

Abstract
K-ras mutations occur in as high as 95% of patients with pancreatic cancer. K-ras activates Rac1-dependent NADPH oxidase, a key source of superoxide. Superoxide plays an important role in pancreatic cancer cell proliferation and scavenging or decreasing the levels of superoxide inhibits pancreatic cancer cell growth both in vitro and in vivo. DNA microarray analysis and RT-PCR has demonstrated that Rac1 is also upregulated in pancreatic cancer. The aim of this study was to determine if inhibiting Rac1 would alter pancreatic tumor cell behavior. Human pancreatic cancer cells with mutant K-ras (MIA PaCa-2), wild-type K-ras (BxPC-3), and the immortal H6c7 cell line (pancreatic ductal epithelium) expressing K-ras oncogene (H6c7eR-KrasT) that is tumorigenic, were infected with a dominant/negative Rac1 construct (AdN17Rac1). In cells with mutant K-ras, AdN17Rac1 decreased rac activity, decreased superoxide levels, and inhibited in vitro growth. However in the BxPC-3 cell line, AdN17Rac1 did not change rac activity, superoxide levels, or in vitro cell growth. Additionally, AdN17Rac1 decreased superoxide levels and inhibited in vitro growth in the KrasT tumorigenic cell line, but had no effect in the immortalized H6c7 cell line. In human pancreatic tumor xenografts, intratumoral injections of AdN17Rac1 inhibited tumor growth. These results suggest that activation of Rac1-dependent superoxide generation leads to pancreatic cancer cell proliferation. In pancreatic cancer inhibition of Rac1 may be a potential therapeutic target.
Keywords: Rac-1, NADPH oxidase, pancreatic cancer, reactive oxygen species
INTRODUCTION
K-ras mutation results in constitutive activation of intracellular signaling pathways, leading to uncontrolled cellular proliferation. Mutations of the K-ras gene occur in 95% of cases with adenocarcinoma of the pancreas (1). Although mutations of K-ras are less common in other cancer types, K-ras mutation has been found in intraductal pancreatic cancer, ductal hyperplasia, and even chronic pancreatitis (2), suggesting that this may be an early event in pancreatic carcinogenesis. Although the entire spectrum of downstream genes regulated by the K-ras activation is not clear, several Ras-mediated signaling pathways and their target proteins have been demonstrated to regulate pancreatic cancer growth and survival. Multiple lines of evidence demonstrate that downstream proteins, such as the G proteins Rac and Rho, are upregulated in pancreatic cancer, and K-ras activated Raf-1 and Rac more effectively than the other ras isoforms, Ha-ras and N-ras (3). Further understanding of these key molecular events in pancreatic carcinogenesis has provided a potential target for novel gene therapies.
Rac may represent an important downstream effecter for Ras activation in many cells (4). In fact, DNA microarray analysis and RT-PCR has demonstrated that Rac1 is also upregulated in pancreatic cancer (5). Rac, a member of the Rho family, is a 21-kDa GTP binding protein. The rho activation cycle regulated by GDP exchange factors and GTPase activating factors catalyze the exchange of inactive GDP bound form to an active GTP bound form (6). There are three isoforms of Rac; Rac1 expression is ubiquitous, but Rac2 is expressed only in hematopoietic cells (7). Rac3 is highly expressed in the brain but in lower levels in a variety of other tissues (8).
Two major functions of Rac1 have been identified including regulation of the organization of the actin cytoskeleton; the other one is controlling the activity of the key enzyme complex NADPH oxidase to mediatesuperoxide production (9). Several groups have reported that Rac1 activation of NADPH oxidase occurs not only in phagocytes, but also in nonphagocytes (10). The subunit composition of the NADPH oxidase complex in the phagocytic cell consists of a plasma membrane flavocytochrome, cytochrome b558, comprised of two subunits gp91phox and p22phox, with additional cytoplasmic components p47phox, p67phox and the Rac1 (3). The NADPH-driven reduction of oxygen to O2•− requires FAD as a cofactor.
The contribution of Rac proteins to the activation of the phagocyte NADPH oxidase has been studied extensively and has recently seen important progress in the analysis in mouse models (11). Several studies have subsequently implicated Rac1-mediated production of reactive oxygen species (ROS) in a variety of cellular responses. Studies by Irani et al., have shown that superoxide production in NIH 3T3 (fibroblastic clonal lines) transformed cells was dependent upon farnesylation of Ras, required Rac1 activity, and the activity of NADPH oxidase complex (12). Using an activated mutant of Rac1, V12Rac1, resulting in increased production of ROS in fibroblasts, Sundaresan has shown the direct link between Rac1 activity and ROS production in nonphagocytic cells (13). Wetering and Moldovan suggested that the constitutive active form of Rac1 increased the production of ROS in endothelial cells, and induced loss of cell–cell adhesion and cytoskeletal reorganization (14, 15). Rac1-mediated production of ROS was also found in HeLa cells and implicated in IL-1ß– mediated activation of NF-κB, further strengthening the idea that Rac1-mediated production of intracellular ROS is instrumental in signal transduction (16). Other studies have shown that downregulation of Rac1 activity by using dominant negative Rac1 mutant, N17Rac1, suppressed NADPH oxidase activity and decreased superoxide production (17). In addition, based on expression of N17Rac1 and the use of the antioxidants, activation of protein tyrosine phosphorylation and extracellular-signal regulated kinase is a Rac1- and ROS-dependent manner (18). Various mechanisms of the Rac1-induced ROS have been proposed. Deem and Cook-Mills suggested that endothelial NADPH oxidase 2 (NOX2) releases ROS extracellularly and stimulates matrix metalloprotease activity (19). Most importantly in pancreatic cancer, Rac activation increases the invasive capacity of pancreatic cancer through NADPH oxidase ROS production (20). Taken together, there is ample evidence for Rac-dependent production of ROS by the NADPH oxidase complex in cellular signaling.
Our present study demonstrates that inhibition of Rac1 activity by using a dominant/negative Rac1 mutant construct inhibited growth in human pancreatic cancer cells with mutant K-ras and in tumorigenic immortalized pancreatic ductal epithelial cells, but not in pancreatic cancer cells with wild-type K-ras or in immortalized pancreatic ductal epithelial cells (non-tumorigenic). The growth inhibitions correlated with decreased steady-state levels of superoxide. Since 95% of pancreatic cancers express mutant K-ras, inhibition of Rac1 may be a potential therapeutic target.
