

Abstract
Background/Objective
Viral infections have been implicated in the pathogenesis of multiple sclerosis (MS). Plasmacytoid dendritic cells (pDCs) are present in peripheral blood, cerebrospinal fluid and brain lesions of MS patients. PDCs sense viral DNA via Toll-like receptor 9 (TLR9) which has to be cleaved from the N-terminal to become functional (TLR9 processing). PDCs activated with TLR9 agonists promote Th1/Th17-responses. In the animal model of MS, TLR9 agonists can induce disease. We hypothesized that pDCs are inhibited by disease-modifying therapy such as IFN-beta, consequently decreasing the frequency of MS attacks.
Methods
We separated pDCs from healthy subjects and patients diagnosed with relapsing-remitting MS and clinically isolated syndrome. Cytokine secretion by pDCs activated with TLR9 agonists was measured by ELISA and multi-analyte profiling. TLR9 gene and protein expression was studied by DNA microarrays and Western blot.
Results
In untreated patients, pDCs activated with TLR9 agonists produced increased levels of IFN-alpha, a Th1 promoting cytokine, as compared to healthy subjects. In IFN-beta treated patients, activated pDCs had decreased ability to produce both IFN-alpha and the pro-inflammatory cytokines IL-6 and TNF-alpha as compared to untreated patients. PDCs separated from IFN-beta treated patients had significantly reduced levels of the processed TLR9 protein but normal levels of the full-length TLR9 protein and TLR9 gene expression as compared to untreated patients.
Interpretation
This finding represents a novel immunomodulatory mechanism of IFN-beta which is inhibition of TLR9 processing. This results in decreased activation of pDCs by viral pathogens and, thus, may affect the frequency of MS exacerbations.
INTRODUCTION
Multiple sclerosis (MS) is considered to be an immune-mediated disorder of the central nervous system 1-2. Although the immunopathogenesis of the disease is not completely understood, both polygenic and environmental factors contribute to disease onset and/or clinical exacerbation 3-5. Viral pathogens have been implicated in the etiology and pathogenesis of MS (reviewed in 6). Among those, strong data implicates Epstein-Barr Virus (EBV), a human DNA virus 7-11. For established MS patients, the risk of disease exacerbation was found to be increased at the time or shortly after clinical viral infections 12-14 (reviewed by Rutschmann et al 15). The antiviral protein IFN-gamma, a T helper type 1 (Th1)-type cytokine produced mainly by NK and T cells, was found to trigger multiple sclerosis in vivo 16-17.
Plasmacytoid dendritic cells (pDCs), characterized as CD11c-CD123++Lin-DR++ cells, produce large amounts of type I IFN in response to viral infection 18-19. Compared to other peripheral blood mononuclear cells, pDCs express a high level of Toll-like receptor 9 (TLR9) 20 which recognizes viral DNA within the early endosomes at the initial phase of viral infection. It has been recently discovered that the full-length TLR9 has to be cleaved from the N-terminal to generate a functional (processed) TLR9 C-terminal 21-22. Activated via TLR9, pDCs can secrete 100-1,000-fold higher levels of interferon (IFN)-alpha, than any other blood cell type 23.
PDCs link innate and adaptive immunity via a number of cytokines implicated in the pathogenesis of demeylination 24. IFN-Type I cytokines induce intracellular signaling in lymphocytes via a transcription factor STAT4 leading to Th1 cell differentiation (reviewed in 25). IL-6 promotes myelin antigen-specific Th17- and Th1-responses in experimental autoimmune encephalomyelitis (EAE) 26. TNF-alpha directly induces oligodendrocyte apoptosis 27 and mediates human neuronal injury after activation with TLR9 agonists 28. Activation of antigen-presenting cells through TLR9 can overcome tolerance and precipitate EAE 29-31. The generation of Th17 cells is decreased in pDC-depleted mice and is associated with less severe clinical and histopathological signs of EAE 32. PDCs are found in the CSF of MS patients 33-36 and accumulate in MS lesions 8,37. Thus, pDCs may serve as a strong link between viral infection and MS exacerbation.
We hypothesize that pDCs may trigger MS exacerbation in response to viral pathogens but are inhibited by disease-modifying therapy such as IFN-beta, consequently decreasing the frequency of MS attacks. Here we describe a new immunodulatory effect of IFN-beta in MS involving the inhibition of TLR9 processing.
MATERIALS AND METHODS
1. Subjects
Patients and healthy donors, 18-60 years old, were enrolled in the study. Patients were diagnosed with clinically definite relapsing-remitting MS (RR MS) or clinically isolated syndrome (CIS) as described 38, and not taking any immuno-modulatory drugs other than IFN-beta based treatment. The typical clinical presentations of patients with CIS were unilateral optic neuritis, hemiparesis or unilateral sensory deficit (confirmed by a symptomatic spinal cord lesion on MRI). Patients with secondary progressive MS and primary progressive MS, patients with EDSS score 6 or higher, or patients who received IV steroids or any other non-IFN-beta immunomodulatory drugs less than 2 months prior to blood drawing were excluded. The patients at the time of clinical attack of MS were excluded. The patients were treated with IFN-beta 1a or IFN-beta 1b in doses approved by FDA and recommended by the drug manufacturers. The patients and healthy donors were enrolled at two MS Centers. The patients and healthy subjects presented in Figures 1 and described in Table 1 were enrolled at the Partners MS Center, Boston, MA. The patients and healthy subjects presented in other figures and described in Table 2 and appropriate figure legends were enrolled at the RWJ Center for MS, New Brunswick, NJ. The participation of patients and healthy subjects in the study was approved by institutional review boards. Informed consent was obtained from all subjects.
