

Abstract
Parkinson’s disease is a neurodegenerative disorder characterized by severe motor deficits mainly due to degeneration of dopaminergic neurons in the substantia nigra. Decreased levels of the cell’s most important anti-oxidant, glutathione, have been detected in nigral neurons of Parkinson patients, but it is unknown if they are the cause or merely the consequence of the disease. To elucidate if glutathione depletion causes selective degeneration of nigral dopaminergic neurons, we down-regulated glutathione synthesis in different brain areas of adult rats by a viral vector-based RNAi approach. Decreased glutathione synthesis resulted in progressive degeneration of nigral dopaminergic neurons, while extra-nigral and striatal neurons were significantly less vulnerable. Degeneration of dopaminergic neurons was accompanied by progressive protein aggregate formation and functional motor deficits and was partially rescued by α-synuclein. That the survival of nigral dopaminergic neurons depends on the precise control of glutathione levels was further demonstrated by significant degeneration induced through moderate overproduction of glutathione. Over-expression of either of the two subunits of glutamate–cysteine ligase induced aberrant glutathiolation of cellular proteins and significant degeneration of dopaminergic neurons. Thus, while glutathione depletion was demonstrated to be a selective trigger for dopaminergic neuron degeneration, a glutathione replacement approach as a potential treatment option for Parkinson’s patients must be considered with great care. In conclusion, our data demonstrate that survival of nigral dopaminergic neurons crucially depends on a tight regulation of their glutathione levels and that the depleted glutathione content detected in the brains of Parkinson’s disease patients can be a causative insult for neuronal degeneration.
Electronic supplementary material
The online version of this article (doi:10.1007/s00401-010-0791-x) contains supplementary material, which is available to authorized users.
Keywords: Glutathione, Parkinson’s disease, α-Synuclein, RNAi
Introduction
Degeneration of nigral dopaminergic (DA) neurons is a major hallmark of Parkinson’s disease (PD), resulting in striatal dopamine loss and inappropriate fine tuning of relevant motor circuitry. About 5% of PD patients develop the disease due to familial mutations while the vast majority of cases are sporadic. No singular pathomechanism initiating PD has been identified so far. Current hypotheses encompass oxidative stress through mitochondrial dysfunction, accumulation of transition metals, dysfunction of protein folding quality control, inflammation, and depletion of neurotrophic support or of anti-oxidant defence mechanisms [34]. Depletion of the major cellular anti-oxidant glutathione (GSH) has been described in several independent studies to take place specifically in the substantia nigra of PD patients [27, 28, 30]. GSH depletion is not detected in patients with multiple system atrophy or progressive supranuclear palsy, despite the degeneration of dopaminergic neurons [29, 33]. In incidental Lewy body disease, which may be considered as a pre-symptomatic form of PD [16] and is present in up to 10% of individuals over the age of 60, GSH depletion is already present in the substantia nigra [18], and thus appears to precede complex I dysfunctions, iron accumulation and striatal dopamine loss [38]. These findings imply decreased GSH levels being an early event in PD aetiology, possibly initiating or facilitating a cascade of further oxidative stress lesions. This view is supported by in vitro and in vivo studies demonstrating that GSH depletion induces oxidative damage to complex I proteins [3, 4].
GSH is a tri-peptide consisting of the amino acids glutamate, cysteine and glycine, the reactive thiol group of the cysteine residue serving as the cells most effective anti-oxidant. The rate-limiting step in GSH synthesis is carried out by glutamate–cysteine ligase (GCL), a dimeric protein composed of a catalytic (GCLc) and a modulatory subunit (GCLm). The modulatory subunit serves to increase the affinity of the catalytic subunit for its substrate glutamate, and renders the holo-enzyme less sensitive to feedback inhibition by GSH. Astrocytes synthesize the vast majority of brain GSH and secrete the peptide, but neurons can take up only the precursors derived from extra-cellular catabolism and must synthesize their own GSH. In non-enzymatic reactions GSH detoxifies NO2 and peroxynitrite, and enzymatically reduces other anti-oxidants as dehydroascorbate and, indirectly, tocopherol and through glutaredoxin maintains sulfhydryl groups of proteins in reduced states. GSH peroxidases use GSH for enzymatic reduction of hydrogen peroxide. Oxidized GSH (GSSG) is recycled into the reduced state by GSH-reductase (GSR) [25].
Whether GSH depletion can be a selective trigger specifically for DA neuron degeneration has not yet been demonstrated experimentally. We thus down-regulated GSH synthesis by viral vector-mediated RNA interference in DA and in non-DA neurons of the adult rat brain. We could demonstrate that depletion of anti-oxidative defence serves as an initiator for progressive DA neuron degeneration, including protein aggregation and functional motor deficits, while non-DA neurons were significantly less vulnerable to GSH depletion-induced degeneration. Dopaminergic neuron degeneration was partially prevented by α-synuclein, a synaptic vesicle-associated protein which forms the major protein component of Lewy bodies, the aggregation entities found in postmortem brains of PD patients [12]. Unexpectedly, over-expression of either of the two subunits of GCL also induced significant degeneration of DA neurons, emphasizing that GSH levels must be maintained within a tightly controlled range and suggesting that GSH substitution as a neuroprotective strategy for PD may pose severe risk.
Methods
shRNAs
ShRNAs were selected using the DEQOR algorithm [13] to target (1) rat glutamate–cysteine ligase catalytic subunit (GCLc, E.C. 6.3.2.2); (2) rat glutamate–cysteine ligase modulatory subunit (GCLm); and (3) rat glutathione reductase (GSR, E.C. 1.8.1.7). Control shRNAs were: EGFP-shRNA, Luciferase-shRNA and scrambled shRNA (Dharmacon-shRNA). Detailed sequences are given in Supplemental Methods. All shRNAs were tested for efficiency of target downregulation in a transient co-transfection approach as described [23] and resulted in at least 80% of target mRNA downregulation.
