

Abstract
Far1 is a bifunctional protein that is required to arrest the cell cycle and establish cell polarity during yeast mating. Here we show that SCFCdc4 ubiquitylates Far1 in the nucleus, which in turn targets the multi-ubiquitylated protein to 26S proteasomes most likely located at the nuclear envelope. In response to mating pheromones, a fraction of Far1 was stabilized after its export into the cytoplasm by Ste21/Msn5. Preventing nuclear export destabilized Far1, while conversely cytoplasmic Far1 was stabilized, although the protein was efficiently phosphorylated in a Cdc28–Cln-dependent manner. The core SCF subunits Cdc53, Hrt1 and Skp1 were distributed in the nucleus and the cytoplasm, whereas the F-box protein Cdc4 was exclusively nuclear. A cytoplasmic form of Cdc4 was unable to complement the growth defect of cdc4-1 cells, but it was sufficient to degrade Far1 in the cytoplasm. Our results illustrate the importance of subcellular localization of F-box proteins, and provide an example of how an extracellular signal regulates protein stability at the level of substrate localization.
Keywords: Cdc4/Far1/Grr1/proteasome/SCF
Introduction
Ubiquitin-dependent degradation is triggered by the covalent attachment of ubiquitin onto lysine residues, which targets substrates for degradation by the 26S proteasome (Ciechanover et al., 2000). Three classes of enzymes are required to mediate these reactions: the E1 ubiquitin-activating enzymes, the E2 ubiquitin-conjugating enzymes and the E3 ubiquitin ligases. In addition, efficient polyubiquitylation may also require an E4-conjugation factor (Koegl et al., 1999). The anaphase-promoting complex (APC) and Skp1–cullin–F-box protein complex (SCF) are multiprotein E3 ligases that regulate the cell cycle (Peters, 1998): the APC promotes entry into anaphase and exit from mitosis, whereas SCF complexes trigger the G1–S transition. In budding yeast, entry into S phase requires ubiquitin-dependent degradation of the cyclin-dependent kinase inhibitor Sic1 (Schwob et al., 1994; Verma et al., 1997), and genetic and biochemical analysis revealed that a high molecular weight complex containing Cdc4, Cdc34, Cdc53, Hrt1/Rbx1 and Skp1 is required for its ubiquitylation (Deshaies, 1999). CDC34 encodes an E2 ubiquitin-conjugating enzyme (Goebl et al., 1988) whereas Cdc4 contains a conserved motif called the F-box, which mediates its interaction with Skp1 (Bai et al., 1996). A large family of proteins containing an F-box motif has been discovered that may function as adaptors to recruit specific substrates to the core ubiquitylation complex (Patton et al., 1998). However, biochemical evidence has only been provided for SCFCdc4, SCFGrr1 and SCFMet30, and it thus remains possible that other F-box-containing proteins may serve functions unlinked to E3 ubiquitin ligase activity (Kaplan et al., 1997). In addition to Sic1, SCFCdc4 is required to degrade Far1, Cdc6 and Gcn4, while ubiquitylation of the G1 cyclins, Cln1 and Cln2, and the bud emergence protein Gic2 is mediated by SCFGrr1 (Deshaies, 1999). Finally, degradation of the Cdk-inhibitory kinase Swe1 (Kaiser et al., 1998), as well as the transcription factor Met4, is dependent on SCFMet30 (Patton et al., 2000; Rouillon et al., 2000). Several SCF substrates need to be phosphorylated for subsequent ubiquitylation to occur and it is thought that phosphorylation of the substrate constitutes at least part of the recognition signal that mediates its binding to the SCF complex (Deshaies, 1999).
Multi-ubiquitylated proteins are recognized and degraded by 26S proteasomes, which are comprised of a 20S catalytic and a 19S regulatory particle (Baumeister et al., 1998). The mature 20S particle forms a cylindrical structure with multiple peptidases in its center, while the 19S particle contains a substrate-unfolding activity and isopeptidases that remove the polyubiquitin chain to recycle ubiquitin. Interestingly, localization of both the 19S and 20S complex in Schizosaccharomyces pombe and Saccharomyces cerevisiae revealed that the majority of the proteasomes are nuclear (Russell et al., 1999) and/or may accumulate at the inner nuclear envelope (NE) and the endoplasmic reticulum (ER) (Enenkel et al., 1998; Wilkinson et al., 1998). In mammalian cells, the localization of proteasomes is less clear: some reports suggest that proteasomes are found in the nucleus, the cytoplasm or both, while others show association with the ER or with centrosomes (Hirsch and Ploegh, 2000). The compartmentalized localization of proteasomes raises the question of whether various substrates are degraded by a specific subset of proteasomes.
We are interested in the temporal and spatial regulation of Far1 during the cell cycle and in response to pheromones. Mating pheromones activate a mitogen-activated protein (MAP) kinase signal transduction pathway that induces changes in gene transcription, alterations of cellular morphology and cell cycle arrest in G1 (Gustin et al., 1998). These responses are initiated by binding of pheromones to a seven-transmembrane receptor that is coupled to a heterotrimeric G-protein. Gβγ then transduces the signal via its effectors Ste5 and Ste20 to a MAP kinase cascade composed of Ste11, Ste7 and Fus3. Fus3 phosphorylates the transcriptional repressors Dig1 and Dig2, resulting in activation of the transcription factor Ste12. In the absence of pheromones, Far1 sequesters the guanine-nucleotide exchange factor Cdc24 in the nucleus of G1 cells; activation of Cdc28–Cln kinase at bud emergence leads to phosphorylation of Far1 at serine 87, which triggers its ubiquitylation by SCFCdc4 and subsequent degradation (O’Shea and Herskowitz, 2000). As a result, Cdc24 is able to localize to the presumptive bud site to activate Cdc42 and trigger bud emergence. During mating, Far1 is required to polarize the actin cytoskeleton along a morphogenetic gradient (Valtz et al., 1995). Mating pheromones trigger nuclear export of a fraction of the Far1–Cdc24 complex thereby targeting Cdc24 to the activated heterotrimeric G-protein (Nern and Arkowitz, 2000; Shimada et al., 2000). Nuclear export of Far1 is mediated by the exportin Msn5/Ste21, which binds to a small motif in the N-terminal domain of Far1 (Blondel et al., 1999). Far1 plays a second role during mating and is required to arrest the cell cycle, presumably by inhibiting cyclin-dependent kinases in the nucleus. The cell cycle arrest function requires phosphorylation of Far1 by Fus3, which promotes binding of Far1 to the Cdc28–Cln complex (Peter et al., 1993; Gartner et al., 1998).
Here we address the spatial regulation of Far1 degradation. We found that Far1 is ubiquitylated by SCFCdc4 in the nucleus, and subsequently degraded by proteasomes, possibly at the nuclear envelope. Cytoplasmic Far1 was stabilized, providing an explanation for its increased half life in response to pheromones. Our results suggest that the subcellular localization of Cdc4 restricts degradation of Far1 to the nucleus, implying that F-box proteins may temporally and spatially control degradation of their targets.
Results
Reduced ubiquitin-dependent degradation of cytoplasmic Far1
During the cell cycle, SCFCdc4 degrades Far1 with a half life of ∼20–25 min (Figure 1; McKinney and Cross, 1995; Henchoz et al., 1997). However, Far1 was slightly, but reproducibly stabilized in cells treated with pheromones and its half life extended to 40 min (Figure 1A). This stabilization was dependent on an intact signaling pathway, and was not observed in cells lacking Fus3 (half life 15 min). Fus3 is involved in nuclear export of Far1 (Blondel et al., 1999), suggesting that pheromones may stabilize Far1 by increasing its cytoplasmic pool. Indeed, the observed stabilization of Far1 in response to α-factor correlated with its nuclear export (Figure 1A, lower blot). To confirm that nuclear localization of Far1 is required for its degradation, we measured the half life of cytoplasmic Far1 containing point mutations in its major nuclear localization signals (NLSs) (Far1-nls1 and Far1-nls1/2; Blondel et al., 1999). As shown in Figure 1B, both Far1-nls1 and Far1-nls1/2 were stabilized and their half life was comparable to Far1-Δ50 (McKinney and Cross, 1995). Moreover, adding the SV40-NLS to the C-terminus of Far1-Δ50 partially relocalized the fusion protein [Far1-Δ50-NLS(SV40)] to the nucleus and correlated with a shorter half life in vivo (t1/2 ∼30 min; Figure 1B), suggesting that the NLS is indeed the degradation signal contained within the N-terminal domain of Far1. Taken together, these results demonstrate that cytoplasmic Far1 is stabilized and suggest that pheromones may increase the half life of Far1 by promoting its nuclear export. Degradation of Far1 is triggered by phosphorylation of serine 87 (Henchoz et al., 1997; Gartner et al., 1998), and as a consequence a non-phosphorylatable residue at this position (Far1-22 has serine 87 mutated to a proline residue) stabilizes the protein, although it is still localized to the nucleus (Figure 1B). In contrast, the Far1-22/nls1 double mutant was predominantly cytoplasmic, and its half life was significantly increased compared with each single mutant. We interpret this additive effect as indicating that phosphorylation of serine 87 and nuclear localization of Far1 contribute separately to its ubiquitin-dependent degradation.

