

Summary
Seizures in the human temporal lobe transiently impair cognition and steadily damage hippocampal circuitry, leading to progressive memory loss. Similarly, the toxic accumulation of Aβ peptides underlying Alzheimer’s Disease triggers synaptic degeneration, circuit remodeling, and abnormal synchronization within the same networks. Since neuronal hyperexcitability amplifies the synaptic release of Aβ, seizures create a vicious spiral that accelerates cell death and cognitive decline in the AD brain. The confluence of hyperexcitability and excitotoxicity, combined with the challenge of seizure detection in the human hippocampus, make epilepsy in these individuals extremely important to correctly diagnose and treat. Emerging clinical evidence reveals an elevated co-morbidity of epilepsy in AD, particularly when linked to mutations in the APP/Aβ gene pathway. Experimental models in genetically engineered mice confirm and extend these findings, highlighting the presence of subclinical seizures and overlapping pathophysiological cascades. There is an urgent need for more clinical and basic investigation to improve the early recognition of hippocampal seizures arising during the course of dementing disorders, and to validate molecular blockers of Aβ-induced aberrant excitability that can slow and potentially reverse the progression of cognitive decline.
Keywords: APP, hippocampal sprouting, hyperexcitability, non-convulsive seizures, Aβ
The molecular pathology underlying neural degeneration in Alzheimer’s Disease has been dramatically clarified over the last decade, however our understanding of the pathogenesis and therapy of human dementia, a foremost clinical problem in our society, is unclear and remains stalled at the level of progressive cell death and altered plasticity at single synapses. Co-morbid conditions such as epilepsy that interfere with memory formation and retrieval in the temporal lobe further complicate the goal of preserving normal cognition. While degenerative processes in the nervous system ultimately result in loss of neural signaling, when active inhibitory mechanisms fail early, the resulting disinhibition may destabilize network oscillatory activity at formative stages of the disease. Critical new evidence implicating cellular hyperexcitablility, hypersynchronous circuit activity, extensive rewiring of hippocampal networks, and subclinical “silent” seizures in the temporal lobe identified in validated mouse models of AD implicates a new level of circuit-based pathophysiology that could lead to the appearance of epilepsy and further aggravate memory loss. Human pathological studies confirm that, much like the positive feedback loop of heat and moisture powering the ‘heat engine’ of a tropical storm, the combination of synaptic hyperactivity with elevated Aβ in AD brain fuels a pathological cascade of cell death and synaptic reorganization within hippocampal networks. These findings identify APP, along with related AD genes leading to aberrant cleavage and toxic accumulation of its Aβ protein fragments, as members of a new gene pathway for temporal lobe epilepsy, and challenge the assumption that the elevated risk of epilepsy in individuals with familial AD is a simple coincidence. The magnitude of the clinical problem justifies a concerted search to unravel the shared mechanisms leading to hyperexcitable networks in AD, and new treatment strategies for dementia focused on stabilizing dynamic network signaling patterns in the AD brain.
Epilepsy and Dementia: Clinical Comorbidity, Diagnostic Uncertainty
The appearance of seizures in individuals with Alzheimer type dementia (AD) has been noted for many years and recently has become the subject of closer epidemiological scrutiny. An increased incidence of seizures is highest among younger persons with early onset dementia (50–60 years), an age when their general incidence in the poulation remains low; even in the more elderly with advanced dementia, seizure incidence remains increased at least threefold over age-matched populations (Figure 1).
Figure 1.
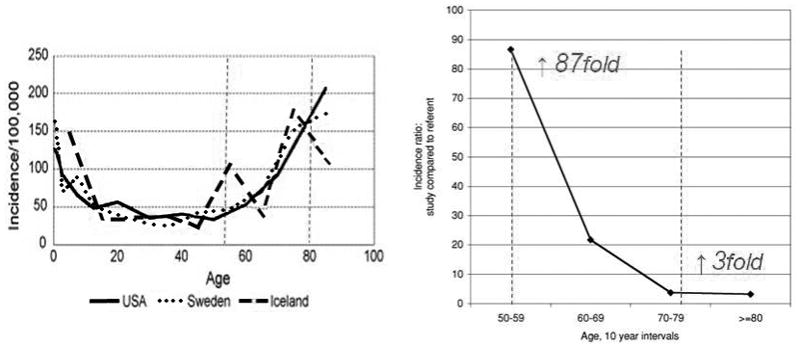
Elevated co-incidence of epilepsy and Alzheimers-type dementia in the elderly. Left: Seizure disorders of all etiologies increase in the second half of life, beginning around the age of 50. The incidence ratio of epilepsy in individuals diagnosed with AD is elevated nearly 87 fold at this age. Seizure incidence at ages beyond 70 remains elevated three-fold over patients of a similar age without AD. Modified from Cloyd et al (2006), and Amatniek et al (2007)
In the majority of AD cases, but particularly in those with early onset, these seizures are classified as complex partial with or without generalized convulsive episodes (Rao et al, 2009). However a variety of factors, rooted in the multifactorial causes, subtle clinical onsets, and episodic phenotype of epilepsy obscure an accurate determination of the co-incidence of these two disorders, leading to difficulties in ascertainment. Temporal lobe epilepsy, when subtle, can masquerade as "pure" dementia for a period of years, and case reports of non-convulsive, subtle seizures point out the diagnostic uncertainty attached to these two diagnoses. Typically an EEG is performed only once clinical symptoms become strongly episodic or following the emergence of overt behavioral seizures, leading to the diagnosis of ‘epileptic pseudodementia’, although this too can generate further error by masking the potential concurrent presence of AD (Tatum et al, 1998; Hogh et al, 2001; Tombini et al, 2005; Ito 2009; Laurentani 2009). Clearly, a dual diagnosis remains tenable.
