

Abstract
Disulfide bond formation in Escherichia coli is a catalyzed reaction accomplished by DsbA. We found that null mutations in a new porin gene, ompL, allowed a total bypass of the DsbA requirement for protein oxidation. These mutations acted as extragenic null suppressors for dsbA, and restored normal folding of alkaline phosphatase and relieved sensitivity to dithiothreitol. ompL dsbA double mutants were completely like wild-type mutants in terms of motility and lack of mucoidy. This suppression was not dependent on DsbC and DsbG, since the oxidation status of these proteins was unaltered in ompL dsbA strains. Purified OmpL allowed diffusion of small solutes, including sugars, but the suppression was not dependent on the carbon sources used. Suppression by ompL null mutations required DsbB, leading us to propose a hypothesis that DsbB oxidizes yet unidentified, low-molecular-weight redox agents in the periplasm. These oxidized agents accumulate and substitute for DsbA if their leakage into the medium is prevented by the absence of OmpL, presumed to form a specific channel for their diffusion.
Keywords: disulfide bond formation/DsbA/DsbB/outer membrane/protein folding
Introduction
In vivo protein folding is an assisted process. In the cytosol of Escherichia coli, molecular chaperones such as GroEL/GroES, DnaK and trigger factor bind to unfolded polypeptides and stabilize their unstable conformers during protein folding. However, no classical ATP-dependent chaperones have been identified in the periplasm of E.coli. Unlike the cytoplasm, the periplasm is separated from the external medium by only a permeable outer membrane, and is, hence, more vulnerable to environmental fluctuations. In recent years, it was established that the periplasm contains two well defined sets of folding catalysts, which overcome two types of rate-limiting steps: peptidyl-prolyl cis–trans isomerases and protein disulfide oxidases, reductases and isomerases (designated as Dsbs), which overall belong to the thioredoxin family. Many periplasmic proteins of E.coli (∼300 proteins) contain two or more cysteine residues and acquire disulfide bonds after their translocation across the cytoplasmic membrane. Up to now, six known dsb genes (dsbA, B, C, D, E and G) have been characterized using a variety of approaches (for review see Raina and Missiakas, 1997). The major catalyst for the disulfide bond formation is DsbA (Bardwell et al., 1991; Kamitani et al., 1992), and during this process its catalytic dithiol motif is reduced. The recycling of DsbA needs DsbB, located in the cytoplasmic membrane (Bardwell et al., 1993; Missiakas et al., 1993). The reduced DsbB is then reoxidized by quinones in the cytoplasmic membrane (Kobayashi et al., 1997; Bader et al., 1999).
When proteins contain more than two cysteine residues, there is a possibility of formation of non-native disulfide bonds. During our search for additional genes that participate in disulfide bond formation, we looked for multicopy suppressors of either dsbA or dsbB null mutants. With this approach, we identified dsbC (Missiakas et al., 1994) and dsbG (Andersen et al., 1997) genes. Both DsbC and DsbG are located in the periplasm. Indeed, DsbC was shown to have an isomerase activity (Zapun et al., 1995), in contrast to DsbA, which is a very potent disulfide oxidase (Wunderlich and Glockshuber, 1993; Zapun et al., 1993).
In yet another genetic approach, we looked for null suppressors of dsbA mutations and identified the dsbD gene (Missiakas et al., 1995). DsbD/DipZ is located in the cytoplasmic membrane, and it reduces DsbC and DsbG. Thus, in dsbD mutants, DsbC and DsbG accumulate in their oxidized forms, and partially substitute for the oxidase function of DsbA in dsbD dsbA double mutants.
To analyze further the mechanism of disulfide bond formation, we used two approaches in this study. One was to look for complete, rather than partial, suppressors of the dsbA phenotype among transposon insertion mutants. The other was to examine in more detail null mutants that produce moderate hypersensitivity to dithiothreitol (DTT). Both of these approaches led to the identification of a new gene, ompL, which codes for a porin. Here we report the unexpected result that the deletion of a porin gene results in a complete suppression of all the examined phenotypes of dsbA mutants. We present a working hypothesis that the periplasm contains, in addition to enzymes involved in disulfide metabolism, low-molecular-weight redox agents, which may play a role in the formation of disulfide bonds in proteins.
Results
Identification and cloning of the ompL gene
Previously, we designed a genetic screen to isolate mutants unable to cope with changes in the redox environment. Whereas wild-type E.coli can grow in the presence of 10–15 mM DTT, Tn10 insertion mutants were isolated for their inability to grow in the presence of 5–7 mM DTT (Missiakas et al., 1993) (Table I). Most of the dsb genes were identified through such screening. Alternatively, a multicopy approach was used, selecting plasmids that allowed E.coli to become more resistant to DTT from an E.coli genomic library. In this manner we identified dsbB (Missiakas et al., 1993) and dsbG (Andersen et al., 1997).
Table I. Restoration of growth on DTT of DsbA null bacteria by the introduction of an ompL null allele.
| Strains | DTT concentration |
||
|---|---|---|---|
| 7 mM | 10 mM | 15 mM | |
| Wild type | + | + | ± |
| ompL::Tn10 Kan | + | ± | – |
| dsbA::Tn10 Tet | – | – | – |
| ompL::Tn10 Kan dsbA::Tn10 Tet | + | + | + |
| dsbB::Tn10 Tet | – | – | – |
| ompL::Tn10 Kan dsbB::Tn10 Tet | – | – | – |
Sensitivity to DTT was assayed as described previously (Missiakas et al., 1993).
An additional strategy is to identify extragenic null suppressors of dsbA mutants, which exhibit a highly pleiotropic phenotype due to the severe defects in disulfide bond formation, including lack of motility, absence of alkaline phosphatase (AP) activity (the native AP has two disulfide bridges), mucoidy, sensitivity to DTT and benzylpenicillin and resistance to phage M13. Earlier we found that mutations in the dsbD/dipZ gene partially suppressed some of the defects of dsbA null mutants. In this study, we looked for extragenic miniTn10-Kan or miniTn10-Tet insertions (Way et al., 1984) that completely suppressed the requirement of DsbA function in the folding of both AP and penicillin-binding protein 4 (which contains two disulfide bonds and its defective folding results in hypersensitivity to benzylpenicillin). To avoid reisolating dsbD mutants, we transformed putative suppressor mutants with a plasmid carrying the dsbD wild-type allele. Among these suppressor mutants, only those that were DTT resistant and fully motile were retained (see Materials and methods). In this way, we obtained one Tn10-Tet (SR2701) and one Tn10-Kan (SR1791) insertion.