AAV vectors
Recombinant vector backbones were constructed so that expression of all shRNAs was driven by the human H1 polymerase III promoter, while expression of the EGFP reporter or of α-synuclein was driven by the human synapsin 1 gene promoter [20], both expression cassettes being in a head-to-head arrangement. The EGFP expression cassette also contains a WPRE for enhanced mRNA stability and a human bovine growth hormone polyadenylation site. While EGFP expression was entirely neuron-restricted by the hSYN promoter, the H1 promoter is putatively active in all cell types. We ruled out that recombinant AAV-2 vectors would result in astrocyte transduction and concomitant expression of shRNAs in these cells, potentially leading to diminished supply of GSH precursors from astrocytes to neurons by constructing an AAV-2 vector driving EGFP expression from an astrocyte-specific GFAP promoter. Injection of this virus into the substantia nigra resulted in EGFP expression in no more than 5–10 astrocytes per whole midbrain, thus confirming the neuron-specific targeting approach (not shown).
Recombinant vectors were prepared as described [24], purified by iodixanol step gradient, heparin sulphate affinity chromatography and dialysis before stored frozen at −80°C in single-use aliquots. Vector genomes were titrated by quantitative PCR. The purity of the preparations was confirmed by SDS gel electrophoresis and silver staining.
Animal procedures
All experimental animal procedures were conducted according to approved experimental animal licenses issued by the responsible animal welfare authority (Niedersächsisches Landesamt für Verbraucherschutz und Lebensmittelsicherheit) and controlled by the local animal welfare committee of the University Medical Center Goettingen. Intracerebral stereotaxic injections into the substantia nigra (coordinates relative to bregma = AP, −5.3; ML, +2.2; DV, −7.7 mm) and striatum (coordinates AP, +1.2; ML, +2.3; DV, −5.0 mm) of 2.5-month-old female Wistar rats, maintenance of animals and killing and perfusion were performed essentially as described [32]. Two microliter of AAV in PBS containing 4 × 109 vector genomes were injected at a speed of 500 nl/min. This application resulted in transduction of 70–75% of nigral DA neurons as judged by EGFP/TH co-labelling (not shown). For numbers of animals used for each individual group please see Table 1 in Supplemental Information.
Functional/behavioural analysis of nigro-striatal projection
Apomorphine-induced circling behaviour was analysed essentially as described for 6-OHDA lesioned rats [31]. Specifically, animals were placed into cylindrical, flat-bottomed chambers of 40 cm diameter, injected i.p. with 0.4 mg apomorphine/kg body weight and their turning behaviour recorded with a video camera for offline analysis. Only complete turns of 360°C were counted.
Quantification of GSH
The glutathione quantification assay is based on the 96-well microplate assay previously reported by Dringen [10]. Details of the procedure are given in Supplemental Methods.
Cell viability assay
Cell viability was measured in 96-well plates using the WST-1 assay according to the protocol provided by the manufacturer (Roche Diagnostics, Germany).
Immunohistochemistry
Details are given in Supplemental Methods.
Thioflavin-S staining
EGFP fluorescence present in the tissue was eliminated by microwave boiling of mounted sections (5 min in 10 mM Na-Citrate, pH 6.0, 0.05% Tween 20; cooled to RT slowly in ca. 1 h) in order to avoid interference with emitted Thio-S fluorescence. Sections were washed in PBS (10 min), in water (5 min), incubated in 0.05% Thio-S in 50% EtOH (filtered through 0.22 μm) for 20 min, washed 3 × 5 min in 80% EtOH, then 3 × 10 min and 1 × 30 min in PBS followed by immunohistochemistry as outlined in Supplemental Methods. Due to the broad emission spectrum of Thio-S we used only Cy 5-coupled secondary antibodies in order to avoid any possible overlap of emission spectra.
Dopamine measurement
Animals were killed by decapitation and their brains quickly removed. Both striata were prepared individually and quick-frozen on dry ice. 143 μl perchloric acid 70% was added per 50 mg of sample and the mixture homogenized by mechanical beads (Precellys 24 homogenizer) and centrifuged twice. 20 μl of the resulting supernatant was then analysed by HPLC with electrochemical detection as described [19].
Statistics
For statistics, the respective groups were evaluated by one-way ANOVA with Bonferroni’s multiple comparison test using GraphPad Prism. Differences between groups were considered statistically significant if p < 0.05. Significances are indicated with *p < 0.05, **p < 0.01, ***p < 0.001. All data are presented as mean ± standard deviation (SD) unless otherwise indicated.
Results
Silencing expression of the catalytic or the modulatory subunit of glutamate–cysteine ligase induces gradual and progressive degeneration of nigral DA neurons
In order to silence glutathione synthesis in neurons of the rat brain, we constructed AAV-2 vectors expressing the fluorescent reporter protein EGFP plus the respective shRNA. Functional verification of the various vectors was performed in cultured primary neurons (see supplemental data for detailed information), since quantification of GSH levels on a per neuron basis could not be performed in tissue samples due to the overriding GSH content of astrocytes. In vitro experiments showed a direct correlation between the extent of decreasing GSH levels and impaired cell viability (Suppl. Fig. 1), proving functionality and specificity of our approach. In vivo, we first confirmed that continuous expression of shRNAs per se did not impact substantially on the survival of nigral DA neurons (Suppl. Fig. 2). Next, we investigated the loss of DA neurons in the substantia nigra of adult rats due to silencing of glutathione synthesis or recycling. DA neuron numbers were quantified by TH and VMAT2 immunohistochemistry, and loss of neurons was also confirmed by NeuN immunohistochemistry (Suppl. Fig. 3). In comparison to expression of the three different control shRNAs, we found a progressive decline in DA neuron numbers after expression of shRNAs targeting the catalytic subunit of glutamate–cysteine ligase (GCLc). At 3, 6 and 9 weeks after vector injections, expression of GCLc-shRNA#2 resulted in loss of 20, 35 and 57% of DA neurons as compared to control shRNAs, while expression of GCLc-shRNA#3 resulted in loss of 6, 21 and 25% of DA neurons (Fig. 1a; Table 1 in Supplement). These data correlate well with the potency of the respective shRNA to decrease GSH levels and to impair viability in vitro (Suppl. Fig. 1). Also in agreement with in vitro data was the finding that targeting glutathione reductase (GSR) did not result in any significant loss of nigral DA neurons when compared to expression of the control shRNAs.
Fig. 1.