Two Disorders or One?
Although their manifold etiologies are well documented, the biological basis for seizures in what has been traditionally held to be a purely neurodegenerative (yet also heterogeneous) disorder has never been clearly explained, and the mechanisms for hyperexcitability in AD brain are largely unknown. A commonly held view, particularly in the dementia research community, is that dementia and seizures represent fundamentally independent disorders, since overt seizures are not reported in most AD cases (Scarameas et al, 2009), nor do all individuals with generalized seizures develop progressive cognitive deficits, although this is certainly the case in temporal lobe epilepsy with hippocampal sclerosis (Helmstaedter and Egler, 2009). Instead, seizures in AD patients could represent the unfortunate co-occurrence of two disease genes or acquired encephalopathies leading to the presence of cortical atrophy. Alternatively, seizures could arise from highly specific neuronal excitability changes related to the molecular pathogenesis of certain subtypes of dementia that give rise to both phenotypes. In fact, seizures are part of the natural history of many pedigrees with autosomal dominant early-onset AD, including those with mutations in presenilin-1, presenilin-2, or the amyloid precursor protein (APP), or with duplications of wild-type APP.
Epilepsy in Human Genotyped AD Cases
In contrast to sporadic dementia, epilepsy is a striking co-morbid phenotype in individuals with genotyped cases of familial AD (Table 1). Multiple studies reveal probands and affected family members with compex partial seizures as a salient neurological co-morbidity of mutations in genes encoding the amyloid precursor protein APP, and the gamma-secretase modulatory genes Presenilin1 (PS1) and Presenilin 2 (PS2). Since individuals with these familial forms of AD present earlier in life, they are likely to account for the striking increase in risk of epilepsy found between 50–60 years of age (Amatniek et al, 2006). It is worth noting that a family history is not the sole criterion for early seizures, since de novo mutations in PSEN1, the most common gene for AD, may also produce dementia phenotypes accompanied by epilepsy (Alberici, 2007).
Table 1.
Human AD Gene Mutations Leading to Elevated Aβ and Epilepsy
| Gene | Mutation | Phenotype | Reference |
|---|---|---|---|
| APP | Val1717gly | seizures | Rossor et al, 1993 |
| APP | Thr714Ala | seizures | Lindquist et al, 2008 |
| APP | duplication | seizures | Cabrejo et al, 2006 |
| APP | Trisomy 21 | seizures | Menedez et al, 2005 |
| Presenilin 1 | M139V | seizures | Fox et al, 1997 |
| Presenilin 1 | 1S169L | seizures | Takao et al, 2001 |
| Presenilin 1 | L420R | seizures | Shrimpton et al, 2007 |
| Presenilin 1 | E280A | seizures | Velez-Pardo et al, 2004 |
| Presenilin 1 | multiple | seizures | Larner, 2010 |
| Presenilin 2 | M239V | seizures | Marcon et al, 2005 |
| Presenilin 2 | N141L | seizures | Jayadev et al, 2010 |
The human evidence strongly implicates a tight genetic association between Aβ overexpression and epilepsy, but does not exclude the chance occurrence of a second pathogenic basis for epilepsy in these families, nor does it prove the seizures arise exclusively from the presence of any particular Aβ peptide fragment, since mutant AD genes may give rise to cleavage patterns in intra- and extracellular peptides of alternative lengths and toxicity, or exert other deleterious effects (LaFerla et al, 2007). Fortunately, experimental mouse models have proven useful for providing further insight into the basic mechanisms of epileptogenesis.
Convulsive Epilepsy in Mouse Models of Abnormal Abeta Expression
Confirmation that accumulation of abnormal AB fragments actually lead to epilepsy was obtained from early studies investigating genetically engineered overexpression of mutant alleles of APP in experimental mouse models. Lowered convulsant thresholds and spontaneous convulsive seizure phenotypes have been observed in AD mouse models for some time, but did not figure strongly in models of the overall pathogenesis of dementia (Laferla et al, 1995; Moechars et al, 1996; Kumar 2000; Lalonde 2005). Interestingly, deletion of APP (Steinbach et al, 1998) and BACE1, the secretase that participates in Aβ release (Hu et al, 2010) also cause an epileptic phenotype, indicating that normal APP signaling is important in the development of hippocampal excitability. In some models, lower thresholds for induced or spontaneous seizures are found even in the absence of amyloid deposits, further implicating the soluble forms of Aβ as the pathogen (Steinbach et al, 1998; Del Vecchio et al, 2004; Kumar et al, 2000).