Figure 1.

IFN-alpha secretion by activated pDCs. pDCs were separated from healthy donors (HD), untreated patients (MS: No Rx), and IFN-beta-treated RR MS patients (MS: IFN-beta). pDCs were activated in vitro with TLR9 agonist for 16 hours. IFN-alpha was measured in culture supernatants by ELISA, as described in Patients and Methods. IFN-alpha production was elevated in untreated patients (mean ± SEM = 8,479 ± 1,898 pg/ml, n = 11) as compared to healthy donors (3,659 ± 891 pg/ml, n = 17), p = 0.0164. MS patients treated with IFN-beta had decreased IFN-alpha production (2,826 ± 1,350 pg/ml, n = 12) as compared to untreated RR MS patients, p = 0.027. No detectable IFN-alpha was seen in control pDC cultures without TLR9 agonist. ELISA = enzyme-linked immunosorbent assay; SEM = standard error of the mean.
Table 1.
Patients presented in Figure 1.
| Subjects: Treatment | Type of MS (number of subjects) | Age (mean ± SD) | Female/Male (% of female subjects) | IFN-beta treatment** (number of patients) |
|---|---|---|---|---|
| Healthy donors | Control (17) | 33.9 ± 6.0 | 10/7 (59%) | None |
| MS: No Rx* | RR MS (11) | 41.9 ± 9.7 | 5/6 (45%) | None |
| MS: IFN-beta* | RR MS (12) | 40.6 ± 13.1 | 9/3 (75%) | Intramuscular IFN-beta-1a (4), Subcutaneous IFN-beta-1b (1), Subcutaneous IFN-beta-1a (7) |
There is no overlap between those two groups of patients.
The duration of IFN-beta treatment was 2.75 ± 1.42 years (mean ± SD)
Table 2.
Patients presented in Figure 4.
| Subjects: Treatment | Type of MS (number of subjects)* | Age (mean ± SD) | Female/Male (% of female subjects) | IFN-beta treatment ** (number of patients) |
|---|---|---|---|---|
| Healthy donors | Control (9) | 34.22 ± 7.6 | 8/1 (89%) | None |
| MS: No Rx | CIS (5), RR MS (9) | 35.7 ± 9.8 | 10/4 (71%) | None |
| MS: IFN-beta | CIS (5), RR MS (7) | 40.1 ± 11.4 | 10/2 (83%) | Intramuscular IFN-beta-1a (5), Subcutaneous IFN-beta-1b (5), Subcutaneous IFN-beta-1a (2) |
The total number of patients is twenty three as three patients were tested both before and after treatment with IFN-beta and therefore were included in both groups
The patients were treated with IFN-beta for 3 months.
2. Blood samples and pDC separation
Peripheral blood mononuclear cells (PBMC) were isolated from 80-100 ml of heparinized blood samples within 4 hours after venipuncture as described 39. Fresh pDCs were separated from PBMC by immunomagnetic sorting and BDCA-4 cell isolation kit (Miltenyi Biotec) with 2 steps of pDC enrichment on magnetic columns. Such sample yields approximately 200,000-250,000 pDCs. More than 90% of separated cells were CD11c-CD123+DR++ as measured by flow cytometry. The separated cells had approximately 100 fold higher level of TLR9 gene expression compared to PBMC (Table 3). The separated cells were immediately used for cell culture experiments, suspended in the RNAlater stabilization reagent (Qiagen, Santa Clarita, CA) for gene expression analysis or frozen at -80°C for Western blot experiments.
Table 3.
TLR9 relative gene expression.
| Sample | TLR9* | IFN-alpha1* | IRF7* |
|---|---|---|---|
| PBMC | 0.024 | 0.67 | 0.08 |
| PBMC depleted from pDCs | 0.015 | 0.40 | 0.077 |
| pDCs | 3.73 | 2.83 | 4.60 |
TLR9 gene expression was compared in freshly isolated PBMC and pDCs by RT-qPCR. IFN-alpha1 and IRF-7 gene expression were tested in the same samples. The human GAPDH, housekeeping gene, was used to normalize gene expression in different samples. The results are representative of three different experiments done with three different healthy subjects.
3. Cytokine secretion by pDCs
Freshly isolated pDC (100,000 cells/ml) were cultured in round-bottomed 96 well plates in a total volume of 0.2 ml/well. Parallel cultures of pDCs were treated with the TLR9 agonist CpG ODN class A (ODN#2336, Coley Pharmaceutical), or control ODN (#2243, Coley Pharmaceutical) at 5 mcg/ml (0.735 mcMol). Cell supernatants were collected after 16 hours and tested by multi-subtype ELISA kit (Cat # 41105-2, PBL Biomedical Laboratories) to measure the total IFN-alpha secretion. The kit is able to recognize 13 different IFN-alpha species. In addition, detection of selected cytokines (IFNα2, TNFα, and IL-6) was provided by the BioMarker Services (Millipore Corporation, St. Charles, MO) using MILLIPLEX Multi-Analyte Profiling with Luminex xMAP Multiplexing Technology (http://www.millipore.com/drugdiscovery/dd3/map_portfolio) with a standard curve range from 3.2 to 10,000 pg/ml.
