

Abstract
The DExH-box NTPase/helicase Prp22p plays two important roles in pre-mRNA splicing. It promotes the second transesterification reaction and then catalyzes the ATP-dependent release of mature mRNA from the spliceosome. Evidence that helicase activity is important emerged from the analysis of Prp22p motif III (SAT) mutations that uncouple the NTPase and helicase activities. We find that S635A and T637A hydrolyse ATP, but are defective in unwinding duplex RNA and releasing mRNA from the spliceosome. The S635A mutation is lethal in vivo at ≤30°C and results in slow growth at 34–37°C. Further insights into helicase action during splicing were gleaned by isolating and characterizing intragenic suppressors of prp22-S635A. Biochemical analysis of the S27 suppressor protein showed that a second mutation of Val539 to Ile in motif Ia revived the helicase activity of the S635A mutant together with the ability to catalyze mRNA release. These findings underscore the tight correlation of RNA unwinding and spliceosome disassembly and demonstrate how suppressor analysis can be used to dissect the subtle internal domain dynamics of helicase action.
Keywords: DExH-box helicases/pre-mRNA splicing/Prp22/spliceosome disassembly
Introduction
Pre-mRNA splicing is catalyzed by the spliceosome, a large ribonucleoprotein complex composed of snRNPs (U1, U2, U5, U4/6 snRNAs plus associated proteins) and non-snRNP protein factors (Guthrie, 1991; Moore et al., 1993; Will and Lührmann, 1997). Splicing entails two sequential transesterification steps in which (i) the 2′-OH at the intron branch point attacks the phosphodiester at the 5′ exon–intron border to form a lariat-intermediate plus the 5′ exon and (ii) the 3′-OH of the 5′ exon then attacks the phosphodiester at the intron–3′ exon junction to form mature mRNA and the excised lariat-intron. The spliceosome components assemble onto the pre-mRNA in an ordered fashion, thereby ensuring accurate recognition of the reactive nucleotides. Conformational changes in the spliceosome involving the formation and remodeling of RNA–RNA and RNA–protein contacts compel and coordinate the chemical steps, the release of the RNA products and the disassembly of the spliceosome complex (Madhani and Guthrie, 1994; Nilsen, 1994). These conformational changes are driven by the energy of NTP hydrolysis catalyzed by proteins that act serially at discrete stages of the splicing pathway (Staley and Guthrie, 1998; Burge et al., 1999).
Among such factors are the non-snRNP splicing factors Prp2p, Prp16p, Prp22p and Prp43p, which are members of the DEAH-box family of nucleic acid-dependent NTPases (Burgess et al., 1990; Chen and Lin, 1990; Company et al., 1991; Arenas and Abelson, 1997). The DExH family is defined by a set of conserved peptide motifs that are required for NTP phosphohydrolase activity (Gorbalenya et al., 1989). The NTPase activities of Prp2p, Prp16p and Prp22p are essential for yeast cell growth (Schwer and Gross, 1998; Edwalds-Gilbert et al., 2000; H.R.Hotz and B.Schwer, unpublished). Prp22p and Prp16p also possess an intrinsic RNA helicase activity, whereby the purified enzymes unwind duplex RNAs in vitro in a reaction requiring NTP hydrolysis (Schwer and Gross, 1998; Wagner et al., 1998; Wang et al., 1998). It is widely presumed that helicases such as Prp22p and Prp16p use the energy of NTP hydrolysis to disrupt or rearrange RNA secondary structures within the spliceosome. Here we subject this hypothesis to a critical test by asking whether the RNA unwinding activity of Prp22p is required for its function in pre-mRNA splicing in vivo and in vitro.