Fig. 1. Cytoplasmic Far1 is stabilized. (A) The half life of Far1 expressed from the GAL promoter was determined by GAL shut-off experiments (see Material and methods) in far1Δ cells (YMP1054) treated (lower blot) or not treated (upper blot) with α-factor (αF) for 2 h. The blots were quantified and the half life (in min) is indicated on the right. The right panels show the localization of Far1–GFP under the same conditions. (B) The half lives (in min) of wild-type and Far1 mutants schematically indicated on the left were determined by GAL shut-off experiments. The localization of the corresponding GFP fusion proteins was visualized by fluorescence microscopy and was overlaid with the phase contrast image.
Preventing nuclear export of Far1 increases its rate of degradation, but does not interfere with its recruitment to specific sites in response to the proteasome inhibitor MG132
To confirm that Far1 is degraded in the nucleus, we measured the half life of wild-type Far1 in msn5/ste21Δ cells (Figure 2A), or conversely Far1-ΔNES in wild-type cells (Figure 2B). In both cases, Far1 was unstable and degraded slightly faster than wild-type Far1 in wild-type cells. Degradation requires phosphorylation of serine 87, as both Far1-22 in msn5/ste21Δ cells and Far1-22/ΔNES in wild-type cells were significantly stabilized (Figure 2A and B). These results imply that the kinase that triggers degradation of Far1 is active in the nucleus. We conclude that nuclear export of Far1 is not needed for its degradation, suggesting that Far1 is ubiquitylated and degraded in the nucleus. These results do not exclude, however, that ubiquitylated Far1 is degraded in the cytoplasm after export by a novel mechanism that may recognize its ubiquitin chains rather than the known NES. To test this possibility, we analyzed the localization of wild-type or mutant Far1 fused to green fluorescent protein (GFP) in erg6Δ cells where proteasomal degradation can be inhibited by the addition of MG132 (Lee and Goldberg, 1998b). In the presence of MG132, Far1–GFP was found in discrete spots, possibly at the nuclear envelope in >90% of the cells (Figure 3A). Occasionally such spots were also detectable in unperturbed cells or cells treated with the solvent dimethylsulfoxide (DMSO) (5% of the cells), suggesting that they are not an artifact of the inhibitor MG132. In contrast, no spots were observed in MG132-treated cells expressing Far1-22–GFP (Figure 3C), indicating that their formation depends on ubiquitylation of Far1. This localization pattern is strikingly similar to proteasomal subunits (Enenkel et al., 1998), suggesting that these spots may represent Far1–GFP interacting with 26S proteasomes at the nuclear envelope. Far1-ΔNES–GFP in wild-type cells (Figure 3B) or wild-type Far1 in msn5Δ/ste21Δ cells (data not shown) similarly accumulated nuclear spots in the presence of MG132, suggesting that nuclear export of Far1 is not required for its ubiquitylation. In contrast, Far1-nls1/nls2 was distributed throughout the cytoplasm and only a few spots were detectable; these spots most likely represent the fraction of Far1-nls1/nls2 that still shuttles through the nucleus (Figure 3D). Taken together these results suggest that Far1 is ubiquitylated in the nucleus, and may be degraded by proteasomes located at the nuclear envelope.
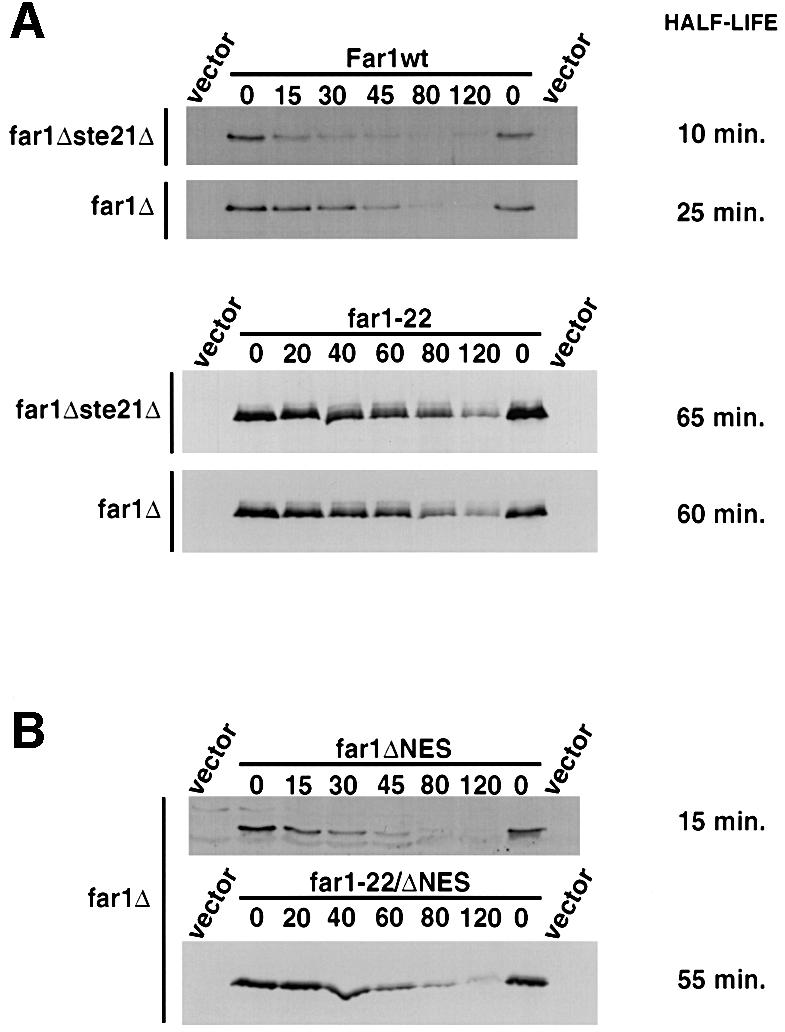
Fig. 2. Preventing nuclear export of Far1 increases its rate of degradation. (A) The half lives of wild-type Far1 (upper panels) or Far1-22 (lower panels) were determined in far1Δste21Δ (YMP1067, upper blots) or for control Δfar1 cells (YMP1054, lower blots) by the GAL shut-off protocol as described. (B) The half lives of Far1-ΔNES (far1ΔNES, upper panel) or Far1-22/ΔNES (far1-22/ΔNES, lower panel) were determined by GAL shut-off experiments in far1Δ cells.

Fig. 3. Ubiquitylated Far1 accumulates at specific sites in the nucleus. (A–D) erg6Δ cells (RH3622) expressing the indicated wild-type or mutant Far1 fused to GFP were treated with the proteasome inhibitor MG132 (+ MG132) or, for control, its solvent DMSO (+ DMSO) and visualized after 3 h by phase contrast (upper rows) or fluorescence microscopy (lower rows). The numbers indicate the percentage of cells that accumulated GFP spots; at least 200 cells were counted for each strain.
Cdc28–Cln can phosphorylate Far1 in the cytoplasm and in the nucleus
Cytoplasmic Far1 may be stable because of a defect in its phosphorylation on serine 87 (Henchoz et al., 1997; Gartner et al., 1998). Far1-nls1 accumulated several modified species that migrated slower on SDS gels and were sensitive to incubation with alkaline phosphatase (Figure 4A), demonstrating that these species are phosphorylated forms of Far1. These modified forms were also detected with Far1-22/nls1, implying that phosphorylation of residues other than serine 87 are responsible for the up-shift. Wild-type or Far1-nls1 were phosphorylated in fus3Δ or fus3-A cells (Figure 4C), implying that the majority of these phosphorylations are not mediated by Fus3. In contrast, cells arrested by depletion of the G1 cyclins did not accumulate phosphorylated forms of wild-type Far1 or Far1-nls1 (Figure 4B, – Cln2), while the same cells grown under conditions that allow expression of Cln2 resulted in efficient phosphorylation of these proteins (+ Cln2). These results suggest that phosphorylation of wild-type or Far1-nls1 is dependent on Cdc28–Cln kinase, and that Far1 may be phosphorylated in both the cytoplasm and the nucleus. Consistent with this notion, Cln1–GFP as well as Cln2–GFP were localized in both the nucleus and the cytoplasm (Figure 4D), as recently described (Miller and Cross, 2000).

