

Abstract
Arrhythmogenic right ventricular cardiomyopathy (ARVC) is characterized by a high incidence of ventricular tachyarrhythmias and sudden cardiac death that appear early in the natural history of the disease and may precede significant ventricular remodeling. The classical pathology of ARVC is degeneration of right ventricular free wall myocardium and its replacement by fat and fibrous tissue. The clinical presentation may be highly variable, however, and genetic penetrance is typically low which makes definitive diagnosis difficult, especially in early stages of the disease. Endomyocardial biopsy (EMB) has not been used widely in the diagnosis of ARVC, in part because pathological changes are usually most conspicuous in the epicardium of the right ventricular free wall and tend to spare the endocardium and interventricular septum. Thus, diagnostic pathological features of ARVC are often not seen in conventional septal biopsies. Diagnostic yield may be increased by sampling the RV free wall or by using 3-dimensional electroanatomic voltage mapping to identify affected areas, but these approaches can carry increased risk and require specialized equipment and experience. Moreover, diagnostic pathological changes may not be apparent in early disease despite an increased risk of arrhythmias and sudden death. This review considers the role of EMB in the diagnosis of ARVC and highlights recent advances in identifying potential tissue biomarkers that arise early in the disease process and occur diffusely throughout the myocardium. Analysis of conventional EMB using such biomarkers could improve diagnostic sensitivity and accuracy but widespread clinical application of this approach requires further validation.
Keywords: arrhythmogenic right ventricular cardiomyopathy, endomyocardial biopsy, intercalated disks, desmosomes, plakoglobin
Introduction
Arrhythmogenic right ventricular cardiomyopathy (ARVC) is a primary myocardial disorder characterized by a high incidence of arrhythmias and sudden cardiac death. The disease name reflects the usual predominant involvement of the right ventricle (RV), but increasing recognition of biventricular or left-dominant forms warrants adoption of the broader term arrhythmogenic cardiomyopathy.1 This review will focus largely on the classical right-dominant form of the disease (ARVC) because it has been studied most extensively.
ARVC has a prevalence of 1:1000 to 1:5000 in the general population2, while in certain parts of the world it is the major cause of sudden death in individuals ≤35 years of age.3 These numbers may be underestimated because wide phenotypic variation, age-related progression and low genetic penetrance may obscure the diagnosis.4 Currently, the diagnosis of ARVC rests upon fulfilling criteria established by an International Task Force (Table 1) which, although relatively specific, are not highly sensitive.5,6
Table 1.
Revised Task Force Criteria for ARVC (modified from Marcus et al.6 with permission).
|
I. Global or regional dysfunction and structural alterations | |
| Major |
By 2D echo:
|
By MRI:
| |
By RV angiography:
| |
| Minor |
By 2D echo:
|
By MRI:
| |
|
II. Tissue characterization of the wall | |
| Major | Residual myocytes <60% by morphometric analysis (or <50% if estimated), with fibrous replacement of the RV free wall myocardium in ≥1 sample, with or without fatty replacement of tissue on endomyocardial biopsy |
| Minor | Residual myocytes 60% to 75% by morphometric analysis (or 50% to 65% if estimated), with fibrous replacement of the RV free wall myocardium in ≥1 sample, with or without fatty replacement of tissue on endomyocardial biopsy |
|
III. Repolarization abnormalities | |
| Major | Inverted T waves in right precordial leads (V1, V2, and V3) or beyond in individuals >14 years of age (in the absence of complete right bundle-branch block QRS ≥120 ms) |
| Minor |
|
|
IV. Depolarization/conduction abnormalities | |
| Major | Epsilon wave (reproducible low-amplitude signals between end of QRS complex to onset of the T wave) in the right precordial leads (V1 to V3) |
| Minor |
|
|
V. Arrhythmias | |
| Major | Nonsustained or sustained ventricular tachycardia of left bundle-branch morphology with superior axis (negative or indeterminate QRS in leads II, III, and aVF and positive in lead aVL) |
| Minor |
|
|
VI. Family history | |
| Major |
|
| Minor |
|
PLAX stands for parasternal long-axis view; RVOT: RV outflow tract; BSA: body surface area; PSAX: parasternal short-axis view; aVF: augmented voltage unipolar left foot lead; and aVL: augmented voltage unipolar left arm lead.
Diagnostic terminology for criteria: definite diagnosis: 2 major or 1 major and 2 minor criteria or 4 minor from different categories; borderline: 1 major and 1 minor or 3 minor criteria from different categories; possible: 1 major or 2 minor criteria from different categories.
A pathogenic mutation is a DNA alteration associated with ARVC that alters or is expected to alter the encoded protein, is unobserved or rare in a large control population, and either alters or is predicted to alter the structure or function of the protein or has demonstrated linkage to the disease phenotype in a conclusive pedigree.