MATERIALS AND METHODS
Cell culture
The following human pancreatic adenocarcinoma cell lines were obtained from American Type Culture Collection (Rockville, MD): BxPC-3 (poorly differentiated, wild-type K-ras), and MIA PaCa-2 (undifferentiated, mutant K-ras). BxPC-3 was maintained in RPMI 1640 with 10% fetal bovine serum. MIAPaCa-2 was maintained in Dulbecco modified Eagle medium supplemented with 10% fetal bovine serum and 2.5% horse serum. All media was obtained from Invitrogen (Carlsbad, CA) and all cell lines were maintained at 37°C in 5% CO2/95% air. In addition, we used an immortalized pancreatic ductal epithelial cell line with near normal genotype and phenotype of pancreatic duct epithelial cells HPV16-E6E7 (H6c7), and the isogenic cell line that expresses K-rasG12V and forms tumors H6c7eR-KrasT (21). These cell lines were maintained in keratinocyte serum free media and supplemented with epidermal growth factor and bovine pituitary extract as previously described (22). Initial work with these cells demonstrated that they did not form colonies. Thus, feeder cells were used and prepared by growing B1 mouse fibroblast cells in DMEM containing 10% FBS and 1% penicillin and streptomycin plus 1x MEM with nonessential amino acids. Cells were then plated in 100-mm dishes and grown to 80% confluence before being irradiated for 37.61 min for a total dose of 30 Gy. The cells were harvest and preserved in growth media containing 10% DMSO and aliquots were frozen in liquid nitrogen. Twenty-four hours before clonogenic assay, irradiated cells were thawed and diluted in keratinocyte serum free media. Cells were then seeded 3 × 105 per well in 6-well plates as described (23).
Adenovirus Gene Transfer
AdN17Rac1-HA (AdN17Rac1) is a “first-generation” adenoviral vector (E1 deleted/E3 partially deleted) encoding HA-epitope tagged cDNA of dominant/negative N17Rac1 containing a substitution at position 17 and was obtained from the University of Iowa Gene Transfer Vector Core. The promoter is a human cytomegalovirus (hCMV) promoter. The AdN17Rac1 was constructed by PCR amplification of cDNA sequences using primers that incorporated an N-terminal HA fusion to Rac1 (17). Approximately 106 MIA PaCa-2 cells were plated in 5 ml of complete media in a 100-mm2 plastic dish and allowed to attach for 24 hours. Cells were then washed three times in serum-and antibiotic-free media. The AdN17Rac1 suspended in 3% sucrose-PBS, was then applied to cells suspended in 5 ml of antibiotic-free media at 0, 10, 25, 50, and 100 MOI (multiplicity of infection). Control cells were treated with 100 MOI of the AdBglII or AdEmpty constructs. AdBglII only has the adenovirus cassette; it does not have any promoter or poly A sequence. The AdEmpty vector has the adenovirus cassette plus the promoter sequence but no transgene. Cells were incubated with the adenovirus constructs for 24 hours. Media was then replaced with 5 ml of complete media.
Cell Homogenization and Protein Determination
Cells were removedfrom the dish using a plastic scraper after washing three times in PBS (pH 7.0), and then suspended in potassium phosphatebuffer (pH 7.8). The cell pellets were sonicated on icethree times for 10 s each using a VibraCell sonicator (Sonicsand Materials Inc., Danbury, CT) at 30% duty cycle. Total protein concentrations were determined using the Bio-Rad (BioRad, Hercules, CA)protein assay kit according to the manufacturer’s instruction.
Immunoblot Analysis
Immunoreactive protein corresponding to Rac1 was identified and quantitated from total cell protein by the specific reaction of the immobilized protein with its antibody. Total protein was electrophoresed in a 12.5% SDS-polyacrylamide running gel and a 5% stacking gel. The proteins were then electotransferred to nitrocellulose sheets. After transfer, the membranes were blocked in 5% nonfat dry milk in TTBS (10 mM Tris-HCl, 150 mM NaCl (pH 8.0), 0.05% Tween 20) at room temperaturefor 1 h. To detect specific proteins, the membrane was incubated with specific antibody. After incubation overnight in 1:1000 dilution of primary antibody to Rac1, MnSOD or CuZnSOD (Cell Signalling Technology, Danvers, MA) at 4 C, membranes are incubated with goat anti-mouse IgG or goat anti-rabbit (1:10 000, Chemicon International, Temecula, CA) at room temperature for 1 h. The washed blot was then treated with SuperSignal West Pico Chemiluminescent substrate (Pierce Biotechnology, Rockford, IL) and exposed to Classic Blue Autoradiography Film (MIDSCITM, St. Louis, MO). Western blots were performed in duplicate. β-actin was used to determine equal loading.
Cell Growth
Cells (1 × 104) were plated in 3 ml complete media in 6-well plates. Cells were trypsinized and then counted everyday for a week using a Coulter counter. Cell population doubling time in hours (DT) was determined using the following equation: DT (hours) = 0.693(t − to)/ln (Nt/No) where to = time at which exponential growth began, t = time in hours, Nt = cell number at time t, and No = initial cell number. For controls the AdBglII or AdEmpty vector constructs were given to equal the viral load of the dominant/negative rac viral construct.
Plating Efficiency
Cells were infected with the AdN17Rac, AdBglII, or AdEmpty vectors and were plated into 6-well plates in complete media. The dishes were maintained in the incubator for 14 days to allow colony formation. The colonies were then fixed and stained with 0.1% crystal violet and 2.1% citric acid, and those colonies containing greater than 50 cells were scored.
Measurement of superoxide
Intracellular levels of superoxide were detected using the oxidant-sensitive probe hydroethidine (Invitrogen, Carlsbad CA). The oxidation products of hydroethidine bind to DNA in the nucleus and give a red fluorescence when oxidized by O2•−. Cells were grown in 60 mm plates and incubated with different treatments as described previously.Forty-eight hours later, the cells were washed and incubatedfor 45 minutes with hydroethidine (10 μM) in HBSS at 37 °C and after incubation the cells were rinsed, cells were collected into Eppendorf tubes, cell pellets were lysed by 1% NP-40. After centrifugation, supernatant were transferred to a 96-well black bottom plate. The intensity of fluorescence (excitation 485 nm, emission 595 nm) was measured using a Tecan Spectra Fluor Plus microplate reader (Tecan, Research Triangle Park, NC). The relative fluorescence intensity wascalculated by dividing the total fluorescence intensity by total protein.
Rac1 activation assay
Rac1 activity was measured in pancreatic cancer cells using a Rac1 activation assay kit (Upstate Biotechnology, Temecula, CA). Cells that were approximately 80% confluent were washed twice with ice-cold PBS and lysed in ice-cold MLB (25 mM HEPES, 150 mM NaCl, 1% Igepal CA-630, 10 mM MgCl2, 1 mM EDTA and 2% glycerol). Cell lysate were centrifuged at 14000 × g for 5 min at 4 °C. Supernatant containing the same amount of protein was incubated with PAK1-PBD agarose for 1 h at 4 °C. Active (GTP-bound) Rac binds specially to the p21-binding domain of p21-activated protein kinase 1. Precipitated GST bound Rac1 were resolved on a 4–20% Tris-HCl Ready Gel (Bio-Rad, Hercules, CA) and immunoblotted using anti-Rac1monoclonal antibody (1:1000, Upstate Biotechnology).