Fig. 4. Cdc28–Cln-dependent phosphorylation of Far1 can occur in the cytoplasm and the nucleus. (A) Far1-nls1 accumulates phosphorylated species (arrowhead). Extracts from far1Δ cells expressing Far1-nls1 were incubated with active (+ CIP, lane 2) or heat-inactivated (– CIP, lane 1) alkaline phosphatase, and immunoblotted with specific antibodies. (B) YMG258 cells (cln1,2,3Δ MET-CLN2) transformed with an empty control plasmid (lanes 3 and 4), or plasmids expressing wild-type (lanes 5 and 6) or mutant Far1 (lanes 7–10) as indicated, were grown in selective media lacking methionine at 30°C to early log phase; the culture was divided and methionine was added to one half to repress Cln2 expression (– Cln2), while no methionine was added to the control half (+ Cln2). Extracts were prepared after 3 h and immunoblotted for Far1. (C) The phosphorylation of wild-type or mutant Far1 was analyzed as above in either far1Δ (YMP1054; lane 19), far1Δ fus3Δ (YMP291, lanes 11–14) or fus3A cells (YMP293, lanes 16–18), transformed with an empty control vector (lanes 11 and 15) or plasmids expressing either wild-type (lanes 12 and 16) or the indicated mutant Far1 (lanes 13, 14, 17–19). (D) Cln1–GFP and Cln2–GFP are localized both in the cytoplasm and in the nucleus. Wild-type cells expressing either Cln1–GFP (left panels) or Cln2–GFP (middle panels) from the GAL promoter were grown at 30°C to early log phase in selective media, and analyzed by fluorescence microscopy. The right panel (no GFP) shows cells expressing untagged Cln2 to control for background fluorescence. (E) Ubiquitylation of wild-type and mutant Far1 by SCFCdc4 in vitro. Wild type (lanes 1–5) or the indicated Far1 mutants (lanes 6–14) were in vitro translated in the presence of [35S]methionine and incubated as indicated with reconstituted SCFCdc4 (SCF), Cdc28–Cln2 (Kinase) and ubiquitin (Ub) as described in Materials and methods. Where indicated, Me-ubiquitin instead of ubiquitin was added as a control (Me-Ub). The arrows point to the position of unphosphorylated (Far1) and phosphorylated Far1 (P-Far1), respectively, while the brackets mark the position of multi-ubiquitylated Far1 (Ubn-Far1).
Cytoplasmic Far1-nls1 and Far1-nls1/nls2 are efficiently ubiquitylated by reconstituted SCFCdc4 in vitro
To exclude the possibility that the mutations introduced into Far1-nls1 and Far1-nls1/nls2 may interfere with its ubiquitylation, we performed in vitro ubiquitylation experiments using reconstituted SCFCdc4 and in vitro-translated 35S-labeled wild-type or mutant forms of Far1 (Figure 4E). Clearly, SCFCdc4 was able to ubiquitylate wild-type Far1 (lanes 1–5), Far1-nls1 (lanes 6–8) and Far1-nls1/2 (lanes 12–14) with similar efficiency. The smear of slower migrating forms represents ubiquitylated species of Far1, because addition of methyl-ubiquitin (Me-Ub), which cannot be extended into multi-ubiquitin chains (Hershko and Heller, 1985), competes with the formation of these high molecular weight forms. As expected, ubiquitylation required the addition of purified Cdc28–Cln2 (kinase) and a phosphorylatable serine 87 (lanes 9–11), confirming that ubiquitylation of Far1-nls1 or Far1-nls1/2 was dependent on phosphorylation of serine 87 by Cdc28–Cln2. We conclude that stabilization of Far1-nls1 and Far1-nls1/nls2 in vivo is not due to a defect in its ubiquitylation caused by these mutations, consistent with our earlier observation that Far1-Δ50 fused to the NLS of SV40 was efficiently degraded (Figure 1B).
Cdc4 is exclusively localized to the cell nucleus
To determine whether the subcellular localization of SCF components may explain the nuclear-specific degradation of Far1, we constructed functional GFP fusions of Cdc53, Skp1, Cdc34 and Hrt1. All these common SCF components were distributed in the cytoplasm and the nucleus (Figure 5E–G; Blondel et al., 2000), at least when overexpressed from the GAL promoter. Thus, the subcellular localization of the core SCF subunits does not exclude cytoplasmic degradation of Far1. In contrast, Cdc4 is found in the nucleus (Choi et al., 1990), raising the possibility that cytoplasmic Far1 may be stable because SCFCdc4 is exclusively localized in the nucleus. To examine the localization of Cdc4 further, we constructed functional Cdc4–GFP and Cdc4–myc proteins (Galan and Peter, 1999), and localized the fusion proteins after expression from the GAL promoter. As shown in Figure 5A, Cdc4–GFP was exclusively nuclear at all stages of the cell cycle as well as in α-factor-arrested cells. Identical results were obtained when Cdc4–myc was localized by indirect immunofluorescence using 9E10 antibodies (data not shown). Interestingly, Cdc4–GFP may also be associated with one of the two spindle poles (arrowhead; C.Lafourcade and M.Peter, unpublished results). Taken together, these results suggest that Far1 may be specifically degraded in the nucleus because of the nuclear localization of Cdc4. In contrast, the F-box protein Grr1 was found both in the nucleus and the cytoplasm at all stages of the cell cycle (Figure 5B) as well as in α-factor-arrested cells (Figure 5C), consistent with the finding that it is required to degrade cytoplasmic Gic2 and the G1 cyclins Cln1 and Cln2. Interestingly, Grr1 localized to the cytokinesis ring late in mitosis (arrowhead), indicating that Grr1 may be involved in degradation of substrates during cytokinesis.

Fig. 5. Cdc4–GFP is exclusively in the nucleus while Grr1–GFP is localized in both nucleus and cytoplasm. (A) Wild-type cells (K699) expressing Cdc4–GFP from the GAL promoter were grown to early log phase in selective media at 30°C, and analyzed by phase (lower rows) and fluorescence microscopy (upper rows). Where indicated, cells were treated with pheromones for 2 h (+ α-factor, right panels). Note that Cdc4–GFP is nuclear at all stages of the cell cycle and in response to pheromones, and may localize to the mother-specific spindle pole body (arrowhead). (B–D) grr1Δ cells (YMP2957) transformed with a control plasmid (D) or a plasmid expressing Grr1–GFP from the GAL promoter were analyzed as described in (A). Grr1–GFP is found at the cytokinesis ring late in mitosis (B, arrowhead). (E–G) Wild-type cells (K699) transformed with plasmids expressing either Cdc34–GFP (E), Cdc53–GFP (F) or Skp1–GFP (G) from the GAL promoter were analyzed as described in (A). Where indicated, α-factor was added for 2 h.
A single monopartite NLS is responsible for nuclear localization of Cdc4
To examine how Cdc4 is targeted to the nucleus we searched for a functional NLS within its coding sequence. We found that a fusion protein between the N-terminal domain of Cdc4 (amino acids 1–106) and GFP localized to the nucleus (Figure 6B), suggesting that it contains a functional NLS. Indeed, amino acids 82–85 resemble a monopartite NLS sequence (Figure 6A; Dingwall and Laskey, 1991), and mutating arginine 83 to glycine strongly reduced the nuclear localization of the GFP fusion protein (Figure 6B). To address the functional importance of this NLS for the localization of full-length Cdc4–GFP, we mutated the basic amino acids in positions 82, 83 and 85 to alanine (Cdc43A). Clearly, the nuclear localization of the mutant protein was strongly reduced (Figure 6C), demonstrating that this motif is indeed part of a functional NLS in vivo.

Fig. 6. Cdc4 contains a monopartite NLS in its N-terminus. (A) Schematic representation of Cdc4. The monopartite NLS (amino acids 82–85; KRVK) and the F-box (black bar) are indicated. The numbers indicate the amino acids starting from the N-terminus (N) to the C-terminus (C) of Cdc4. (B) Wild-type cells (K699) expressing from the GAL promoter GFP fused to the N-terminal 106 amino acids of wild-type (Cdc41–106–GFP, left panels) or mutant Cdc4 (Cdc41–106(R83G)–GFP, right panels) were analyzed by fluorescence microscopy. (C) Wild-type cells (K699) expressing from the GAL promoter either wild-type Cdc4–GFP (Cdc4wt, left panels) or a mutant Cdc4–GFP harboring three changes (K82A, R83A and K85A) in the putative monopartite NLS (Cdc43A, right panels). (D) cdc4-1 cells expressing GFP alone (GFP), Cdc43A–GFP or wild-type Cdc4–GFP from the GAL promoter were grown for 3 days at 37°C on media containing galactose (GAL).