One feature that distinguishes ARVC from other primary heart muscle disorders associated with sudden death is the early occurrence of arrhythmias out of proportion to the degree of structural damage and contractile derangement. The so-called “concealed phase” of ARVC, in which electrophysiological abnormalities emerge before the onset of apparent pathological changes, is unique among the primary cardiomyopathies. In hypertrophic cardiomyopathy, for example, the risk of arrhythmias appears to be linked to the underlying substrate of myocyte disarray, hypertrophy and fibrosis. In dilated cardiomyopathy, arrhythmic events generally occur in the context of LV dilatation and contractile dysfunction. In contrast, life-threatening arrhythmias may appear early in the natural history of ARVC often preceding significant structural remodeling of the myocardium. In this sense, the early concealed phase of ARVC is reminiscent of the ion channelopathies in which arrhythmias develop in structurally normal hearts. Although there is usually no difficulty in distinguishing ARVC from ion channel diseases, these two groups of diseases share the common feature of a highly arrhythmogenic phenotype that can arise without apparent structural changes. Despite the clear risk of sudden death during the “concealed phase”, the diagnosis of ARVC and risk stratification can be particular difficult.7
Structural changes develop as the disease progresses and may become pronounced. The earliest evidence of myocyte degeneration and accumulation of fibrofatty scar tissue typically occurs in the posterior RV free wall, the outflow tract and the apex, areas that have come to be known as the “triangle of dysplasia”.1 Myocyte degeneration begins in the epicardial-most muscle and progresses towards the endocardium, typically sparing a thin lining of endocardial muscle. This pattern of injury is distinctly different than in others types of cardiomyopathy or in ischemic or pressure overload injury in which pathological changes are more prominent in endocardial muscle. Mononuclear inflammatory infiltrates are often seen in affected areas although it is unclear whether this is a primary manifestation of disease or develops as a secondary response to myocyte injury. In either scenario, inflammation could play a pathogenic role in tissue injury and arrhythmogenesis, although this potential mechanism remains largely unexplored. Ongoing disease progression can lead to diffuse RV free wall and LV involvement, but even in advanced disease, the interventricular septum tends to be spared. A small subset of patients develops biventricular heart failure as a late complication.1 In general, tissue samples from patients with established ARVC may show a heterogeneous picture with variable degrees of myocardial injury and repair including acute necrosis with inflammatory infiltrates, subacute damage with active fibrosis and adipocytes replacing myocytes, and chronic damage with mature fibrous tissue and adipocytes surrounding residual surviving myocytes (Fig 1).8
Figure 1.
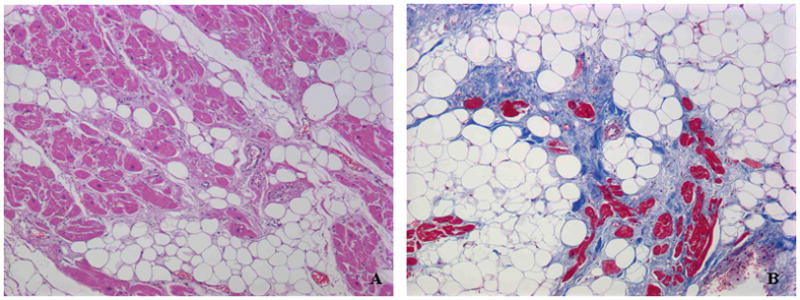
Myocyte degeneration and fibrofatty replacement in right ventricular free wall biopsy samples from patients with ARVC. A: hematoxylin/eosin stain; B: Masson’s trichrome stain.
The genetic basis of ARVC
ARVC is a familial disease in ~50% of cases and most commonly follows an autosomal dominant pattern of inheritance, although recessive forms have been recognized.4 Initial insights into disease mutations came from analysis of Naxos disease and Carvajal syndrome, recessively inherited cardiocutaneous conditions characterized by cardiomyopathy combined with palmoplantar keratoderma and “woolly” hair.9,10 Unambiguous identification of affected individuals with these rare recessive forms facilitated identification of truncation mutations in genes encoding the desmosomal proteins plakoglobin (Naxos disease)9 and desmoplakin (Carvajal syndrome),10 which provided the initial insight that ARVC is related to potential defects in desmosomes. Desmosomes are intercellular adhesion junctions. They are particularly abundant in tissues subjected to mechanical stress, such as the heart and the epidermis.11 They are composed of membrane-spanning desmosomal cadherins (desmocollins and desmogleins) and intracellular linker proteins of the armadillo (plakoglobin and plakophilins) and plakin (desmoplakin) families that connect adhesion junctions to intermediate filaments of the cytoskeleton (Fig 2).12 Mutations in genes encoding all five desmosomal proteins have been implicated in ARVC and can be identified in ~50% of cases that fulfill the Task Force criteria.4 In some cases, a single sequence variant in a desmosomal protein gene, such as that encoding plakophilin2, may be insufficient to cause ARVC by itself but may contribute to disease expression when combined with another sequence variant in the same gene (compound heterozygosity) or with a sequence variant in another desmosomal protein gene (digenic heterozygosity).13,14 Mutations in nondesmosomal genes have also been associated with ARVC, the most convincing of which include those encoding transmembrane protein4315 and the intermediate filament protein desmin.16 Others include transforming growth factor beta-317 and the cardiac ryanodine receptor, although the latter has been more strongly linked to another distinct entity, catecholaminergic polymorphic ventricular tachycardia.18
Figure 2.