Prior studies showed that yeast Prp22p acts at two stages during pre-mRNA splicing; it is needed for the second transesterification reaction and for the release of mature mRNA during spliceosome disassembly. Splicing extracts depleted of Prp22p accumulate 5′ exon and lariat-intermediate, and are arrested prior to the second chemical step (Schwer and Gross, 1998). Step 1-arrested spliceosomes are complemented for step 2 by adding back purified Prp22p. Remarkably, Prp22p action in promoting step 2 is independent of its ATPase activity. For example, mutant proteins Prp22-K512Ap and Prp22-K512Ep, in which the conserved Lys in motif I (GKT) is replaced by either Ala or Gln, are defective for ATP hydrolysis, but are nonetheless capable of complementing step 2 chemistry in vitro. In contrast, ATP hydrolysis is essential for Prp22p action in releasing the mature mRNA product from the spliceosome after step 2 is complete (Schwer and Gross, 1998; Wagner et al., 1998). The lethality of the prp22-K512A allele implies that Prp22p catalysis of NTP-dependent spliceosome disassembly is essential in vivo (Schwer and Gross, 1998).
Here we show that the NTP hydrolysis and helicase activities of Prp22p are not obligately coupled. We identify and characterize two Prp22p mutants, S635A and T637A, which hydrolyse ATP but do not unwind double-stranded (ds) RNA. S635A and T637A complement step 2 transesterification in vitro, but are defective in catalyzing mRNA release from the spliceosome. To our knowledge, this is the first evidence that helicase activity is essential for any stage of pre-mRNA splicing. The S635A mutation in motif III (SAT) elicits conditional lethality at ≤30°C, yet prp22-S635A cells grow at 37°C, suggesting that thermal energy may compensate in vivo to weaken macromolecular contacts within the spliceosome that are normally resolved by the Prp22p helicase.
The internal dynamics of helicase action were explored by isolating and characterizing intragenic suppressors of prp22-S635A. A pseudorevertant within motif III (AAT→TAT) and single amino acid substitutions at 11 different positions outside motif III restored growth at 30°C. Ten of the suppressor mutations mapped within or between the conserved motifs of the DExH family. Biochemical characterization of one of the suppressors showed that a rather subtle second mutation of Val539 to Ile restored helicase activity to the S635A protein, together with the ability to catalyze mRNA release from the spliceosome at 30°C. These experiments underscore the critical role of domain dynamics in coupling chemical energy to RNA unwinding by DExH family members.
Results
Conserved residues in motifs II, III and VI are important for Prp22p function in vivo
Members of the DExH family of nucleic acid-dependent NTPases contain six conserved motifs arrayed in a collinear fashion (Gorbalenya et al., 1989; de la Cruz et al., 1999; Jankowsky and Jankowsky, 2000). Extensive mutational analyses of the virus-encoded DExH-box proteins NPH-I, NPH-II and NS3 have established that conserved amino acid side chains in motifs I (GKT), II (DExH), III ([S/T]AT) and VI (QRxGRxGR) are essential for NTP hydrolysis or for the coupling of NTP hydrolysis to duplex unwinding (Gross and Shuman, 1995, 1996; Heilek and Peterson, 1997; Kim et al., 1997; Martins et al., 1999). Ala-scanning mutagenesis of the yeast splicing factor Prp16p showed that the motif I Lys, motif II Asp and Glu, and motif VI Arg side chains were essential for Prp16p function in vivo (Hotz and Schwer, 1998). Preliminary mutational analysis of yeast Prp22p established an essential role for Lys512 in motif I in vivo and in spliceosome disassembly in vitro. Here we extended the Ala scan of Prp22p to 11 other conserved positions in motifs II, III and VI (Figure 1).

Fig. 1. Alanine-substitution mutations of PRP22. The sequences of conserved motifs I, II, III and VI of Prp22p are shown with the sites of Ala substitution underlined. The K512A motif I mutation has been described previously (Schwer and Gross, 1998). The PRP22-Ala alleles on TRP1 CEN plasmids were tested by plasmid shuffle for complementation of a prp22Δ strain. Lethal mutations were those that yielded no 5-FOA-resistant colonies at 14, 25, 30 or 37°C. Cold-sensitive mutations were those that yielded colonies on FOA at 37°C, but were growth defective or lethal at lower temperatures.