Cdc4 fused to a nuclear export signal (NES) is predominantly cytoplasmic
To test the idea that nuclear localization of Cdc4 is responsible for compartmentalized degradation of Far1, we examined the phenotype of cells expressing Cdc4 in the cytoplasm. For this purpose, we fused the leucine-rich NES sequence of human PKI (Figure 7A) to the N-terminus of Cdc4–GFP (NES–Cdc4); this NES sequence is able to function efficiently as an NES in budding yeast, presumably by binding to the export receptor Crm1. For control, Cdc4–GFP was fused to a mutated NES where all four leucines were replaced by alanine residues (NES4A–Cdc4). All Cdc4 fusion proteins were expressed from the inducible GAL promoter or the constitutive TEF promoter (Ronicke et al., 1997), and were present at similar levels (Figure 8D). Interestingly, NES–Cdc4–GFP was predominantly cytoplasmic, and the diffuse nuclear staining of NES–Cdc4–GFP was virtually absent (Figure 7B). In contrast, NES4A–Cdc4–GFP was exclusively nuclear (Figure 7C) and indistinguishable from Cdc4–GFP (Figures 5A and 6C; data not shown), confirming that indeed the function of the NES and not adverse effects of the N-terminal fusion was responsible for the redistribution of Cdc4 to the cytoplasm.

Fig. 7. A fusion of Cdc4 with the NES of human PKI is predominantly cytoplasmic. (A) Amino acid sequence of the wild-type (NES–Cdc4) or mutant NES sequence (NES4A–Cdc4) fused to the N-terminus of Cdc4. (B and C) GFP fluorescence (upper rows) and phase microscopy images (lower rows) of wild-type cells (K699) expressing NES–Cdc4–GFP (B) or NES4A–Cdc4–GFP (C) from the inducible GAL promoter. Cells were grown at 30°C to early log phase in selective media containing 2% raffinose, at which time expression of NES–Cdc4–GFP or NES4A–Cdc4–GFP was induced for 3 h by addition of 2% galactose.

Fig. 8. Expression of NES–Cdc4 is not able to bypass the G1 arrest of cdc4-1 mutant cells. (A) cdc4-1 cells (YMT668) were transformed with an empty control vector or plasmids expressing from the constitutive TEF promoter either NES4A–Cdc4, NES–Cdc4 or wild-type Cdc4, and grown for 3 days on selective media at either 25°C (left panel) or 37°C (right panel). (B and C) To examine the arrest phenotype, the cdc4-1 transformants described above were grown at 25°C to early log phase in selective media, at which time the cultures were shifted to 37°C. After 3 h, the cells were analyzed by phase contrast microscopy (B) or FACS analysis (C). (D) An aliquot of the cultures described in (B) was analyzed by immunoblotting for the presence of Cdc4 (upper blot) and Sic1 (lower blot). The asterisk marks the position of a protein that cross reacts with the Cdc4 antibody. (E) GFP fluorescence (upper rows) and phase microscopy images (lower rows) of wild-type cells (K699) expressing Sic1–GFP.
Nuclear localization of Cdc4 is required for its essential function in vivo
We next determined the functional consequences of cytoplasmic Cdc4. Wild-type or different forms of Cdc4 expressed from the TEF promoter were analyzed for their ability to restore growth of a temperature-sensitive cdc4-1 strain (Figure 8A). Interestingly, both Cdc43A as well as NES–Cdc4 failed to restore growth of cdc4-1 cells at 37°C (Figures 6D and 8A). In contrast, cdc4-1 cells expressing wild-type Cdc4 or NES4A–Cdc4 grew efficiently (Figure 8A). While we cannot exclude the possibility that the mutations in Cdc43A may interfere with its function by means other than localization, this is unlikely to be the case for NES–Cdc4, because no mutations have been introduced into the coding sequence of Cdc4 and N-terminal fusions do not interfere with its function. Thus, these results strongly suggest that cytoplasmic Cdc4 is defective for its essential function in vivo. At the restrictive temperature, cdc4-1 cells carrying an empty vector or expressing cytoplasmic Cdc4 arrest with elongated buds (Figure 8B), G1 DNA content (Figure 8C) and increased levels of Sic1 (Figure 8D), suggesting that nuclear Cdc4 is required to degrade Sic1 at the G1–S transition. Consistent with this notion, a functional Sic1–GFP fusion protein was localized in the nucleus (Figure 8E).
Expression of cytoplasmic Cdc4 in wild-type cells from its endogenous promoter, or the constitutive TEF- or CYC promoters, had no obvious effect on cell morphology or growth (data not shown). In contrast, overexpression of NES–Cdc4, but not NES4A–Cdc4, from the GAL promoter was toxic (Figure 9A), possibly because cytoplasmic Cdc4 may induce ectopic degradation of proteins that are not normally accessible to SCFCdc4. Interestingly, SCFCdc4 was able to ubiquitylate Cln2 in vitro (Figure 9B), and Cln2 was weakly stabilized in cdc4-1 mutant cells in vivo (t1/2 = 4 min in wild type, ∼7.5 min in cdc4-1 cells). Cln2 is mainly ubiquitylated by SCFGrr1 (Barral et al., 1995; Seol et al., 1999; Skowyra et al., 1999), indicating that the subcellular localization of Cdc4 and Grr1 may contribute to their substrate specificity in vivo. However, Cln2 is not the only substrate that may be degraded by overexpressed cytoplasmic Cdc4, because cells expressing a non-phosphorylatable and thus stable Cln2 (Cln2-4T3S; Lanker et al., 1996) were still sensitive to overexpression of NES–Cdc4 (data not shown). Nevertheless, these results suggest that compartmentalization of Cdc4 may contribute to its substrate specificity in vivo.
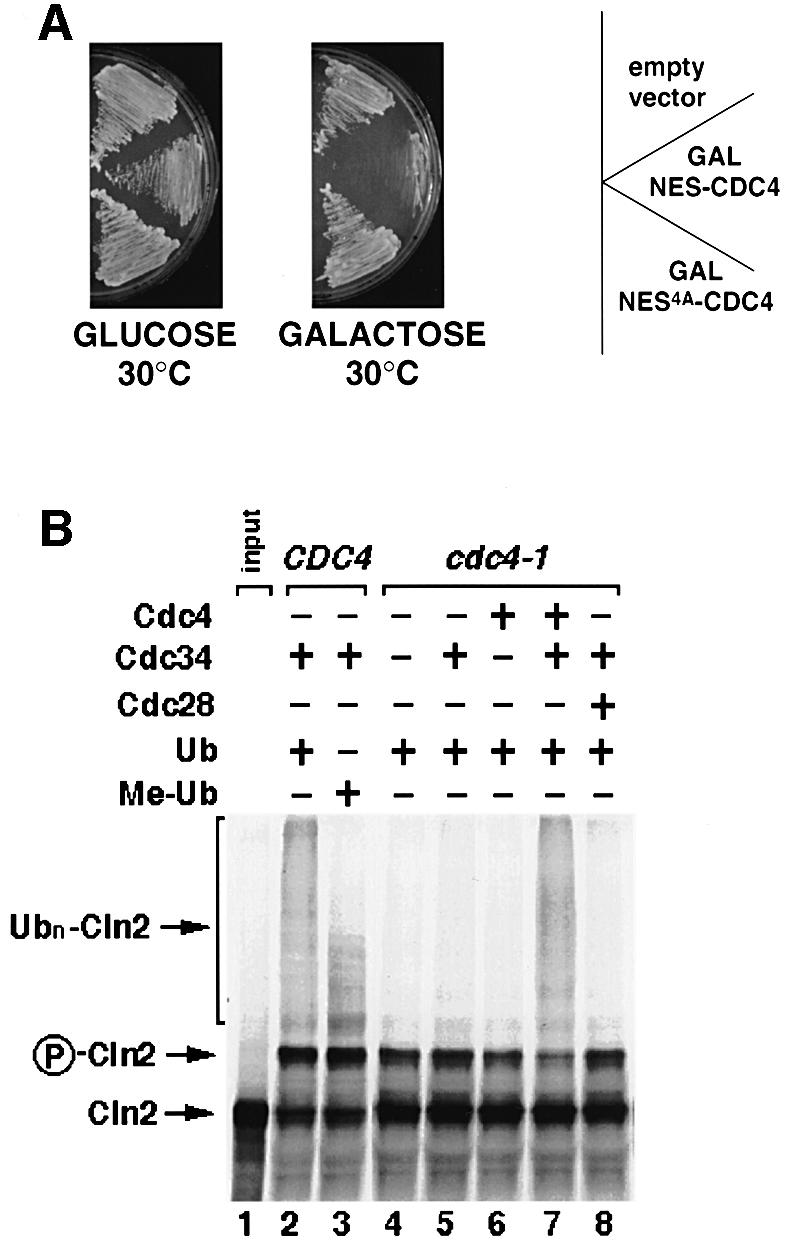
Fig. 9. Subcellular localization may contribute to substrate specificity of Cdc4 in vivo. (A) Wild-type cells (K699) were transformed with a control vector (empty vector), or plasmids expressing either NES–Cdc4 or NES4A–Cdc4 from the inducible GAL promoter. Individual transformants were grown at 30°C on medium containing galactose (right plate, GAL promoter on) or glucose (left plate, GAL promoter off), and photographed after 3 days. (B) SCFCdc4 is able to ubiquitylate Cln2 in vitro. In vitro-translated [35S]methionine-labeled Cln2 (lane 1) was incubated in DEAE-extracts prepared from wild-type (lanes 2 and 3) or cdc4-1 cells (lanes 4–8), complemented where indicated (+) with ubiquitin (Ub), and bacculo-expressed Cdc34, Cdc4 and Cdc28. In lane 3, methylated ubiquitin (Me-Ub) was added instead of ubiquitin to compete for polyubiquitylation. The arrow points to unphosphorylated (Cln2) or phosphorylated Cln2 (P-Cln2), while the bracket marks the position of polyubiquitylated Cln2 (Ubn-Cln2).