Non-convulsive Seizures in AD Mouse Models
The presence of subtle seizure phenotypes in AD mouse models and hippocampal pathology characteristic of epilepsy was not appreciated until videoEEG and molecular pathology studies were performed. To demonstrate the presence of neuronal hypersynchronous discharge activity, chronic EEG monitoring was performed in our laboratory in a mouse model engineered to overexpress a gene containing a human familial Alzheimers Disease (FAD) mutation, the J20 mouse (Palop et al, 2007). These recordings revealed the first evidence of abnormal synchronous interictal EEG discharges, both cortical and hippocampal, as well as clinical seizure activity of two types.
The most frequent seizures were purely electrographic, i.e. non-convulsive without behavioral arrest (Figure 2); in addition, rare and unprovoked motor convulsions were also observed. These transgenic mice therefore demonstrate that simple genetic overexpression of a human mutant form of Aβ is sufficient to create non-convulsive seizure semiology, and their appearance also raised the question of whether abnormal hippocampal neuronal synchronization might go clinically undetected in human AD patients, and indeed whether this network level abnormality might contribute to the more rapid cognitive decline in at least a subset of patients with genotyped familial AD.
Figure 2.
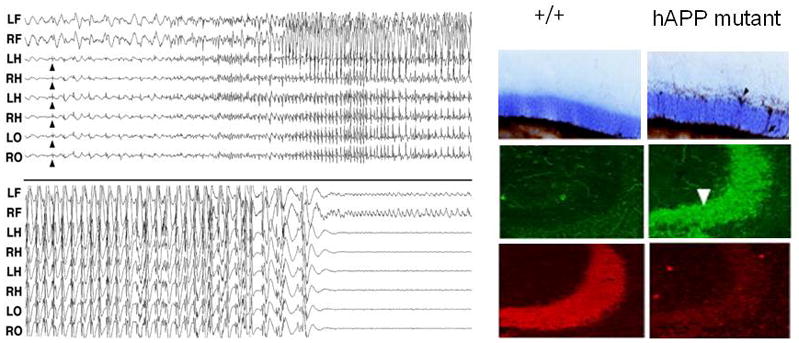
Non-convulsive hippocampal seizures and network remodeling in transgenic mouse model of AD. Left: spontaneous seizure with first appearance of epileptiform discharges (arrows) in hippocampal depth electrodes and subsequent generalization in the hAPP J20 mouse. No behavioral signs were evident during or after the seizure episode. Right: seizure-related plasticity absent in wild type hippocampus is clearly demonstrated in hippocampal circuitry of hAPP J20 mice, including mossy fiber sprouting (upper), ectopic NPY expression in mossy fibers (center), and loss of calbindin staining (lower) in dentate granule cells. Modified from Palop et al. (2007).
In patients with sporadic Alzheimer’s disease, scalp-recorded EEG patterns typically show abnormalities predominantly composed of slowed background rhythms, and cortical interictal discharges are unusual in routine EEG scalp recordings from individuals with idiopathic dementia. Even in patients with confirmed temporal lobe epilepsy, scalp-recorded interictal discharges are seen in fewer than half the cases (Nilsson et al, 2009). Three factors may lead to this apparent lack of evidence for cerebral hyperexcitability in AD patients in contrast to mouse AD models. First, EEG studies are not routinely performed in individuals with cognitive decline, and in any event would typically consist of a brief sample of activity rather than prolonged monitoring. Second, most AD cases are sporadic, and true monogenic familial cases comprise a rare cause of AD. Thus few individuals with dementia who are examined by EEG have been genotypically proven to have mutations in the APP gene. Third, unlike the ~1 mm proximity of mouse hippocampus to the cortical recording surface, detection of seizure activity originating in the human medial temporal lobe is difficult without intracranial if not depth electrode recordings (Nilsson et al, 2009). Unfortunately, other non-invasive measures of human temporal lobe ictal activity such as magnetoencephalography are relatively insensitive to aberrant electrical activity in the medial temporal lobe (Shigeto et al, 2002).
Network Remodeling in AD and TLE mouse models
Based on a growing appreciation of network level dysfunction in AD, Palop (Palop et al, 2007) examined the hippocampus in J20 mice and discovered striking evidence for hippocampal network axon remodeling that is similar, but not identical to, the changes identified in both patients with temporal lobe epilepsy and experimental models of hippocampal seizures (Figure 2). The cellular changes included ectopic sprouting of dentate granule cell mossy fibres and sprouting of fibers containing the inhibitory neurotransmitter NPY, two structural alterations long known to be present in human and experimental models of temporal lobe epilepsy and believed to be the direct result of excess glutamate toxicity and seizure-induced cell death (de Lanerolle et al, 1989; Sutula and Dudek, 2007). An additional feature noted in the AD models was loss of calbindin, a calcium binding protein, staining in granule cell bodies. Convulsive seizures with associated hippocampal network plasticity have been confirmed in other AD mouse models (Minkevienne et al 2008).
Recent examination of the progenitor population of dividing cells responsible for adult neurogenesis in the dentate gyrus in several AD mouse models has revealed a further similarity with epilepsy models (Parent and Murphy, 2008), namely an increased number of immature newborn cells at early stages of the disease (Gan et al 2008; Jin et al 2004a; Sun et al 2009). Interestingly, fewer of these cells are likely to mature into gabaergic neurons, and impairments of GABA transmission in the J20 brain provide an interesting basis for epileptogenesis in these models. Increased adult neurogenesis is found in both human AD and TLE cases (Jin et al, 2004b; Sutula and Dudek, 2007), can be replicated in vitro by selective exposure to AB42 (Lopez-Toledano and Shelanski, 2004), and figures prominently in the ‘gating’ function of the dentate gyrus with its central role in hippocampal epileptogenesis (Scharfman, 2007).