In a complementary approach, we screened for mutants that are weakly hypersensitive to DTT and not mapping to any known dsb gene. Seven such Tn10 insertions, which did not show severe growth defects at <10 mM DTT and were resistant to phage M13, were transduced by phage P1 into a dsbA::Tn10Tet background (SR2262) or dsbA::Tn10Kan (SR1790). We examined whether any of these double null mutants also totally restored the disulfide bond formation. Interestingly, one such mutant (SR2772) was similar to the wild-type bacteria in the various phenotypes mentioned above.
All three Tn10 insertions were transduced into a wild-type background. All three insertion mutations were found to confer the following phenotypes in this background. (i) Slow growth at 37°C with a doubling time of 70 min compared with the isogenic wild type (45 min); (ii) mild sensitivity to DTT (these mutants grew up to the level of 10 mM DTT, compared with the wild type that grew up to 15 mM DTT and to dsbA or dsbB mutants that could not grow even at 7 mM); (iii) inability to plate phage M13; and (iv) no defect per se in disulfide bond formation. We used a cosmid library to complement the two phenotypes of the mutants, the slow growth and the resistance to phage M13. Only those cosmid clones that had the ability to suppress all of the three Tn10 insertions were retained. Using standard subcloning techniques, we constructed a clone containing a 2.5 kb KpnI–NdeI fragment (pSR4485), which restored the sensitivity to M13. This fragment was subsequently transferred to different vectors (Figure 1) and shown to be able to suppress the three Tn10 insertions.

Fig. 1. Restriction map of the ompL gene and surrounding DNA sequences. Columns on the right hand indicate the ability of the different clones to complement for M13 resistance and to complement for dsbA mutant phenotypes (sensitivity to DTT and AP–). The strains SR2701, SR2772 and SR1791 correspond to single Tn10 insertions at positions 495, 537 and 553, respectively.
To map exactly the mutations, the Tn10 junctions were sequenced using a synthetic 24mer oligonucleotide. This experiment revealed that Tn10 disrupted a hypothetical open reading frame (ORF), called yshA (predicted to encode a 230 residue polypeptide), at positions 495, 537 and 553 in SR2701, SR2772 and SR1791, respectively (Figure 1).
YshA does not contain even a single cysteine residue. Analysis of the sequence by program pSORT and PhD_ Sec (Profile network prediction at Heidelberg) predicted it to contain a cleavable signal sequence and to be an outer membrane protein (OMP). The predicted score for the outer membrane localization was 0.937 compared with 0.946 for OmpC, 0.944 for OmpF, 0.928 for OmpG and 0.488 for OmpA. We confirmed the outer membrane localization for subcellular fractionation. Hence we named the gene ompL. The OmpL protein, expressed from a T7 promoter in the vector pET-22b, was indeed found in the outer membrane fraction (Figure 2, lane labelled ‘OMPs’). The presence of signal sequence was confirmed by N-terminal sequencing using the purified OmpL protein. Protein samples were subjected to Edman degradation on aProcise cLC 492 from Applied Biosystems. The un ambiguous sequence is Gly-Ala-Tyr-Val-Glu-. Therefore, the cleavage site is clearly at Ala20 (bold arrow in Figure 3).
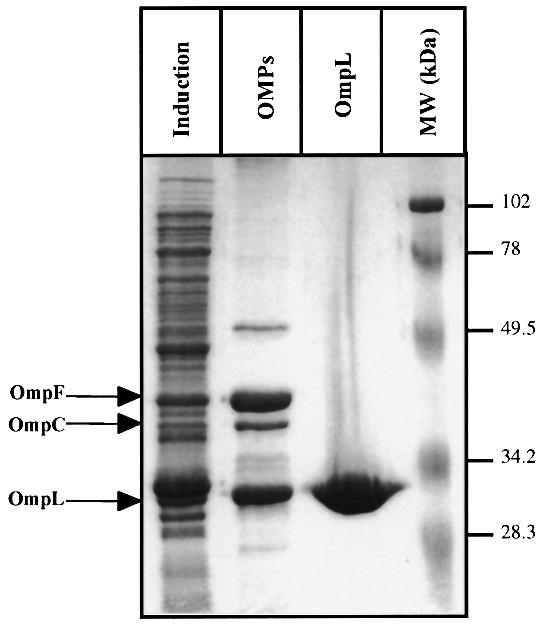
Fig. 2. Subcellular fractionation of OmpL and its purification. Cultures of BL21(DE3) containing ompL gene (pCD695), cloned in pET-22b under transcription of the T7 polymerase promoter, were grown in M9 minimal medium at 37°C. Expression of T7 RNA polymerase was induced by the addition of 1 mM IPTG for 4 h. Lane 1 (‘Induction’) is the total extract from induced bacteria carrying pCD695 (ompL gene), lane 2 (‘OMPs’) represents the outer-membrane fractions after LDS extraction and lane 3 (‘OmpL’) contains purified OmpL protein after LDS extraction made from BL21(DE3) ompA– ompC– ompF– lamB– followed by gel filtration. Proteins were resolved by SDS–PAGE (12.5%). The gel was stained with Coomassie Brilliant Blue. The arrows on the left represent the position of OmpF, OmpC and OmpL. On the right are molecular weight size standards (Bio-Rad).

Fig. 3. Predicted topology of OmpL. Using the Chou and Fasman (1974) program, the secondary structure prediction was carried out. The amino acid residues in bold indicate potential β-sheets. The signal sequence cleavage site is marked with a downward arrow. E, predicted extended sheets; T, turns. The highly conserved G and F at the C-terminus, which are present in β-barrel OMPs, are indicated in big bold letters and underlined.
OmpL is a porin
At its C-terminus, OmpL has the highly conserved Gly and Phe residues (shown in bold in Figure 3) found in β-barrel OMPs such as porins OmpF, OmpC, LamB etc. The secondary structure prediction using the Chou–Fasman program (Chou and Fasman, 1974) predicts 12 β-strands, which should be sufficient to form a channel (Figure 3).
To test whether OmpL is a porin it was purified from the E.coli B derivative BL21(DE3). Escherichia coli B naturally lacks one of the major porins, OmpC. Escherichia coli B is also crp–, and does not produce LamB. To avoid contamination from other porins, ompF and ompA null alleles carrying Tn10 insertions were introduced by P1 transduction into BL21(DE3). The ompL gene was expressed using the T7 promoter, and OmpL was purified to homogeneity, using lithium lauryl sulfate extraction followed by gel filtration (Sugawara and Nikaido, 1992). The gel filtration conditions are described in Materials and methods.