4. RT-qPCR
Total RNA was prepared from 1.0 × 105 cell sample using the RNeasy Mini Kit (Qiagen, Santa Clarita, CA) according to the manufacturer’s protocol 40 and processed for cDNA synthesis using the TaqMan Reverse Transcription Kit (Cat No N808-0234, Applied Biosystems, Foster City, CA). For TaqMan RT-PCR, Master Mix Kit (Cat No 4304437) and TaqMan® Gene Expression Assays (the set of primers and probes) for human genes such as GAPDH, IFN-alpha1, IRF7, TLR9, were purchased from Applied Biosystems. RT-qPCR was performed according to the manufacturer’s protocol using iCycler iQ Real-Time detection System (Bio-Rad Laboratories, Hercules, CA). The human GAPDH, housekeeping gene, was used to normalize each sample and each gene.
5. Affymetrix microarray analysis of TLR9 gene expression
Total RNA was prepared from 1.0 × 105 pDC sample using Trizol reagent (Invitrogen, USA) and further purified with Qiagen RNAeasy columns with DNase treatment (Qiagen, Valencia, CA). RNA quality was assessed by capillary electrophoresis using the Agilent Bioanalyzer 2100 and spectrophotometric analysis prior to cDNA synthesis. Fifty nanograms of total RNA from each sample were amplified to cDNA, fragmentated and biotinylated using the Nugen kits WT Ovation Pico (NuGen, San Carlos, CA) and FL Ovation Biotin (NuGen). The labeled samples were hybridized to Affymetrix GeneChip® Human Genome U133A 2.0 Arrays according to manufacturer’s recommendations for hybridization, washing and scanning. Data analysis was done for each pair of patient by GeneSpring software to get the fold change of relative gene expression.
6. Western Blot
100,000 cells were sonicated in Nupage LDS Sample buffer (4X) (Invitrogen) with 50mM DTT and heated at 70°C for 10 minutes. Cell lysates were loaded on Nupage 4-12% Bis-Tris gels (Invitrogen), electrophoresed and then transferred to PVDF membrane (Biorad) by electroblotting. The membranes were blocked in Tris/Tween-20 buffer with 5% Non-fat dry milk (Biorad) for 1hr at room temperature and then probed with 1.5 mcg/ml of rabbit polyclonal anti-human TLR9 antibodies (Ab) (Santa Cruz Biothechnology) for overnight at 4°C to detect the C–terminal of TLR9 (processed TLR9). Membranes were washed three times with 50mM Tris/Tween-20 buffer (pH7.4) with 150mM NaCl and 0.1% Tween-20 and probed with anti-rabbit IgG-HRP (1:25,000 final dilution, Sigma-Aldrich) for one hour at room temperature. The Pierce ECL Western Blotting Substrate was used to detect proteins by chemiluminescence. The intensities of protein bands were measured with Image J software (NIH). Membranes were stripped with Restore Western Blot Stripping Buffer (Pierce) and re-probed with 2 mcg/ml of mouse monoclonal anti-human TLR9 Ab (Imgenex) to detect the full length TLR9 followed by anti-mouse IgG-HRP (1:1,000 final dilution, Sigma-Aldrich). Membranes were stripped again and re-probed with mouse monoclonal anti-Beta-actin Ab (1:5,000 final dilution, Sigma-Aldrich) followed by anti-mouse IgG-HRP (1:10,000 final dilution, Sigma-Aldrich) for protein normalization.
6. Statistical analysis
Within patient change from baseline (Figure 2), laboratory measures were tested by a paired t-test. The difference between groups (Figures 1 and 4) was tested by unpaired t-test.
Figure 2.
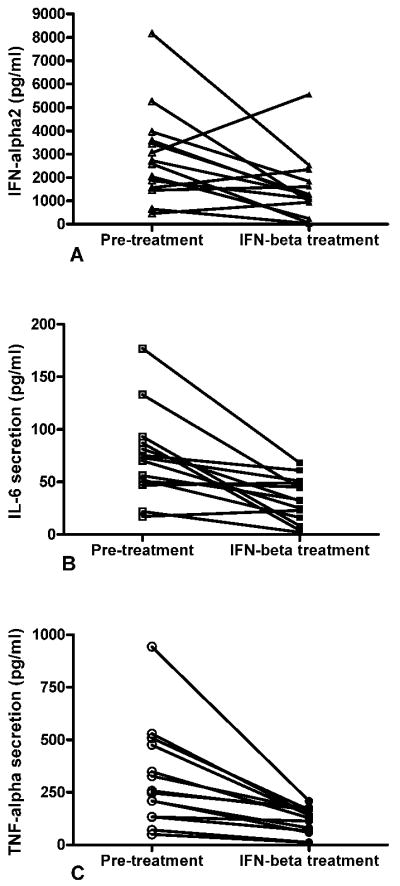
IFN-beta treatment inhibits IFN-alpha, IL-6, and TNF-alpha secretion by activated pDCs. pDCs were separated from the same MS/CIS patients before (pretreatment) and after a 3-month course of treatment (IFN-beta treatment) with recombinant human IFN-beta-1b (subcutaneous, n = 8) or recombinant human IFN-beta-1a (intramuscular, n = 6). The average age of patients was 36.3 years; 9 females and 5 males. pDCs were activated in vitro with TLR9 agonist for 16 hours. (A) IFN-alpha2, (B) IL-6, and (C) TNF-alpha were measured in the same cell supernatants by MILLIPLEX Multi-Analyte Profiling as described in Patients and Methods. IFN-beta-treated patients had decreased levels of IFN-alpha2 (mean ± SEM = 1,498 ± 372 pg/ml vs 2,917 ± 535; p = 0.0246), IL6 (33 ± 6 pg/ml vs 74 ± 11; p = 0.0012), and TNF-alpha (117 ± 17 pg/ml vs 317 ± 63; p = 0.0021) when compared to untreated patients. SEM = standard error of the mean.
