DOI] [PubMed] [Google Scholar]
- Brünger A.T. et al. (1998) Crystallography and NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr. D, 54, 905–921. [DOI] [PubMed] [Google Scholar]
- Collaborative Computational Project Number 4 (1994) The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D, 50, 760–763. [DOI] [PubMed] [Google Scholar]
- Culver G., McCraith,S., Zillmann,M., Kierzeck,R., Michaud,N., LaReau,R., Turner,D. and Phizicky,E. (1993) An NAD derivative produced during transfer RNA splicing: ADP-ribose-1′′,2′′-cyclic-phosphate. Science, 261, 206–208. [DOI] [PubMed] [Google Scholar]
- Culver G., Consaul,S., Tykowski,K., Filipowicz,W. and Phizicky,E. (1994) tRNA splicing in yeast and wheat germ. A cyclic phosphodiesterase implicated in the metabolism of ADP-ribose. J. Biol. Chem., 269, 24928–24934. [PubMed] [Google Scholar]
- Culver G.M., McCraith,S.M., Consaul,S.A., Stanford,D.R. and Phizicki,E.M. (1997) A 2′-phosphotransferase implicated in tRNA splicing is essential in Saccharomyces cerevisiae. J. Biol. Chem., 272, 13203–13210. [DOI] [PubMed] [Google Scholar]
- de la Fortelle E. and Bricogne,G. (1997) Maximum-likelihood heavy-atom parameter refinement for multiple isomorphous replacement and multiwavelength anomalous diffraction methods. Methods Enzymol., 276, 472–494. [DOI] [PubMed] [Google Scholar]
- Esnouf R.M. (1999) Further additions to MolScript version 1.4, including reading and contouring of electron-density maps. Acta Crystallogr. D, 55, 938–940. [DOI] [PubMed] [Google Scholar]
- Feese M., Faber,R., Bystrom,C., Pettigrew,D. and Remington,S. (1998) Glycerol kinase from Escherichia coli and an Ala65Thr mutant: the crystal structures reveal conformational changes with implications for allosteric regulations. Structure, 6, 1407–1418. [DOI] [PubMed] [Google Scholar]
- Filipowicz W. and Shatkin,A. (1983) Origin of splice junction phosphate tRNAs processed by HeLa cell extract. Cell, 32, 547–557. [DOI] [PubMed] [Google Scholar]
- Furey W. and Swaminathan,S. (1995) PHASES-95: a program package for the processing and analysis of diffraction data from macromolecules. Methods Enzymol., 277, 590–619. [DOI] [PubMed] [Google Scholar]
- Genschik P., Hall,J. and Filipowicz,W. (1997a) Cloning and characterisation of the Arabidopsis cyclic phosphodiesterase which hydrolyses ADP-ribose-1″,2″-cyclic-phosphate and nucleoside-2′,3′-cyclic-phosphates. J. Biol. Chem., 272, 13211–13219. [DOI] [PubMed] [Google Scholar]
- Genschik P., Billy,E., Swianiewicz,M. and Filipowicz,W. (1997b) The human RNA 3′-terminal phosphate cyclase is a member of a new family of proteins conserved in Eucarya, Bacteria and Archaea. EMBO J., 16, 2955–2967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez T.N., Sidrauski,C., Dorfler,S. and Walter,P. (1999) Mechanism of non-spliceosomal mRNA splicing in the unfolded protein response pathway. EMBO J., 18, 3119–3132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janin J. (1997) Specific versus non-specific contacts in protein crystals. Nature Struct. Biol., 4, 973–974. [DOI] [PubMed] [Google Scholar]
- Jones T.A., Zou,J.Y., Cowan,S. and Kjeldgaard,M. (1991) Improved methods for building protein models in electron density maps and location of errors in these models. Acta Crystallogr. A, 47, 110–119. [DOI] [PubMed] [Google Scholar]
- Kleywegt G.J. and Jones,T.L. (1997) Detecting folding motifs and similaritites in protein structures. Methods Enzymol., 277, 525–545. [DOI] [PubMed] [Google Scholar]
- Kraulis P.J. (1991) MOLSCRIPT: a program to produce both detailed and schematic plots of protein structures. J. Appl. Crystallogr., 24, 946–950. [Google Scholar]
- Laski F.A., Fire,A.Z., RajBhandary,U.L. and Sharp,P.A. (1983) Characterization of tRNA precursor splicing in mammalian extracts. J. Biol. Chem., 258, 11974–11980. [PubMed] [Google Scholar]
- Laskowski R.A., MacArthur,M.W., Moss,D.S. and Thornton,J.M. (1993) PROCHECK: program to check the stereochemical quality of protein structures. J. Appl. Crystallogr., 26, 283–291. [Google Scholar]
- March J. (1985) Advanced Organic Chemistry: Reactions, Mechanisms and Structure. John Wiley & Sons, New York, NY. [Google Scholar]
- Martzen M.R., McCraith,S.M., Spinelli,S.L., Torres,F.M., Fields,S., Grayhack,E.J. and Phizicki,E.M. (1999) A biochemical genomics approach for identifying genes by the activity of their products. Science, 286, 1153–1155. [DOI] [PubMed] [Google Scholar]
- Matthews B.W. (1968) Solvent content of protein crystals. J. Mol. Biol., 33, 491–497. [DOI] [PubMed] [Google Scholar]