NES–Cdc4 is able to degrade cytoplasmic Far1-nls1
If cytoplasmic Far1 were indeed stable because Cdc4 is exclusively nuclear, we would predict that NES–Cdc4 should trigger degradation of Far1-nls1. Indeed, the steady-state levels of Far1-nls1 were greatly reduced in wild-type cells expressing NES–Cdc4 from the TEF promoter, and the phosphorylated forms of Far1-nls1 were virtually undetectable (data not shown). This reduction in Far1-nls1 levels was due to increased degradation, because its half life decreased to ∼25 min (Figure 10A). In contrast, expression of NES4A–Cdc4 (Figure 10A) or wild-type Cdc4 (data not shown) had no effect. Cytoplasmic degradation of Far1-nls1 was still dependent on phosphorylation of serine 87 (Figure 10B), indicating that it is mediated by the SCFNES–Cdc4 complex. However, degradation of Far1-nls1 by NES–Cdc4 required overexpression and was only weakly observed if NES–Cdc4 was expressed from the endogenous promoters (data not shown). Taken together, these results show that targeting Cdc4 to the cytoplasm is necessary and sufficient to induce degradation of Far1-nls1, suggesting that the subcellular localization of Cdc4 restricts degradation of Far1 to the nucleus.

Fig. 10. Wild-type cells expressing NES–Cdc4 are able to degrade cytoplasmic Far1-nls1. (A and B) The half life of Far1-nls1 (A) or Far1-22/nls1 (B) was determined by GAL-shut-off experiments in far1Δ cells (YMP1054) containing either an empty vector or plasmids expressing NES–Cdc4 or NES4A–Cdc4 from the TEF promoter. The blots were quantified and the half lives (in min) are shown on the right. Note that cytoplasmic NES–Cdc4 is able to degrade cytoplasmic Far1-nls1 in a manner dependent on a phosphorylatable serine 87.
Discussion
We show here that Far1 was rapidly ubiquitylated and degraded in the nucleus, while cytoplasmic Far1 was stabilized. Activation of the MAP kinase pathway during yeast mating stabilizes a fraction of Far1, most likely by promoting its nuclear export. Compartmentalized degradation of Far1 was mediated by nuclear localization of the F-box protein Cdc4, and targeting Cdc4 to the cytoplasm was sufficient to promote degradation of cytoplasmic Far1. Thus, internal and external signals may regulate degradation of key proteins by changing their subcellular localization.
Ubiquitylation of SCFCdc4 substrates in the nucleus
Several lines of evidence strongly suggest that Far1 is ubiquitylated and degraded only in the nucleus. First, nuclear Far1 unable to be exported is rapidly degraded, while conversely cytoplasmic Far1 is stabilized. Secondly, Cdc4 is localized in the nucleus, and relocalization of Cdc4 to the cytoplasm was sufficient to induce degradation of cytoplasmic Far1. We conclude that the localization of Cdc4 restricts degradation of Far1 to the nucleus. Besides Far1, SCFCdc4 complexes also govern ubiquitylation of Cdc6, Sic1 and Gcn4 (Deshaies, 1999). Interestingly, cytoplasmic mutants of Cdc6 (cdc6-nls and cdc6Δ23–36) are significantly stabilized, although the proteins interact efficiently with Cdc4 (Elsasser et al., 1999). In addition, cdc4-1 cells expressing NES–Cdc4 failed to degrade Sic1, suggesting that most, if not all, substrates of SCFCdc4 are exclusively ubiquitylated in the nucleus.
Degradation of Far1 by 26S proteasomes located at the nuclear envelope?
Strikingly, ubiquitylated Far1 concentrated at specific sites around the nucleus in the presence of the proteasome inhibitor MG132. It was estimated that ∼80% of the yeast proteasomes are located at the NE or ER (Enenkel et al., 1998), suggesting that most of the ubiquitylated proteins are degraded in this cellular compartment. By immuno-electron microscopy, a large fraction of S.pombe proteasomes has been shown to reside at the inner nuclear envelope (Wilkinson et al., 1998), implying that nuclear proteins do not need to be exported into the cytoplasm for degradation. We thus speculate that the spots observed in MG132-treated cells represent ubiquitylated Far1, which may accumulate at proteasomes located at the nuclear envelope. However, we cannot exclude the possibility that ubiquitylated Far1 aggregates unspecifically in the nucleus in response to MG132, possibly because inhibition of proteasomal degradation may trigger a stress response (Lee and Goldberg, 1998a; Meriin et al., 1998) or interfere with nuclear transport mechanisms (Tomoda et al., 1999). In contrast to Far1, Gic2 was distributed throughout the cytoplasm in MG132-treated cells, while the F-box protein Grr1 accumulated at both the NE and in the cytoplasm (data not shown), suggesting that only nuclear proteins accumulate in discrete spots when proteasomal degradation is inhibited. Thus, cytoplasmic proteins may be degraded by cytoplasmic proteasomes, implying that distinct proteasomes degrade proteins ubiquitylated in the nucleus or the cytoplasm. Because Grr1 is found in both populations, it is unlikely that substrates contain intrinsic signals that target them to specific subsets of proteasomes, although differences in ubiquitin chains or linkage cannot be excluded. We speculate that the subcellular compartment where proteins are ubiquitylated determines by which proteasomes they will subsequently be degraded.
Specific subcellular localization of F-box proteins
The common core subunits of yeast SCF complexes, including Cdc53, Skp1 and Hrt1 (Blondel et al., 2000), are distributed in the nucleus and cytoplasm, and likewise Cdc34 is found in both compartments (Goebl et al., 1994). In contrast, several F-box proteins localize to specific subcellular locations. Cdc4 and Met30 are nuclear (Rouillon et al., 2000), while Grr1 is nuclear and cytoplasmic. In addition, Cdc4 may associate with one of the two spindle pole bodies, raising the possibility that Cdc4 may ubiquitylate an unknown target involved in spindle function or spindle pole body maturation and/or duplication (Freed et al., 1999). Grr1 also localized to the actin ring in telophase, and thus may play a role in cytokinesis. Thus, the localization of F-box proteins may be responsible, at least in part, for the subcellular compartmentalization of SCF-mediated degradation. The localization and abundance of F-box proteins may also be regulated during the cell cycle or in response to signals. For example, the mammalian F-box protein, Skp2, is predominantly nuclear, but is expressed in a cell cycle-dependent manner and is rapidly degraded when cells exit from the cell cycle (Wirbelauer et al., 2000). Intracellular and extracellular signals may thus regulate both expression and localization of F-box proteins.
In most cases, binding of substrates to their F-box protein is dependent on site-specific phosphorylation (Deshaies, 1999), providing the possibility of controlling protein degradation by regulating the activity of the kinase. Several SCFCdc4 substrates, including Far1, Sic1 and Cdc6, are phosphorylated by cyclin-dependent kinases (Schneider et al., 1996; Henchoz et al., 1997; Elsasser et al., 1999), which can function in the nucleus as well as the cytoplasm (Miller and Cross, 2000). Nevertheless, cytoplasmic Far1 was stabilized at all stages of the cell cycle, suggesting that phosphorylation is not the only mechanism to temporally control degradation. On the other hand, phosphorylation of Gic2 is mediated by a kinase, which responds to Cdc42-GTP and may be active only after bud emergence. Thus, spatial and temporal degradation of SCF targets may be regulated by the expression and localization of F-box proteins, together with activation of protein kinases that phosphorylate substrates to increase their binding affinity for F-box proteins.