Overlapping Regional Pathologies in Human AD and TLE
While many detailed cellular abnormalities remain to be described in cortical microcircuits contributing to dementia and temporal lobe epilepsy, imaging studies of human cases demonstrate regional overlap in AD and TLE pathology (Figure 3). While the boundaries are dynamic, and ‘averaging’ across cases to obtain representative disease territories obscures the variation due to individual differences in age, disease duration, severity, and treatment course, digital analyses using MRI-based volummetry in both disorders reveal cortical volume loss that predominates in mesial, lateral temporal and frontal cortices at all stages of the disease, corresponding with the patterns seen in temporal lobe epilepsy (Thompson et al, 2003; Berhardt et al, 2008).
Figure 3.
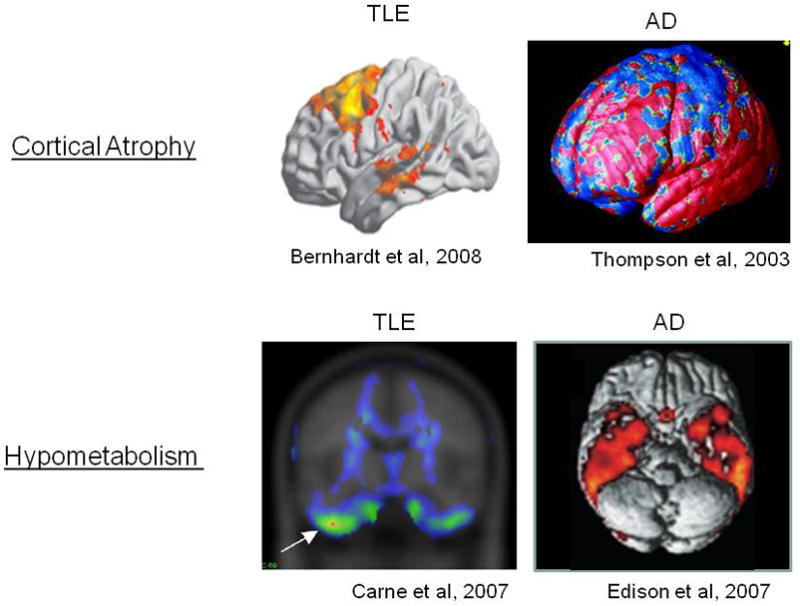
Confluence of paths of epilepsy and early Alzheimer’s Disease in the human temporal lobe. Above: MRI-based volumetric analysis of TLE cases reveals atrophy in both lateral temporal and frontal cortices, overlapping regions affected in AD. Below: Regional PET based imaging demonstrates hypometabolism in the basal temporal lobe in both disorders.
Regional brain metabolic studies using PET tracer imaging show hypometabolism in inferior temporal regions in both diseases (Carne et al, 2007; Edison et al, 2007). Without serial longitudinal studies, it remains unclear which of these two structural and functional biomarkers has temporal precedence, as well as their relationships to aberrant excitability patterns. Nevertheless, their regional confluence is sufficient evidence of risk to presume a major effect of hippocampal epilepsy in aggavating ongoing AD pathology.
Epileptic Excitotoxicity and AD Pathology
The multiple lines of evidence discussed above show that soluble forms of Aβ are cytotoxic, induce the appearance of aberrant excitatory neuronal network activity in vivo, and trigger complex molecular and cellular patterns of compensatory inhibitory and excitatory mechanisms in hippocampal circuitry. While many additional biological signaling pathways contribute to the emergence of this complex lesion, at least two positive feedback mechanisms intersecting with seizure-related pathophysiology have been identifed that synergistically enhance the further release of Aβ by seizures. The first is that the release of Aβ from synapses is activity-dependent, and has been shown to share the same presynaptic exocytotic mechanisms as vesicular glutamate release. In vivo microdialysis studies reveal that kainate-induced seizures actively elevate soluble AB levels in the hippocampal extracellular space and can be blocked by presynaptic inhibitors of vesicular transmitter release and clathrin-mediated endocytosis (Cirrito et al, 2005, 2008). The second is that excessive glutamate release activates NMDA receptors which in turn stimulate a shift from α- to β-secretase cleavage of the amyloid precursor protein APP, facilitating Aβ release (Lesne et al, 2005). Both pre- and postsynaptic mechanisms provide a molecular basis for the model of an unchecked vicious cycle at the neuronal network level capable of accelerating AD pathology progression (Figure 4).
Figure 4.