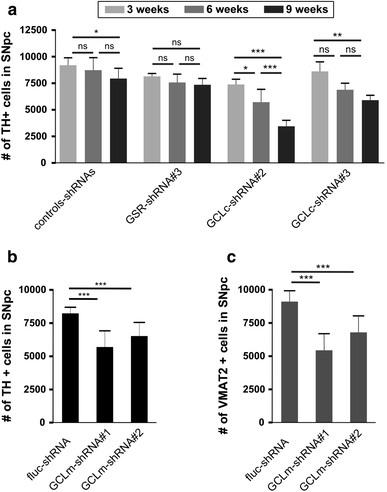
Silencing of glutathione synthesis induces progressive degeneration of nigral dopaminergic neurons in the adult rat brain. a AAV vectors expressing either one of three different control shRNAS (for details see Suppl Fig 1), a shRNA targeting glutathione reductase (GSR), or either one of two shRNAs targeting the catalytic subunit of glutamate–cysteine ligase (GCLc) were injected into rat SNpc; dopaminergic neurons were counted by TH and VMAT2 immunohistochemistry at 3 weeks (light grey bars), 6 weeks (dark grey bars) and 9 weeks (black bars) after transduction. Quantification of surviving DA neurons in SNpc at 9 weeks after targeting the modulatory subunit of glutamate–cysteine ligase (GCLm) with two different shRNAs is shown in b for TH immunohistochemistry and c for VMAT-2 immunohistochemistry, exemplarily demonstrating equal cell numbers with both stains. Bars mean ± SD
In order to evaluate if downregulation of the modulatory subunit of GCL might also result in degeneration of DA neurons, we expressed two different shRNAs targeting GCLm, and achieved moderate but significant loss of DA neurons at 9 weeks after transduction of 29 and 18%, respectively, compared to control shRNAs. As for GCLc, cell counts for TH and VMAT resulted in almost identical numbers (Fig. 1b, c).
These results demonstrated that targeting GSH production by silencing the expression of GCLc or GCLm is sufficient to induce a slowly progressive loss of nigral DA neurons, whereas targeting GSH recycling by silencing the expression of GSR is insufficient to induce such neuronal loss.
Depleted GSH synthesis causes progressive motor imbalance
Next, we analysed impairments of motor control induced by the targeting of GSH synthesis in order to evaluate if DA neuron loss could be documented by behavioural tests. The DA agonist apomorphine induces contralateral rotation behaviour in animals with unilateral de-afferentiation of nigral DA neurons, whereas ipsilateral rotations in unilateral lesions are mainly due to loss of striatal termini of DA neurons. Already 3 weeks after the expression of the GCLc-shRNA#2, at a time when there is only minor DA neuron loss in the SNpc, a significant preference for turning towards the ipsilateral side was evident (Fig. 2a). Both frequency and duration of ipsilateral rotations increased through the course of the experiment, while we detected no significant preference for turning behaviour at any time after expression of the control shRNA. After analysis of rotation behaviour at 9 weeks, the animals were killed and ipsilateral and contralateral striata were quantified for their content of DA and its metabolites DOPAC and HVA (Fig. 2b). After expression of GCLc-shRNA#2, DA levels were found to be reduced to 55% in ipsilateral versus contralateral striatum. Levels of metabolites were found to be reduced to only about 80%, indicating a higher rate of DA metabolism in these brains, as exemplified by a significantly enhanced striatal DA turnover rate (DOPAC/DA = 0.29 ± 0.04 for controls, but 0.42 ± 0.06 after GCLc-shRNA#2 expression; p < 0.05). Expression of control shRNA did not result in any decrease of DA or metabolites.
Fig. 2.
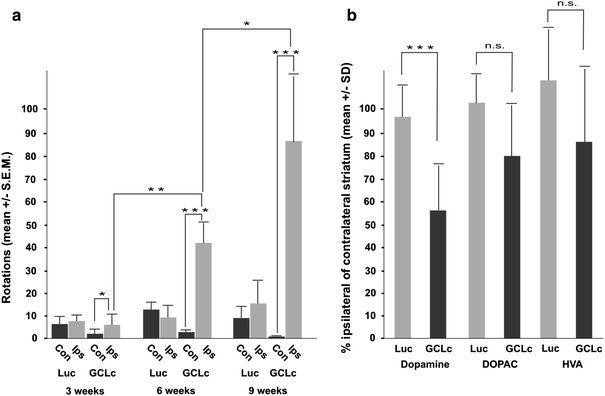
Progressive motor impairments develop after silencing of glutathione synthesis. a Apomorphine-induced turning behaviour was recorded to monitor functional striatal imbalance in DA transmission. At 3, 6 and 9 weeks after administration of an AAV vector expressing a control shRNA (Luc) or GCLc-shRNA#2 (GCLc) the number of 360° rotations to the contralateral side (Con, black bars) or the ipsilateral side (Ips, grey bars) was quantified. b After killing of these animals at 9 weeks after vector administrations, ipsilateral and contralateral striatal levels of DA, DOPAC, and HVA were measured by HPLC and are shown as the percentage of ipsilateral versus contralateral level for control (Luc, grey bars) and GCLc-shRNA#2 (GCLc, dark bars)
Nigral DA neurons are more vulnerable to GSH depletion than extranigral or striatal neurons
All shRNA-expressing viral vectors used in this study also express EGFP as a reporter protein. This allowed us to monitor neuronal transduction and survival independently from cell type specific markers. Injection of AAV-2 vectors into the substantia nigra demonstrated some preference for transduction of DA neurons, but transduced neurons were also readily detected dorsally (in the parabrachial pigmented nucleus) and ventrally (in the pars reticulata) to the pars compacta (Fig. 3a, b). Neurons transduced outside the pars compacta appeared to be morphologically fully intact after 9 weeks of expression of GSH-targeting shRNAs (see insets in Fig. 3a, b). When counting EGFP+/TH− neurons in sections with similar rostro-caudal position at 9 weeks after expression of control or GSH-targeting shRNAs, we found no difference in cell numbers (Fig. 3c), suggesting a selective vulnerability of DA neurons to GSH depletion.
Fig. 3.