Adenovirus Vector-Mediated N17Rac1 in vivo Gene Transfer
Thirty-day-old athymic nude mice (Nu/NU) were obtained from Harlan Sprague-Dawley (Indianapolis, IN). The nude mice protocol was reviewed and approved by the Animal Care and Use Committee of the University of Iowa and were in compliance with The U.S. Public Health Service Policy on Humane Care and Use of Laboratory Animals (NIH). The animals were housed four to a cage and fed a sterile commercial stock diet and tap water, ad libitum. Animals were allowed to acclimate in the unit for one week before any manipulations were performed. MIA PaCa-2 tumor cells (3 × 106) were delivered subcutaneously into the flank region of the mice with a 1-cc tuberculin syringe equipped with a 25-gauge needle. The tumors were allowed to grow until they reached between 3 mm and 4 mm in greatest dimension (2 weeks), at which time they were treated with adenovirus. The adenovirus constructs were delivered through two injection sites in the tumor. Approximately 1 × 109 plaque-forming units (PFU) in 50 μL of PBS of the AdN17Rac1 constructs were delivered to the tumor by means of a 25-gauge needle attached to a 1-cc tuberculin syringe on days 1, 7, and 14 for a total of 3 injections. Control tumors received PBS, or adenovirus containing no gene (AdEmpty) in similar volumes and PFUs at the same time points. There were 7–8 mice in each treatment group. Tumor size was measured every two to three days by means of a vernier caliper, and tumor volume was estimated according to the following formula: tumor volume = π/6 × L × W2, where L is the greatest dimension of the tumor, and W is the dimension of the tumor in the perpendicular direction (24). Animals were killed by CO2 asphyxiation when the tumors reached a predetermined size of 1000 mm3 and this was considered the time to sacrifice.
Statistical Analysis
Statistical analysis for the in vitro studies was performed using SYSTAT. A single factor ANOVA, followed by post-hoc Tukey test, was used to determine statistical differences between means. All means were calculated from three experiments, and error bars represent standard error of the mean (SEM). All western blots, activity assays, and activity gel assays were repeated at least twice. For the in vivo studies, the statistical analyses focused on the effects of different treatments on cancer progression. The primary outcome of interest was tumor growth over time. Once tumors were visible, the mice were then randomly assigned to a treatment group and followed until death or until the experiment was terminated. Tumor sizes (mm3) were measured throughout the experiments, resulting in repeated measurements across time for each mouse. Linear mixed effects regression models were used to estimate and compare the group-specific tumor growth curves. In the growth curve analyses, statistically significant global tests of equality across groups were followed up with pairwise comparisons to identify specific group differences. All tests were two-sided and carried out at the 5% level of significance. Analyses were performed with the SAS and R statistical software packages.
RESULTS
Infection of mutant K-ras pancreatic cancer cells with AdN17Rac1 increases Rac1 protein, decreases Rac1 activity, and inhibits growth
MIA PaCa-2 cells that express mutant K-ras were transduced with AdN17Rac1 and showed a dose-dependent increase in expression of the N17 Rac1 immunoreactivity at 48 h after the infection with the adenovirus (Figure 1A). To ensure equal proteinloading, actin expression was measured. There is a significant increase in Rac1 immunoreactive protein with 25 MOI of AdN17Rac1 when compared to cells treated with the AdBglII (100 MOI) vector. The maximum increase in Rac1 immunoreactive protein appears to occur at 100 MOI of the AdN17Rac1 vector. In contrast, Rac activity decreased with increasing N17 Rac1 immunoreactivity (Figure 1B). The Rac-GTP binding assay demonstrated that rac activity decreased with increasing viral titer. The maximum decrease in rac activity was seen with 100 MOI of the AdN17Rac1 vector. The increase in Rac1 immunoreactivity and decrease in rac activity inhibited cell growth. Figure 1C demonstrates significant inhibition of cell growth after infection of MIA PaCa-2 cells with 25, 50, and 100 MOI of the AdN17Rac1 vector. At day 6, cell number decreased by 73% with the 100 MOI AdN17 Rac1compared with AdBglII 100 MOI. In addition, cell doubling time increased significantly. Cell doubling time in controls and cells infected with the AdBglII vector were 22 ± 1 and 22 ± 1 hour, respectively. Infection with the AdN17Rac1 vector significantly increased doubling time to 25 ± 1* with 25 MOI, 27 ± 2* with 50 MOI and 32 ± 2* hours with 100 MOI (n =3, *P < 0.05 vs. AdBglII, Means ± SEM).
Figure 1.
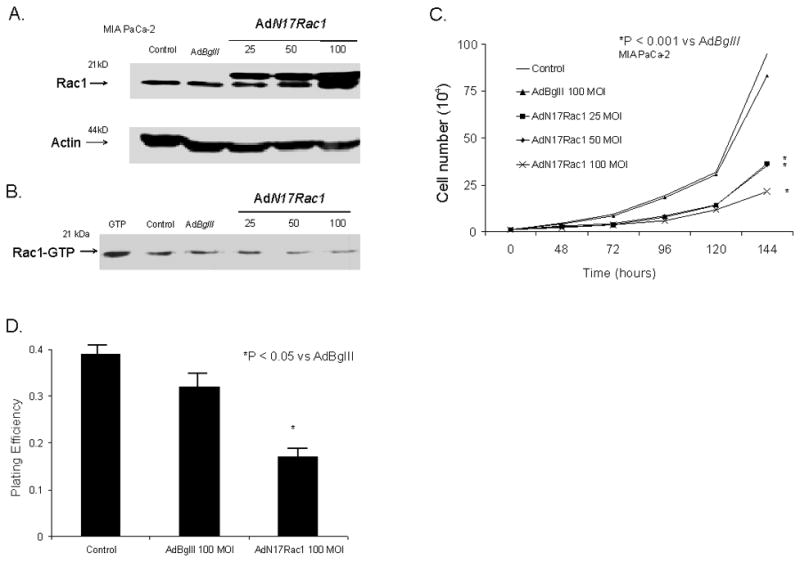
AdRac1N17 infection increased Rac1 protein levels, decreased Rac activity, and inhibited growth in MIAPaCa-2 cells. A. Cells transduced with 0-100 MOI AdRac1N17 demonstrated increases in Rac1 immunoreactivity with increasing viral titer. No difference was seen with AdBglII transfection compared with parental cells. For total Rac1 measurement, 30 μg cellular protein per well was used for western. All of the western blots were repeated to confirm the findings.
B. Representative western blot showing dose-dependent decreases in Rac1 activation in AdRac1N17-transduced MIAPaCa-2 cells. AdBglII served as a control. Cell lysates were immunoprecipitated with PAK-1 PBD-agarose. Associated GTP-bound Rac1 was determined by immunoblotting with anti-Rac1 antibody. 10 nM GTP was added to untreated cells as the positive control.