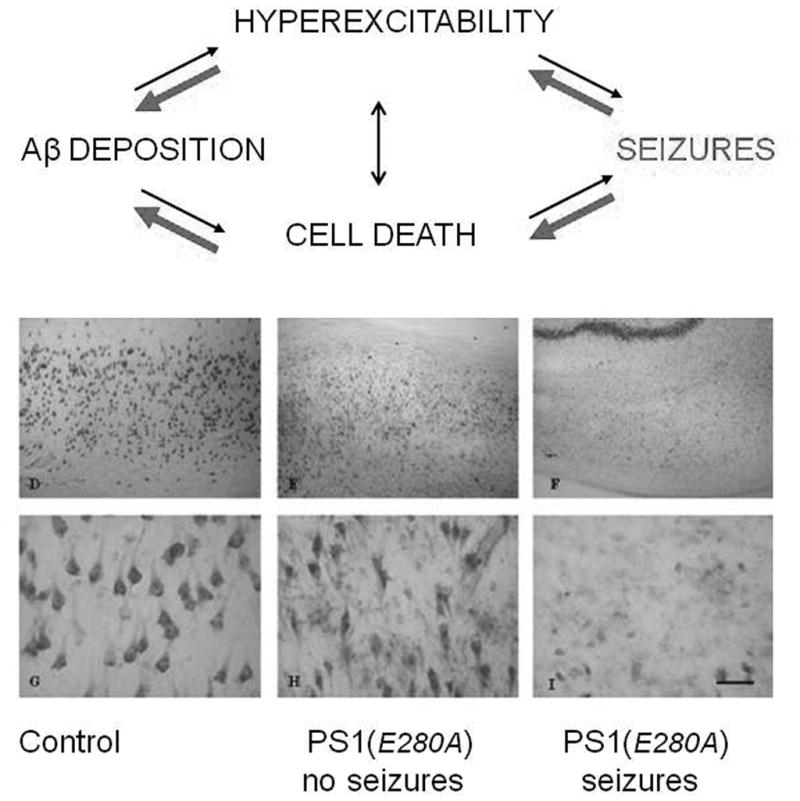
Potentiating cytopathic effects of seizures in Alzheimer’s Disease. Above: Model depicting self-amplifying neurodegenerative cascade leading to hyperexcitability and networking reorganization. Damage is accelerated by linked effects of seizure excitotoxicity on hippocampal circuitry and augmented release of toxic Aβ peptides. Below: Comparison of hippocampal cell death at postmortem examination of genotyped PSEN1 AD patients with no overt seizure history (middle), and those with positive seizure history (right). Seizures were accompanied by extensive cell death, consistent with positive feedback model above. Modified form Velez-Pardo, (2004).
Pathological evidence supporting this model has been demonstrated in a key study comparing the cellular pathology of human hippocampal autopsy specmens obtained from individuals bearing PSEN1 E280A mutations (Velez-Pardo et al, 2007). When such cases were compared between individuals with, and without a reported seizure history, a clear aggravation of hippocampal pyramidal cell death was found in AD individuals with a history of seizures (Figure 4). This exacerbation not only supports the cyclical model of excitotoxic neurodegeneration, but offers an explanation of why dementia onset is earlier in inherited forms of AD, where the linked expression of seizures is highest.
Future Perspectives
Whether it is subconvulsive seizures or other derangements of normal oscillatory activity patterns due to plasticity within hippocampal circuitry (Chauviere et al, 2009) that contribute to the memory impairment of AD, new insights into the neurobiology of epilepsy and Alzheimers Disease are coalescing into an understanding that the two share major pathogenic elements of interest in the search for new avenues of disease modification. While these two maladaptive reactive cascades may not coincide in every patient presenting with AD, the incendiary co-expression of hyperexcitability and seizures within overlapping brain circuits in temporal and extratemporal regions critical for cognitive processing poses an enormous risk to memory function in those with familial forms of the disorder. Since this pathogenic cycle amplifies dysfunction within the neural circuit, it is intuitively clear that preventing or interrupting epilepsy could prove to be an important new approach to slowing the progression of cognitive decline in individuals with genotyped Aβ pathology. Further translational exploration of these issues in animals and human studies over the coming decade will determine which patients may benefit from new therapeutic approaches directed at selectively suppressing aberrant network excitability in the temporal lobe.
Acknowledgments
The author would like to thank the NIH and the Blue Bird Circle Foundation for generous support.
Footnotes
Disclosure The author has no conflicts to disclose.