The PRP22-Ala alleles were cloned into CEN TRP1 vectors under the control of the PRP22 promoter, transformed into a prp22Δ strain containing a CEN PRP22 URA3 plasmid, and then tested for function by plasmid shuffle. Six of the new mutations were lethal, i.e. the TRP1 transformants were unable to form colonies on medium containing 5-fluoroorotic acid (5-FOA) at either 14, 25, 30 or 37°C. The lethal mutations of Prp22p were D603A and E604A in motif II and Q804A, G807A, R808A and R811A in motif VI (Figure 1). Ala substitutions of the equivalent positions of Prp16p were also lethal in vivo (Hotz and Schwer, 1998). Ala mutations at the equivalent positions of NPH-I and NPH-II eliminated the nucleic acid-dependent NTPase activities of those enzymes (Gross and Shuman, 1995, 1996; Martins et al., 1999). Thus, it is likely that loss of ATPase activity is responsible for the lethality of the Prp22p mutations. The purification and biochemical characterization of the lethal Prp22 mutant proteins D603A, E604A, Q804A and R808A will be described elsewhere (S.Schneider and B.Schwer, manuscript in preparation).
Five of the new mutant alleles yielded 5-FOA-resistant colonies when the transformants were plated at 37°C. These mutants (H606A, S635A, T637A, R805A and G810A) all formed colonies on YPD medium at 37°C, but were inviable at 14°C (Figure 2). The severity of the cold-sensitive (cs) growth phenotype differed for each mutation. The most severe effects were elicited by the motif III mutations S635A, which slowed growth at 37 and 34°C and was lethal at 30 and 25°C (Figure 2), and T637A, which slowed growth at 34°C and did not support growth at 25°C (Figure 2). The motif III mutants and the His mutant in the DExH-box were of particular interest in light of studies of NPH-II, NS3 and eIF4A, suggesting that these side chains are responsible for coupling NTP hydrolysis and duplex unwinding (Pause and Sonenberg, 1992; Heilek and Peterson, 1997; Gross and Shuman, 1998).

Fig. 2. Cold-sensitive growth phenotypes of viable PRP22-Ala mutants. Mutants that supported growth of prp22Δ cells on 5-FOA at 37°C were streaked onto YPD agar medium. The plates were photographed after incubation for 4 days at 14°C, 3 days at 25°C or 2 days at 30, 34 and 37°C.
Non-functional Prp22 mutants exert dominant-negative effects in vivo
Wild-type Prp22p and the ATPase/helicase-defective K512A mutant bind equally well to the spliceosome in vitro in the absence of ATP (Schwer and Gross, 1998). This finding raised the question of whether the non-functional and conditional Prp22 mutants described above retain the ability to bind to the spliceosome in vivo and thereby inhibit growth by competing with wild-type Prp22p. To address this issue, PRP22 and 11 of the lethal or cs mutant alleles were placed under the transcriptional control of the GAL1 promoter in CEN TRP1 plasmids and then transformed into PRP22 cells. Trp+ transformants were selected and grown in glucose-containing liquid medium at 30°C. Equivalent numbers of cells were then spotted onto plates containing either glucose or galactose as the carbon source. All of the strains grew readily on glucose when expression of the PRP22 gene on the TRP1 plasmid was repressed (Figure 3, glucose). Galactose induction of the expression of the six lethal mutants (K512A, D603A, E604A, Q804A, G807A, R808A and R811A) resulted in dominant-negative inhibition of growth at all temperatures tested from 14 to 37°C (Figure 3, galactose). Induction of wild-type PRP22 expression had no effect. Galactose-induced expression of cs mutants H606A, S635A, T637A and R805A exerted a dominant-negative effect on growth at 14°C (where they are not functional in vivo in a prp22Δ background), but did not inhibit growth at their permissive temperatures of 34–37°C (Figure 3). Galactose induction of S635A had the most severe conditional dominant-negative effect, completely blocking growth of PRP22 cells at 30°C (Figure 3). The finding that lethal and conditional mutants elicited dominant-negative effects in a PRP22 background argues that the lethal and conditional phenotypes cannot simply be accounted for by failure to produce the mutant polypeptides in vivo. We surmise based on these results that the mutant proteins exerted their dominant-negative effects by binding to the spliceosome and blocking the function of wild-type Prp22p. Northern blot analysis confirmed that pre-mRNA splicing was inhibited when cells were grown transiently in galactose to induce overexpression of the lethal mutants (not shown).