C. Overexpression of AdRac1N17 delayed MIAPaCa-2 cell growth. MIAPaCa-2 cells transduced with 0 100MOI AdRac1N17 or 100 MOI AdBglII demonstrated reductions in cell growth with AdRac1N17100 MOI. No significant changes were seen with AdBglII transduction compared with parental cells. Mean in vitro cell growth of control, AdRac1N17-or AdBglII-transduced MIAPaCa-2 cells are shown. Each point represents the mean values, n= 3. *P < 0.05 vs. 100 MOI AdBglII.
D. Plating efficiency was decreased with increased AdRac1N17 transduction in MIAPaCa-2 cells. MIAPaCa-2 cells were seeded at 5 × 105 cells / plate overnight until attached, transduced by AdRac1N17 or AdBglII for 24 hours, recovered in full media without adenovirus for another 24 hours, and were then trypsinized and plated for clonogenic survival. AdRac1N17 transduced cells decreased plating efficiency at 100 MOI. No significant changes were seen with AdBglII transfer compared with parental cells. Each point represents the mean values, n = 3. *P < 0.05 vs.100 MOI AdBglII.
Next, we tested the ability of the AdN17Rac1-transduced MIA PaCa-2 cells to form colonies. Plating efficiency also decreased with infection of the AdN17Rac1 vector. Figure 1D. Plating efficiency decreased by 47% with infection of 100 MOI of the AdN17Rac1 vector compared to infection with 100 MOI AdBglII (n =3, P < 0.05 vs. AdBglII, Means ± SEM). Thus, infection of mutant K-ras pancreatic cancer cells with a dominant/negative rac construct increased rac1 immunoreactive protein, decreased rac activity and inhibited in vitro cell growth characteristics.
Infection of wild-type K-ras pancreatic cancer cells with AdN17Rac1 increases immunoreactive protein but does not inhibit rac activity and does not affect growth
BxPC-3 cells that express wild-type K-ras were transduced with AdN17Rac1 and showed a dose-dependent increase in expression of the N17 Rac1 immunoreactivity at 48 h after the infection with the adenovirus (Figure 2A). There was a significant increase in Rac1 immunoreactive protein with 25 MOI of AdN17Rac1 when compared to cells treated with the AdBglII (100 MOI) vector. The maximum increase in Rac1 immunoreactive protein appeared to occur at 100 MOI of the AdN17Rac1 vector. Unlike the MIA PaCa-2 cells that express mutant K-ras, the increase in Rac1 immunoreactivity did not affect cell growth in the BxPC-3 cell line. Figure 2B demonstrates no changes in cell growth after infection of BxPC-3 cells with 25, 50, and 100 MOI of the AdN17Rac1 vector. In addition, cell doubling time did not change. Cell doubling time in controls and cells infected with the AdBglII vector were 52 ± 1 and 52 ± 2 hours, respectively. Infection with the AdN17Rac1 vector changed little with doubling times of 53 ± 1 with 25 MOI, 53 ± 2 with 50 MOI and 53 ± 2 with 100 MOI (n = 3, P > 0.05 vs. AdBglII, Means ± SEM).
Figure 2.
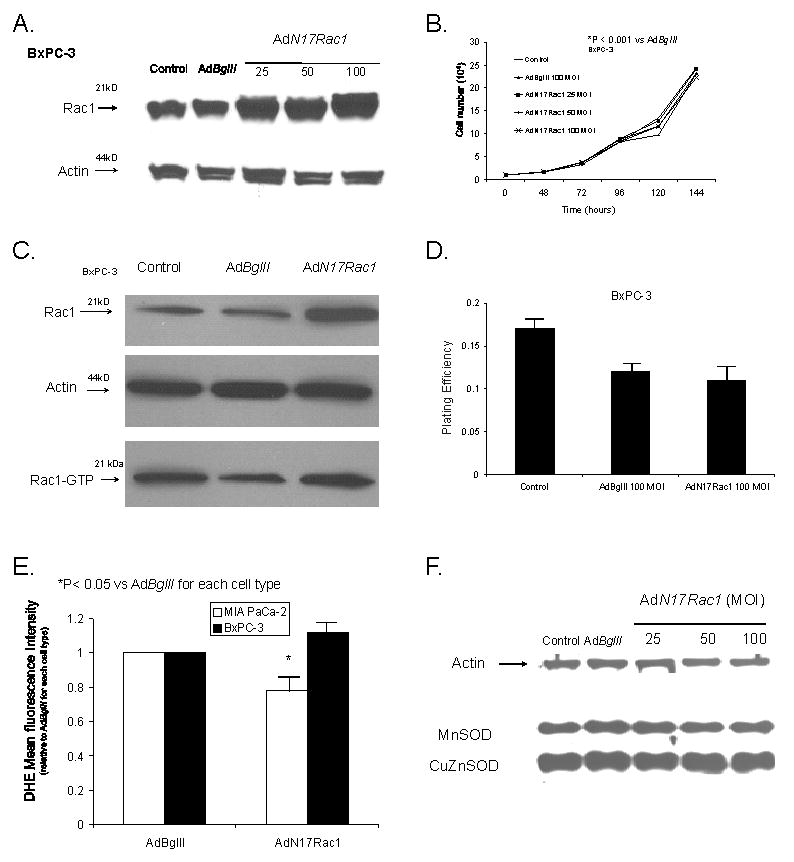
AdRac1N17 infection increased Rac1 protein levels but had no effect on growth in BxPC-3 cells. A. Cells transduced with 0–100 MOI AdRac1N17 demonstrated increases in Rac1 immunoreactivity with increasing viral titer. No difference was seen with AdBglII transfer compared with parental cells.
B. Overexpression of AdRac1N17 had no effect on BxPC-3 cell growth. BxPC-3 cells transduced with 0 100MOI AdRac1N17 or 100 MOI AdBglII demonstrated no changes in cell growth with AdRac1N17100 MOI. No significant changes were seen with AdBglII transduction compared with parental cells. Mean in vitro cell growth of control, AdRac1N17-or AdBglII-transduced BxPC-3 cells are shown. Each point represents the mean values, n= 3. *P < 0.05 vs. 100 MOI AdBglII.
C. Infection with AdRac1N1 increased Rac1 immunoreactivity but did not decrease Rac1 activity. Representative western blot showing increases in Rac1 protein but no change in Rac1 activation in AdRac1N17-transduced BxPC-3 cells.
D. Plating efficiency was unchanged with AdRac1N17 infection in BxPC-3 cells. BxPC-3 cells were infected with AdRac1N17 or AdBglII and then plated for clonogenic survival. AdRac1N17 infected cells did not change plating efficiency at 100 MOI when compared to AdBglII infected cells. Each point represents the mean values, n = 3.