The channel-forming functions of the porins can be tested by the diffusion of test solutes, such as sugars, with reconstituted proteoliposomes (Nikaido et al., 1991). Reconstitution was carried out as described in Materials and methods, and the proteoliposomes were diluted in iso-osmotic solutions of various sugars. The permeation rates of sugars were measured by the osmotic swelling of liposomes, which causes a decrease in optical density (OD). As seen in Table II, OmpL allowed efficient diffusion of small sugars such as l-arabinose (150 g/mol), d-glucose (180 g/mol) and d-galactose (180 g/mol). However, the diffusion rate declined with larger sugars such as N-acetyl-d-glucosamine (222 g/mol) and no detect able permeation was observed with maltose (342 g/mol). These data suggest that the channel size must be relatively small. The rate of arabinose diffusion by OmpL-containing liposomes was roughly one-fifth of those containing the same amount of OmpF. When tetraglycine was tested as the solute, the tetrapeptide diffused through OmpL rapidly (Table II), suggesting that OmpL is not a sugar-specific channel.
Table II. Diffusion rate of sugars through the OmpL channel.
| Sugar | Specific activitya |
|---|---|
| Glucose | 81 ± 10 |
| Arabinose | 70 ± 18 |
| Galactose | 76 ± 7 |
| N-acetyl glucosamine | 12 ± 3 |
| Maltose | 2 ± 1 |
| Sucrose | 2 ± 1 |
| Stachyose | 0 |
| Gly (4) peptide | 148 ± 21 |
Purified OmpL protein (1 µg) was reconstituted with 2.4 µmol of phosphatidylcholine and 0.2 µmol of diacetylphosphate and resuspended in 300 µl of 15% Dextran T-40. Seventeen microlitres of proteoliposome suspension were diluted into 0.6 ml of an iso-osmotic solution of sugar. The complete details of the proteoliposome construction are as described previously (Nikaido et al., 1991). The initial rate of decrease of optical density was measured.
aOD change ×1000/min. The standard deviation is from four experiments carried out under identical conditions.
We also examined whether the diffusion rates of sugars were linear with respect to the amount of purified OmpL added. As can be seen, the diffusion rates of glucose and arabinose were proportional to the amount of OmpL used in the reconstitution (Figure 4).

Fig. 4. Linear uptake of sugars by the OmpL porin. Here we show the dependence of diffusion rates of two sugars on the concentration of OmpL porin in proteoliposomes. The permeation rates of arabinose and glucose were measured using the reconstituted proteoliposome assay with different amounts of OmpL. Proteoliposomes were prepared as described earlier (Nikaido et al., 1991).
Because OmpL was suspected to function as a channel for low-molecular-weight redox agents present in the periplasm (see below), we examined the diffusion of several common compounds containing -SH and -SS groups. These compounds were charged, necessitating the use of proteoliposomes reconstituted in a solution containing stachyose and NAD+ (Nikaido and Rosenberg, 1983). The results showed that OmpL allowed rapid permeation of cysteine and reduced glutathione at rates expected from the sizes of these compounds. Furthermore, OmpL also allowed a slow but steady influx of oxidized glutathione (613 Da) but not that of stachyose (666 Da) or NAD+ (613 Da).
Introduction of ompL null mutation bypasses the requirement of DsbA function
We studied the double dsbA ompL null mutant first for the oxidation status of a disulfide-containing protein, AP (Table III). We found, remarkably, that AP was completely oxidized in the double mutant, and was as active in the double mutant as in the wild-type strain, in contrast to the enzyme in the dsbA single mutant, which had only ∼30% of the full activity. Western blot analysis also showed that AP, which normally contains two disulfide bonds, accumulated in a reduced state in the dsbA single mutant, but this defect was completely corrected by the additional presence of ompL null mutation (Figure 5). When the sulfhydryl groups of the periplasmic proteins, generated because of the impaired formation of disulfides, were titrated by using DTNB [5,5′-dithiobis-(2- nitrobenzoic acid)], the level in the double null mutant was as low as in the wild type, in striking contrast to the high level found in the dsbA single mutant (Table III). The double mutant was non-mucoid, unlike the dsbA mutant (Table III). Finally, the double dsbA ompL mutants showed total suppression of the motility defect found in the dsbA single mutants, caused by defects in P-ring formation during flagellar assembly (Table III).
Table III. Restoration of oxidizing conditions in the periplasm of dsbA mutant strains by the introduction of ompL null mutation.
| Genotype | Free thiolsa (nmol of periplasmic thiol/g of bacteria) | AP activity (unit/mg of bacteria) | Mucoidy | Motility |
|---|---|---|---|---|
| Wild type | 120 ± 20 | 0.92 | – | + |
| dsbA::Tn10 Tet | 960 ± 47 | 0.27 | + | – |
| ompL::Tn10 Kan | 170 ± 15 | 0.80 | – | ± |
| dsbA::Tn10 Tet ompL::Tn10 Kan | 150 ± 20 | 0.87 | – | + |
aThese values were determined using Ellman’s reagent DTNB (14 150 M–1cm–1 is the molar extinction coefficient at 412 nm) and 2.5 mg of cell extract for each measure were used.
Results are averages of experiments done in triplicate. The AP activity was measured as described previously (Missiakas et al., 1995). Motility assays were performed by stabbing a single colony of isogenic strains onto a soft agar lawn of 0.4 LB-agar. The zone of growth was measured. For mucoidy, – is for non-mucoid phenotype and + for mucoid strains.