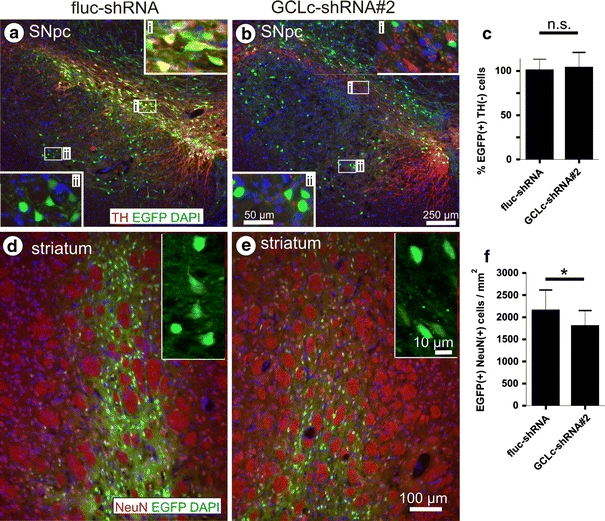
Nigral DA neurons are selectively vulnerable to glutathione depletion. Transduction of SNpc (a, b) and striatum (d, e) by AAV vectors expressing control shRNA (a, d) or GCLc-shRNA#2 (b, e). Pictures are shown as overlays of EGFP fluorescence (green) with TH immunoreactivity (a, b) or NeuN immunoreactivity (d, e) (red) and nuclear DAPI stain (blue). High magnification insets in a and b are taken from areas inside the SNpc (i) and outside the SNpc (parabrachial pigmented nucleus, substantia nigra pars reticulate) (ii). Scale bars are 250 μm in (a, b) (50 μm in insets) and 100 μm in (d, e). Note that exposure times for EGFP were 1,000 ms in d, but 4,000 ms in e. Quantification of EGFP(+)/TH(−) neurons in SNpc is shown in c, quantification of EGFP-positive neurons in striatum after control (fluc-shRNA) or targeting of GSH synthesis (GCLc-shRNA#2) are shown in f as mean ± SD
We also injected vectors encoding control shRNA or shRNA targeting GCLc (GCLc-shRNA#2) into the striatum and recorded EGFP fluorescence at 9 weeks after transduction. We counted 2,148 ± 468 EGFP-positive neurons per mm2 in control and 1,799 ± 352 EGFP-positive neurons per mm2 in GCLc-shRNA expressing brains (Fig. 3d–f). This difference was statistically significant, representing a 16% loss of striatal neurons through GSH depletion compared to control shRNA expression. Since during the same time period 57% of nigral DA neurons degenerated, we conclude the latter to be significantly more vulnerable to GSH depletion. We detected no morphological differences in striatal neurons expressing control or GCLc-targeting shRNAs; however, striatal neurons clearly reacted to a targeting of GSH synthesis in that they displayed a substantially reduced (about tenfold) EGFP fluorescence compared to controls.
Silencing neuronal GSH synthesis induces aggregate formation and moderate gliosis
Thioflavin-S staining for accumulated β-sheet structures revealed several small and uniformly distributed positive signals in DA neurons of the SNpc under all conditions (non-injected, control shRNA expressing and contralateral nigra, Fig. 4a) but not in DA neurons of the VTA (Suppl. Fig. 4). After 3 weeks of expression of GCLc-shRNA#2 many DA neurons displayed Thio-S-positive inclusions just as the controls did (Fig. 4b), but we also detected what appeared to be a progressive formation of aggregated material: several DA neurons showed larger Thio-S positive inclusions (arrow in Fig. 4d) and inclusions within neurites (Fig. 4c), and some cells already demonstrated large Thio-S positive bodies (Fig. 4e, f). The latter cells displayed inhomogeneous or even no TH immunoreactivity. In no case did we detect fragmented nuclei in the vicinity of Thio-S accumulations, arguing against a major contribution of classical apoptosis in the degeneration of these DA neurons, an assumption strengthened by the near absence of activated caspases (Suppl. Fig. 6). At 9 weeks after expression of GCLc-shRNA#2 we mainly detected large peri-nuclear Thio-S positive structures within substantia nigra but not in controls (Fig. 4g, h). It should be noted that we did not detect α-synuclein or ubiquitin immunoreactivity in these aggregates. Neurites which were still TH immunoreactive demonstrated large numbers of substantial swellings (Fig. 4h) after GCLc-shRNA#2 expression, which were almost absent in controls (Fig. 4g).
Fig. 4.
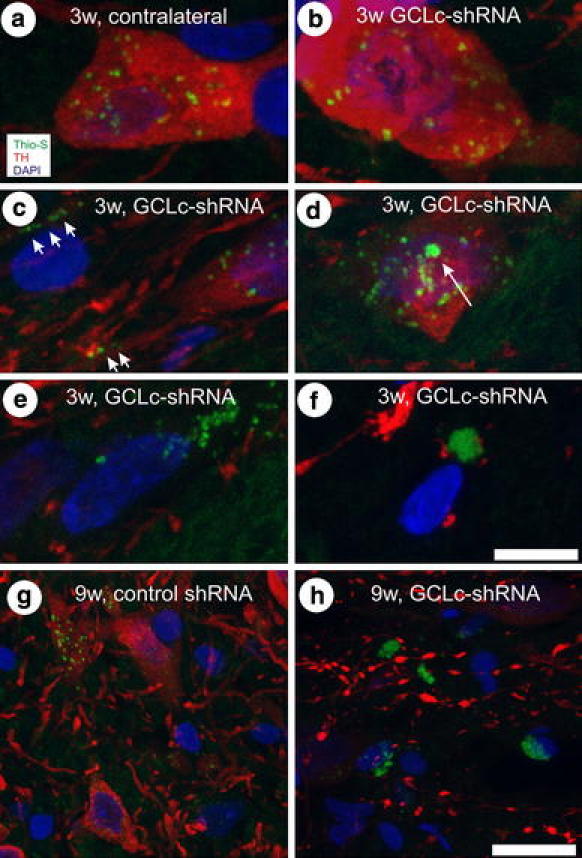
Accumulation of Thioflavin-S-positive protein aggregates in nigral DA neurons. Confocal Z-stacks covering a depth of 15 μm were recorded and combined into a single image, showing Thio-S stain in green, TH immunoreactivity in red and nuclear DAPI stain in blue. Images were recorded at three (a–f) or 9 weeks after transduction (g, h). Arrowheads in c point to neuritic inclusions, arrow in d points to larger cytoplasmic inclusion. In g neurite architecture is intact, while predominantly swollen neurites are seen in h. Scale bar 5 μm in a–f and 20 μm in g, h
GSH depletion-initiated neurodegeneration was accompanied by glial responses. We detected increased immunoreactivity for astrocytic GFAP and microglial CD11b in GCLc-shRNA#2 transduced nigrae (Suppl. Fig. 5b, d) compared to control shRNA-expressing nigrae (Suppl. Fig. 5a, c). All CD11b-positive microglial cells presented ramified morphology, we did not detect cells which had differentiated into macrophages.