E. Superoxide levels decreased in MIA PaCa-2 cells after infection with the AdN17Rac1 construct without changes in antioxidant proteins that scavenge superoxide. Intracellular hydroethidine fluorescence decreased in MIA PaCa-2 cells but not BxPC-3 cells infected with AdN17Rac1. Intracellular superoxide levels as measured by DHE decreasedsignificantly 48 hours after infection with AdN17Rac1 100 MOI compared to MIA PaCa-2 pancreatic carcinoma cells infected with the AdBglII vector. *p < 0.05 vs AdBglII, means ± SEM, n=3.
F. Overexpression of AdN17Rac1 did not change immunoreactive protein for MnSOD or CuZnSOD in MIA PaCa-2 cells. MIA PaCa-2 cells were transfected with 100 multiplicity of infection (MOI) of AdBglII as a negative control or 0 to 100 MOI of AdN17Rac1. After 48 hr of transfection, total cell lysate was prepared and immunoblotted for the proteins with cellular actin as a loading control. Total protein was electrophoresed in a 12.5% SDS-polyacrylamide running gel and a 5% stacking gel. After blocking in 20% fetal bovine serum for 1 hr, the sheets were washed and then treated with antibodies to MnSOD and CuZnSOD.
Next, we tested the ability of the AdN17Rac1-transduced BxPC-3 cells to form colonies. Once again, infection with the AdN17Rac1 vector increased immunoreactive protein (Figure 2C). However, unlike the MIA PaCa-2 cells, the BxPC-3 cells infected with AdN17Rac1 100 MOI had little change in Rac1 activity (Figure 2C). There were no significant changes in plating efficiency with infection of the AdN17Rac1 vector (Figure 2D) when compared to infection with 100 MOI AdBglII. Thus, infection of wild-type K-ras pancreatic cancer cells with a dominant/negative rac construct increased rac1 immunoreactive protein, but did not change rac activity or in vitro cell growth characteristics.
Superoxide levels decreased in MIA PaCa-2 cells after infection with the AdN17Rac1 construct without changes in antioxidant proteins that scavenge superoxide
One of the major functions of Rac1 includes controlling the activity of NADPH oxidase to mediate superoxide production (14). Intracellular levels of superoxide were detected following infection of the MIA PaCa-2 and BxPC-3 cell lines with AdN17Rac1. As a positive control, we also used AdMnSOD and DPI, a non-specific inhibitor of flavin oxidases like NADPH oxidase. AdN17 Rac1 (100 MOI) decreased hydroethidine fluorescence by 18% ± 2 in MIA PaCa-2 cells when compared to the same cells treated with the AdBglII vector (100 MOI) (Figure 2E). In addition, treatment of cells with AdMnSOD (100 MOI) also decreased hydroethidine fluorescence 14% ± 5, while DPI 15 μM decreased hydroethidine fluorescence by 32% ± 6 (data not shown, means ± SEM, n = 3, P < 0.05 vs AdBglII). Hydroethidine fluorescence did not change significantly when BxPC-3 cells were infected with AdN17Rac1 100 MOI when compared to the same cells infected with the AdBglII vector (Figure 2E). Next we wanted to determine if the change in hydroethidine fluorescence was mediated by Rac1 inhibition, and not increases in either CuZnSOD or MnSOD, the two superoxide dismutase enzymes that scavenge superoxide. Western blots for MnSOD and CuZnSOD demonstrated no change in immunoreactive protein in MIA PaCa-2 cells infected with the AdN17Rac1 construct (25–100 MOI) when compared to the parental cell line or cells infected with the AdBglII vector (Figure 2F).
Infection of immortalized H6c7 cell line with AdN17Rac1 has no effect on growth, while infection of the isogenic cell line that expresses K-ras with AdN17Rac1 increases Rac1 protein and inhibits growth
To further characterize the mechanism of Rac1 activity in the growth of pancreatic cancer, we used an immortalized pancreatic ductal epithelial cell line with near normal genotype and phenotype of pancreatic duct epithelial cells HPV16-E6E7 (H6c7), and the isogenic cell line that expresses K-rasG12V and forms tumors H6c7eR-KrasT (KrasT) (21). Infection of these cell lines with the AdN17Rac vector (100 MOI) demonstrated increased immunoreactive protein when compared to the parental cell lines and cells infected with the AdEmpty vector (100 MOI) (Figure 3A). We then tested the ability of these cells to form colonies after infection with the AdN17Rac1 vector. In the H6c7 cell lines, infection with AdN17Rac1 100 MOI did not change plating efficiency when compared to infection with 100 MOI AdEmpty (means ± SEM, n = 3, P > 0.05 vs. AdEmpty). (Figure 3B). However, infection with 100 MOI AdN17Rac1 significantly decreased plating efficiency in the KrasT cells (means ± SEM, n = 3, P < 0.05 vs. AdEmpty). (Figure 3B). Thus, infection of immortalized pancreatic ductal epithelial cells with a dominant/negative rac construct increased rac1 immunoreactive protein, but did not significantly change in vitro cell growth characteristics. However, infection of the isogeneic cell line that expresses K-ras with a dominant/negative rac construct also increased rac1 protein and inhibited cell growth.
Figure 3.
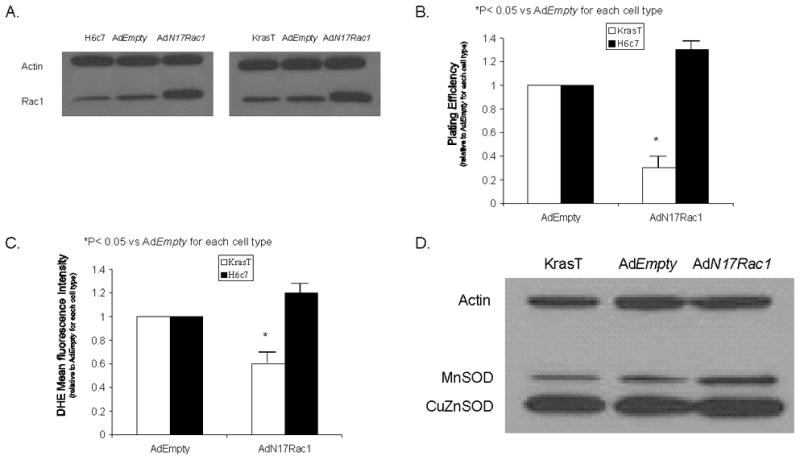
AdRac1N17 infection increases Rac1 protein levels, inhibits growth, and decreases superoxide levels in tumorigenic pancreatic ductal epithelial cells that express K-ras.
A. Cells transduced with 100 MOI AdRac1N17 demonstrated increases in Rac1 immunoreactivity with increasing viral titer. No difference was seen with AdEmpty 100 MOI transfection compared with parental cells.