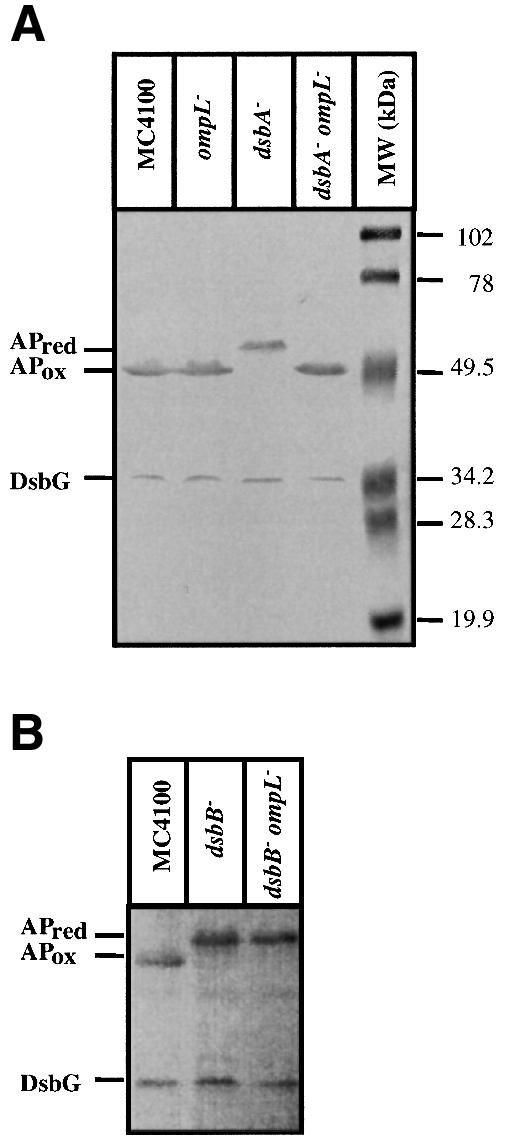
Fig. 5. Restoration of folding status of AP in vivo by introduction of ompL or null mutation in dsbA– background (A). Isogenic cultures of wild type, dsbA::Tn10, ompL::Tn10 and their various double mutants were grown in LB medium up to an OD of 0.2 at 595 nm. Cultures were spun, washed and resuspended in low phosphate M63 medium. Bacterial cells were harvested at 0.5 OD, TCA precipitated and resuspended in buffer containing 1% SDS and 10 mM AMS. Samples were run on 12% SDS–PAGE and proteins visualized using antibodies against AP. To check the folding status of DsbG at the same time, antibodies against DsbG were used in the immunoblot assay. The positions of reduced (red) and oxidized (ox) forms are indicated. Note that there is no change in reduced status of DsbG. (B) The dependence on functional copy of dsbB for the suppression noted with the lack of ompL. As can be seen in lane 2 (‘dsbB–’) and lane 3 (‘dsbB–ompL–’), there is no restoration of AP into the oxidized state as compared with dsbA–ompL– (A).
Previously, it was reported that null mutation in ompA could suppress, very weakly, a dsbA null mutation. OmpA, however, has a disulfide bridge, and is a major substrate of DsbA (Rietsch et al., 1996). The partial suppression is thus accounted for by the lack of a major substrate, and the mechanism here is totally different from the suppression by mutations in ompL described in this study.
Null mutations in ompL do not change the redox status of DsbC and DsbG
Suppression of dsbA by the absence of OmpL is not dependent on the nature of the carbon source. We have described above that a null mutation in porin gene ompL caused the complete and partial suppression of dsbA phenotypes (Tables I and III). One possible interpretation was that the absence of OmpL porin leads to a decreased influx of sugars, such as glucose, from the medium, and these sugars could have made the periplasm more reducing. In such a case, there should be no suppression in the absence of sugars. For ompL, the data in Table IV are clearly inconsistent with this prediction, as the ompL dsbA double mutant behaved like wild type in terms of AP activity and DTT resistance, even when non-reducing compounds such as mannitol and sorbitol were used as the carbon source. Thus, we conclude that it is not the change of redox environment by the carbon source that leads to correct oxidative folding in the ompL dsbA double mutant.
Table IV. Restoration of AP activity by introduction of ompL null allele in a dsbA mutant is not dependent.
| Strain | Carbon source in M9 minimal medium |
|||
|---|---|---|---|---|
| Glucose | Mannitol | Sorbitol | Lactate | |
| Wild type | + | + | + | + |
| ompL::Tn10 Kan | + | + | + | + |
| dsbA::Tn10 Tet | – | – | – | – |
| ompL::Tn10 Kan dsbA::Tn10 Tet | + | + | + | + |
| dsbB::Tn10 Tet | – | – | – | – |
| ompL::Tn10 Kan dsbB::Tn10 Tet | – | – | – | – |
The dependence on the presence of reducing sugars with ompF, but not with ompL, is illustrated. Also note the total requirement of DsbB function in the case of ompL suppression. All the carbon sources used were at a concentration of 0.2% in low phosphate medium supplemented by 40 µg/ml XP, which acts as a substrate for AP.
Suppression of dsbA by ompL null mutations is DsbB dependent
DsbA activity is dependent on DsbB, which reoxidizes the reduced DsbA. As presented in the Discussion, the results of Table IV led us to suspect that OmpL may control the leakage, from periplasm to medium, of hitherto unidentified, low-molecular-weight redox agents. In the absence of OmpL porin, these low-molecular-weight compounds accumulate in the periplasm, and if they are in the oxidized form, they may allow the formation of disulfide bonds even in the absence of DsbA. We suspected that DsbB might also oxidize such compounds present in the periplasm. To test such a hypothesis, we introduced ompL null mutation into a dsbB null strain (SR2264) by P1 transduction. The double dsbB ompL strain (SR4612) was compared with the isogenic dsbB mutant and the wild type. The data presented in Tables I and IV and in Figure 5B clearly show the essential role of DsbB in restoration of the oxidative folding in the absence of OmpL porin. The dsbB ompL mutant behaved phenotypically quite like the single dsbB mutant (and the single dsbA mutant), in sharp contrast to the dsbA ompL double mutant. These findings present an additional role for DsbB and suggest that its substrates are not limited to DsbA.
Overproduction of OmpL does not induce rpoE transcription
Overproduction of OMPs often induces the expression of the alternate sigma factor RpoE (Mecsas et al., 1993; Raina et al., 1995). However, when the level of OmpL alone was elevated, there was no induction of the rpoE regulon (Table V). The rpoE activity was reflected by the lack of induction of either phtrA-lacZ or rpoHP3-lacZ activity. It has previously been established that htrA and rpoHP3 promoters are transcribed by EσE polymerase (Mecsas et al., 1993; Raina et al., 1995). This observation is the first of its kind to date on the non-induction of rpoE expression by overproducing an outer membrane protein.
Table V. Overexpression of OmpL does not induce rpoE regulon.
| Strain | β-galactosidase (Miller units) |
|---|---|
| ΦphtrA-lacZ + vector | 123 ± 9 |
| ΦphtrA-lacZ + pompL | 119 ± 12 |
| ΦprpoHP3-lacZ + vector | 69 ± 7 |
| ΦprpoHP3-lacZ + pompL | 73 ± 11 |
β-galactosidase activity was measured as described previously (Raina et al., 1995), these values representing the average of triplicate experiments carried out under identical conditions.
Discussion
A decade ago nothing was known about the mechanism of protein folding in the bacterial periplasm. Since periplasm lacks ATP, classical ATP-dependent chaperones were not expected to exist in the periplasm. However, in recent years, a variety of folding catalysts and chaperones have been identified in this compartment (reviewed by Missiakas and Raina, 1997). The periplasm is relatively oxidizing and contains a strong sulfhydryl-oxidizing catalyst DsbA. Such an environment facilitates the disulfide bond formation in substrates such as OmpA, AP and penicillin-binding protein 4. Previously, we looked for extragenic suppressor mutations for dsbA, an effort that led to the identification of the dsbD gene. We and others have shown that DsbD reduces disulfide reductases/isomerases DsbC and DsbG, and that the oxidized forms of DsbC and DsbG, present in dsbD mutants, partially substituted for the disulfide oxidase function of DsbA. However, suppression by dsbD mutation was only partial.