Figure 4.

IFN-beta treatment inhibits TLR9 processing in MS. Plasmacytoid dendritic cells (pDCs) were separated from healthy donors (HD), nontreated MS/CIS patients (MS: No Rx), and MS/CIS patients treated with IFN-beta–based medications (MS: IFN-beta) for at least 3 months. The patient’s characteristics are presented in Table 2. The level of the (A) full-length and (B) processed TLR9 protein expression was determined in the same patients by western blot, as described in Patients and Methods. IFN-beta-treated CIS/MS patients had a similar level of the full-length TLR9 (mean ± SEM = 0.108 ± 0.025 relative units, n = 12) compared to untreated patients (0.112 ± 0.015 relative units, n = 14). However, IFN beta-treated patients had a significantly decreased level of the processed TLR9 C-terminal when compared to untreated patients (mean ± SEM = 0.924 ± 0.063 vs 1.445 ± 0.106 relative units, respectively; p = 0.0005). The level of the TLR9 C-terminal in healthy subjects was 1.189 ± 0.1385 relative units (n = 9), which was less than, but not statistically different from, untreated patients. SEM = standard error of the mean.
RESULTS
1. Cytokine production by activated pDCs in RR MS
a) Increased level of IFN-alpha production in RR MS patients is inhibited in IFN-beta treated patients
PDCs are the major source of the Th1-promoting cytokine IFN-alpha. We compared IFN-alpha production by activated pDCs in healthy donors (HD), untreated and treated RR MS patients. As shown in Figure 1, IFN-alpha production was significantly elevated in untreated RR MS patients as compared to healthy subjects, p = 0.0164. RR MS patients treated with IFN-beta based medications had significantly decreased IFN-alpha production as compared to untreated RR MS patients, p = 0.027. There was no statistically significant difference between healthy subjects and IFN-beta treated MS patients, p > 0.05.
b) IFN-beta inhibits TLR9-dependent intracellular signaling pathways
Two major TLR9-dependent intracellular signaling pathways were described. Both pathways share a key adaptor MyD88 (myeloid differentiation primary-responsible gene 88). The first pathway leads to production of IFN-type I cytokines and requires phosphorylation and translocation of IRF7 to the nucleus promoting IFN-alpha and IFN-beta transcription. In contrast, the gene expression of pro-inflammatory cytokines, such as TNF-alpha and IL-6, is dependent on activation of NF-κB and ATF2-c-Jun 19 41. Therefore we studied IL-6, TNF-alpha and IFN-alpha production in the same cultures of pDCs activated with TLR9 agonists (Figure 2). In addition, to avoid genetic heterogeneity between tested groups and to minimize the possible effect of IFN-beta neutralizing antibodies which usually do not appear until 6 months after IFN-beta is initiated 42, each MS patient was tested prior to and after a 3month course of IFN-beta treatment. The patients had no prior history of being treated with IFN-beta-based drugs. As shown on Figure 2, compared to pre-treatment, the average cytokine production was significantly reduced by 49% (IFN-alpha2), 55% (IL-6) and 63% (TNF-alpha) in IFN-beta treated patients. Among fourteen patients tested, we could identify three groups of patients based on their response to IFN-beta. The first group (ten patients) had all cytokines decreased after treatment with IFN-beta. The second group (two patients) had increased IFN-alpha2 secretion and decreased IL-6 and TNF-alpha secretion. The third group (two patients) had both IFN-alpha and IL-6 increased while TNF-alpha secretion was decreased. In part, it could be linked to different intracellular signaling mechanisms involved in regulation of IFN-type I cytokines versus other pro-inflammatory cytokines, such as TNF-alpha and IL-6 as described above.
Although disease modifying treatment was offered to all patients diagnosed with MS or CIS, two patients declined treatment after being diagnosed with CIS. They agreed to provide 2 blood samples 3 months apart. The mean cytokine production for those patients at two different time points was 2873±1707 and 3220±1045 pg/ml (IFN-alpha2), 150±93 and 172.5±74.5 pg/ml (IL-6), 418±212 and 380±87 pg/ml (TNF-alpha). In summary, the results presented in Figures 1 and 2 strongly suggest that IFN-beta treatment inhibits TLR9-mediated intracellular signaling pathways in activated pDCs from MS patients.
2. IFN-beta does not affect TLR9 gene expression
TLR9, a unique receptor able to recognize DNA sequences typical of DNA viruses, is expressed mostly by pDCs and B cells in human peripheral blood 43. We have compared TLR9, IRF7 and IFN-alpha gene expression in PBMC and pDCs by QRT-PCR. As expected, expression of TLR9 was approximately 100 fold elevated in pDCs compared to PBMC (Table 3). Please note that TLR9 is expressed in the population of PBMC depleted from pDCs due to the presence of TLR9-positive B cells. IRF7 is constitutively expressed in pDCs at high levels 44-45.