Schematic diagram of a desmosome.
Pathological changes in endomyocardial biopsy (EMB)
The first widely recognized clinical description of ARVC was reported in 1982,19 but it was not until the late 1980’s that pathological features of the disease were systematically assessed.20 In an autopsy study of sudden deaths from the Veneto region of Italy, Thiene and colleagues described two distinct patterns – a predominantly fatty (lipomatous) pattern characterized by infiltration of muscle by mature adipocytes only, and a fibrofatty (fibrolipomatous) pattern characterized by both fat and fibrous tissue. Both patterns extended from the epicardium to the endocardium and spared the trabecular myocardium.20 The fatty variant typically involved the RV anterolateral and infundibular regions, whereas the posteroinferior wall, the septum and the LV appeared normal. In the fibrofatty variant, the RV free wall appeared thinner and focally translucent, with saccular aneurysms of the apex, the infundibulum and the posteroinferior wall (the so-called triangle of dysplasia).20 In a subsequent study, the same group described pathological changes in 18 hearts showing the fibrofatty pattern. The RV was affected in all cases, whereas fibrofatty tissue accumulation in the LV was observed in 14 and septal involvement was seen in 6.21 Histologically, affected areas showed myocyte degeneration associated with extensive accumulation of fat and fibrous tissue. A majority of cases exhibited inflammatory cell infiltrates associated with focal myocyte necrosis.21 This was one of the first descriptions of LV involvement in “classical” ARVC, a pattern that has since become widely recognized. Although these authors reported septal involvement in roughly a third of the specimens, they did not provide details about the amount and specific location of fibrofatty tissue accumulation in the interventricular septum.21 In general, however, the septum tends to be the least frequently affected area in ARVC.
Since the original description of these two pathological variants, it has been recognized that individuals with purely fatty infiltration of the RV are older, tend not to have a family history of sudden death and do not appear to be at increased risk of sudden death during sleep or nonstrenuous activity.22 Fat is normally present on the epicardial surface of the RV and typically surrounds intramural coronary vessels.22 Small foci of intramyocardial fat also occur within the RV in healthy individuals and may be increased in association with chronic alcohol abuse or inherited myopathies.23 Intramyocardial fat has been observed in 85% of individuals who died of noncardiac causes, with greater amounts in older subjects and women.24 The lateral RV wall is most commonly affected, followed by the anterior wall, with the least amount of fat present posteriorly.24 Taken together, these observations suggest that infiltration of the myocardium by adipose tissue without demonstrable myocyte degeneration and fibrosis does not fall within the pathological spectrum of arrhythmogenic cardiomyopathy.24 For this reason, fibrofatty replacement, but not pure fatty infiltration, of the myocardium on EMB was considered to be a major diagnostic criterion in the original ARVC Task Force recommendations.5 However, this was included as a purely qualitative determination without an attempt to link diagnostic predictive power to the extent of this pathological change in a conventional septal biopsy.
Recently, Basso et al. analyzed “simulated biopsies” obtained using a clinical bioptome in explanted hearts from patients with ARVC, dilated cardiomyopathy and pure fatty infiltration of the RV to establish quantitative morphometric criteria for the diagnosis of ARVC.25 They sampled each heart in various locations and measured the amount of total biopsy section area occupied by fat, fibrosis, and “residual myocardium”. The most reliable indicator of ARVC was the amount of residual myocardium in biopsies that also contained a combination of fat and fibrous tissue. A diagnostic specific of 95% and sensitivity of 80% was achieved in biopsies containing <59% residual myocardium, with the remaining area occupied by a variable mixture of fat and fibrous tissue. However, the diagnostic yield varied considerably in biopsies obtained from different sites and the interventricular septum was among the least informative areas. The presence of fat without fibrosis was not considered of diagnostic value in ARVC.25 These observations, which have been incorporated in the updated Task Force Criteria,6 underscore the importance of sampling location as a factor in the diagnostic yield of EMB in ARVC. It should also be stressed that this morphometric study was limited to analysis of ARVC hearts in which diffuse or segmental pathological changes were well developed. These quantitative criteria would not be expected to be fulfilled in patients with early ARVC including those exhibiting frequent arrhythmias.