α-Synuclein partially protects from GSH-depletion-induced neurodegeneration
In rodents the AAV-2-mediated over-expression of α-synuclein-A53T results in a slowly progressive degeneration of the nigro-striatal projection, with significant impact on axonal and synaptic functionality but without overt loss of DA cell bodies at 9 weeks after vector application [5]. This delayed degeneration is most probably due to the fact that for rodents’ threonine at position 53 represents the wild-type amino acid. In order to investigate if such moderate α-synuclein-mediated neurotoxicity would aggravate degeneration of DA neurons initiated by GSH depletion, we co-expressed α-synuclein-A53T together with either a control shRNA or GCLc-shRNA #2 in DA neurons. α-Synuclein-A53T co-expression with the control shRNA did not result in loss of DA neurons while GCLc-shRNA #2 expression alone resulted in pronounced DA neuron loss. Unexpectedly, we detected a moderate but significant protection from DA neuron loss by co-expression of α-synuclein-A53T with GCLc-shRNA #2, demonstrating that at least under the experimental conditions used in this study α-synuclein can protect DA neurons from GSH depletion-initiated degeneration (Fig. 5a).
Fig. 5.
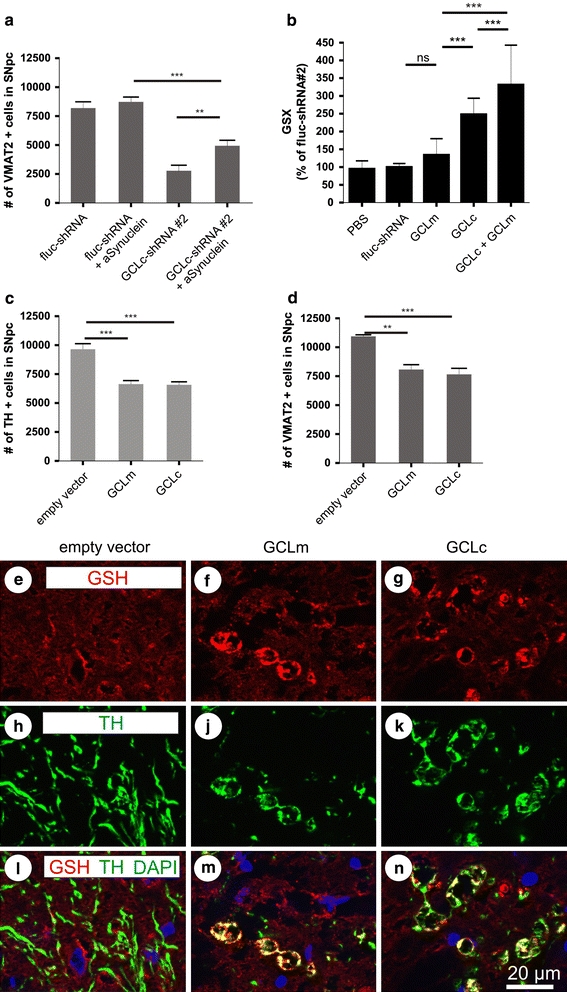
α-Synuclein partially prevents GSH-induced degeneration and overproduction of GSH induces degeneration of nigral DA neurons. Quantification of nigral dopaminergic neurons after co-expression of α-synuclein with control shRNA or GCLc-targeting shRNA is shown in a. GSH overproduction in cultured neurons was quantified after transduction with AAV vectors expressing a control shRNA, the catalytic or the modulatory subunit of GCL, or both subunits together (b). Quantification of SNpc TH+ (c) or VMAT-2+ (d) cells is shown at 9 weeks after transduction with AAV vectors expressing GCLm or GCLc constructs. Immunohistochemistry for GSH-protein adducts (e–g), TH (h, j, k) and their superposition with nuclear DAPI stain (l–n) are shown in tissue sections obtained at 9 weeks after transduction with a control vector (an AAV expressing no transgene, empty vector), AAV-GCLc or AAV-GCLm
Over-expression of GCLc or GCLm leads to aberrant glutathiolation of neuronal proteins and induces neurodegeneration
We furthermore set out to perform a rescue experiment, in which a shRNA can downregulate expression of its cellular target protein, but a non-targetable “rescue protein” is co-expressed to restore the RNAi-induced phenotype. Such a rescue protein has the same amino acid sequence as its cellular counterpart, but is coded by a cDNA in which the binding site for the shRNA is eliminated by nucleotide exchanges. Enhancement of GSH production obtained through over-expression of these rescue proteins was quantified in cultured neurons and demonstrated that a moderate increase in GSH was obtained from over-expression of the human GCLm (120% of control), while about 2.2-fold more GSH than in controls was produced by over-expression of the GCLc-rescue protein. Co-expression of both proteins resulted in approximately threefold higher levels of GSH (Fig. 5b). The respective rescue proteins were then co-expressed in substantia nigra together with shRNAs targeting the endogenous GCLc (GCLc-shRNA#2) or GCLm (GCLm shRNA#1). Surprisingly, we did not find any amelioration of DA neuron death through this rescue approach (data not shown). The unravelling of this paradox came from the finding that over-expression of both subunits of GCL per se, in the absence of any shRNA, induced degeneration of DA neurons (Fig. 5c, d; Table 1 in Supplement). We detected large numbers of distorted neurites emanating from surviving DA neurons, presenting with pearl necklace-like swellings. These swellings stained for TH but also for GSH adducts on proteins, indicating an aberrant glutathiolation of cellular proteins (Fig. 5e–n). These results indicated that even relatively moderate increase of GSH production can serve as a trigger for DA neuron degeneration, suggesting that GSH levels in nigral DA neurons must be intimately maintained within a narrow range.