Finally, the subcellular localization of F-box proteins may also contribute to their substrate specificity in vivo. Although Cln2 is predominantly ubiquitylated by SCFGrr1 in vivo (Barral et al., 1995), SCFCdc4 was able to ubiquitylate Cln2 in vitro (Figure 9B). Moreover, overexpression of cytoplasmic NES–Cdc4 was toxic in wild-type cells, suggesting that F-box proteins may be sequestered in subcellular compartments where they have access to only a subset of their potential targets.
Regulating protein degradation by changing the subcellular localization
Compartmentalized degradation provides the possibility of controlling degradation of specific proteins by regulating their subcellular localization. Far1 is a striking example because it functions both in the nucleus and the cytoplasm during yeast mating (Blondel et al., 1999). Activation of the mating pathway triggers export of Far1 into the cytoplasm, thereby stabilizing the cytoplasmic fraction of Far1. Degradation of Far1 in the cytoplasm may be unfavorable because cells need to assemble large complexes at the cell cortex that organize the actin cytoskeleton. However, cells expressing NES–Cdc4 did not exhibit a severe mating defect (data not shown), perhaps because cytoplasmic Far1 may not be easily accessible to SCFCdc4 complexes.
In contrast, nuclear Far1 is rapidly degraded even when cells are arrested by pheromones. This is surprising at first, because Cdc28–Cln activity, which is thought to trigger degradation of Far1 at bud emergence by phosphorylating serine 87, is inhibited in response to pheromones (Wittenberg and Reed, 1988; Peter and Herskowitz, 1994). Nevertheless, degradation of Far1 in α-factor-treated cells requires phosphorylation of serine 87, implying that a kinase other than Cdc28–Cln may phosphorylate Far1 in response to pheromones. Interestingly, Pho85-Pcl2 is able to phosphorylate Far1 (Huang et al., 1998), and has been proposed to contribute to the degradation of Sic1 in vivo. Degradation of nuclear Far1 in response to pheromones may allow cells to resume cell cycle progression rapidly if the pheromone signal ceases.
The localization of the SCFCdc4 target, Cdc6, is cell cycle regulated: Cdc6 is predominantly cytoplasmic in the G1 phase, but enters the nucleus at START where it stays until the end of the next M phase (Jong et al., 1996). Nuclear Cdc6 is required to load Mcm proteins onto DNA after inactivation of Cdc28–Clb kinase, thereby establishing competence for DNA replication after mitosis (Diffley, 1998). Cdc6 levels increase during G1 (Detweiler and Li, 1997), thus correlating with its cytoplasmic localization. Stable cytoplasmic Cdc6 may provide the possibility of re-importing Cdc6 after a prolonged G1 arrest during mating. Thus, compartmentalized degradation of regulatory proteins may contribute to their rapid activation by relocalizing these proteins from a compartment where they are stable to the site of action.
Compartmentalized degradation of proteins is not unique to yeast. Nuclear export of the mammalian tumor suppressor protein p53 to the cytoplasm is required for its degradation (Freedman and Levine, 1998), and it has been shown that various stress signals activate p53 by regulating its subcellular localization (Ashcroft et al., 2000). Consistent with this model, inhibiting nuclear export by addition of leptomycin B stabilizes p53, thereby increasing its transcriptional activity. Likewise, degradation of the CKI p27 requires its nuclear export by the signalosome component Jab1 (Tomoda et al., 1999), perhaps by association with the pore component mNPAP60 (Muller et al., 2000). Finally, degradation of cyclin B at metaphase starts at the mitotic spindle, and only at later stages occurs all over the nucleoplasm (Clute and Pines, 1999; Huang and Raff, 1999). The reasons for these differences are not fully understood, but in analogy to F-box proteins may reflect distinct localization of the substrate-specific APC adaptors, Cdc20 and Cdh1. Taken together, local degradation of key proteins may be a general mechanism to regulate various biological processes in a spatial and temporal manner.
Materials and methods
Yeast strains and genetic manipulations
Yeast strains are described in Supplementary table I, available at The EMBO Journal Online. All strains are derived from W303: ade2-1, trp1-1, can1-100, leu2-3,112, his3-11,15, ura3, GAL+, psi+, ssd1-d2 or S288c: ade2-101, ura3-52, lys2-801, trp1-Δ1, his3Δ200, leu2-Δ. Standard yeast growth conditions and genetic manipulations were used (Guthrie and Fink, 1991).
DNA manipulations
Plasmids and oligonucleotides (synthesized by Genset) are listed in Supplementary tables II and III. Detailed descriptions of the plasmid constructions are provided upon request. PCRs were performed with the Expand polymerase kit as recommended by the manufacturer (Boehringer Mannheim), and confirmed by sequencing. The following oligo pairs were used to construct the GFP fusions with the vector pMJ200 (Jaquenoud et al., 1998). Cdc4–GFP: oTP718/oTP719; Grr1–GFP: oTP720/oTP721; Cdc53–GFP: oTP834/oTP833; Skp1–GFP: oTP831/oTP856; Cln1–GFP; oTP772/oTP773; Cln2–GFP: oTP770/oTP771. Wild-type or the mutant NES sequence was fused to CDC4 by PCR using oTP760 and oTP719, respectively. The K82A, R83A and K85A substitutions were introduced into Cdc4 (cdc43A–GFP) by oligo-directed mutagenesis using the primers oTP718, oTP781, oTP780 and oTP719. To express Far1-22/Δ338–382 from the inducible GAL promoter, pTP62 was digested with XhoI and SphI, and ligated with the XhoI–SpeI fragment from pTP63 and the SpeI–SphI fragment from pBM77 to yield pBM125. pBM3 was constructed by ligating the NotI–XhoI, XhoI–SphI, SphI–EcoRI and NotI–XhoI fragments from pTP487 together with the EcoRI–BamHI fragment from pRS316-NLS(SV40)–GFP to yield pBM3.
Western blots and antibodies
Standard procedures were used for yeast cell extract preparation and immunoblotting. Antibodies against the N- or C-terminal domain of Cdc4 were used as recommended by the manufacturer (Santa Cruz antibody company). Polyclonal antibodies against Far1 were used as described (Blondel et al., 1999). P.Silver kindly provided antibodies against GFP.
In vitro ubiquitylation assays
In vitro ubiquitylation assays using either cell free extracts from budding yeast (Henchoz et al., 1997) or reconstituted SCFCdc4 (Seol et al., 1999) have been described previously. Substrates were generated by in vitro transcription and translation of linearized plasmids in the presence of [35S]methionine (Promega). In a complete reaction, the yeast extracts were replaced by purified Uba1 (50 ng), Cdc34 (100 ng) and SCFCdc4 complex (50–100 ng). Cdc28–Cln2 kinase activity was supplied as insect lysate (2 µg total protein) containing expressed Cln2myc, GST–Cdc28, Cks1 and Cak1 (Seol et al., 1999). Insect lysate containing expressed Cln2 alone was used to represent absence of Cdc28–Cln2 kinase activity. SCFCdc4 complex (PyHACdc4–Cdc53–Skp1–Hrt1) was expressed in SF9 insect cells and bound to α-polyoma beads as described (Seol et al., 1999). The α-polyoma beads were eluted with polyoma peptide and the eluate was used to supply SCFCdc4 activity.
Microscopy and FACS analysis
GFP-tagged proteins were visualized using a Chroma GFPII filter (excitation 440–470 nm), photographed with a Photometrics CCD camera and analyzed with Photoshop 4.0 software (Adobe). Cells expressing GFP fusion proteins from the inducible GAL promoter were grown to early log phase at 25°C in selective media containing 2% raffinose, at which time 2% galactose was added for 5 h.
The DNA content of cdc4-1 mutant strains harboring either wild-type or mutant forms of CDC4 was monitored by fluorescence-activated cell sorting (FACS) analysis as described (Epstein and Cross, 1992).
Determination of half life
Half lives were determined as described previously (Henchoz et al., 1997). Briefly, far1Δ cells (YMP1054) were grown at 30°C to early log phase in medium containing 2% raffinose, at which time expression of wild-type or mutant Far1 from the GAL promoter was induced by adding 2% galactose. Where indicated, α-factor was added to a final concentration of 25 µg/ml. After 2 h, 2% glucose was added, aliquots were collected after the times indicated and the Far1 protein levels were analyzed by immunoblotting. The half lives were determined after quantitation of the immunoblots using the NIH image program. The stability of Gic2 was also determined in erg6Δ cells (RH3622) treated or not treated with MG132 (Sigma, 50 µM final concentration, diluted from 50 mM stock solution in DMSO) added at the same time as glucose.