Fig. 3. Dominant-negative phenotypes. Wild-type PRP22 cells were transformed with TRP1 CEN plasmids carrying the indicated PRP22 genes under the transcriptional control of the GAL1 promoter. GAL-PRP22 cells were spotted onto glucose-containing agar and the plate was photographed after incubation for 3 days at 30°C (glucose). Equivalent aliquots of GAL-PRP22 cells were spotted onto galactose-containing agar. The plates were photographed after incubation for 4 days at 30, 34 and 37°C, 5 days at 25°C, or 15 days at 14°C (galactose).
The S635A and T637A mutations in motif III uncouple ATP hydrolysis and RNA unwinding
The motif III S635A and T637A mutants of Prp22p were produced in bacteria as His-tagged fusion proteins and purified from soluble bacterial lysates by Ni–agarose affinity chromatography. Wild-type Prp22p was purified in parallel. The recombinant Prp22 proteins were eluted from the nickel resin with imidazole and then purified further by phosphocellulose chromatography and glycerol gradient sedimentation. The peak glycerol gradient fractions were virtually homogeneous with respect to the 130 kDa Prp22p polypeptide (Figure 4A). Wild-type Prp22p, S635A and T637A were assayed for ATP hydrolysis in the presence of an RNA cofactor. The extent of ATP hydrolysis during a 30 min incubation at 30°C was proportional to the amount of input enzyme (Figure 4B). The specific activities of the S635A mutant (6.4 molecules of ATP hydrolyzed per enzyme per second) and T637A (7.5/s) were slightly higher than that of wild-type Prp22p (5.2/s) (Figure 4B). Thus, it is clear that the motif III Ser and Thr residues are not required for ATP hydrolysis. The ATPase activities of S635A and T637A were stimulated to the same extent by RNA as the wild-type Prp22p (not shown).

Fig. 4. Effects of motif III mutations on ATP hydrolysis. (A) Aliquots (1 µg) of the glycerol gradient preparations of wild-type (WT) Prp22p and the Ala substitution mutants S635A and T637A were analyzed by electrophoresis in an 8% polyacrylamide gel containing 0.1% SDS. Polypeptides were visualized by staining with Coomassie Blue. The asterisk marks the position of the Prp22 proteins. The positions and sizes (in kDa) of marker proteins are indicated on the left. (B) The ATPase activity of WT Prp22p and mutant S635A and T637A was assayed as described in Materials and methods and is plotted as a function of input Prp22p.
Helicase activity was tested using a dsRNA substrate formed by annealing a 107-nucleotide RNA strand to a 103-nucleotide radiolabeled RNA strand to produce a 25 bp duplex with 82- and 78-nucleotide 3′ tails (Figure 5A) (Lee and Hurwitz, 1992). Wild-type Prp22p unwound the dsRNA molecule in the presence of ATP (Figure 5B). The labeled product of the helicase reaction co-migrated during electrophoresis with the single-stranded species released by thermal denaturation of the substrate (Figure 5B, lane ΔT). The extent of RNA unwinding by wild-type Prp22p was proportional to the level of input protein (Figure 5C). The specific activity of the S635A protein was 3.5% of the wild-type activity (Figure 5C). T637A was 6% as active as wild-type Prp22p in unwinding dsRNA (data not shown).

Fig. 5. RNA helicase activity. (A) 3′-tailed RNA duplex helicase substrate. (B) Helicase reaction mixtures contained 25 fmol of the RNA substrate and the indicated amounts of recombinant Prp22p proteins. Prp22p was omitted from a control mixture (lane –). The reaction products were resolved by native PAGE; an autoradiograph of the gel is shown with the positions of the duplex substrate RNA (ds) and the displaced single-stranded product (ss) denoted on the left. An aliquot of the duplex substrate that was heat denatured at 95°C for 5 min and then quenched on ice is analyzed in lane ΔT. (C) The extent of RNA unwinding in (B) is plotted as a function of input Prp22p.
We conclude from these experiments that the NTPase activity of Prp22p is insufficient to elicit strand displacement. Chemical energy must be harnessed or coupled to helicase action and motif III (SAT) plays an essential role in this coupling process.