Diagnostic yield versus risk
Typically, ARVC spares the septum and even in advanced cases usually does not involve the entire thickness of the RV.25 It is not surprising, therefore, that a biopsy obtained from the right side of the interventricular septum is unlikely to be diagnostic.25 Sampling the RV free wall, especially in the most frequently involved regions that make up the “triangle of dysplasia”, may increase diagnostic yield but also carries increased risk of ventricular perforation and tamponade.1 Moreover, even this approach is limited by the patchy nature of the disease. Consequently, a negative biopsy is of no value in excluding ARVC. One potential strategy to improve diagnostic yield is to use electroanatomic voltage mapping (EAM) to guide sampling of areas most likely to show pathological features. One study examined 11 patients with overt and 11 with suspected ARVC. All subjects underwent EMB sampling at RV areas of low-voltage areas identified by EAM. In 81% of cases, voltage mapping-guided EMB was diagnostic for ARVC suggesting a high diagnostic sensitivity with this approach.26 Complications were unusual; only 2 patients showed minimal pericardial effusion following the procedure.26 In another study, 32 patients who fulfilled Task Force criteria after noninvasive clinical evaluation were subjected to EAM and EMB to validate the diagnosis.27 Low-voltage areas were generally associated with the presence of myocardial degeneration and fibrofatty replacement. No RV free wall perforations or other complications were reported. Interestingly, a subset of patients who fulfilled clinical criteria for ARVC showed inflammation without fibrofatty replacement in their biopsies, consistent with an inflammatory cardiomyopathy mimicking ARVC.27 More recently, Pieroni et al. studied 30 patients with a clinical diagnosis of ARVC, 29 of whom showed an abnormal voltage map.28 EAM-guided EMB confirmed the diagnosis of ARVC in 15 patients, while the remaining patients had active myocarditis fulfilling the Dallas criteria. Taken together, there studies demonstrate that EAM can improve diagnostic yield in EMB in patients who fulfill clinical criteria for the disease. They also focus renewed attention on the relationship between inflammation and clinical manifestations of ARVC and emphasize the need for additional work on this question. Although complication rates were low, there remain concerns about the hazards of sampling the RV free wall, particularly in nonreferral centers lacking extensive experience. Furthermore, EAM guidance is probably of limited value in early disease in which there may be a significant risk of serious arrhythmias with minimal or absent structural abnormalities.
Beyond histology -- characterizing the molecular pathology of ARVC
To better understand the pathogenesis of ARVC in patients, we have used immunohisto-chemistry to characterize the distribution of mechanical junction molecules at intercalated disks in diseased human myocardium. Because ARVC is typically caused by autosomal dominant mutations, we first asked whether mutant desmosomal proteins are expressed in the myocardium and, if so, whether they reside within cell-cell junctions at intercalated disks. Given the complex binding interactions of many proteins within desmosomes, we also asked whether a dominant mutation in a single desmosomal protein could affect the distribution of other (nonmutant) binding partners within the intercalated disk.29
Our initial studies were focused on Naxos disease, a rare recessive condition caused by truncation of the C-terminal domain of the desmosomal protein plakoglobin (γ-catenin).9 We observed that the mutant protein, plakoglobin, failed to localize normally at cardiac intercalated disks, whereas Western blots of whole tissue lysates showed clearly that it was expressed in the myocardium.30 Additional studies of Carvajal syndrome, a related recessive cardiocutaneous syndrome caused by truncation of the desmosomal protein desmoplakin,10 showed that both the mutant protein (desmoplakin) and one of its binding partners, plakoglobin, failed to localize at cell-cell junctions.31 This observation showed that a mutation in one desmosomal protein may affect the distribution or localization of another (non-mutant) protein of the desmosome.
More recent studies of patients with nonsyndromic ARVC have also shown diminished immunoreactive signal for plakoglobin at intercalated disks (Fig 3).32 This consistent finding occurred in patients with documented mutations in desmoplakin, plakophilin-2, desmoglein-2 and plakoglobin, but also in cases of pathologically documented ARVC in which genetic screening had not identified a mutation in any of the known candidate genes. Plakoglobin signal at cell-cell junctions was reduced not only in RV regions showing typical pathological changes of fibrofatty replacement, but also in the LV free wall and interventricular septum (including the subendocardium) which appeared histologically unremarkable.32 Reduced plakoglobin signal at intercalated seemed to be specific for ARVC, at least based on the observation that signal appeared normal in 15 cases of end-stage heart failure caused by ischemic, dilated or hypertrophic cardiomyopathy.32
Figure 3.