Phosphatase assays and analysis of Far phosphorylation
Extracts prepared from far1Δ cells expressing Far1-nls1 were resuspended in 100 µl of alkaline phosphatase (CIP) buffer and split in two parts. One half was incubated with 1 U of CIP for 15 min at 37°C, whereas the other half was incubated under the same conditions but with 1 U of heat inactivated (10 min at 100°C) CIP. Proteins were precipitated with trichloroacetic acid (10% final concentration), resuspended in gel sample buffer and analyzed by immunoblotting with Far1 antibodies.
YMG258 cells were transformed with plasmids expressing either wild-type or mutant Far1 and grown at 30°C to early log phase in selective media lacking methionine. The culture was divided, and methionine was added to one half to repress expression of Cln2. Cells were harvested after 2 h and immunoblotted with Far1 antibodies.
Localization of GFP-tagged proteins with the proteasome inhibitor MG132
erg6Δ cells (RH3622) transformed with plasmids expressing the indicated GFP-tagged proteins from the inducible GAL promoter were grown at 30°C in selective media containing 2% raffinose to mid log phase, at which time 2% galactose was added for 2 h. MG132, or for control an equal amount of its solvent DMSO, was added to a final concentration of 50 µM, and the cells were analyzed after 2 h by fluorescence microscopy.
Supplementary data
Supplementary data for this paper are available at The EMBO Journal Online.
Acknowledgments
Acknowledgements
We thank M.Tyers, M.Jaquenoud, P.Silver and C.Mann for providing antibodies, plasmids and strains. We are grateful to R.Feldman and C.Correll for providing several bacculovirus-expressed proteins. We thank G.Ammerer and C.Dargemont for helpful suggestions, and W.Krek for communicating unpublished results. N.Perrinjaquet and P.Pagé are acknowledged for excellent technical assistance, members of the laboratory for discussion, and R.Iggo and J.Pines for critical reading of the manuscript. M.B. and J.-M.G. are supported by EMBO postdoctoral fellowships and a Roche fellowship to M.B. M.P. is supported by the Swiss National Science Foundation, the Swiss Cancer League and a Helmut Horten Incentive Award. Part of this work was also supported by an HSFP fellowship.
References
- Ashcroft M., Taya,Y. and Vousden,K.H. (2000) Stress signals utilize multiple pathways to stabilize p53. Mol. Cell. Biol., 20, 3224–3233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai C., Sen,P., Hofmann,K., Ma,L., Goebl,M., Harper,J.W. and Elledge,S.J. (1996) SKP1 connects cell cycle regulators to the ubiquitin proteolysis machinery through a novel motif, the F-box. Cell, 86, 263–274. [DOI] [PubMed] [Google Scholar]
- Barral Y., Jentsch,S. and Mann,C. (1995) G1 cyclin turnover and nutrient uptake are controlled by a common pathway in yeast. Genes Dev., 9, 399–409. [DOI] [PubMed] [Google Scholar]
- Baumeister W., Walz,J., Zuhl,F. and Seemuller,E. (1998) The proteasome: paradigm of a self-compartmentalizing protease. Cell, 92, 367–380. [DOI] [PubMed] [Google Scholar]
- Blondel M., Alepuz,P.M., Huang,L.S., Shaham,S., Ammerer,G. and Peter,M. (1999) Nuclear export of Far1p in response to pheromones requires the export receptor Msn5p/Ste21p. Genes Dev., 13, 2284–2300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blondel M., Galan,J.M. and Peter,M. (2000) Isolation and characteriz ation of HRT1 using a genetic screen for mutants unable to degrade Gic2p in Saccharomyces cerevisiae. Genetics, 155, 1033–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi W.J., Clark,M.W., Chen,J.X. and Jong,A.Y. (1990) The CDC4 gene product is associated with the yeast nuclear skeleton. Biochem. Biophys. Res. Commun., 172, 1324–1330. [DOI] [PubMed] [Google Scholar]
- Ciechanover A., Orian,A. and Schwartz,A.L. (2000) Ubiquitin-mediated proteolysis: biological regulation via destruction. BioEssays, 22, 442–451. [DOI] [PubMed] [Google Scholar]
- Clute P. and Pines,J. (1999) Temporal and spatial control of cyclin B1 destruction in metaphase. Nature Cell Biol., 1, 82–87. [DOI] [PubMed] [Google Scholar]
- Deshaies R.J. (1999) SCF and Cullin/Ring H2-based ubiquitin ligases. Annu. Rev. Cell Dev. Biol., 15, 435–467. [DOI] [PubMed] [Google Scholar]
- Detweiler C.S. and Li,J.J. (1997) Cdc6p establishes and maintains a state of replication competence during G1 phase. J. Cell Sci., 110, 753–763. [DOI] [PubMed] [Google Scholar]
- Diffley J.F. (1998) Replication control: choreographing replication origins. Curr. Biol., 8, R771–R773. [DOI] [PubMed] [Google Scholar]
- Dingwall C. and Laskey,R.A. (1991) Nuclear targeting sequences—a consensus? Trends Biochem. Sci., 16, 478–481. [DOI] [PubMed] [Google Scholar]
- Elsasser S., Chi,Y., Yang,P. and Campbell,J.L. (1999) Phosphorylation controls timing of Cdc6p destruction: a biochemical analysis. Mol. Biol. Cell, 10, 3263–3277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enenkel C., Lehmann,A. and Kloetzel,P.M. (1998) Subcellular distribution of proteasomes implicates a major location of protein degradation in the nuclear envelope–ER network in yeast. EMBO J., 17, 6144–6154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epstein C.B. and Cross,F.R. (1992) CLB5: a novel B cyclin from budding yeast with a role in S phase. Genes Dev., 6, 1695–1706. [DOI] [PubMed] [Google Scholar]
- Freed E., Lacey,K.R., Huie,P., Lyapina,S.A., Deshaies,R.J., Stearns,T. and Jackson,P.K. (1999) Components of an SCF ubiquitin ligase localize to the centrosome and regulate the centrosome duplication cycle. Genes Dev., 13, 2242–2257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freedman D.A. and Levine,A.J. (1998) Nuclear export is required for degradation of endogenous p53 by MDM2 and human papillomavirus E6. Mol. Cell. Biol., 18, 7288–7293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galan J.M. and Peter,M. (1999) Ubiquitin-dependent degradation of multiple F-box proteins by an autocatalytic mechanism. Proc. Natl Acad. Sci. USA, 96, 9124–9129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gartner A., Jovanovic,A., Jeoung,D.I., Bourlat,S., Cross,F.R. and Ammerer,G. (1998) Pheromone-dependent G1 cell cycle arrest requires Far1 phosphorylation, but may not involve inhibition of Cdc28-Cln2 kinase, in vivo. Mol. Cell. Biol., 18, 3681–3691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goebl M.G., Yochem,J., Jentsch,S., McGrath,J.P., Varshavsky,A. and Byers,B. (1988) The yeast cell cycle gene CDC34 encodes a ubiquitin-conjugating enzyme. Science, 241, 1331–1335. [DOI] [PubMed] [Google Scholar]
- Goebl M.G., Goetsch,L. and Byers,B. (1994) The Ubc3 (Cdc34) ubiquitin-conjugating enzyme is ubiquitinated and phosphorylated in vivo. Mol. Cell. Biol., 14, 3022–3029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustin M.C., Albertyn,J., Alexander,M. and Davenport,K. (1998) MAP kinase pathways in the yeast Saccharomyces cerevisiae. Microbiol. Mol. Biol. Rev., 62, 1264–1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guthrie C. and Fink,G.R. (1991) Guide to Yeast Genetics and Molecular Biology. Academic Press Inc., San Diego, CA. [Google Scholar]
- Henchoz S., Chi,Y., Catarin,B., Herskowitz,I., Deshaies,R.J. and Peter,M. (1997) Phosphorylation- and ubiquitin-dependent degradation of the cyclin-dependent kinase inhibitor Far1p in budding yeast. Genes Dev., 11, 3046–3060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hershko A. and Heller,H. (1985) Occurrence of a polyubiquitin structure in ubiquitin-protein conjugates. Biochem. Biophys. Res. Commun., 128, 1079–1086. [DOI] [PubMed] [Google Scholar]
- Hirsch C. and Ploegh,H.L. (2000) Intracellular targeting of the proteasome. Trends Cell Biol., 10, 268–272. [DOI] [PubMed] [Google Scholar]