- Nasr F. and Filipowicz,W. (2000) Characterization of the Saccharomyces cerevisiae cyclic nucleotide phosphodiesterase involved in the metabolism of ADP-ribose 1′′,2′′-cyclic phosphate. Nucleic Acids Res., 28, 1676–1683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholls A. (1992) GRASP: Graphical Representation and Analysis of Surface Properties. Columbia University, New York, NY. [Google Scholar]
- Niwa M., Sidrauski,C., Kaufman,R.J. and Walter,P. (1999) A role for presenilin-1 in nuclear accumulation of Ire1 fragments and induction of the mammalian unfolded protein response. Cell, 99, 691–702. [DOI] [PubMed] [Google Scholar]
- Otwinowski Z. and Minor,W. (1997) Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol., 276, 307–326. [DOI] [PubMed] [Google Scholar]
- Perkins E.L., Sterling,J.F., Hashem,V.I. and Resnick,M.A. (1999) Yeast and human genes that affect the Escherichia coli SOS response. Proc. Natl Acad. Sci. USA, 96, 2204–2209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phizicky E.M. and Greer,C. (1993) Pre-tRNA splicing: variation on a theme or exception to the rule? Trends Biochem. Sci., 18, 31–34. [DOI] [PubMed] [Google Scholar]
- Phizicky E., Schwartz,R. and Abelson,J. (1986) Saccharomyces cerevisiae tRNA ligase. J. Biol. Chem., 261, 2978–2986. [PubMed] [Google Scholar]
- Sheldrick G. (1991) Tutorial on automated Patterson interpretation to find heavy atoms. In Moras,D., Podjarny,A. and Thierry,J. (eds), Crystallographic Computing 5. Oxford University Press, Oxford, UK, pp. 145–157. [Google Scholar]
- Spinelli S.L., Malik,H.S., Consaul,S.H. and Phizicki,E.M. (1998) A functional homolog of a yeast tRNA splicing enzyme is conserved in higher eukaryotes and in Escherichia coli.Proc. Natl Acad. Sci. USA, 95, 14136–14141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sprinkle T.J. (1989) 2′,3′-Cyclic nucleotide 3′-phosphodiesterase, an oligodendrocyte-Schwann cell and myelin-associated enzyme of the nervous system. CRC Crit. Rev. Neurobiol., 4, 235–301. [PubMed] [Google Scholar]
- Terwilliger T.C. and Berendzen,J. (1999) Automated MAD and MIR structure solution. Acta Crystallogr. D, 55, 849–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong L. and Rossmann,M. (1990) The locked rotation function. Acta Crystallogr. A, 46, 783–792. [DOI] [PubMed] [Google Scholar]
- Tyc K., Kellenberger,C. and Filipowicz,W. (1987) Purification and characterisation of wheat germ 2′,3′-cyclic-nucleotide-3′-phospho diesterase. J. Biol. Chem., 262, 12994–13000. [PubMed] [Google Scholar]
- Van Duyne G., Standaert,R., Karplus,A., Schreiber,S. and Clardy,J. (1993) Atomic structures of the human immunophilin FKBP-12 complexes with FK506 and rapamycin. J. Mol. Biol., 229, 105–124. [DOI] [PubMed] [Google Scholar]
- Varani G. (1998) RNA–protein recognition during RNA processing and maturation. In Eggleston,D. (ed.), The Many Faces of RNA. Academic Press, Cambridge, UK, pp. 145–156. [Google Scholar]
- Weeks C. and Miller,R. (1999) The design and implementation of SnB v2.0. J. Appl. Crystallogr., 32, 120–124. [Google Scholar]
- Westaway S.K. and Abelson,J. (1995) Splicing of tRNA precursors. In Söll,D. and RajBhandary,U.L. (eds), tRNA: Structure, Biosynthesis and Function. American Society for Microbiology, Washington, DC, pp. 79–92. [Google Scholar]
- Wlodawer A. (1985) Structure of bovine pancreatic ribonuclease by X-ray and neutron diffraction. In Jurnak,F.A. and McPherson,A. (eds), Biological Macromolecules and Assemblies. John Wiley & Sons, New York, NY, pp. 393–439. [Google Scholar]
- Wlodawer A. and Sjolin,L. (1983) Structure of ribonuclease A: results of joint neutron and X-ray refinement at 2.0 Å resolution. Biochemistry, 22, 2720–2728. [DOI] [PubMed] [Google Scholar]
- Xu Q., Teplow,D., Lee,T.D. and Abelson,J. (1990) Domain structure in yeast tRNA ligase. Biochemistry, 29, 6132–6138. [DOI] [PubMed] [Google Scholar]
- Yang W., Hendrickson,W., Kalman,E. and Crouch,R. (1990) Expression, purification and crystallisation of natural and selenomethionyl recombinant ribonuclease H from E.coli. J. Biol. Chem., 265, 13553–13559. [PubMed] [Google Scholar]
- Zamyatnin A. (1984) Amino acid, peptide, and protein volume in solution. Annu. Rev. Biophys. Bioeng., 13, 145–165. [DOI] [PubMed] [Google Scholar]
- Zillmann M., Gorovsky,M.A. and Phizicky,E.M. (1991) Conserved mechanism of tRNA splicing in eukaryotes. Mol. Cell. Biol., 11, 5410–5416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zillmann M., Gorovsky,M. and Phizicky,E. (1992) HeLa cells contain a 2′-phosphate-specific phosphotransferase similar to an yeast enzyme implicated in tRNA splicing. J. Biol. Chem., 267, 10289–10294. [PubMed] [Google Scholar]