S635A and T637A promote step 2 transesterification but are defective in catalyzing mRNA release from the spliceosome
We exploited the S635A and T637A mutants to probe the role of ATP-dependent RNA unwinding during pre-mRNA splicing in vitro. Yeast whole-cell extracts depleted of Prp22p were reacted with radiolabeled ACT1 pre-mRNA for 10 min at 23°C. The reaction mixtures were then supplemented with purified recombinant wild-type Prp22p, the S635A mutant or a buffer control and incubation was continued for 15 min at 23 or 30°C. The labeled reaction products were then analyzed by PAGE (Figure 6). The depleted extract efficiently catalyzed the first transesterification reaction, leading to the accumulation of the products of the lariat-intermediate and exon 1 (Figure 6, lane –). Little mature RNA was formed. Supplementation of the extract with wild-type Prp22p reversed the second step arrest and promoted the reaction of 5′ exon with the lariat-intermediate to form the mature spliced mRNA (Figure 6, lane WT). The S635A mutant also restored splicing activity to the depleted extract at 23 and 30°C. Thus, the helicase activity of Prp22p is not required for step 2 transesterification. This result is consistent with the earlier demonstration that ATP hydrolysis by Prp22p is not required for step 2 (Schwer and Gross, 1998).
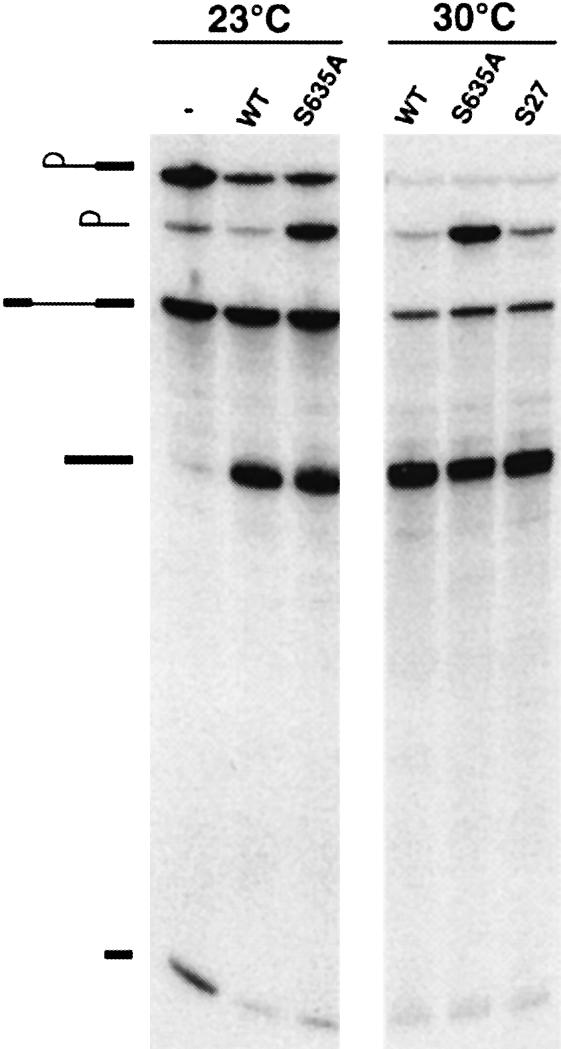
Fig. 6. Splicing in vitro. Step 1 of splicing of radiolabeled ACT1 pre-mRNA was catalyzed by Prp22p-depleted extract during a 10 min reaction at 23°C. Aliquots of the reaction mixture were supplemented with either buffer (–) or recombinant WT Prp22p, mutant S635A, or suppressor mutant S27. Incubation was continued for 15 min at either 23 or 30°C. The reaction products were resolved by denaturing PAGE and visualized by autoradiography. The symbols on the left indicate the positions of the following labeled RNA species, proceeding from the top to bottom of the gel: lariat-intermediate; lariat-intron; pre-mRNA substrate; mature spliced mRNA; 5′ exon.
Higher amounts of excised lariat-intron accumulated in the splicing reactions containing S635A (Figure 6). The lariat-intron is normally debranched and degraded in the yeast extract after being released from the spliceosome but is protected when held within the spliceosome. Thus, the S635A mutant may be compromised in its ability to catalyze mRNA release.