- Huang D., Moffat,J., Wilson,W.A., Moore,L., Cheng,C., Roach,P.J. and Andrews,B. (1998) Cyclin partners determine Pho85 protein kinase substrate specificity in vitro and in vivo: control of glycogen biosynthesis by Pcl8 and Pcl10. Mol. Cell. Biol., 18, 3289–3299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J. and Raff,J.W. (1999) The disappearance of cyclin B at the end of mitosis is regulated spatially in Drosophila cells. EMBO J., 18, 2184–2195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaquenoud M., Gulli,M.P., Peter,K. and Peter,M. (1998) The Cdc42p effector Gic2p is targeted for ubiquitin-dependent degradation by the SCFGrr1 complex. EMBO J., 17, 5360–5373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jong A., Young,M., Chen,G.C., Zhang,S.Q. and Chan,C. (1996) Intracellular location of the Saccharomyces cerevisiae CDC6 gene product. DNA Cell. Biol., 15, 883–895. [DOI] [PubMed] [Google Scholar]
- Kaiser P., Sia,R.A., Bardes,E.G., Lew,D.J. and Reed,S.I. (1998) Cdc34 and the F-box protein Met30 are required for degradation of the Cdk-inhibitory kinase Swe1. Genes Dev., 12, 2587–2597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan K.B., Hyman,A.A. and Sorger,P.K. (1997) Regulating the yeast kinetochore by ubiquitin-dependent degradation and Skp1p-mediated phosphorylation. Cell, 91, 491–500. [DOI] [PubMed] [Google Scholar]
- Koegl M., Hoppe,T., Schlenker,S., Ulrich,H.D., Mayer,T.U. and Jentsch,S. (1999) A novel ubiquitination factor, E4, is involved in multiubiquitin chain assembly. Cell, 96, 635–644. [DOI] [PubMed] [Google Scholar]
- Lanker S., Valdivieso,M.H. and Wittenberg,C. (1996) Rapid degradation of the G1 cyclin Cln2 induced by Cdk-dependent phosphorylation. Science, 271, 1597–1601. [DOI] [PubMed] [Google Scholar]
- Lee D.H. and Goldberg,A.L. (1998a) Proteasome inhibitors cause induction of heat shock proteins and trehalose, which together confer thermotolerance in Saccharomyces cerevisiae. Mol. Cell. Biol., 18, 30–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee D.H. and Goldberg,A.L. (1998b) Proteasome inhibitors: valuable new tools for cell biologists. Trends Cell Biol., 8, 397–403. [DOI] [PubMed] [Google Scholar]
- McKinney J.D. and Cross,F.R. (1995) FAR1 and the G1 phase specificity of cell cycle arrest by mating factor in Saccharomyces cerevisiae. Mol. Cell. Biol., 15, 2509–2516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meriin A.B., Gabai,V.L., Yaglom,J., Shifrin,V.I. and Sherman,M.Y. (1998) Proteasome inhibitors activate stress kinases and induce Hsp72. Diverse effects on apoptosis. J. Biol. Chem., 273, 6373–6379. [DOI] [PubMed] [Google Scholar]
- Miller M.E. and Cross,F.R. (2000) Distinct subcellular localization patterns contribute to functional specificity of the Cln2 and Cln3 cyclins of Saccharomyces cerevisiae. Mol. Cell. Biol., 20, 542–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller D., Thieke,K., Burgin,A., Dickmanns,A. and Eilers,M. (2000) Cyclin E-mediated elimination of p27 requires its interaction with the nuclear pore-associated protein mNPAP60. EMBO J., 19, 2168–2180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nern A. and Arkowitz,R.A. (2000) Nucleocytoplasmic shuttling of the Cdc42p exchange factor Cdc24p. J. Cell Biol., 148, 1115–1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Shea E.K. and Herskowitz,I. (2000) The ins and outs of cell-polarity decisions. Nature Cell Biol., 2, E39–E41. [DOI] [PubMed] [Google Scholar]
- Patton E.E., Willems,A.R. and Tyers,M. (1998) Combinatorial control in ubiquitin-dependent proteolysis: don’t Skp the F-box hypothesis. Trends Genet., 14, 236–243. [DOI] [PubMed] [Google Scholar]
- Patton E.E., Peyraud,C., Rouillon,A., Surdin-Kerjan,Y., Tyers,M. and Thomas,D. (2000) SCFMet30-mediated control of the transcriptional activator Met4 is required for the G1–S transition. EMBO J., 19, 1613–1624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peter M. and Herskowitz,I. (1994) Direct inhibition of the yeast cyclin-dependent kinase Cdc28-Cln by Far1. Science, 265, 1228–1231. [DOI] [PubMed] [Google Scholar]
- Peter M., Gartner,A., Horecka,J., Ammerer,G. and Herskowitz,I. (1993) FAR1 links the signal transduction pathway to the cell cycle machinery in yeast. Cell, 73, 747–760. [DOI] [PubMed] [Google Scholar]
- Peters J.M. (1998) SCF and APC: the Yin and Yang of cell cycle regulated proteolysis. Curr. Opin. Cell Biol., 10, 759–768. [DOI] [PubMed] [Google Scholar]
- Ronicke V., Graulich,W., Mumberg,D., Muller,R. and Funk,M. (1997) Use of conditional promoters for expression of heterologous proteins in Saccharomyces cerevisiae. Methods Enzymol., 283, 313–322. [DOI] [PubMed] [Google Scholar]
- Rouillon A., Barbey,R., Patton,E.E., Tyers,M. and Thomas,D. (2000) Feedback-regulated degradation of the transcriptional activator Met4 is triggered by the SCFMet30 complex. EMBO J., 19, 282–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell S.J., Steger,K.A. and Johnston,S.A. (1999) Subcellular localization, stoichiometry and protein levels of 26 S proteasome subunits in yeast. J. Biol. Chem., 274, 21943–21952. [DOI] [PubMed] [Google Scholar]
- Schneider B.L., Yang,Q.H. and Futcher,A.B. (1996) Linkage of replication to start by the Cdk inhibitor Sic1. Science, 272, 560–562. [DOI] [PubMed] [Google Scholar]
- Schwob E., Bohm,T., Mendenhall,M.D. and Nasmyth,K. (1994) The B-type cyclin kinase inhibitor p40SIC1 controls the G1 to S transition in S.cerevisiae. Cell, 79, 233–244. [DOI] [PubMed] [Google Scholar]
- Seol J.H. et al. (1999) Cdc53/cullin and the essential Hrt1 RING-H2 subunit of SCF define a ubiquitin ligase module that activates the E2 enzyme Cdc34. Genes Dev., 13, 1614–1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimada Y., Gulli,M.-P. and Peter,M. (2000) Nuclear sequestration of the exchange factor Cdc24 by Far1 regulates cell polarity during yeast mating. Nature Cell Biol., 2, 117–124. [DOI] [PubMed] [Google Scholar]
- Skowyra D., Koepp,D.M., Kamura,T., Conrad,M.N., Conaway,R.C., Conaway,J.W., Elledge,S.J. and Harper,J.W. (1999) Reconstitution of G1 cyclin ubiquitination with complexes containing SCFGrr1 and Rbx1. Science, 284, 662–665. [DOI] [PubMed] [Google Scholar]
- Tomoda K., Kubota,Y. and Kato,J. (1999) Degradation of the cyclin-dependent-kinase inhibitor p27Kip1 is instigated by Jab1. Nature, 398, 160–165. [DOI] [PubMed] [Google Scholar]
- Valtz N., Peter,M. and Herskowitz,I. (1995) FAR1 is required for oriented polarization of yeast cells in response to mating pheromones. J. Cell Biol., 131, 863–873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verma R., Annan,R.S., Huddleston,M.J., Carr,S.A., Reynard,G. and Deshaies,R.J. (1997) Phosphorylation of Sic1p by G1 Cdk required for its degradation and entry into S phase. Science, 278, 455–460. [DOI] [PubMed] [Google Scholar]
- Wilkinson C.R., Wallace,M., Morphew,M., Perry,P., Allshire,R., Javerzat,J.P., McIntosh,J.R. and Gordon,C. (1998) Localization of the 26S proteasome during mitosis and meiosis in fission yeast. EMBO J., 17, 6465–6476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wirbelauer C., Sutterlüty,H., Blondel,M., Gstaiger,M., Peter,M., Reymond,F. and Krek,W. (2000) The F-box protein Skp2 is a ubiquitination target of a Cul1-based core ubiquitin ligase complex: evidence for a role of Cul1 in the suppression of Skp2 expression in quiescent fibroblasts. EMBO J., 19, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittenberg C. and Reed,S.I. (1988) Control of the yeast cell cycle is associated with assembly/disassembly of the Cdc28 protein kinase complex. Cell, 54, 1061–1072. [DOI] [PubMed] [Google Scholar]