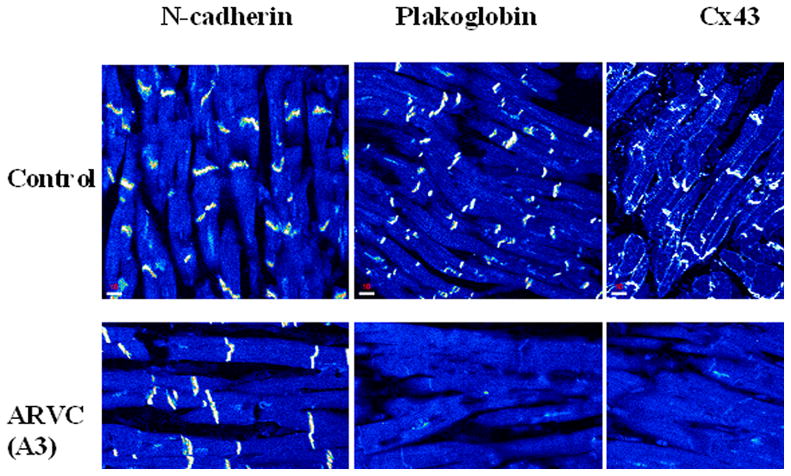
Representative myocardial immunofluorescence images in a control and two examples of autosomal dominant ARVC. Specific immunoreactive signal for all intercalated disk proteins appears as bright white structures at apparent cell-cell junctions against the darker (lower intensity) background of myocytes cut in a longitudinal plane of section. Signal for plakoglobin at intercalated disks is reduced in both cases of ARVC, compared to controls, whereas the amount of signal at cell-cell junctions for other desmosomal proteins including desmoplakin and plakophilin2 is reduced in some cases of ARVC but not in others. In contrast, signal at intercalated disks for the nondesmosomal adhesion molecule N-cadherin is normal in ARVC. The apparent absence of specific junctional signal for a given protein in an ARVC does not prove that the protein is absent from the intercalated disks but only that it is below the threshold of detection by indirect immunofluorescence microscopy.
These observations suggest that reduced immunoreactive signal for plakoglobin in a heart biopsy may be useful in the diagnosis of ARVC. Further studies will be required to validate the specificity and sensitivity of this test but, based on these initial observations, it appears that reduced plakoglobin signal at cell-cell junctions occurs early in the disease (perhaps during the concealed phase) and diffusely throughout the myocardium. Thus, it may be possible in the future to aid in the diagnosis of ARVC by identifying changes in the distribution of desmosomal proteins in conventional RV septal biopsies that otherwise show no evidence of myocyte degeneration or fibrofatty tissue accumulation. It must be remembered, however, that the cause of ARVC is undetermined in the roughly 50% of patients with no known desmosomal gene mutation. Whether loss of plakoglobin signal from junctional pools occurs in these cases remains unknown and requires additional study.29 Thus far, immunohistochemical studies have been confined to examples of typical right-sided ARVC. Its potential diagnostic utility in other forms of arrhythmogenic cardiomyopathy including the recently recognized left-dominant form33 is unknown. There has also been no systematic analysis of asymptomatic individuals or family members of probands, largely because there has been no compelling medical indication for them to undergo EMB. And, there has been no systematic analysis of some other types of heart disease associated with arrhythmias and sudden death such as ion channel diseases, idiopathic ventricular tachycardia and various inflammatory diseases. Thus, the sensitivity and specificity of this marker in identifying early disease or its ability to distinguish penetrant and non-penetrant gene carriers remains to be determined.
As a member of the catenin family of proteins, plakoglobin fulfills dual roles as both an adhesion junction protein and a nuclear signaling molecule that can participate in Wnt signaling pathways. Wnt pathways are involved in fundamental biological processes such as normal cardiac development and differentiation, cardiac hypertrophy, heart failure and aging.34 Wnt pathways regulate cell adhesion and morphogenetic movements by mediating changes in cell cycling, gene expression, cytoskeletal rearrangements and calcium homeostasis. A major component of the canonical Wnt pathway is β-catenin which, like plakoglobin (γ-catenin), resides in cell-cell junctions and is also a signaling molecule.34 The precise role of plakoglobin in Wnt signaling is still incompletely understood, but some evidence suggests that redistribution of plakoglobin from junctional to intracellular and/or intranuclear sites can perturb Wnt signaling pathways.35 The fact that changes in junctional plakoglobin signal occur consistently in nonsyndromic ARVC with or without documented desmosomal gene mutations raises the possibility that altered Wnt signaling mediated by redistribution of plakoglobin may be part of a final common pathway in disease pathogenesis.36
Conclusions
ARVC has an unusually prominent arrhythmogenic phenotype that is manifest early in the natural history of the disease, often preceding the development of ventricular remodeling or contractile dysfunction. Accurate and timely diagnosis is, therefore, pivotal in preventing sudden, unexpected cardiac death. Although the pathological demonstration of myocardial degeneration and fibrofatty replacement is regarded as the diagnostic “gold standard”, endomyocardial biopsy has not been consistently helpful in recognizing the disease partly owing to its patchy nature and lack of histological abnormalities at early stages. Recently, novel molecular markers have been identified that could prove useful if adopted in clinical practice, but further research is required to confirm their diagnostic usefulness.
Acknowledgments
Drs. Saffitz and Asimaki are supported by research grant NIH R01 HL 102361-01. Dr. Asimaki was supported by a Fellowship from the Heart Rhythm Society.
Footnotes
No disclosures.