In the present work, we looked for extragenic null suppressors of dsbA phenotype, rather than point mutations. In addition, we retained only those mutants that produced a total suppression of various phenotypic defects of dsbA, i.e. total restoration of AP activity, DTT resistance, motility and the lack of mucoidy. Using these stringent conditions, we identified mutations in a new gene designated ompL. Surprisingly, the ompL gene product is an outer membrane protein, which contains no cysteine residue. OmpL protein, purified to homogeneity and reconstituted into proteoliposomes, allowed an efficient diffusion of low-molecular-weight solutes such as small sugars and tetraglycine. In addition, the oxidation status of DsbC and DsbG was unchanged in ompL mutants, showing that the mechanism of suppression by ompL involves a mechanism different from that found in dsbD mutants.
Since OmpL is not a major porin, we wondered whether null mutations in other porin genes lead to a similar suppression of dsbA phenotype. We have found that a null mutation in ompF leads to a weak suppression of the dsbA phenotype (data not shown). Nevertheless, we did not pick up ompF in our initial screening for suppressor null mutants. This was presumably because the double ompF dsbA mutants were still DTT hypersensitive, non-motile and mucoid, just like the dsbA single mutants.
The partial suppression of dsbA phenotypes by the ompF null mutation occurs only when glucose is the carbon source in a minimal medium. An interesting hypothesis would be that the decreased uptake of glucose across the outer membrane would alter the oxidative status of the periplasm, glucose acting as the direct reductant in some way. This explanation, however, is not entirely satisfactory, because the ompF mutants should still be able to produce the OmpC porin, which allows a very rapid diffusion of small sugars such as glucose (Nikaido and Rosenberg, 1983). This system obviously requires further study, also because the suppression is incomplete and weak.
How does the ompL null mutation suppress dsbA phenotypes?
The ompL null mutations completely restore all of the tested phenotypic defects in a dsbA mutant. The suppression occurs irrespective of the nature of carbon sources in the growth medium. In addition, OmpL is only a minor porin, and the diffusion rate of small sugars through OmpL was only about one-fifth of that through the major porin OmpF. These observations indicate that the suppression effect cannot be explained by the slower influx of sugars.
We believe that the most plausible hypothesis involves the presence, in the periplasm, of hypothetical low-molecular-weight redox agents (X-SH and X-SS-X of Figure 6). Although we could not detect any evidence for the specific diffusion of common sulfhydryl (and disulfide) compounds through the OmpL channel, these results do not rule out the possibility that the hypothetical periplasmic oxidant is other than those tested, and indeed is the specific ligand recognized by the OmpL channel. This assumption is not unreasonable, as glutathione has been known to be secreted or leaked out into the periplasm. Suppression by null mutations in ompL required the presence of functional DsbB. Thus, these compounds are oxidized by DsbB, and presumably make the periplasm oxidizing enough to effect a complete suppression of dsbA phenotype (Figure 6C). We assume that OmpL is a specific porin-like channel for these redox buffer compounds. Specific channels such as LamB allow the non-specific diffusion of general solutes at a modest rate, and yet are extremely efficient in facilitating the diffusion of their specific substrates (Luckey and Nikaido, 1980). This explains the diffusion of small solutes through the OmpL channel in proteoliposome reconstitution assays, and its strong suppression effect despite its modest expression level. We thus assume that in ompL dsbA double mutants, oxidation of X-SH by DsbB produces X-SS-X, which accumulates in the periplasm because it is too large to diffuse away through the general porins OmpF/C, and because OmpL, the specific channel for its diffusion, is absent (Figure 6C). In contrast, the periplasm remains more reducing in dsbA single mutants, because X-SS-X leaks out into the medium through OmpL (Figure 6B).

Fig. 6. The proposed model for restoration of oxidative environment in the absence of OmpL porin. We postulate the presence of one or more species of reduced molecules in the periplasm that is/are a substrate for DsbB. Under normal conditions it is pumped out by the OmpL channel and with the presence of DsbA periplasm stays oxidizing, AP+ (A). (B) In the absence of DsbA oxidant, the periplasm is relatively reduced and hence AP–. (C) Here the DsbB is free to oxidize the reducing compounds and these compounds are not pumped out via the OmpL channel, hence periplasm stays oxidizing, being AP+. In the absence of OmpL and DsbB (D), such a reducing molecule as well as DsbA is not oxidized and thus the periplasm stays reducing, leading to an AP– phenotype.
The ompL single mutants paradoxically showed a modest DTT hypersensitivity, in spite of the complete DTT resistance of ompL dsbA double mutants. However, our model is perfectly consistent with this observation. Thus, DTT reduces the hypothetical redox buffer compound rapidly to X-SH, and in the ompL single mutants X-SH accumulates, competing with the DsbA disulfide oxidase, resulting in some hypersensitivity to DTT. In contrast, in the wild-type ompL+ strain, X-SH will rapidly diffuse away from the periplasm via OmpL. These considerations suggest that OmpL probably acts as a safety valve, useful when the cells encounter strong reducing agents. Escherichia coli is known to have a mechanism that allows its survival during the burst of cysteine synthesis by actively pumping out cysteine and its precursors (Dassler et al., 2000), and it would not be surprising for it to possess a specific porin that prevents the excessive accumulation of periplasmic oxido/reductant molecules.
Another dimension to this work is that DsbB re oxidizes not only DsbA but also additional substrates. We have already shown that synthesis of c-type cytochrome requires functional DsbA as well as DsbB (Metheringham et al., 1996). However, and quite unlike dsbA mutants, dsbB mutant bacteria are also defective in periplasmic nitrate reductase (Metheringham et al., 1996). The current study also suggests that DsbB oxidizes hitherto unidentified low-molecular-weight redox compounds in the periplasm. Interestingly, overproduction of DsbB alone can make E.coli hyperresistant to DTT (Missiakas et al., 1993). The function of DsbB may be similar to that of yeast EroI, which was recently shown to oxidize not only protein sulfhydryl groups but also reduced glutathione (Cuozzo and Kaiser, 1999). Here, we propose that a molecule, probably larger than glutathione, may diffuse or be translocated in the periplasm. This molecule is a substrate for DsbB and its oxidized and/or reduced form is effluxed by the OmpL channel. However, at this point we can not rule out the possibility that disulfide-bond-containing periplasmic proteins may tolerate slower folding if the concentration of this putative X-SS-X substrate is high enough. We are designing additional genetic strategies to find this reducing molecule or its generation.