Discussion
It has long been debated whether GSH depletion as found in PD patients’ substantia nigra is a cause or a consequence of DA neuron degeneration, and if DA neurons would indeed be specifically vulnerable to GSH depletion. Targeting of the catalytic subunit of the rate-limiting enzyme in GSH synthesis, glutamate–cysteine ligase (GCLc) has been the traditional approach to elucidate this issue. Pharmacological inhibition by infusion of the GCLc inhibitor buthionine sulfoxamine (BSO) into the adult rat brain did not induce degeneration of DA neurons [35]. While constitutive knock-out of GCLc was embryonically lethal [7], a conditionally GCLc anti-sense RNA-expressing mouse demonstrated oxidative damage of mitochondrial complex I as a consequence of GSH depletion and showed a minor loss of DA neurons in aged animals [4]. However, given the possible off-target effects of expression of the anti-sense RNA and the lack of any control for such effects [1], this model must be considered with caution. Furthermore, no effects of GSH depletion in non-DA neurons could be assessed, and no behavioural readout could be obtained. We now demonstrate that by an RNAi-mediated approach we could unequivocally induce progressive DA neuron degeneration by targeting the expression of GCLc or GCLm. While nigral DA neurons degenerated progressively over time in a pace depending on potency of the respective shRNA to decrease GSH levels as determined in vitro, extra-nigral non-DA neurons and striatal neurons were apparently less vulnerable. This finding contradicts data obtained from cultured dopaminergic neurons, which were more resistant to BSO application than non-dopaminergic midbrain neurons [26]. In AAV-GCLc-shRNA#2 transduced striatal neurons we found an almost tenfold reduction of fluorescence of the EGFP reporter compared to controls, which might be explained by the strong quenching of the EGFP fluorophore by reactive oxygen species like singlet oxygen derived from lipid peroxidation [11] under conditions of depleted anti-oxidant defence capabilities. This quenching of the EGFP reporter co-expressed with the GCLc-targeting shRNA demonstrates substantial impact on striatal neuron physiology, which, however, degenerated to a much lesser extent than nigral DA neurons. These results for the first time suggest a selective vulnerability of DA neurons towards depletion of anti-oxidant defence, which might be an explanation for their preferential death in PD patients.
GSH depletion induced an apparent continuum of formation of Thio-S positive aggregate structures within affected DA neurons. The morphology of these aggregates resembled the so-called pale bodies (PB) which are thought to be a precursor stage of Lewy bodies (LB), and the obvious morphogenesis of these structures recapitulates the human pathology revealed by immunohistochemical methods in postmortem tissue [21]. However, since they are negative for α-synuclein and ubiquitin immunoreactivity, their composition appeared to be quite different from Lewy bodies. Thus, while GSH depletion can be a selective trigger for DA neurons to degenerate, further impacts must occur in patients to cause α-synuclein aggregation. Intriguingly, our attempts to foster GSH-induced degeneration by over-expression of the familial α-synuclein mutant A53T demonstrated that α-synuclein partially ameliorated DA neuron survival. It should be emphasized that for rodent α-synuclein T53 is the wild-type amino acid, and that AAV-2-mediated over-expression of α-synuclein-A53T in the rat nigra did not result in DA cell body loss at 9 weeks in an independent study [5]. Thus, this protection may not be representative for other α-synuclein mutants or more enforced expression of α-synuclein. However, this finding is reminiscent of α-synuclein’s neuroprotective effect in CSP-α-deficient mice [2], which is mediated through interaction with synaptic vesicles. Together with the early appearance of ipsilateral rotation behaviour and the prevalence of Fluoro-Jade positive neurites rather than cell bodies, protection achieved by α-synuclein suggest that GSH-depletion-induced neurodegeneration might be initiated at synaptic or axonal sites.
While it has been demonstrated in vitro and in vivo that depletion of GSH leads to nitric oxide-mediated oxidative damage of mitochondrial complex I components [4, 8], the mechanism of neuronal cell loss is less clear. Cultured cortical neurons which were depleted by GCLc or GCLm demonstrated features of apoptotic cell death [8], but in concordance with results obtained in postmortem tissue [22], we did not detect significant activation of caspases in nigral DA neurons and no fragmented nuclei by GSH depletion. Activation of astrocytes was moderate, while staining for activated microglia showed more robust increase, as seen in postmortem PD brains [14]. However, microglia morphology remained ramified and no cells with spherical macrophage-like shape appeared, indicating that classical necrosis might not be a prominent feature of DA neuronal death after GSH depletion.
GSH depletion induced a progressive decline in motor control, allowing us to follow the degeneration of the nigro-striatal system by simple behavioural assessment. Thus, this animal model is well suited for testing drugs aimed at protecting DA neurons from degeneration in functional assays, with the option to be transferred to brains of non-human primates. As it is based on a patho-physiological principle detected in brains of sporadic PD patients, it may be a valuable alternative to toxin-based models of PD currently employed, since compounds tested successfully in MPTP and 6-OHDA models have met with very limited benefit in clinical trials [17].
Increase of GSH production by GCLc over-expression was protective against oxidative stress in cultured cortical neurons [8]. The obvious strategy to increase GSH production in DA neurons as a means to protect them from degeneration, however, led to significant cell death in our in vivo study, presumably due to formation of GSH adducts on cellular proteins as detected by a GSH-adduct-specific antibody. In recent years, it has become evident that GSH has important functions besides its reducing activity. In general, thiolation or dethiolation of cellular proteins by GSH may either increase or decrease their levels of activity or binding to other cellular structures [6, 36, 37]. Specifically within the CNS, GSH has been demonstrated to be able to modify NMDA receptor activity and leukotriene metabolism, and thus is directly involved in neuromodulatory events [9, 15]. Due to the overriding GSH content of astrocytes it is not possible to reliably quantify the extent to which GSH was reduced or elevated in individual neurons in vivo, but in cultured neurons over-expressing either the regulatory or the catalytic subunit of GCL we measured only moderate increases in GSH levels, in the range of 1.5- to 2.5-fold to controls. Thus, our results suggest that even relatively moderate increases in GSH can initiate degeneration of DA neurons, arguing against delivery of GCLc or GCLm to the brain, e.g. by gene therapeutic attempts, as a potential treatment strategy for PD.