References
- 1.Sen-Chowdhry S, Morgan RD, Chambers JC, McKenna WJ. Arrhythmogenic cardiomyopathy: etiology, diagnosis and treatment. Ann Rev Med. 2010;61:233–253. doi: 10.1146/annurev.med.052208.130419. [DOI] [PubMed] [Google Scholar]
- 2.Peters S, Trummel M, Meyners W. Prevalence of right ventricular dysplasia-cardiomyopathy in a nonreferral hospital. Int J Cardiol. 2004;97:499–501. doi: 10.1016/j.ijcard.2003.10.037. [DOI] [PubMed] [Google Scholar]
- 3.Basso C, Corrado D, Thiene G. Cardiovascular causes of sudden death in young individuals including athletes. Cardiol Rev. 1999;7:127–135. doi: 10.1097/00045415-199905000-00009. [DOI] [PubMed] [Google Scholar]
- 4.Sen-Chowdhry S, Surris P, Ward D, Asimaki A, Sevdalis E, McKenna WJ. Clinical and genetic characterization of families with arrhythmogenic right ventricular dysplasia/cardiomyopathy provides novel insights into patterns of disease expression. Circulation. 2007;115:1710–1720. doi: 10.1161/CIRCULATIONAHA.106.660241. [DOI] [PubMed] [Google Scholar]
- 5.McKenna WJ, Thiene G, Nava A, Fontaliran F, Blomstrom-Lungvist C, Fontaine G, Camerini F. Diagnosis of arrhythmogenic right ventricular dysplasia/cardiomyopathy. Task Force of the Working Group Myocardial and Pericardial Disease of the European Society of Cardiology and of the Scientific Council on Cardiomyopathies of the International Society and Federation of Cardiology. Br Heart J. 1994;71:215–218. doi: 10.1136/hrt.71.3.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Marcus FI, McKenna WJ, Sherrill D, Basso C, Bauce B, Bluemke DA, Calkins H, Corrado D, Cox MGPJ, Daubert JP, Fontaine G, Gear K, Hauer R, Nava A, Picard MH, Protonotarios N, Saffitz JE, Yoerger Sanborn DM, Steinberg JS, Tandri H, Thiene G, Towbin JA, Tsatsopoulou A, Wichter T, Zareba W. Diagnosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia: proposed modification of the Task Force Criteria. Circulation. 2010;121:1533–1541. doi: 10.1161/CIRCULATIONAHA.108.840827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Towbin JA. Arrhythmogenic right ventricular cardiomyopathy: a paradigm of overlapping disorders. Ann Noninvasive Electrocardiol. 2008;13:325–326. doi: 10.1111/j.1542-474X.2008.00241.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Corrado D, Basso C, Thiene G, McKenna WJ, Davies MJ, Fontaliran F, Nava A, Silvestri F, Blomstrom-Lundqvist C, Wlodarska EK, Fontaine G, Camerini F. Spectrum of clinicopathological manifestations of arrhythmogenic right ventricular cardiomyopathy/dysplasia: a multicenter study. J Am Coll Cardiol. 1997;30:1512–1520. doi: 10.1016/s0735-1097(97)00332-x. [DOI] [PubMed] [Google Scholar]
- 9.McKoy G, Protonotarios N, Crosby A, Tsatsopoulou A, Anastasakis A, Coonar A, Norman M, Baboonian C, Jeffery S, McKenna WJ. Identification of a deletion in plakoglobin in arrhythmogenic right ventricular cardiomyopathy with palmoplantar keratoderma and woolly hair (Naxos disease) Lancet. 2000;355:2119–2124. doi: 10.1016/S0140-6736(00)02379-5. [DOI] [PubMed] [Google Scholar]
- 10.Norgett EE, Hatsell SJ, Carvajal-Huerta L, Cabezas JC, Common J, Purkis PE, Whittock N, Leigh IM, Stevens HP, Kelsell DP. Recessive mutation in desmoplakin disrupts desmoplakin-intermediate filament interactions and causes dilated cardiomyopathy, woolly hair and keratoderma. Hum Mol Genet. 2000;9:2761–2766. doi: 10.1093/hmg/9.18.2761. [DOI] [PubMed] [Google Scholar]
- 11.North AJ, Bardsley WG, Hyam J, Bornslaeger EA, Cordingley HC, Trinnaman B, Hatzfield M, Green KJ, Magee AI, Garrod DR. Molecular map of the desmosomal plaque. J Cell Sci. 1999;112:4325–4336. doi: 10.1242/jcs.112.23.4325. [DOI] [PubMed] [Google Scholar]
- 12.Getsios S, Huen AC, Green KJ. Working out the strength and flexibility of desmosomes. Nat Rev Mol Cell Biol. 2004;5:271–281. doi: 10.1038/nrm1356. [DOI] [PubMed] [Google Scholar]