References
- Alberici A, Bonato C, Borroni B, Cotelli M, Mattioli F, Binetti G, Gennarelli M, Luca MD, Simonati A, Perani D, Rossini P, Padovani A. Dementia, delusions and seizures: storage disease or genetic AD? Eur J Neurol. 2007;14:1057–9. doi: 10.1111/j.1468-1331.2007.01664.x. [DOI] [PubMed] [Google Scholar]
- Amatniek JC, Hauser WA, DelCastillo-Castaneda C, Jacobs DM, Marder K, Bell K, Albert M, Brandt J, Stern Y. Incidence and predictors of seizures in patients with Alzheimer’s disease. Epilepsia. 2006;475:867–872. doi: 10.1111/j.1528-1167.2006.00554.x. [DOI] [PubMed] [Google Scholar]
- Bernhardt BC, Worsley KJ, Besson P, Concha L, Lerch JP, Evans AC, Bernasconi N. Mapping limbic network organization in temporal lobe epilepsy using morphometric correlations: insights on the relation between mesiotemporal connectivity and cortical atrophy. Neuroimage. 2008;42:515–24. doi: 10.1016/j.neuroimage.2008.04.261. [DOI] [PubMed] [Google Scholar]
- Cabrejo L, Guyant-Maréchal L, Laquerrière A, Vercelletto M, De la Fournière F, Thomas-Antérion C, Verny C, Letournel F, Pasquier F, Vital A, Checler F, Frebourg T, Campion D, Hannequin D. Phenotype associated with APP duplication in five families. Brain. 2006;129:2966–2976. doi: 10.1093/brain/awl237. [DOI] [PubMed] [Google Scholar]
- Carne RP, Cook MJ, MacGregor LR, Kilpatrick CJ, Hicks RJ, O’Brien TJ. Magnetic Resonance Imaging Negative Positron Emission Tomography Positive Temporal Lobe Epilepsy: FDG-PET Pattern Differs from Mesial Temporal Lobe Epilepsy. Molecular Imaging and Biology. 2007;9:32–42. doi: 10.1007/s11307-006-0073-0. [DOI] [PubMed] [Google Scholar]
- Chauvière L, Rafrafi N, Thinus-Blanc C, Bartolomei F, Esclapez M, Bernard C. Early deficits in spatial memory and theta rhythm in experimental temporal lobe epilepsy. J Neurosci 2009. 2009;29:5402–10. doi: 10.1523/JNEUROSCI.4699-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cirrito JR, Yamada KA, Finn MB, Sloviter RS, Bales KR, May PC, Schoepp DD, Paul SM, Mennerick S, Holtzman DM. Synaptic activity regulates interstitial fluid amyloid-beta levels in vivo. Neuron. 2005;48:913–22. doi: 10.1016/j.neuron.2005.10.028. [DOI] [PubMed] [Google Scholar]
- Cirrito JR, Kang JE, Lee J, Stewart FR, Verges DK, Silverio LM, Bu G, Mennerick S, Holtzman DM. Endocytosis is required for synaptic activity-dependent release of amyloid-beta in vivo. Neuron. 2008;58:42–51. doi: 10.1016/j.neuron.2008.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cloyd J, Hauser W, Towne A, Ramsay R, Mattson R, Gilliam F, Walczak T. Epidemiological and medical aspects of epilepsy in the elderly. Epilepsy Res. 2006;68(Suppl 1):S39–48. doi: 10.1016/j.eplepsyres.2005.07.016. [DOI] [PubMed] [Google Scholar]
- Del Vecchio R, Gold LH, Novick SJ, Wong G, Hyde LA. Increased seizure threshold and severity in young transgenic CRND8 mice. Neuroscience Letters. 2004;367:164–167. doi: 10.1016/j.neulet.2004.05.107. [DOI] [PubMed] [Google Scholar]
- de Lanerolle NC, Kim JH, Robbins RJ, Spencer DD. Hippocampal interneuron loss and plasticity in human temporal lobe epilepsy. Brain Research. 1989;495:387–395. doi: 10.1016/0006-8993(89)90234-5. [DOI] [PubMed] [Google Scholar]
- Edison P, Archer HA, Hinz R, Hammers A, Pavese N, Tai YF, Hotton G, Cutler D, Fox N, Kennedy A, Rossor M, Brooks DJ. Amyloid, hypometabolism, and cognition in Alzheimer disease: an [11C]PIB and [18F]FDG PET study. Neurology. 2007;68:501–8. doi: 10.1212/01.wnl.0000244749.20056.d4. [DOI] [PubMed] [Google Scholar]
- Fox NC, Kennedy AM, Harvey RJ, Lantos PL, Roques PK, Collinge J, Hardy J, Hutton M, Stevens JM, Warrington EK, Rossor MN. Clinicopathological features of familial Alzheimer's disease associated with the M139V mutation in the presenilin 1 gene. Pedigree but not mutation specific age at onset provides evidence for a further genetic factor. Brain. 1997;120:491–501. doi: 10.1093/brain/120.3.491. [DOI] [PubMed] [Google Scholar]
- Gan L, Qiao S, Lan X, Chi L, Luo C, Lien L, Yan Liu Q, Liu R. Neurogenic responses to amyloid-beta plaques in the brain of Alzheimer’s disease-like transgenic (pPDGF-APPSw,Ind) mice. Neurobiol Dis. 2008;29:71–80. doi: 10.1016/j.nbd.2007.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helmstaedter C, Elger CE. Chronic temporal lobe epilepsy: a neurodevelopmental or progressively dementing disease? Brain. 2009;132:2822–30. doi: 10.1093/brain/awp182. [DOI] [PubMed] [Google Scholar]