Next we studied if IFN-beta treatment affects TLR9 gene expression in MS patients. TLR9 gene expression was studied by oligo microarrays in paired pDC samples separated from 8 MS patients before and after a 3 month course of treatment with recombinant human IFN-beta-1b (subcutaneous, n=4) or recombinant human IFN-beta-1a (intramuscular, n=4). The average age of patients was 37 ± 12 years old (Mean ±SD), 6 females and 2 males. Based on paired t test, there was no statistically significant effect of IFN-beta treatment on TLR9 gene expression. The fold change of relative TLR9 gene expression after treatment was 1.205 ± 0.286 (Mean ±SD), p = 0.082. The analysis of the same oligo microarrays before and after treatment with the imposed minimal threshold of 1.5 fold change from the baseline and the p value of 0.05 or less did not show a significant effect of IFN-beta treatment on gene expression for the following molecules involved in TLR9 intracellular pathway as depicted in Kegg Toll-like receptor signaling pathway (http://www.genome.jp/kegg-bin/show_pathway?hsa04620): MyD88, IRAK-1, IRAK-4, TRAF6, IRF5, IRF7, CHUK (IKKα), CHUK (IKKα), CHUK (IKKα), NFKB1 (NFkB), NFKBIA(IkBα) (data not shown).
3. IFN-beta treatment inhibits TLR9 processing in MS patients
It has been recently discovered that a full length TLR9 (100 to 150 kDa depending on the level of protein glycosylation) is in fact non-functional and needs to be cleaved to generate a functional C-terminal TLR9 with a molecular weight between 65 to 80 kDa depending on the source of TLR9-positive cells 21-22. Therefore, to study the processing of TLR9, we applied Western Blot analysis and probing with the C terminal-specific Abs. We were able to detect a 65 kDa band corresponding to the processed C-terminal TLR9 in all pDC samples (Figure 3). As expected, the processed TLR9 was expressed mostly in pDCs. The expression of the processed TLR9 in B cells was approximately 20-50 fold less as compared to pDCs and was extremely low in the total population of PBMC (Figure 3). We also proved the specific interaction between a 65 kDa protein and CpG oligonucleotide (TLR9 agonist) by co-culture of biotinylated CpG oligonucleotides with pDCs followed by protein immunoprecipitation with Streptavidin Dynabeads followed by Western Blot of the precipitate with C-terminal anti-TLR9 Abs (Supplementary Figure 1). We then analyzed TLR9 processing in pDCs isolated from healthy subjects and MS patients. As shown in Figure 4A, there was no significant difference in the level of full-length unprocessed TLR9 between treated (n = 14) and untreated (n = 12) patients. However, IFN beta-treated CIS/RR MS patients had significantly decreased level of the processed (functionally active) TLR9 C-terminal as compared to untreated patients, p = 0.0005. Therefore, it was not the full-length TLR9 protein synthesis but the processing of the TLR9 protein which was significantly inhibited in IFN-beta treated patients. TLR9 processing was also analyzed separately in patients with CIS and patients with clinically definite RR MS. Among patients with RR MS, TLR9 processing was also significantly decreased (p = 0.003) in IFN-beta treated patients (n = 7) as compared to untreated patients (n=9) but did not reach statistical significance for CIS patients (n = 5 in each group), which could be due to the small number of subjects tested.
Figure 3.

TLR9 processing in pDCs and B cells. PBMCs, pDCs, and B cells were obtained from a healthy subject. The levels of the full-length TLR9 with the expected molecular weight of 115kDa (which may be higher due to protein glycosylation) and the C-terminal of TLR9 (processed TLR9) with the expected molecular weight of 65–80kDa were determined by western blot. The beta-actin level in the same sample was used for protein normalization. Note that more PBMCs and B cells were used for each experiment (1 × 106) than pDCs (1 × 105). The results are representative of 3 independent experiments with 3 different healthy subjects.
DISCUSSION
Although many IFN-alpha species and IFN-beta bind to the same Type I IFN receptor, it is not quite clear why there is so much variation in their biological activity 46.
Our initial results (Figure 1) suggested that activated pDCs produce increased level of IFN-alpha, a Th1 promoting cytokine, in non-treated MS patients as compared to healthy subjects. In addition, IFN-alpha secretion was significantly decreased in MS patients treated with recombinant IFN-beta as compared to non-treated patients. Several studies examined the function of pDCs in MS 35,36. IFN-beta treatment did not affect the concentration of pDCs in peripheral blood from MS patients 35,37, but inhibited expression of MHC class I and BDCA-2 and upregulated CD38, B7H1 37 and CD123 35 on pDCs. TLR9 ligand-induced IFN-alpha production by unfractionated peripheral blood mononuclear cells was decreased in MS patients but was not studied in enriched pDCs 36. Recently, Bayas et al. studied TLR9-agonist activated IFN-alpha secretion by pDCs in MS. Bayas et al. used methods for pDC separation, culture condition and IFN-alpha detection different from our protocol. The difference between MS patients and healthy subjects was not statistically significant for the strong IFN-alpha inducer (CpG Type A oligo which is similar to TLR9 agonist used in our study) 47. IFN-beta treatment inhibited IFN-alpha production by pDCs separated from healthy subjects but was not studied in MS patients 37.
In our experiments, IFN-beta treated patients had decreased production of IFN-alpha, IL-6 and TNF-alpha by activated pDCs (Figure 2). Those cytokines are regulated by two different TLR9-dependent pathways 19,41. Therefore, it led us to hypothesize that IFN-beta may modulate TLR9 expression.
However, IFN-beta did not affect TLR9 gene expression or the level of unprocessed TLR9 protein (Figure 4a). Therefore, we hypothesized that IFN-beta treatment may inhibit the effect of viral agonists on pDCs by reducing the processing of TLR9. Ex-vivo experiments with pDCs separated from CIS/MS patients confirmed this hypothesis (Figure 4b).