- 13.Xu T, Yang Z, Vatta M, Rampazzo A, Beffagna G, Pilichou K, Scherer SE, Saffitz J, Kravitz J, Zareba W, Danieli GA, Lorenzon A, Nava A, Bauce B, Thiene G, Basso C, Calkins H, Gear K, Marcus F, Towbin JA. Multidisciplinary Study of Right Ventricular Dysplasia Investigators. Compound and digenic heterozygosity contributes to arrhythmogenic right ventricular cardiomyopathy. J Am Coll Cardiol. 2010;55:587–597. doi: 10.1016/j.jacc.2009.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bhuiyan ZA, Jongbloed JD, van der Smagt J, Lombardi PM, Wiesfeld AC, Nelen M, Schouten M, Jongbloed R, Cox MG, van Wolferen M, Rodriguez LM, van Gelder IC, Bikker H, Suurmeijer AJ, van den Berg MP, Mannens MM, Hauer RN, Wilde AA, van Tintelen JP. Desmoglein-2 and desmocollins-2 mutations in Dutch arrhythmogenic right ventricular dysplasia/cardiomyopathy patients: results from a multicenter study. Circ Cardiovasc Genet. 2009;2:418–427. doi: 10.1161/CIRCGENETICS.108.839829. [DOI] [PubMed] [Google Scholar]
- 15.Merner ND, Hodgkinson KA, Haywood AF, Connors S, French VM, Drenckhahn JD, Kupprion C, Ramadanova K, Thierfelder L, McKenna WJ, Gallagher B, Morris-Larkin L, Bassett AS, Parfrey PS, Young TL. Arrhythmogenic right ventricular cardiomyopathy type 5 is a fully penetrant, lethal arrhythmic disorder caused by a missense mutation in the TMEM43 gene. Am J Hum Genet. 2008;82:809–821. doi: 10.1016/j.ajhg.2008.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Otten E, Asimaki A, Maass A, van Langen IM, van der Wal A, de Jonge N, van den Berg MP, Saffitz JE, Wilde AA, Jongbloed JD, van Tintelen JP. Desmin mutations as a cause of right ventricular heart failure affect the intercalated disks. Heart Rhythm. 2010;7:1058–1064. doi: 10.1016/j.hrthm.2010.04.023. [DOI] [PubMed] [Google Scholar]
- 17.Beffagna G, Occhi G, Nava A, Vitiello L, Ditadi A, Basso C, Bauce B, Carraro G, Thiene G, Towbin JA, Danieli GA, Rampazzo A. Regulatory mutations in transforming growth factor-beta3 gene cause arrhythmogenic right ventricular cardiomyopathy type 1. Cardiovasc Res. 2005;65:366–373. doi: 10.1016/j.cardiores.2004.10.005. [DOI] [PubMed] [Google Scholar]
- 18.Bauce B, Rampazzo A, Basso C, Bagattin A, Daliento L, Tiso N, Turrini P, Thiene G, Danieli GA, Nava A. Screening for ryanodine receptor type 2 mutations in families with effort-induced polymorphic ventricular arrhythmias and sudden death: early diagnosis of asymptomatic carriers. J Am Coll Cardiol. 2002;40:341–349. doi: 10.1016/s0735-1097(02)01946-0. [DOI] [PubMed] [Google Scholar]
- 19.Marcus FI, Fontaine G, Guiraudon G, Frank R, Laurenceau JL, Malergue S, Groshogeat Y. Right ventricular dysplasia: A report of 24 adult cases. Circulation. 1982;65:384–398. doi: 10.1161/01.cir.65.2.384. [DOI] [PubMed] [Google Scholar]
- 20.Thiene G, Nava A, Corrado D, Rossi L, Pennelli N. Right ventricular cardiomyopathy and sudden death in young people. N Engl J Med. 1988;318:129–133. doi: 10.1056/NEJM198801213180301. [DOI] [PubMed] [Google Scholar]
- 21.Basso C, Thiene G, Corrado D, Angelini A, Nava A, Valente M. Arrhythmogenic right ventricular cardiomyopathy: Dysplasia, dystrophy or myocarditis? Circulation. 1996;94:983–991. doi: 10.1161/01.cir.94.5.983. [DOI] [PubMed] [Google Scholar]
- 22.Schejbal V. Epicardial fatty tissue of the right ventricle-morphology, morphometry and functional significance. Pneumonologie. 1989;43:490–499. [PubMed] [Google Scholar]
- 23.Burke AP, Farb SA, Tashko G, Virmani R. Arrhythmogenic right ventricular cardiomyopathy and fatty replacement of the right ventricular myocardium. Circulation. 1998;97:1571–1580. doi: 10.1161/01.cir.97.16.1571. [DOI] [PubMed] [Google Scholar]
- 24.Tansey DK, Aly Z, Sheppard MN. Fat in the right ventricle of the normal heart. Histopathology. 2005;46:98–104. doi: 10.1111/j.1365-2559.2005.02054.x. [DOI] [PubMed] [Google Scholar]