In conclusion, our results demonstrate the distinct dependence of nigral DA neurons on tight control of GSH levels, suggesting that disturbed GSH homeostasis can be a trigger in the initiation of DA neuron degeneration in PD.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgments
We are grateful to Ulrike Schöll for excellent technical assistance with primary neuron preparations, Erin Butler and Anja Drinkut for help with HPLC, and Cathy Ludwig for critically reading the manuscript. This work was supported by the European Community’s Seventh Framework Programme FP7/2007-2013 under grant agreement no. HEALTH-F5-2008-222925, by the German Research Council-funded Center of Molecular Physiology of the Brain (CMPB) and by the European Research Training Network 2004–2007 under contract no. MRTN-CT-2003-504636.
Conflict of interest
The authors declare that they have no conflict of interest.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Abbreviations
- AAV-2
Adeno-associated virus serotype 2
- DA
Dopamine
- EGFP
Enhanced green fluorescent protein
- GCL-c
Catalytic subunit of glutamate-cysteine ligase
- GCL-m
Modulatory subunit of glutamate-cysteine ligase
- GSH
Glutathione, reduced state
- GSSG
Oxidized glutathione
- GSX
Total glutathione
- shRNA
Small helical RNA
- TH
Tyrosine hydroxylase
- PD
Parkinson’s disease
- VMAT-2
Vesicular mono amine transporter 2
References
- 1.Achenbach TV, Brunner B, Heermeier K. Oligonucleotide-based knockdown technologies: antisense versus RNA interference. Chembiochem. 2003;4:928–935. doi: 10.1002/cbic.200300708. [DOI] [PubMed] [Google Scholar]
- 2.Chandra S, Gallardo G, Fernandez-Chacon R, Schlüter OM, Südhof TC. Alpha-synuclein cooperates with CSPalpha in preventing neurodegeneration. Cell. 2005;123:383–396. doi: 10.1016/j.cell.2005.09.028. [DOI] [PubMed] [Google Scholar]
- 3.Chinta SJ, Andersen JK. Reversible inhibition of mitochondrial complex I activity following chronic dopaminergic glutathione depletion in vitro: implications for Parkinson’s disease. Free Radic Biol Med. 2006;41:1442–1448. doi: 10.1016/j.freeradbiomed.2006.08.002. [DOI] [PubMed] [Google Scholar]
- 4.Chinta SJ, Kumar MJ, Hsu M, et al. Inducible alterations of glutathione levels in adult dopaminergic midbrain neurons result in nigrostriatal degeneration. J Neurosci. 2007;27:13997–14006. doi: 10.1523/JNEUROSCI.3885-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chung CY, Koprich JB, Siddiqi H, Isacson O. Dynamic changes in presynaptic and axonal transport proteins combined with striatal neuroinflammation precede dopaminergic neuronal loss in a rat model of AAV alpha-synucleinopathy. J Neurosci. 2009;29:3365–3373. doi: 10.1523/JNEUROSCI.5427-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dalle-Donne I, Rossi R, Colombo G, Giustarini D, Milzani A. Protein S-glutathionylation: a regulatory device from bacteria to humans. Trends Biochem Sci. 2009;34:85–96. doi: 10.1016/j.tibs.2008.11.002. [DOI] [PubMed] [Google Scholar]
- 7.Dalton TP, Dieter MZ, Yang Y, Shertzer HG, Nebert DW. Knockout of the mouse glutamate cysteine ligase catalytic subunit (Gclc) gene: embryonic lethal when homozygous, and proposed model for moderate glutathione deficiency when heterozygous. Biochem Biophys Res Commun. 2000;279:324–329. doi: 10.1006/bbrc.2000.3930. [DOI] [PubMed] [Google Scholar]
- 8.Diaz-Hernandez JI, Almeida A, Delgado-Esteban M, Fernandez E, Bolanos JP. Knockdown of glutamate–cysteine ligase by small hairpin RNA reveals that both catalytic and modulatory subunits are essential for the survival of primary neurons. J Biol Chem. 2005;280:38992–39001. doi: 10.1074/jbc.M507065200. [DOI] [PubMed] [Google Scholar]
- 9.Dringen R. Metabolism and functions of glutathione in brain. Prog Neurobiol. 2000;62:649–671. doi: 10.1016/S0301-0082(99)00060-X. [DOI] [PubMed] [Google Scholar]
- 10.Dringen R, Hamprecht B. Glutathione content as an indicator for the presence of metabolic pathways of amino acids in astroglial cultures. J Neurochem. 1996;67:1375–1382. doi: 10.1046/j.1471-4159.1996.67041375.x. [DOI] [PubMed] [Google Scholar]
- 11.Greenbaum L, Rothmann C, Lavie R, Malik Z. Green fluorescent protein photobleaching: a model for protein damage by endogenous and exogenous singlet oxygen. Biol Chem. 2000;381:1251–1258. doi: 10.1515/BC.2000.153. [DOI] [PubMed] [Google Scholar]
- 12.Hardy J, Lewis P, Revesz T, Lees A, Paisan-Ruiz C. The genetics of Parkinson’s syndromes: a critical review. Curr Opin Genet Dev. 2009;19:254–265. doi: 10.1016/j.gde.2009.03.008. [DOI] [PubMed] [Google Scholar]
- 13.Henschel A, Buchholz F, Habermann B. DEQOR: a web-based tool for the design and quality control of siRNAs. Nucleic Acids Res. 2004;32:W113–W120. doi: 10.1093/nar/gkh408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hirsch EC, Hunot S. Neuroinflammation in Parkinson’s disease: a target for neuroprotection? Lancet Neurol. 2009;8:382–397. doi: 10.1016/S1474-4422(09)70062-6. [DOI] [PubMed] [Google Scholar]
- 15.Janaky R, Ogita K, Pasqualotto BA, et al. Glutathione and signal transduction in the mammalian CNS. J Neurochem. 1999;73:889–902. doi: 10.1046/j.1471-4159.1999.0730889.x. [DOI] [PubMed] [Google Scholar]
- 16.Jellinger KA. A critical reappraisal of current staging of Lewy-related pathology in human brain. Acta Neuropathol. 2008;116:1–16. doi: 10.1007/s00401-008-0406-y. [DOI] [PubMed] [Google Scholar]