TLR9 processing has been recently discovered in two independent laboratories 21-22. To our knowledge, TLR9 processing has not been studied in human diseases. The exact mechanism of modulation of TLR9 processing is not clear at this time and may be influenced by the effect of IFN-beta on other immunomodulatory proteins. Protease inhibitors were shown to prevent TLR9 processing; however, proteases responsible for cleavage of TLR9 have not been definitively identified 21-22. Matsumoto et al reported that cathepsins B and L inhibitor block TLR9 responses 48. Interestingly, recombinant human IFN-beta was also shown to suppress the cathepsin B activity in a dose-dependent manner 49. However, Ewald et al. suggested that in addition to cathepsins, other proteases are capable to cleave TLR9 as selective cathepsin inhibitors could not abolishTLR9 processing completely 21-22.
The finding of decreased TLR9 processing in IFN-beta treated MS patients establishes a new mechanism of disease-modifying treatment in MS. TLR9 is the only member of the family of TLRs able to sense DNA viruses implicated in the pathogenesis of MS. If confirmed by others and linked to clinical disease activity, modulation of TLR9 processing may prove to be an important target for a new generation of MS immunomodulatory drugs such as TLR antagonists.
Supplementary Material
Acknowledgments
K.E.B. is supported by grant number K23NS052553 from the National Institute of Neurological Disorders and Stroke and grants from the National Multiple Sclerosis Society and Bayer Healthcare. We thank Joan Moore and Sheila Reaves for assistance in preparing this manuscript.
Footnotes
- Konstanatin E. Balashov has served as a consultant for Biogen, TEVA Neuroscience, Bayer Healthcare and EMD Serono.
- Suhayl Dhib-Jalbut has served as a consultant for Biogen, TEVA Neuroscience, Bayer Healthcare and EMD Serono.
- Howard L. Weiner has served as a consultant and participated on advisory boards for Bayer Healthcare, EMD Serono and Biogen Idec.
- Latt Latt Aung and Adi Vaknin-Dembinsky have nothing to disclose.
References
- 1.McFarland HF, Martin R. Multiple sclerosis: a complicated picture of autoimmunity. Nat Immunol. 2007;8(9):913–9. doi: 10.1038/ni1507. [DOI] [PubMed] [Google Scholar]
- 2.Sospedra M, Martin R. Immunology of multiple sclerosis. Annu Rev Immunol. 2005;23:683–747. doi: 10.1146/annurev.immunol.23.021704.115707. [DOI] [PubMed] [Google Scholar]
- 3.Hafler DA, Compston A, Sawcer S, Lander ES, Daly MJ, De Jager PL, et al. Risk alleles for multiple sclerosis identified by a genomewide study. N Engl J Med. 2007;357(9):851–62. doi: 10.1056/NEJMoa073493. [DOI] [PubMed] [Google Scholar]
- 4.Noseworthy J, Lucchinetti C, Rodriguez M, Weinshenker B. Multiple Sclerosis. New England Journal of Medicine. 2000;343(13):938–52. doi: 10.1056/NEJM200009283431307. [DOI] [PubMed] [Google Scholar]
- 5.Compston A, Coles A. Multiple Sclerosis. Lancet. 2002;359:1221–31. doi: 10.1016/S0140-6736(02)08220-X. [DOI] [PubMed] [Google Scholar]
- 6.Cook S, Rohowsky-Kochan C, Bansil S, Dowling P. Evidence for a viral etiology of multiple sclerosis. In: Cook S, editor. Handbook of multiple sclerosis. 2. New York: Marcel Dekker, Inc; 1996. pp. 97–118. [Google Scholar]
- 7.Haahr S, Hollsberg P. Multiple sclerosis is linked to Epstein-Barr virus infection. Rev Med Virol. 2006;16(5):297–310. doi: 10.1002/rmv.503. [DOI] [PubMed] [Google Scholar]
- 8.Serafini B, Rosicarelli B, Franciotta D, Magliozzi R, Reynolds R, Cinque P, et al. Dysregulated Epstein-Barr virus infection in the multiple sclerosis brain. J Exp Med. 2007;204(12):2899–912. doi: 10.1084/jem.20071030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Giovannoni G, Cutter GR, Lunemann J, Martin R, Munz C, Sriram S, et al. Infectious causes of multiple sclerosis. Lancet Neurol. 2006;5(10):887–94. doi: 10.1016/S1474-4422(06)70577-4. [DOI] [PubMed] [Google Scholar]
- 10.Levin LI, Munger KL, Rubertone MV, Peck CA, Lennette ET, Spiegelman D, et al. Multiple sclerosis and Epstein-Barr virus. Jama. 2003;289(12):1533–6. doi: 10.1001/jama.289.12.1533. [DOI] [PubMed] [Google Scholar]
- 11.Ascherio A, Munger K. Epidemiology of multiple sclerosis: from risk factors to prevention. Semin Neurol. 2008;28(1):17–28. doi: 10.1055/s-2007-1019126. [DOI] [PubMed] [Google Scholar]