- Høgh P, Smith SJ, Scahill RI, Chan D, Harvey RJ, Fox NC, Rossor MN. Epilepsy presenting as AD: neuroimaging, electroclinical features, and response to treatment. Neurology. 2002;58:298–301. doi: 10.1212/wnl.58.2.298. [DOI] [PubMed] [Google Scholar]
- Hu X, Zhou X, He W, Yang J, Xiong W, Wong P, Wilson CG, Yan R. BACE1 deficiency causes altered neuronal activity and neurodegeneration. J Neurosci. 2010;30:8819–29. doi: 10.1523/JNEUROSCI.1334-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito M, Echizenya N, Nemoto D, Kase M. A case series of epilepsy-derived memory impairment resembling Alzheimer disease. Alzheimer Dis Assoc Disord. 2009;23:406–9. doi: 10.1097/WAD.0b013e31819fe7bd. [DOI] [PubMed] [Google Scholar]
- Jayadev S, Leverenz JB, Steinbart E, Stahl J, Klunk W, Yu CE, Bird TD. Alzheimer's disease phenotypes and genotypes associated with mutations in presenilin 2. Brain. 2010;133:1143–54. doi: 10.1093/brain/awq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin K, Galvan V, Xie L, Mao XO, Gorostiza OF, Bredesen DE, Greenberg DA. Enhanced neurogenesis in Alzheimer’s disease transgenic (PDGF-APPSw,Ind) mice. Proc Natl Acad Sci USA. 2004a;101:13363–13367. doi: 10.1073/pnas.0403678101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin K, Peel AL, Mao XO, Xie L, Cottrell BA, Henshall DC, Greenberg DA. Increased hippocampal neurogenesis in Alzheimer’s disease. Proc Natl Acad Sci USA. 2004b;101:343–347. doi: 10.1073/pnas.2634794100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar-Singh S, Dewachter I, Moechars D, Lübke U, De Jonghe C, Ceuterick C, Checler F, Naidu A, Cordell B, Cras P, Van Broeckhoven C, Van Leuven F. Behavioral disturbances without amyloid deposits in mice overexpressing human amyloid precursor protein with Flemish (A692G) or Dutch (E693Q) mutation. Neurobiol Dis. 2000;7:9–22. doi: 10.1006/nbdi.1999.0272. [DOI] [PubMed] [Google Scholar]
- LaFerla FM, Tinkle BT, Bieberich CJ, Haudenschild CC, Jay G. The Alzheimer’s A beta peptide induces neurodegeneration and apoptotic cell death in transgenic mice. Nat Genet. 1995;9:21–30. doi: 10.1038/ng0195-21. [DOI] [PubMed] [Google Scholar]
- LaFerla FM, Green KN, Oddo S. Intracellular amyloid-beta in Alzheimer's disease. Nat Rev Neurosci. 2007;8:499–509. doi: 10.1038/nrn2168. [DOI] [PubMed] [Google Scholar]
- Lalonde R, Dumont M, Staufenbiel M, Strazielle C. Neurobehavioral characterization of APP23 transgenic mice with the SHIRPA primary screen. Behav Brain Res. 2005;157:91–98. doi: 10.1016/j.bbr.2004.06.020. [DOI] [PubMed] [Google Scholar]
- Larner AJ. Epileptic seizures in AD patients. Neuromolecular Med. 2010;12:71–7. doi: 10.1007/s12017-009-8076-z. [DOI] [PubMed] [Google Scholar]
- Lauretani F, Maggio M, Nardelli A, Saccavini M, Ceda GP. Is Non-Convulsive Status Epilepticus (NCSE) undertreated in patients affected by dementia? Aging Clin Exp Res. 2009;21:363–4. doi: 10.1007/BF03324929. [DOI] [PubMed] [Google Scholar]
- Lesné S, Ali C, Gabriel C, Croci N, MacKenzie ET, Glabe CG, Plotkine M, Marchand-Verrecchia C, Vivien D, Buisson A. NMDA receptor activation inhibits alpha-secretase and promotes neuronal amyloid-beta production. J Neurosci. 2005;25:9367–77. doi: 10.1523/JNEUROSCI.0849-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindquist SG, Nielsen JE, Stokholm J, Schwartz M, Batbayli M, Ballegaard M, Erdal J, Krabbe K, Waldemar G. Atypical early-onset Alzheimer's disease caused by the Iranian APP mutation. J Neurol Sci. 2008;268:124–30. doi: 10.1016/j.jns.2007.11.021. [DOI] [PubMed] [Google Scholar]
- Lopez-Toledano MA, Shelanski M. Neurogenic Effect of β-Amyloid Peptide in the Development of Neural Stem Cells. J Neuroscience. 2004;24:5439–5444. doi: 10.1523/JNEUROSCI.0974-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menendez M. Down syndrome, Alzheimer's disease and seizures. Brain Dev. 2005;27:246–52. doi: 10.1016/j.braindev.2004.07.008. [DOI] [PubMed] [Google Scholar]
- Minkeviciene R, Rheims S, Dobszay MB, Zilberter M, Hartikainen J, Fülöp L, Penke B, Zilberter Y, Harkany T, Pitkänen A, Tanila H. Amyloid beta-induced neuronal hyperexcitability triggers progressive epilepsy. J Neurosci. 2009;29:3453–3462. doi: 10.1523/JNEUROSCI.5215-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moechars D, Lorent K, De Strooper B, Dewachter I, Van Leuven F. Expression in brain of amyloid precursor protein mutated in the a-secretase site causes disturbed behavior, neuronal degeneration and premature death in transgenic mice. EMBO Journal. 1996;15:1265–1274. [PMC free article] [PubMed] [Google Scholar]