Materials and methods
Bacterial strains and plasmids
The bacterial strains and plasmids used in this study are listed in Table VI.
Table VI. Bacterial strains and plasmids.
| Relevant characteristics | Reference or source | |
|---|---|---|
| Strains | ||
| BL21 | ompT lon– | our collection |
| CA8000 | HfrH thi | our collection |
| MC4100 | F– araD139 Δ(argF-lac) U169 | our collection |
| CD726 | BL21 ompL : :Tn10Kanr | this study |
| CD676 | BL21 ompA : :Tn10Tetr | this study |
| CD677 | BL21 ompF : :Tn10Kanr | this study |
| CD687 | BL21 ompA : :Tn10Tetr ompF : :Tn10Kanr | this study |
| CD706 | MC4100 Φ(ompL-lacZ) | this study |
| CG3122 | B178 ompA : :Tn10Tetr | C.Georgopoulos, unpublished |
| CG3127 | B178 ompF : :Tn10Kanr | C.Georgopoulos, unpublished |
| SR1458 | MC4100 Φ(htrA-lacZ) | Raina et al. (1995) |
| SR1710 | MC4100 Φ(rpoHP3-lacZ) | Raina et al. (1995) |
| SR1790 | MC4100 dsbA : :Tn10Kanr | Missiakas et al. (1993) |
| SR1791 | CA8000 ompL : :Tn10Kanr | this study |
| SR2262 | MC4100 dsbA : :Tn10Tetr | this study |
| SR2264 | MC4100 dsbB : :Tn10Tetr | this study |
| SR2701 | CA8000 ompL : :Tn10Tetr | this study |
| SR2772 | CA8000 ompL : :Tn10Kanr | this study |
| SR4444 | MC4100 dsbA : :Tn10Tetr ompL : :Tn10Kanr | this study |
| SR4612 | MC4100 dsbB : :Tn10Tetr ompL : :Tn10Kanr | this study |
| Plasmids | ||
| pKO3 | Allelic replacement vector | Link et al. (1997) |
| pCD695 | pET ompL+ (NdeI–EcoRI) | this study |
| pCD700 | pRS550 ΦompL | this study |
| pDM1463 | pWSK dsbD+ (3.6 kbp PstI-StyI) | Missiakas et al. (1995) |
| pSR1865 | pOK12 dsbA+ (1.8 kbp Sau3A) | Missiakas et al. (1993) |
| pSR4485 | pKS ompL+ (2.5 kbp KpnI–NdeI) | this study |
Media and chemicals
Luria–Bertani (LB) broth, LB agar and M9 minimal media were prepared as described by Miller (1992). When necessary, the media were supplemented with ampicillin (Amp, 100 µg/ml), tetracycline (Tet, 15 µg/ml), kanamycin (kan, 50 µg/ml), streptomycin (Str, 100 µg/ml) or chloramphenicol (Cm, 20 µg/ml). Whenever needed, the media were supplemented with different concentrations of DTT purchased from Sigma. For AP assay, the XP glucose minimal medium was prepared as described by Miller (1992) and XP (5-bromo 4-chloro 3-indolyl phosphate) was used at 40 µg/ml.
Selection strategies and cloning of the ompL gene
Since dsbA null mutants do not plate at concentrations in the range of 5–10 mM DTT, we looked for suppressors that are resistant to DTT and can grow well up to 15 mM concentration. This was achieved by using SR2262 dsbA::Tn10Tet or 1790 dsbA::Tn10Kan strains and mutagenized with λminiTn10-Kan or λminiTn10-Tet and plated on LB-agar supplemented with either 10 or 15 mM DTT. The DTT-resistant colonies were pooled and transformed with a low copy plasmid pSR containing the wild-type dsbD gene. This was necessitated to avoid isolating dsbD gene again in this suppression selection. The resulting 70 insertion mutants isolated were transduced back into dsbA– strains using appropriate antibiotics and plated on minimal phosphate medium supplemented with 20 µg/ml XP substrate for AP activity. The individual colonies, which were AP+ and non-mucoid were retained. It is known that on minimal medium, dsbA mutants are highly mucoid due to induction of cps genes, which make colonic acid. To make sure that there is a complete suppression of dsbA mutants these double mutants TetR KanR were replica plated on LB medium supplemented with 30 µg/ml benzylpenicillin. At this concentration a wild-type strain can form colonies, but not dsbA null mutants. From the DTTR, AP+ and BP+, 127 mutants were obtained. These were tooth picked and checked for motility on LB-agar medium with 3% agar at 37°C. The dsbA mutants are non-motile. The restoration of motility defect was checked by the zone of spread. On the same plate, we also incubated wild-type strain CA8000 and isogenic dsbA mutant bacteria. Twenty-three dsbA extragenic null suppressor strains were selected since they became completely wild type for motility, just like for DTTR, AP+ and BP+ phenotypes. All of these were transduced in fresh wild-type background and used to make further complementation groups. To our surprise, we found that all the suppressors were linked ∼70% to the dsbA gene. We retained two SR2701Tn10Kan and SR2772Tn10Tet strains. We also checked whether these Tn10 insertions conferred any phenotype. Such insertions grew at 10 mM DTT but slowly and were resistant to filamentous bacteriophage M13. We used the M13 resistance phenotype to clone the wild-type gene by first isolating cosmids, which enable plating of M13. To be sure that the cosmids bred true we checked for recombination of Tn10 elements onto these cosmids. Further subcloning was performed using M13mp18 vector taking advantage of the plaque forming ability of M13 phage by complementing clone. One such clone containing an ∼2.5 kb KpnI–NdeI DNA fragment was found to perfectly complement (pSR4485). This fragment was transferred by standard molecular biology techniques into different vectors. A minimal clone carrying ∼1.2 kb of DNA fragment was found to complete at the same level as the original clone pSR4490 and M13mp18-derived plasmid. The sequence analysis showed that it contains a single ORF, which corresponded to the yshA gene (from the E.coli genome project). Based on the sequence prediction and other results, we found it to encode for an outer membrane protein and designated the gene as ompL. We also sequenced all the three Tn10 insertion sequences by recombination of Tn10Tet or Tn10Kan on the plasmid. By digesting with HindIII, which cuts in the polylinker and once inside the Tn10, we removed one half of Tn10. In this way, we could sequence directly from such subclones using them as the template. The exact junction was sequenced by using the Tn10-specific primer as described previously (Raina et al., 1991). In a complementary genetic approach, we looked into our old collection of Tn10 insertions, which were DTT sensitive (Missiakas et al., 1993). In this screen, we obtained dsbA and dsbB, because of their hypersensitivity to DTT. Based on the identification of two extragenic null suppressors with Tn10 for circumventing the need of dsbA gene, we looked for those mutants, which were still M13 resistant and mildly sensitive to DTT growing up to 10 mM concentration of DTT. We examined seven independent Tn10Kan insertion mutants with the M13 sensitivity phenotype, but not mapping to either dsbA or dsbB genes. These Tn10Kan insertions were transduced into dsbA::Tn10Tet (SR2262) and selected for TetR and KanR. One such cross led to the identification of strain SR1791, the mutation that when crossed into dsbA background gave the same phenotype as for SR2701 and SR2772.