To gauge directly the release of mature RNA from the spliceosome, the splicing reaction mixtures reconstituted with wild-type Prp22p or S635A at 30°C were analyzed by zonal velocity sedimentation through 15–40% glycerol gradients. In splicing reactions containing wild-type Prp22p, the mature ACT1 mRNA sedimented as a discrete peak in fractions 7–13 near the top of the gradient, whereas in the reactions containing S635A, the mature mRNA was retained in the spliceosome and sedimented as a discrete peak in fractions 19–25 near the bottom of the gradient, together with the lariat-intron (Figure 7).

Fig. 7. Motif III mutations affect Prp22p-catalyzed release of spliced mRNA from spliceosomes. Step 1 splicing reactions performed in Prp22p-depleted extract were chased by the addition of recombinant WT Prp22p, mutant S635A, or suppressor mutant S27. The reaction mixtures were analyzed by zonal velocity sedimentation. RNA was isolated from aliquots of the odd-numbered fractions (3–29) of each glycerol gradient, analyzed by denaturing PAGE and visualized by autoradiography. The positions of the lariat-intermediate, lariat-intron, pre-mRNA, and spliced mRNA and are indicated at the left of each gel.
The T637A mutant protein also restored step 2 splicing activity to Prp22-depleted spliceosomes at 23°C, the restrictive temperature for T637A in vivo (data not shown), but the mature mRNAs produced in the in vitro reaction remained associated with the spliceosome, whereas the mRNA produced by reactions containing wild-type Prp22p was completely released (data not shown).
We conclude from these experiments that: (i) the Ser and Thr hydroxyls of motif III are essential for Prp22p-catalyzed release of spliced mRNA from the spliceosome; (ii) ATP hydrolysis by Prp22p does not suffice for mRNA release; and (iii) the RNA helicase activity of Prp22p is required for mRNA release during spliceosome disassembly.
Intragenic missense mutations suppress the cold-sensitive sensitive growth of prp22-S635A
Directional unwinding of duplex nucleic acids by monomeric helicases is proposed to occur via a ratchet-like translocation of protein bound to a single-strand tail of the molecule being unwound. To accomplish this movement, the helicase executes a graceful series of conformational changes coupled to the binding of ATP, hydrolysis to ADP and Pi, and release of ADP (Kim et al., 1998; Velankar et al., 1999). The conformational changes entail back and forth movements of protein domains relative to flexible interdomain linker segments. In the DExH and DEAD-box proteins, motif III is located in a region that connects one domain containing the GKT and DExH elements to a second domain containing motif VI. It is thought that motif III plays a role in transmitting the NTP-dependent conformational switch. Little is known about the interdomain dynamics of duplex unwinding or the nature of the atomic interaction of the motif III amino acids. To explore this issue genetically, we performed a screen for second-site mutations of Prp22p that would suppress the cs growth phenotype caused by the S635A mutation in motif III. The rationale was that such mutations might define other structural elements of Prp22p that either interact with motif III or otherwise facilitate the conformational steps required for helicase activity.
Spontaneous suppressor mutations were selected after inoculation of 80 independent cultures with single colonies of prp22Δ [CEN URA3 prp22-S635A] cells and subsequent growth of the cultures in liquid medium at 30°C. Thirty-three of the cultures showed visible growth after incubation for 3–6 days at 30°C. An aliquot of each was tested for growth on YPD agar at 30°C; 26 of the selected strains formed colonies after 3–7 days. To determine whether the suppressor mutations were intragenic or extragenic, each of the 26 strains was transformed with a CEN TRP1 prp22-S635A plasmid and Trp+ derivatives were plated at 37°C on medium containing 5-FOA to select cells that had lost the original CEN URA3 prp22-S635A plasmid. The 5-FOA survivors were then tested for growth on YPD agar at 30 and 37°C. Eighteen of the strains failed to form colonies at 30°C, indicating that the suppressor mutation was linked to the URA3 prp22-S635A plasmid. We recovered the plasmids from these 18 yeast strains and sequenced the PRP22 gene. In every case, we identified a nucleotide change that resulted in a single amino acid substitution within the S635A polypeptide. The results are summarized in Figure 8. None of the suppressor mutations entailed reversion of Ala635 to Ser. However, one of the suppressor alleles, prp22-S14, was a pseudorevertant, in which Ala635 was changed to Thr. This result highlights the importance of the side-chain hydroxyl in motif III. Indeed, the first position of motif III is often occupied by a Thr in other members of the DExH-box family.