- Nilsson D, Fohlen M, Jalin C, Dorfmuller G, Bulteau C, Delalande O. Foramen ovale electrodes in the preoperative evaluation of temporal lobe epilepsy in children. Epilepsia. 2009;50:2085–96. doi: 10.1111/j.1528-1167.2009.02135.x. [DOI] [PubMed] [Google Scholar]
- Palop JJ, Chin J, Roberson ED, Wang J, Thwin MT, Bien-Ly N, Yoo J, Ho KO, Yu GQ, Kreitzer A, Finkbeiner S, Noebels JL, Mucke L. Aberrant excitatory neuronal activity and compensatory remodeling of inhibitory hippocampal circuits in mouse models of Alzheimer's disease. Neuron. 2007;55:697–711. doi: 10.1016/j.neuron.2007.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palop JJ, Mucke L. Epilepsy and cognitive impairments in Alzheimer disease. Arch Neurol. 2009;66:435–40. doi: 10.1001/archneurol.2009.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parent JM, Murphy GG. Mechanisms and functional significance of aberrant seizure-induced hippocampal neurogenesis. Epilepsia. 2008;49(Suppl 5):19–25. doi: 10.1111/j.1528-1167.2008.01634.x. [DOI] [PubMed] [Google Scholar]
- Rao SC, Dove G, Cascino GD, Petersen RC. Recurrent seizures in patients with dementia: frequency, seizure types, and treatment outcome. Epilepsy Behav. 2009;14:118–20. doi: 10.1016/j.yebeh.2008.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossor MN, Newman S, Frackowiak RS, Lantos P, Kennedy AM. Alzheimer's disease families with amyloid precursor protein mutations. Ann N Y Acad Sci. 1993;695:198–202. doi: 10.1111/j.1749-6632.1993.tb23052.x. [DOI] [PubMed] [Google Scholar]
- Scarameas N, Honig LS, Choi H, Cantero J, Brandt J, Blacker D, Albert M, Amatniek JC, Marder K, Bell K, Hauser WA, Stern Y. Seizures in Alzheimer disease: who, when, and how common? Arch Neurol. 2009;66:992–7. doi: 10.1001/archneurol.2009.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scharfman HE. The CA3 "backprojection" to the dentate gyrus. Prog Brain Res. 2007;163:627–37. doi: 10.1016/S0079-6123(07)63034-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shigeto H, Morioka T, Hisada K, Nishio S, Ishibashi H, Kira D, Tobimatsu S, Kato M. Feasibility and limitations of magnetoencephalographic detection of epileptic discharges: simultaneous recording of magnetic fields and electrocorticography. Neurol Res. 2002;24:531–6. doi: 10.1179/016164102101200492. [DOI] [PubMed] [Google Scholar]
- Shrimpton AE, Schelper RL, Linke RP, Hardy J, Crook R, Dickson DW, Ishizawa T, Davis RL. A presenilin 1 mutation (L420R) in a family with early onset Alzheimer disease, seizures and cotton wool plaques, but not spastic paraparesis. Neuropathology. 2007;27:228–32. doi: 10.1111/j.1440-1789.2007.00766.x. [DOI] [PubMed] [Google Scholar]
- Steinbach JP, Müller U, Leist M, Li ZW, Nicotera P, Aguzzi A. Hypersensitivity to seizures in beta-amyloid precursor protein deficient mice. Cell Death Differ. 1998;5:858–66. doi: 10.1038/sj.cdd.4400391. [DOI] [PubMed] [Google Scholar]
- Sun B, Halabisky B, Zhou Y, Palop JJ, Yu G, Mucke L, Gan L. Imbalance between GABAergic and Glutamatergic Transmission Impairs Adult Neurogenesis in an Animal Model of Alzheimer's Disease. Cell Stem Cell. 2009;5:624–33. doi: 10.1016/j.stem.2009.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutula TP, Dudek FE. Unmasking recurrent excitation generated by mossy fiber sprouting in the epileptic dentate gyrus: an emergent property of a complex system. Prog Brain Res. 2007;163:541–63. doi: 10.1016/S0079-6123(07)63029-5. [DOI] [PubMed] [Google Scholar]
- Tatum WO, 4th, Ross J, Cole AJ. Epileptic pseudodementia. Neurology. 1998;50:1472–5. doi: 10.1212/wnl.50.5.1472. [DOI] [PubMed] [Google Scholar]
- Thompson PM, Hayashi KM, de Zubicaray G, Janke AL, Rose SE, Semple J, Herman D, Hong MS, Dittmer SS, Doddrell DM, Toga AW. Dynamics of gray matter loss in Alzheimer's disease. J Neurosci. 2003;23:994–1005. doi: 10.1523/JNEUROSCI.23-03-00994.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tombini M, Koch G, Placidi F, Sancesario G, Marciani MG, Bernardi G. Temporal lobe epileptic activity mimicking dementia: a case report. Eur J Neurol. 2005;12:805–6. doi: 10.1111/j.1468-1331.2005.01078.x. [DOI] [PubMed] [Google Scholar]
- Velez-Pardo C, Arellano JI, Cardona-Gomez P, Jimenez Del Rio M, Lopera F, De Felipe J. CA1 hippocampal neuronal loss in familial Alzheimer's disease presenilin-1 E280A mutation is related to epilepsy. Epilepsia. 2004;45:751–6. doi: 10.1111/j.0013-9580.2004.55403.x. [DOI] [PubMed] [Google Scholar]