- 25.Basso C, Ronco F, Marcus F, Abudureheman A, Rizzo S, Frigo AC, Bauce B, Maddalena F, Nava A, Corrado D, Grigoletto F, Thiene G. Quantitative assessment of endomyocardial biopsy in arrhythmogenic right ventricular cardiomyopathy/dysplasia: an in vitro validation of diagnostic criteria. Euro Heart J. 2008;29:2760–2771. doi: 10.1093/eurheartj/ehn415. [DOI] [PubMed] [Google Scholar]
- 26.Avella A, D’Amati G, Pappalardo A, Re F, Silenzi PF, Laurenzi F, De Girolamo P, Pelargonio G, Dello Russo A, Baratta P, Messina G, Zecchi P, Zachara E, Tondo C. Diagnostic value of endomyocardial biopsy guided by electroanatomic voltage mapping in arrhythmogenic right ventricular cardiomyopathy/dysplasia. J Cardiovasc Electrophysiol. 2008;19:1127–1134. doi: 10.1111/j.1540-8167.2008.01228.x. [DOI] [PubMed] [Google Scholar]
- 27.Corrado D, Basso C, Leoni L, Tokajuk B, Bauce B, Frigo G, Tarantini G, Napodano M, Turrini P, Ramondo A, Daliento L, Nava A, Buja G, Iliceto S, Thiene G. Three-dimensional electroanatomic voltage mapping increases accuracy of diagnosing arrhythmogenic right ventricular cardiomyopathy/dysplasia. Circulation. 2005;111:3042–3050. doi: 10.1161/CIRCULATIONAHA.104.486977. [DOI] [PubMed] [Google Scholar]
- 28.Pieroni M, Dello Russo A, Marzo F, Pelargonio G, Casella M, Bellocci F, Crea F. High prevalence of myocarditis mimicking arrhythmogenic right ventricular cardiomyopathy differential diagnosis by electroanatomic mapping-guided endomyocardial biopsy. J Am Coll Cardiol. 2009;53:681–689. doi: 10.1016/j.jacc.2008.11.017. [DOI] [PubMed] [Google Scholar]
- 29.Saffitz JE, Asimaki A, Huang H. Arrhythmogenic right ventricular cardiomyopathy: new insights into disease mechanisms and diagnosis. J Invest Med. 2009;57:861–864. doi: 10.2310/JIM.0b013e3181c5e631. [DOI] [PubMed] [Google Scholar]
- 30.Kaplan SR, Gard JJ, Protonotarios N, Tsatsopoulou A, Spiliopoulou C, Anastasakis A, Squarcioni CP, McKenna WJ, Thiene G, Basso C, Brousse N, Fontaine G, Saffitz JE. Remodeling of myocyte gap junctions in arrhythmogenic right ventricular cardiomyopathy due to a deletion in plakoglobin (Naxos disease) Heart Rhythm. 2004;1:3–11. doi: 10.1016/j.hrthm.2004.01.001. [DOI] [PubMed] [Google Scholar]
- 31.Kaplan SR, Gard JJ, Carvajal-Huerta L, Ruiz-Cabezas JC, Thiene G, Saffitz JE. Structural and molecular pathology of the heart in Carvajal syndrome. Cardiovasc Pathol. 2004;13:26–32. doi: 10.1016/S1054-8807(03)00107-8. [DOI] [PubMed] [Google Scholar]
- 32.Asimaki A, Tandri H, Huang H, Halushka MK, Gautam S, Basso C, Thiene G, Tsatsopoulou A, Protonotarios N, McKenna WJ, Calkins H, Saffitz JE. A new diagnostic test for arrhythmogenic right ventricular cardiomyopathy. N Engl J Med. 2009;360:1075–1084. doi: 10.1056/NEJMoa0808138. [DOI] [PubMed] [Google Scholar]
- 33.Sen-Chowdhry S, Syrris P, Prasad SK, Hughes SE, Merrifield R, Ward D, Pennell DJ, McKenna WJ. Left-dominant arrhythmogenic cardiomyopathy: an under-recognized clinical entity. J Am Coll Cardiol. 2008;52:2175–2187. doi: 10.1016/j.jacc.2008.09.019. [DOI] [PubMed] [Google Scholar]
- 34.Rao TP, Kuhl M. An updated overview on Wnt signaling pathways. A prelude for more. Circ Res. 2010;106:1798–1806. doi: 10.1161/CIRCRESAHA.110.219840. [DOI] [PubMed] [Google Scholar]
- 35.Garcia-Gras E, Lombardi R, Giocondo MJ, Willerson JT, Schneider MD, Khoury DS, Marian AJ. Suppression of canonical Wnt/betacatenin signaling by nuclear plakoglobin recapitulates phenotype of arrhythmogenic right ventricular cardiomyopathy. J Clin Invest. 2006;116:2012–2021. doi: 10.1172/JCI27751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Van Tintelen JP, Hauer RNW. New test for arrhythmogenic right ventricular cardiomyopathy. Nat Rev Cardiol. 2009;6:450–451. doi: 10.1038/nrcardio.2009.97. [DOI] [PubMed] [Google Scholar]