- 12.Edwards S, Zvartu M, Clarke H, Irving W, Blumhardt L. Clinical relapses and disease activity on magnetic resonance imaging associated with viral upper respiratory tract infections in multiple sclerosis. Journal of Neurology, Neurosurgery and Psychiatry. 1998;64:736–41. doi: 10.1136/jnnp.64.6.736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sibley W, Bamford C, Clark L. Clinical viral infections and multiple sclerosis. Lancet. 1985;1:1313–15. doi: 10.1016/S0140-6736(85)92801-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Panitch HS. Influence of infection on exacerbations of multiple sclerosis. Ann Neurol. 1994;36(Suppl):S25–8. doi: 10.1002/ana.410360709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rutschmann OT, McCrory DC, Matchar DB. Immunization and MS: a summary of published evidence and recommendations. Neurology. 2002;59(12):1837–43. doi: 10.1212/wnl.59.12.1837. [DOI] [PubMed] [Google Scholar]
- 16.Panitch HS, Hirsch RL, Schindler J, Johnson KP. Treatment of multiple sclerosis with gamma interferon: exacerbations associated with activation of the immune system. Neurology. 1987;7:1097–102. doi: 10.1212/wnl.37.7.1097. [DOI] [PubMed] [Google Scholar]
- 17.Panitch HS, Hirsch RL, Haley AS, Johnson KP. Exacerbations of multiple sclerosis in patients treated with gamma interferon. Lancet. 1987;1(8538):893–5. doi: 10.1016/s0140-6736(87)92863-7. [DOI] [PubMed] [Google Scholar]
- 18.Colonna M, Trinchieri G, Liu Y. Plasmacytoid dendritic cells in immunity. Nature Immunology. 2004;5(12):1219–26. doi: 10.1038/ni1141. [DOI] [PubMed] [Google Scholar]
- 19.Gilliet M, Cao W, Liu YJ. Plasmacytoid dendritic cells: sensing nucleic acids in viral infection and autoimmune diseases. Nat Rev Immunol. 2008;8(8):594–606. doi: 10.1038/nri2358. [DOI] [PubMed] [Google Scholar]
- 20.Liu YJ. IPC: professional type 1 interferon-producing cells and plasmacytoid dendritic cell precursors. Annu Rev Immunol. 2005;23:275–306. doi: 10.1146/annurev.immunol.23.021704.115633. [DOI] [PubMed] [Google Scholar]
- 21.Ewald SE, Lee BL, Lau L, Wickliffe KE, Shi GP, Chapman HA, et al. The ectodomain of Toll-like receptor 9 is cleaved to generate a functional receptor. Nature. 2008;456(7222):658–62. doi: 10.1038/nature07405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Park B, Brinkmann MM, Spooner E, Lee CC, Kim YM, Ploegh HL. Proteolytic cleavage in an endolysosomal compartment is required for activation of Toll-like receptor 9. Nat Immunol. 2008;9(12):1407–14. doi: 10.1038/ni.1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Siegal F, Kadowaki N, Shodell M, Fitzgerald-Bocarsly P, Shah K, Ho S, et al. The nature of the principal type 1 interferon-producing cells in human blood. Science. 1999;284:1835–37. doi: 10.1126/science.284.5421.1835. [DOI] [PubMed] [Google Scholar]
- 24.McKenna K, Beignon AS, Bhardwaj N. Plasmacytoid dendritic cells: linking innate and adaptive immunity. J Virol. 2005;79(1):17–27. doi: 10.1128/JVI.79.1.17-27.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Korman BD, Kastner DL, Gregersen PK, Remmers EF. STAT4: genetics, mechanisms, and implications for autoimmunity. Curr Allergy Asthma Rep. 2008;8(5):398–403. doi: 10.1007/s11882-008-0077-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Serada S, Fujimoto M, Mihara M, Koike N, Ohsugi Y, Nomura S, et al. IL-6 blockade inhibits the induction of myelin antigen-specific Th17 cells and Th1 cells in experimental autoimmune encephalomyelitis. Proc Natl Acad Sci U S A. 2008;105(26):9041–6. doi: 10.1073/pnas.0802218105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Akassoglou K, Bauer J, Kassiotis G, Pasparakis M, Lassmann H, Kollias G, et al. Oligodendrocyte apoptosis and primary demyelination induced by local TNF/p55TNF receptor signaling in the central nervous system of transgenic mice: models for multiple sclerosis with primary oligodendrogliopathy. Am J Pathol. 1998;153(3):801–13. doi: 10.1016/S0002-9440(10)65622-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Iliev AI, Stringaris AK, Nau R, Neumann H. Neuronal injury mediated via stimulation of microglial toll-like receptor-9 (TLR9) Faseb J. 2004;18(2):412–4. doi: 10.1096/fj.03-0670fje. [DOI] [PubMed] [Google Scholar]
- 29.Segal BM, Chang JT, Shevach EM. CpG oligonucleotides are potent adjuvants for the activation of autoreactive encephalitogenic T cells in vivo. J Immunol. 2000;164(11):5683–8. doi: 10.4049/jimmunol.164.11.5683. [DOI] [PubMed] [Google Scholar]
- 30.Ichikawa HT, Williams LP, Segal BM. Activation of APCs through CD40 or Toll-like receptor 9 overcomes tolerance and precipitates autoimmune disease. J Immunol. 2002;169(5):2781–7. doi: 10.4049/jimmunol.169.5.2781. [DOI] [PubMed] [Google Scholar]
- 31.Waldner H, Collins M, Kuchroo VK. Activation of antigen-presenting cells by microbial products breaks self tolerance and induces autoimmune disease. Journal of Clinical Investigation. 2004;113(7):990–97. doi: 10.1172/JCI19388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Isaksson M, Ardesjo B, Ronnblom L, Kampe O, Lassmann H, Eloranta ML, et al. Plasmacytoid DC promote priming of autoimmune Th17 cells and EAE. Eur J Immunol. 2009;39(10):2925–35. doi: 10.1002/eji.200839179. [DOI] [PubMed] [Google Scholar]