OmpL purification and biochemical assays
The minimal ompL ORF was PCR amplified using primers 5′-AATGGACTTCATATGAAAAAG-3′ and 5′-GCTTGATCTCTTGGTCCGCCA-3′. The PCR product was digested with NdeI and EcoRI restriction enzymes and cloned into pET-22b using the same cloning sites. The cloned ompL gene (pCD695) was transformed into the BL21 (DE3) (ompA–, ompC–, ompF–, lamB–) derivative strain. Bacterial cultures were induced with 1 mM isopropyl-β-d-thiogalactopyranoside (IPTG), at an OD of 0.4 at 595 nm for 4 h. Cultures were centrifuged at 4°C for 10 min at 7K. The pellet was resuspended in buffer A (100 mM Tris–HCl pH 8, 10 mM EDTA) with lysozyme (100 µg/ml) for 20 min at 4°C. MgCl2 (10 mM) and DnaseI were added for 20 min on ice, then cells were lysed by sonication. The lysate was centrifuged at 35 000 r.p.m. for 45 min at 4°C to remove the soluble proteins. To remove the inner membrane fraction, the pellet was resuspended in 20 mM Na2HPO4 pH 7 containing 0.5% sarkosyl to solubilize the inner membrane incubated at room temperature for 30 min. The samples were mixed several times until the pellet solubilized. This mixture was then spun again for 45 min at 35K. The pellet was resuspended in buffer B containing 0.15% lithium lauryl sulfate (LDS) and kept on ice for 30 min and resuspended. This fraction was centrifuged again for 1 h at 35K. The pellet was then resuspended in buffer B supplemented with 3% LDS, which solubilized the outer membranes. The samples were centrifuged for an additional 1 h at 35K. The soluble fraction was equilibrated by an equal volume of buffer C (0.08% dodecylamine, 0.4 M NaCl, 1 mM EDTA, 10 mM Tris–HCl pH 8, 3 mM NaN3) and loaded onto a gel filtration column (Pharmacia). Fractions were analyzed by SDS–PAGE, stained with Coomassie Brilliant Blue. Purified OmpL eluted as a single peak and was found to migrate as a 27 kDa species. The fact that it was indeed OmpL was verified by the N-terminal sequencing by Edman degradation on a Procise cLC 492 from Applied Biosystems. The unambiguous sequence is Gly-Ala-Tyr-Val-Glu-, which shows that it has a signal sequence and is processed after Ala20. The N-terminal sequence was performed at the University of Bern.
For the initial subcellular localization of OmpL, we followed the same procedure as described above to prepare outer membranes. However, in this case it should be noted that we used normal BL21(DE3), which has wild-type OmpF/OmpA porins. The cloned ompL gene (pCD695) in pET-22b was induced with IPTG. This was done to show that OmpL purifies with the major outer membranes like OmpF, OmpA, etc.
Assays using Ellman’s reagent were performed as described earlier, using a molar extinction coefficient of 14 150 M–1 at 412 nm (Missiakas et al., 1993). To assay for AP activity, toluenized extracts were prepared as described previously (Manoil and Beckwith, 1985).
Folding studies with alkaline phosphatase
One millilitre cultures were grown to OD (595 nm) 0.5, trichloroacetic acid (TCA)-precipitated, acetone washed and resuspended in Tris–HCl buffer pH 7.5 containing 1% SDS and 10 mM AMS (4-acetamido-4′-maleimidylstilbene-2,2′-disulfonic acid disodium salt). Alkylation of free sulfhydryl groups with AMS introduces an additional 500 Da. Samples were run on 12% SDS–PAGE and proteins were visualized by immunoblotting using the appropriate antibody. The antisera against AP were a kind gift from Dr J.M.Betton. Rabbit polyclonal antisera against DsbC and DsbG have been described previously (Missiakas et al., 1995; Chung et al., 2000).
Preparation of proteoliposomes and measurement of their osmotic swelling rate
These procedures were as described earlier (Nikaido et al., 1991). A mixture of 2.4 µmol of acetone-washed egg phosphatidylcholine (Type IV; Sigma) and 0.2 µmol of diacetylphosphate was dried under a stream of N2. The lipids were dissolved in anhydrous benzene and dried to remove traces of water and were then dissolved in ethylether and dried to produce a thin, even film. Then the lipid film was resuspended in 0.2 ml of water or water containing the purified protein. The resuspension was completed by vortexing and finally a brief (1–2 min) sonication (in a bath-type sonicator). The lipid/protein mixture was then dried. The dried protein/lipid film was resuspended in 5 mM Tris–HCl pH 8 containing 15% (w/v) of dextran T-40 (Pharmacia). Proteoliposomes were used after being left at room temperature for 0.5–1 h. Seventeen microlitres of proteoliposome were diluted in 0.6 ml of an iso-osmotic solution of sugar. The permeation rates were measured by the initial rate decrease of the optical density of the proteoliposome suspension. Swelling assays with sulfhydryl and disulfide compounds were carried out in a similar way, but with proteoliposomes prepared as described by Nikaido and Rosenberg (1983).
Acknowledgments
Acknowledgements
We are grateful to Drs Robert Freedman and Rudi Glockshuber for helpful suggestions. We thank Dr Costa Georgopoulos for providing ompF and ompA mutants. We also thank E.Sugawara from H.Nikaido’s laboratory for help in the preparation of proteoliposomes. This work was supported by a grant from the Fond National Scientifique Suisse (FN31-42429-94 and FN3100-059131.99/1) to S.R. and by a US Public Health Service grant (AI-09644) to H.N.