B. Plating efficiency was unchanged with AdRac1N17 infection in H6c7 cells but decreased in KrasT cells. Feeder cells were used by irradiating B1 mouse fibroblasts Twenty-four hours before clonogenic assay, irradiated cells were thawed and diluted in keratinocyte serum free media. Cells were then seeded 3 × 105/ plate overnight until attached, transduced by AdRac1N17 or AdEmpty for 24 hours, recovered in full media without adenovirus for another 24 hours, and then trypsinized and plated for clonogenic survival. No significant changes were seen plating efficiency in H6c7 cells infected with the AdRac1N17mpty vector. However, KrasT cells infected with AdRac1N17 decreased plating efficiency. Each point represents the mean values, n = 3. *P < 0.05 vs.100 MOI AdEmpty.
C. Superoxide levels decreased in KrasT cells after infection with the AdN17Rac1 construct. Intracellular hydroethidine fluorescence decreased in KrasT cells but not in H6c7 cells infected with AdN17Rac1. Intracellular superoxide levels as measured by DHE decreased significantly 48 hours after infection with AdN17Rac1 100 MOI compared to KrasT cells infected with the AdEmpty vector. *p < 0.05 vs Adempty, means ± SEM, n=3.
D. Overexpression of AdN17Rac1 did not change immunoreactive protein for MnSOD or CuZnSOD in KrasT cells. KrasT cells were transfected with 100 multiplicity of infection (MOI) of AdEmpty as a negative control or 100 MOI of AdN17Rac1. After 48 hr of transfection, total cell lysate was prepared and immunoblotted for MnSOD and CuZnSOD.
Next we also measured hydroethidine fluorescence in the H6c7 and KrasT cells. Forty-eight hours after infection with AdEmpty or AdN17Rac1 vectors, H6c7 and KrasT cells were incubatedfor 45 minutes with hydroethidine (10 μM) and the intensity of fluorescence was measured using flow cytometry. AdN17 Rac1 (100 MOI) decreased hydroethidine fluorescence by 40% ± 6 in KrasT cells when compared to the same cells treated with the AdEmpty vector (100 MOI) (Figure 3C). In contrast, hydroethidine fluorescence did not change significantly when H6c7 cells were infected with AdN17 Rac1 100 MOI when compared to the same cells infected with the AdEmpty vector (Figure 3C). As seen in the other pancreatic cancer cell lines, Western blots for MnSOD and CuZnSOD demonstrated no change in immunoreactive protein in the KrasT cells infected with the AdN17Rac1 construct (100 MOI) when compared to the parental cell line or cells infected with the AdEmpty vector (Figure 3D).
AdN17Rac1 decreases in vivo tumor growth
To determine if AdN17Rac1 could reduce tumor growth in vivo, the AdN17Rac1 or AdEmpty constructs were delivered to pre-established tumors (3– 4 mm) on days 1, 7, and 14 for a total of 3 injections. On day 1 when the adenoviral injections were started the mean tumor volume in the controls was 47 ± 10, 76 ± 25 in the AdEmpty group and 47 ± 9 in the AdN17 Rac1 group (Means ± SEM, P > 0.05 between groups). When the AdN17Rac1 construct was given, a slower growth in tumor was observed in comparison to the control group as well as the AdEmpty injected group as seen in the estimated growth curves in Figure 4. For example, on Day 17, the control and the AdEmpty group had mean tumor volumes of 745 mm3 and 582 mm3, respectively, while the AdN17Rac1 group had a mean tumor volume of 47 mm3 (P = 0.001 vs AdEmpty, n = 7 – 8 mice/group).
Figure 4.
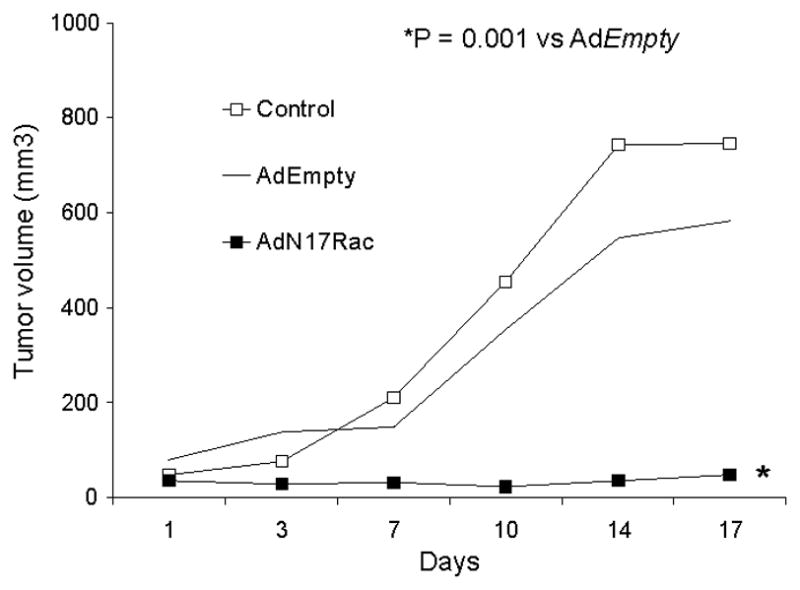
AdN17Rac1 injections decreased MIA PaCa-2 tumor growth in nude mice. The AdN17Rac1group had significantly slower tumor growth when compared to the control and AdEmpty, (p < 0.001, n = 7–8/group). MIA PaCa-2 tumor cells (3 × 106) were delivered subcutaneously into the flank region of nude mice. Controls received serum-free media in similar volumes. 1 × 109 PFUs of the AdN17Rac1 or AdEmpty constructs were delivered to the tumor on day 1, 7, and 14 of the experiment. On day 17 there was a 10-fold decrease in tumor growth in animals receiving the AdN17Rac1 vector when compared to treatment with the AdEmpty control vector.
DISCUSSION
Reactive oxygen species (ROS) like superoxide have been linked to pancreatic cancer. Superoxide is generated during normal aerobic metabolism and increased levels of these species are produced during various forms of oxidative stress. The net intracellular concentration of superoxide is the result of the production and the ability of antioxidants to remove it. MacMillan-Crow and colleagues have demonstrated 21- to 97-fold increases in levels of nitrotyrosine, a footprint of the reactive nitrogen species peroxynitrite (formed by the reaction of superoxide and nitric oxide), in pancreatic cancer specimens compared to normal pancreas (25). Findings from our laboratory add to MacMillan-Crowe’s studies by demonstrating that pancreatic cancer cell lines have increased intracellular superoxide levels compared to other cell lines studied as measured by hydroethidine fluorescence (26). One reason why pancreatic cancer is so aggressive and unresponsive to treatments is its resistance to apoptosis. Vaquero and colleagues have recently demonstrated that ROS are prosurvival, antiapoptotic factors in pancreatic cancer (27). Their study showed that growth factors stimulate ROS generation by activation of membrane nonmitochondrial NAD(P)H oxidase and that inhibiting ROS by different approaches stimulates apoptosis in pancreatic cancer cells. Thus the prosurvival effect of superoxide may be an important mechanism of pancreatic cancer cell resistance to therapy. Our current study demonstrates that Rac1 plays a role in growth of pancreatic adenocarcinoma via a superoxide-mediated mechanism. Previous studies from our laboratory have also implicated superoxide as a mitogenic signaling molecule in pancreatic cancer (24, 28). Scavenging of superoxide with manganese superoxide dismutase (24), copper/zinc superoxide dismutase (28), or extracellular superoxide dismutase (28) all have similar effects in decreasing superoxide levels and inhibiting in vitro growth.