- 33.Pashenkov M, Huang Y, Kostulas V, Haglund M, Soderstrom M, Link H. Two subsets of dendritic cells are present in human cerebrospinal fluid. Brain. 2001;124:480–92. doi: 10.1093/brain/124.3.480. [DOI] [PubMed] [Google Scholar]
- 34.Pashenkov M, Teleshova N, Kouwenhoven M, Kostulas V, Huang Y, Soderstrom M, et al. Elevated expression of CCR5 by myeloid (CD11c+) blood dendritic cells in multiple sclerosis and acute optic neuritis. Clinical and Experimental Immunology. 2002;127:519–26. doi: 10.1046/j.1365-2249.2002.01779.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lopez C, Comabella M, Al-zayat H, Tintore M, Montalban X. Altered maturation of circulating dendritic cells in primary progressive MS patients. J Neuroimmunol. 2006;175(1-2):183–91. doi: 10.1016/j.jneuroim.2006.03.010. [DOI] [PubMed] [Google Scholar]
- 36.Stasiolek M, Bayas A, Kruse N, Wieczarkowiecz A, Toyka KV, Gold R, et al. Impaired maturation and altered regulatory function of plasmacytoid dendritic cells in multiple sclerosis. Brain. 2006;129(Pt 5):1293–305. doi: 10.1093/brain/awl043. [DOI] [PubMed] [Google Scholar]
- 37.Lande R, Gafa V, Serafini B, Giacomini E, Visconti A, Remoli ME, et al. Plasmacytoid dendritic cells in multiple sclerosis: intracerebral recruitment and impaired maturation in response to interferon-beta. J Neuropathol Exp Neurol. 2008;67(5):388–401. doi: 10.1097/NEN.0b013e31816fc975. [DOI] [PubMed] [Google Scholar]
- 38.Jacobs LD, Beck RW, Simon JH, Kinkel RP, Brownscheidle CM, Murray TJ, et al. Intramuscular interferon beta-1a therapy initiated during a first demyelinating event in multiple sclerosis. CHAMPS Study Group. N Engl J Med. 2000;343(13):898–904. doi: 10.1056/NEJM200009283431301. [DOI] [PubMed] [Google Scholar]
- 39.Balashov KE, Rottman JB, Weiner HL, Hancock WW. CCR5(+) and CXCR3(+) T cells are increased in multiple sclerosis and their ligands MIP-1alpha and IP-10 are expressed in demyelinating brain lesions. Proc Natl Acad Sci U S A. 1999;96(12):6873–8. doi: 10.1073/pnas.96.12.6873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wandinger K, Sturzebecher C, Bielekova B, Detore G, Rosenwald A, Staudt L, et al. Complex immunomodulatory effects of interferon-beta in multiple sclerosis include the upregulation of T helper 1-associated marker genes. Annals of Neurology. 2001;50:349–57. doi: 10.1002/ana.1096. [DOI] [PubMed] [Google Scholar]
- 41.Kawai T, Akira S. Innate immune recognition of viral infection. Nat Immunol. 2006;7(2):131–7. doi: 10.1038/ni1303. [DOI] [PubMed] [Google Scholar]
- 42.Giovannoni G, Goodman A. Neutralizing anti-IFN-beta antibodies: how much more evidence do we need to use them in practice? Neurology. 2005;65(1):6–8. doi: 10.1212/01.wnl.0000172080.54415.f1. [DOI] [PubMed] [Google Scholar]
- 43.Ito T, Wang YH, Liu YJ. Plasmacytoid dendritic cell precursors/type I interferon-producing cells sense viral infection by Toll-like receptor (TLR) 7 and TLR9. Springer Semin Immunopathol. 2005;26(3):221–9. doi: 10.1007/s00281-004-0180-4. [DOI] [PubMed] [Google Scholar]
- 44.Izaguirre A, Barnes BJ, Amrute S, Yeow WS, Megjugorac N, Dai J, et al. Comparative analysis of IRF and IFN-alpha expression in human plasmacytoid and monocyte-derived dendritic cells. J Leukoc Biol. 2003;74(6):1125–38. doi: 10.1189/jlb.0603255. [DOI] [PubMed] [Google Scholar]
- 45.Dai J, Megjugorac NJ, Amrute SB, Fitzgerald-Bocarsly P. Regulation of IFN regulatory factor-7 and IFN-alpha production by enveloped virus and lipopolysaccharide in human plasmacytoid dendritic cells. J Immunol. 2004;173(3):1535–48. doi: 10.4049/jimmunol.173.3.1535. [DOI] [PubMed] [Google Scholar]
- 46.Ortaldo JR, Herberman RB, Harvey C, Osheroff P, Pan YC, Kelder B, et al. A species of human alpha interferon that lacks the ability to boost human natural killer activity. Proc Natl Acad Sci U S A. 1984;81(15):4926–9. doi: 10.1073/pnas.81.15.4926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bayas A, Stasiolek M, Kruse N, Toyka KV, Selmaj K, Gold R. Altered innate immune response of plasmacytoid dendritic cells in multiple sclerosis. Clin Exp Immunol. 2009;157(3):332–42. doi: 10.1111/j.1365-2249.2009.03964.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Matsumoto F, Saitoh S, Fukui R, Kobayashi T, Tanimura N, Konno K, et al. Cathepsins are required for Toll-like receptor 9 responses. Biochem Biophys Res Commun. 2008;367(3):693–9. doi: 10.1016/j.bbrc.2007.12.130. [DOI] [PubMed] [Google Scholar]
- 49.Rollag H, Morland B. The effect of recombinant interferons on cathepsin B activity in human monocytes. Scand J Rheumatol Suppl. 1988;76:79–83. doi: 10.3109/03009748809102956. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.