References
- Andersen C., Matthey-Dupraz,A., Missiakas,D. and Raina,S. (1997) A new Escherichia coli gene, dsbG, encodes a periplasmic protein involved in disulphide bond formation, required for recycling DsbA/DsbB and DsbC redox proteins. Mol. Microbiol., 26, 121–132. [DOI] [PubMed] [Google Scholar]
- Bader M., Muse,W., Ballou,D.P., Gassner,C. and Bardwell,J.C. (1999) Oxidative protein folding is driven by the electron transport system. Cell, 98, 217–227. [DOI] [PubMed] [Google Scholar]
- Bardwell J.C.A., McGovern,K. and Beckwith,J. (1991) Identification of a protein required for disulfide bond formation in vivo. Cell, 67, 581–589. [DOI] [PubMed] [Google Scholar]
- Bardwell J.C.A., Lee,J.-O., Jander,G., Martin,N., Belin,D. and Beckwith,J. (1993) A pathway for disulphide bond formation in vivo. Proc. Natl Acad. Sci. USA, 90, 1038–1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bessette P., Cotto,J., Gilbert,H. and Georgiou,G. (1999) In vivo and in vitro function of the Escherichia coli periplasmic cysteine oxidoreductase DsbG. J. Biol. Chem., 274, 7784–7792. [DOI] [PubMed] [Google Scholar]
- Chou P.Y. and Fasman,G.D. (1974) Prediction of protein conformation. Biochemistry, 13, 222–245. [DOI] [PubMed] [Google Scholar]
- Chung J., Chen,T. and Missiakas,D. (2000) Transfer of electrons across the cytoplasmic membrane by DsbD, a membrane protein involved in thiol–disulphide exchange and protein folding in the bacterial periplasm. Mol. Microbiol., 35, 1099–1109. [DOI] [PubMed] [Google Scholar]
- Cuozzo J.W. and Kaiser,C.A. (1999) Competition between glutathione and protein thiols for disulphide-bond formation. Nature Cell Biol., 1, 130–135. [DOI] [PubMed] [Google Scholar]
- Dassler T., Maier,T., Winterhalter,C. and Bock,A. (2000) Identification of a major facilitator protein from Escherichia coli involved in efflux of metabolites of the cysteine pathway. Mol. Microbiol., 36, 1101–1112. [DOI] [PubMed] [Google Scholar]
- Kamitani S., Akiyama,Y. and Ito,K. (1992) Identification and characterization of an Escherichia coli gene required for the formation of correctly folded alkaline phosphatase, a periplasmic enzyme. EMBO J., 11, 57–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi T., Kishigami,S., Sone,M., Inokuchi,H., Mogi,T. and Ito,K. (1997) Respiratory chain is required to maintain oxidized states of the DsbA–DsbB disulphide bond formation system in aerobically growing Escherichia coli cells. Proc. Natl Acad. Sci. USA, 94, 11857–11862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luckey M. and Nikaido,H. (1980) Specificity of diffusion channels produced by lambda phage receptor protein of Escherichia coli. Proc. Natl Acad. Sci. USA, 77, 167–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manoil C. and Beckwith,J. (1985) TnphoA: a transposon probe for protein export signals. Proc. Natl Acad. Sci. USA, 82, 8129–8133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mecsas J., Rouviere,P.E., Erickson,J.W., Donohue,T.J. and Gross,C.A. (1993) The activity of σE, an Escherichia coli heat-inducible sigma factor, is modulated by expression of outer membrane proteins. Genes Dev., 7, 2618–2628. [DOI] [PubMed] [Google Scholar]
- Metheringham R., Tyson,K., Crooke,H., Missiakas,D., Raina,S. and Cole,J.A. (1996) Effects of mutations in genes for proteins involved in disulphide bond in the periplasm on the activities of anaerobically induced electron transfer chains in Escherichia coli K12. Mol. Gen. Genet., 253, 95–102. [DOI] [PubMed] [Google Scholar]
- Miller J. (1992) A Short Course in Bacterial Genetics: A Laboratory Manual and Handbook for Escherichia coli and Related Bacteria. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- Missiakas D. and Raina,S. (1997) Protein folding in the bacterial periplasm. J. Bacteriol., 179, 2465–2471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Missiakas D., Georgopoulos,C. and Raina,S. (1993) Identification and characterization of the Escherichia coli gene dsbB, whose product is involved in the formation of disulfide bonds in vivo. Proc. Natl Acad. Sci. USA, 90, 7084–7088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Missiakas D., Georgopoulos,C. and Raina,S. (1994) The Escherichia coli dsbC (xprA) gene encodes a periplasmic protein involved in disulfide bond formation. EMBO J., 13, 2013–2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Missiakas D., Schwager,F. and Raina,S. (1995) Identification and characterization of a new disulfide-isomerase like protein (DsbD) in Escherichia coli. EMBO J., 14, 3415–3424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikaido H. and Rosenberg,E.Y. (1983) Porin channels in Escherichia coli: studies with liposomes reconstituted from purified proteins. J. Bacteriol., 153, 241–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikaido H., Nikaido,K. and Harayama,S. (1991) Identification and characterization of porins in Pseudomonas aeruginosa. J. Biol. Chem., 266, 770–779. [PubMed] [Google Scholar]
- Raina S. and Missiakas,D. (1997) Making and breaking of disulfide bonds. Annu. Rev. Microbiol., 51, 179–202. [DOI] [PubMed] [Google Scholar]
- Raina S., Mabey,L. and Georgopoulos,C. (1991) The Escherichia coli htrP gene product is essential for bacterial growth at high temperature: mapping, cloning, sequencing and transcriptional regulation of htrP. J. Bacteriol., 173, 5999–6008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raina S., Missiakas,D. and Georgopoulos,C. (1995) The rpoE gene encoding the σE (σ24) heat shock sigma factor of Escherichia coli. EMBO J., 14, 1043–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rietsch A., Belin,D., Martin,N. and Beckwith,J. (1996) An in vivo pathway for disulphide bond isomerization in Escherichia coli. Proc. Natl Acad. Sci. USA, 93, 13048–13053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugawara E. and Nikaido,H. (1992) Pore-forming activity of OmpA protein of Escherichia coli. J. Biol. Chem., 267, 2507–2511. [PubMed] [Google Scholar]
- Way J.C., Davis,M.A., Morisato,D., Roberts,D.E and Kleckner,N. (1984) New Tn10 derivatives for transposon mutagenesis and for construction of lacZ operon fusions by transposition. Gene, 32, 369–379. [DOI] [PubMed] [Google Scholar]
- Wunderlich M. and Glockshuber,R. (1993) Redox properties of protein disulphide isomerase (DsbA) from Escherichia coli. Protein Sci., 2, 717–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zapun A., Bardwell,J.C.A. and Creighton,T.E. (1993) The reactive and destabilizing disulfide bond of DsbA, a protein required for protein disulfide bond formation in vivo. Biochemistry, 32, 5083–5092. [DOI] [PubMed] [Google Scholar]
- Zapun A., Missiakas,D., Raina,S. and Creighton,T.E. (1995) Structural and functional characterization of DsbC, a protein involved in disulfide bond formation in Escherichia coli. Biochemistry, 34, 5075–5089. [DOI] [PubMed] [Google Scholar]