In a lung cancer model of tumorigenesis, K-ras activation combined with Rac1 deletion caused a profound decrease in proliferation (29). As a downstream effector protein of Ras, Rac1 activity is also regulated by Ras. There is growing evidence for Rac1 regulation of ROS in non-phagocytic cells. For example, stimuli that increase Rac1-GTP in gastric epithelial cells increase ROS production(30). Also, mutationally activated Rac1 induces ROS formation (31), and second site mutations showed that Rac1 activation ofROS production correlates with mitogenic stimulation, but not with actin polymerization or JNK activation (31).
Our findings demonstrate that Rac1 may be involved in the activation of NADPH oxidase leading to superoxide generation in some pancreatic cancer cells, it is still unclear about the specific structure of this enzyme complex. Recently, Nox (nonphagocytic NAD(P)H oxidase) proteins, homologous to gp91phox, have been identified in a variety of cell types, including pancreatic cancer cells (32, 33). Vaquero et al., showed that different Nox isoforms are expressed in various pancreatic cancer cells, for example, Nox3 and Nox4 are expressed in MIAPaCa-2 cells, while Nox4 is expressed in Panc-1 cells (27). However, the modulation of Rac1 and the subsequent changes in superoxide and ROS generation through the NADPH oxidase pathway has remained uncharacterized in pancreatic cells.
In summary, in pancreatic cancer cells with mutant K-ras, a dominant/negative rac adenoviral construct decreased rac activity, decreased superoxide levels, and inhibited in vitro growth but had no effect in pancreatic cancer cells with wild-type K-ras.. Additionally, the same construct had no effect on an immortalized pancreatic ductal epithelial cell line, but decreased superoxide levels and inhibited in vitro growth in the isogeneic tumorigenic cell line that expresses K-ras. In addition, the AdN17Rac construct inhibited in vivo tumor growth. These results suggest that activation of Rac1-dependent superoxide generation leads to pancreatic cancer cell proliferation. In pancreatic cancer inhibition of Rac1 may be a potential therapeutic target.
Supplementary Material
Acknowledgments
Supported by NIH grants CA115785, CA66081, the Susan L. Bader Foundation of Hope, and a Merit Review grant from the Medical Research Service, Department of Veterans Affairs
Footnotes
Conflict of Interest
There are no financial or other interests with regard to the submitted manuscript that might be construed as a conflict of interest.
References
- 1.Uemura T, Hibi K, Kaneko T, Takeda S, Inoue S, Okochi O, et al. Detection of K-ras mutations in the plasma DNA of pancreatic cancer patients. J Gastroenterol. 2004;39:56–60. doi: 10.1007/s00535-003-1245-1. [DOI] [PubMed] [Google Scholar]
- 2.Maisonneuve P, Lowenfels AB. Chronic pancreatitis and pancreatic cancer. Dig Dis. 2002;20:32–37. doi: 10.1159/000063165. [DOI] [PubMed] [Google Scholar]
- 3.Matallanas D, Arozarena I, Berciano MT, Aaronson DS, Pellicer A, Lafarga M, et al. Differences on the inhibitory specificities of H-Ras, K-Ras, and N-Ras (N17) dominant negative mutants are related to their membrane microlocalization. J Biol Chem. 2003;278:4572–4581. doi: 10.1074/jbc.M209807200. [DOI] [PubMed] [Google Scholar]
- 4.Milasincic DJ, Calera MR, Farmer SR, Pilch PF. Stimulation of C2C12 myoblast growth by basic fibroblast growth factor and insulin-like growth factor 1 can occur via mitogen activated protein kinase-dependent and -independent pathways. Mol Cell Biol. 1996;16:5964–5973. doi: 10.1128/mcb.16.11.5964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Crnogorac-Jurcevic T, Efthimiou E, Capelli P, Blaveri E, Baron A, Terris B, et al. Gene expression profiles of pancreatic cancer and stromal desmoplasia. Oncogene. 2001;20:7437–7446. doi: 10.1038/sj.onc.1204935. [DOI] [PubMed] [Google Scholar]
- 6.Bokoch GM, Diebold BA. Curent molecular models for NADPH oxidase regulation by Rac GTPase. Blood. 2002;100:2692–2696. doi: 10.1182/blood-2002-04-1149. [DOI] [PubMed] [Google Scholar]
- 7.Didsbury J, Weber RF, Bokoch GM, Evans T, Snyderman R. Rac, a novel ras-related family of proteins that are botulinum toxin substrates. J Biol Chem. 1989;264:16378–16382. [PubMed] [Google Scholar]
- 8.Chan AY, Coniglio SJ, Chuang YY, Michaelson D, Knaus UG, Philips MR, et al. Roles of the Rac1 and Rac3 GTPases in human tumor cell invasion. Oncogene. 2005;24:7821–7829. doi: 10.1038/sj.onc.1208909. [DOI] [PubMed] [Google Scholar]
- 9.Gregg D, Rauscher FM, Goldschmidt-Clermont PJ. Rac regulates cardiovascular superoxide through diverse molecular interactions: more than a binary GTP switch. Am J Physiol, Cell Physiol. 2003;285:723–734. doi: 10.1152/ajpcell.00230.2003. [DOI] [PubMed] [Google Scholar]
- 10.Sahai E, Marshall CJ. RHO-GTPases and cancer. Nat Rev Cancer. 2002;2:133–142. doi: 10.1038/nrc725. [DOI] [PubMed] [Google Scholar]
- 11.Hordijk PL. Regulation of NADPH oxidases: the role of Rac proteins. Circ Res. 2006;98:453–462. doi: 10.1161/01.RES.0000204727.46710.5e. [DOI] [PubMed] [Google Scholar]
- 12.Irani K, Xia Y, Zweier JL, Sollott SJ, Der CJ, Fearon ER, et al. Mitogenic signaling mediated by oxidants in Ras-transformed fibroblasts. Science. 1997;275:1649–1652. doi: 10.1126/science.275.5306.1649. [DOI] [PubMed] [Google Scholar]