- 17.Jenner P. Functional models of Parkinson’s disease: a valuable tool in the development of novel therapies. Ann Neurol. 2008;64(Suppl 2):S16–S29. doi: 10.1002/ana.21489. [DOI] [PubMed] [Google Scholar]
- 18.Jenner P, Dexter DT, Sian J, Schapira AH, Marsden CD. Oxidative stress as a cause of nigral cell death in Parkinson’s disease and incidental Lewy body disease. The Royal Kings and Queens Parkinson’s Disease Research Group. Ann Neurol. 1992;32(Suppl):S82–S87. doi: 10.1002/ana.410320714. [DOI] [PubMed] [Google Scholar]
- 19.Kowsky S, Poppelmeyer C, Kramer ER, et al. RET signaling does not modulate MPTP toxicity but is required for regeneration of dopaminergic axon terminals. Proc Natl Acad Sci USA. 2007;104:20049–20054. doi: 10.1073/pnas.0706177104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kügler S, Lingor P, Schöll U, Zolotukhin S, Bähr M. Differential transgene expression in brain cells in vivo and in vitro from AAV-2 vectors with small transcriptional control units. Virology. 2003;311:89–95. doi: 10.1016/S0042-6822(03)00162-4. [DOI] [PubMed] [Google Scholar]
- 21.Kuusisto E, Parkkinen L, Alafuzoff I. Morphogenesis of Lewy bodies: dissimilar incorporation of alpha-synuclein, ubiquitin, and p62. J Neuropathol Exp Neurol. 2003;62:1241–1253. doi: 10.1093/jnen/62.12.1241. [DOI] [PubMed] [Google Scholar]
- 22.Levy OA, Malagelada C, Greene LA. Cell death pathways in Parkinson’s disease: proximal triggers, distal effectors, and final steps. Apoptosis. 2009;14:478–500. doi: 10.1007/s10495-008-0309-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Malik I, Garrido M, Bähr M, Kügler S, Michel U. Comparison of test systems for RNA interference. Biochem Biophys Res Commun. 2006;341:245–253. doi: 10.1016/j.bbrc.2005.12.173. [DOI] [PubMed] [Google Scholar]
- 24.Malik JM, Shevtsova Z, Bähr M, Kügler S. Long-term in vivo inhibition of CNS neurodegeneration by Bcl-XL gene transfer. Mol Ther. 2005;11:373–381. doi: 10.1016/j.ymthe.2004.11.014. [DOI] [PubMed] [Google Scholar]
- 25.Martin HL, Teismann P. Glutathione—a review on its role and significance in Parkinson’s disease. FASEB J. 2009;23:3263–3272. doi: 10.1096/fj.08-125443. [DOI] [PubMed] [Google Scholar]
- 26.Nakamura K, Wright DA, Wiatr T, et al. Preferential resistance of dopaminergic neurons to the toxicity of glutathione depletion is independent of cellular glutathione peroxidase and is mediated by tetrahydrobiopterin. J Neurochem. 2000;74:2305–2314. doi: 10.1046/j.1471-4159.2000.0742305.x. [DOI] [PubMed] [Google Scholar]
- 27.Pearce RK, Owen A, Daniel S, Jenner P, Marsden CD. Alterations in the distribution of glutathione in the substantia nigra in Parkinson’s disease. J Neural Transm. 1997;104:661–677. doi: 10.1007/BF01291884. [DOI] [PubMed] [Google Scholar]
- 28.Perry TL, Godin DV, Hansen S. Parkinson’s disease: a disorder due to nigral glutathione deficiency? Neurosci Lett. 1982;33:305–310. doi: 10.1016/0304-3940(82)90390-1. [DOI] [PubMed] [Google Scholar]
- 29.Perry TL, Yong VW. Idiopathic Parkinson’s disease, progressive supranuclear palsy and glutathione metabolism in the substantia nigra of patients. Neurosci Lett. 1986;67:269–274. doi: 10.1016/0304-3940(86)90320-4. [DOI] [PubMed] [Google Scholar]
- 30.Riederer P, Sofic E, Rausch WD, et al. Transition metals, ferritin, glutathione, and ascorbic acid in parkinsonian brains. J Neurochem. 1989;52:515–520. doi: 10.1111/j.1471-4159.1989.tb09150.x. [DOI] [PubMed] [Google Scholar]
- 31.Schwarting RK, Huston JP. The unilateral 6-hydroxydopamine lesion model in behavioral brain research. Analysis of functional deficits, recovery and treatments. Prog Neurobiol. 1996;50:275–331. doi: 10.1016/S0301-0082(96)00040-8. [DOI] [PubMed] [Google Scholar]
- 32.Shevtsova Z, Malik I, Garrido M, Schöll U, Bähr M, Kügler S. Potentiation of in vivo neuroprotection by BclX(L) and GDNF co-expression depends on post-lesion time in deafferentiated CNS neurons. Gene Ther. 2006;13:1569–1578. doi: 10.1038/sj.gt.3302822. [DOI] [PubMed] [Google Scholar]
- 33.Sian J, Dexter DT, Lees AJ, et al. Alterations in glutathione levels in Parkinson’s disease and other neurodegenerative disorders affecting basal ganglia. Ann Neurol. 1994;36:348–355. doi: 10.1002/ana.410360305. [DOI] [PubMed] [Google Scholar]
- 34.Thomas B, Beal MF (2007) Parkinson’s disease. Hum Mol Genet 16(Spec No. 2):R183–R194 [DOI] [PubMed]
- 35.Toffa S, Kunikowska GM, Zeng BY, Jenner P, Marsden CD. Glutathione depletion in rat brain does not cause nigrostriatal pathway degeneration. J Neural Transm. 1997;104:67–75. doi: 10.1007/BF01271295. [DOI] [PubMed] [Google Scholar]
- 36.Wang J, Boja ES, Tan W, et al. Reversible glutathionylation regulates actin polymerization in A431 cells. J Biol Chem. 2001;276:47763–47766. doi: 10.1074/jbc.C100415200. [DOI] [PubMed] [Google Scholar]
- 37.Ward NE, Stewart JR, Ioannides CG, O’Brian CA. Oxidant-induced S-glutathiolation inactivates protein kinase C-alpha (PKC-alpha): a potential mechanism of PKC isozyme regulation. Biochemistry. 2000;39:10319–10329. doi: 10.1021/bi000781g. [DOI] [PubMed] [Google Scholar]
- 38.Zeevalk GD, Razmpour R, Bernard LP. Glutathione and Parkinson’s disease: is this the elephant in the room? Biomed Pharmacother. 2008;62:236–249. doi: 10.1016/j.biopha.2008.01.017. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.