- 13.Sundaresan M, Yu ZX, Ferrans VJ, Sulciner DJ, Gutkind JS, Irani K, et al. Regulation of reactive-oxygen-species generation in fibroblasts by Rac1. Biochem J. 1996;318:379–382. doi: 10.1042/bj3180379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Moldovan L, Irani K, Moldovan NI, Finkel T, Goldschmidt-Clermont PJ. The actin cytoskeleton reorganization induced by Rac1 requires the production of superoxide. Antioxid Redox Signal. 1999;1:29–43. doi: 10.1089/ars.1999.1.1-29. [DOI] [PubMed] [Google Scholar]
- 15.Wetering S, Buul JD, Quik S, Mul FP, Anthony EC, Klooster JP, et al. Reactive oxygen species mediate Rac-induced loss of cell–cell adhesion in primary human endothelial cells. J Cell Sci. 2002;115:1837–1846. doi: 10.1242/jcs.115.9.1837. [DOI] [PubMed] [Google Scholar]
- 16.Sulciner DJ, Irani K, Yu ZX, Ferrans VJ, Goldschmidt-Clermont P, Finkel T. Rac1 regulates a cytokine-stimulated, redox-dependent pathway necessary for NF-kappaB activation. Mol Cell Biol. 1996;16:7115–7121. doi: 10.1128/mcb.16.12.7115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zimmerman MC, Dunlay RP, Lazartigues E, Zhang Y, Sharma RV, Engelhardt JF, et al. Requirement for Rac1-dependent NADPH oxidase in the cardiovascular and dipsogenic actions of angiotensin II in the brain. Circ Res. 2004;95:532–539. doi: 10.1161/01.RES.0000139957.22530.b9. [DOI] [PubMed] [Google Scholar]
- 18.Yeh LH, Park YJ, Hansalia RJ, Ahmed IS, Deshpande SS, Goldschmidt-Clermont PJ, et al. Shear-induced tyrosine phosphorylation in endothelial cells requires Rac-1-dependent production of ROS. Am J Physiol. 1999;276:C838–C847. doi: 10.1152/ajpcell.1999.276.4.C838. [DOI] [PubMed] [Google Scholar]
- 19.Deem TL, Cook-Mills JM. Vascular cell adhesion molecule 1 (VCAM-1) activation of endothelial cell matrix metalloproteinases: role of reactive oxygen species. Blood. 2004;104:2385–2393. doi: 10.1182/blood-2004-02-0665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Binker MG, Binker-Cosen AA, Richards D, Oliver B, Cosen-Binker LI. EGF promotes invasion by PANC-1 cells through Rac1/ROS-dependent secretion and activation of MMP-2. Biochem Biophys Res Commun. 2009;379:445–450. doi: 10.1016/j.bbrc.2008.12.080. [DOI] [PubMed] [Google Scholar]
- 21.Qian J, Niu J, Li M, Chiao PJ, Tsao MS. In vitro modeling of human pancreatic duct epithelial cell transformation defines gene expression changes induced by K-ras oncogenic activation in pancreatic carcinogenesis. Cancer Res. 2005;65:5045–5053. doi: 10.1158/0008-5472.CAN-04-3208. [DOI] [PubMed] [Google Scholar]
- 22.Lewis A, Ough M, Du J, Tsao MS, Oberley LW, Cullen JJ. Targeting NAD(P)H:quinone oxidoreductase (NQO1) in pancreatic cancer. Molecular Carcinogenesis. 2005;43:215–224. doi: 10.1002/mc.20107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jessop NW, Hay RJ. Preparation, preservation, recovery and use of irradiated feeder layers in cell culture research. TCA Manual. 1979;5:1137–1139. [Google Scholar]
- 24.Wydert C, Roling B, Liu J, Ritchie JM, Oberley LW, Cullen JJ. Suppression of the malignant phenotype in human pancreatic cancer cells by the overexpression of manganese superoxide dismutase. Mol Cancer Ther. 2003;2:361–369. [PubMed] [Google Scholar]
- 25.MacMillan-Crow LA, Greendorfer JS, Vickers SM, Thompson JA. Tyrosine nitration of c-SRC tyrosine kinase in human pancreatic ductal adenocarcinoma. Arch Biochem Biophys. 2000;377:350–356. doi: 10.1006/abbi.2000.1799. [DOI] [PubMed] [Google Scholar]
- 26.Grady M, Gaut AW, Liu J, Zhang YP, Zhang HJ, Weydert C, et al. K-ras mutations in pancreatic cancer. Free Radic Biol Med. 2002;33:S440. [Google Scholar]
- 27.Vaquero EC, Edderkaoui M, Pandol SJ, Gukovsky I, Gukovskaya AS. Reactive oxygen species produced by NAD(P)H oxidase inhibit apoptosis in pancreatic cancer cells. J Biol Chem. 2004;279:34643–34654. doi: 10.1074/jbc.M400078200. [DOI] [PubMed] [Google Scholar]
- 28.Teoh MLT, Sun W, Smith BJ, Oberley LW, Cullen JJ. Modulation of reactive oxygen species (ROS) in pancreatic cancer: Insight into tumor growth suppression by the superoxide dismutases. Clin Cancer Res. 2007;13:7441–7450. doi: 10.1158/1078-0432.CCR-07-0851. [DOI] [PubMed] [Google Scholar]
- 29.Kissil JL, Walmsley MJ, Hanlon L, Haigis KM, Kim CF, Sweet-Cordero A, et al. Requirement for Rac1 in a K-ras-induced lung cancer in a mouse. Cancer Res. 2007;67:8089–8094. doi: 10.1158/0008-5472.CAN-07-2300. [DOI] [PubMed] [Google Scholar]
- 30.Kawahara T, Kiohjima M, Kuwano Y, Mino H, Teshima-Kondo S, Takeya R, et al. Helicobacter pylori lipopolysaccharide activates Rac1 and transcription of NADPH oxidase Nox1 and its organizer NOXO1 in guinea pig gastric mucosal cells. Am J Physiol. 2005;288:C450–C457. doi: 10.1152/ajpcell.00319.2004. [DOI] [PubMed] [Google Scholar]
- 31.Joneson T, Bar-Sagi D. A Rac1 effector site controlling mitogenesis through superoxide production. J Biol Chem. 1998;273:17991–17994. doi: 10.1074/jbc.273.29.17991. [DOI] [PubMed] [Google Scholar]
- 32.Wang S, Pogue R, Morre DM, Morre DJ. NADH oxidase activity (NOX) and enlargement of HeLa cells oscillate with two different temperature-compensated period lengths of 22 and 24 minutes corresponding to different NOX forms. Biochim Biophys Acta. 2001;1539:192–204. doi: 10.1016/s0167-4889(01)00107-0. [DOI] [PubMed] [Google Scholar]
- 33.Lim SD, Sun C, Lambeth JD, Marshall F, Amin M, Chung L, et al. Increased Nox1 and hydrogen peroxide in prostate cancer. Prostate. 2005;62:200–207. doi: 10.1002/pros.20137. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.