Fig. 8. Intragenic suppressor mutations of S635A. The coding changes of 18 independent intragenic suppressors of the cs growth of S635A are listed in the table on the left and highlighted above the amino acid sequence of S635A on the right. Motifs I, II, III and VI are in shaded boxes. Positions that were mutated in more than one suppressor allele are underlined.
The remaining 17 intragenic suppressor alleles embraced 14 different amino acid substitutions involving 11 positions of Prp22p (Figure 8). Eleven of the 14 different suppressor mutations were located within the core NTPase domain extending from motifs I to VI, and two of the suppressor mutations (A809S and K816E) mapped within or immediately adjacent to motif VI. The finding of a cluster of two suppressor mutations (M853I and G854V) at vicinal residues downstream of motif VI and a single suppressor even further downstream (S1058L) may suggest a role for the carboxyl-domain in RNA unwinding. In contrast, no suppressor mutations were identified in the N-terminal domain proximal to motif I.
The prp22-S alleles were cloned into CEN TRP1 plasmids and tested by plasmid shuffle for complementation of a prp22Δ strain. Following selection for 5-FOA resistance at 37°C, the prp22-S cells were grown in liquid medium and aliquots of serial dilutions were spotted onto YPD agar plates and assayed for growth at 37, 30 and 25°C (Figure 9). PRP22 cells grew at all temperatures, whereas S635A grew only at 37°C. All the suppressor mutants formed colonies at 30°C. The strength of suppression varied for different intragenic mutations, as gauged by their ability to sustain growth at 25°C. S4, S13, S14, S24, S25 and S27 cells grew well at 25°C, whereas S3, S15, S16, S20 and S31 did not (Figure 9). Testing for growth at 14°C showed that S4 cells formed small colonies after 7 days, whereas the other prp22-S strains failed to grow, even the pseudorevertant S14 (not shown).

Fig. 9. Intragenic suppression of cold-sensitive growth. prp22Δ cells carrying wild-type PRP22, S635A, or the indicated intragenic suppressors were grown in liquid medium at 37°C. The A600 was adjusted to 0.1. Aliquots (3 µl) of serial 10-fold dilutions were spotted onto YPD agar medium. The plates were photographed after incubation for 3 days at 37 and 30°C, or 4 days at 25°C.
A subset of the suppressor mutations was examined for their phenotypic effects per se, by introducing the amino acid changes into a Prp22 protein that contained a wild-type SAT sequence in motif III. Single mutants D581V, A760V, A809S, K816E and S1058L grew as well as wild-type PRP22 cells at all temperatures tested, whereas the G692R and G692D mutations conferred a temperature-sensitive growth defect at 37°C (not shown).
The S27 suppressor (V539I) is a gain-of-function mutation that restores RNA unwinding and spliceosome disassembly in vitro
The molecular basis for suppression of the cs growth phenotype of S635A cells was examined by purifying one of the suppressor proteins, Prp22-S27p, and characterizing its enzymatic activity. Prp22-S27p is a double-mutant containing the original S635A change in motif III and a second mutation V539I located in the segment between motifs I and II (Figure 8). This subtle Val to Ile change in S27 restored growth at 30 and 25°C. The S27 protein was produced in bacteria as a His-tagged fusion and purified by Ni–agarose and phosphocellulose chromatography, and glycerol gradient sedimentation. The peak glycerol gradient fraction of S27 contained a single 130 kDa polypeptide (Figure 10A). The specific ATPase of S27 was 50% of the specific activity of wild-type Prp22p (Figure 10B). ATP hydrolysis by S27 was stimulated to the same extent by RNA as wild-type Prp22p (not shown). The V539I suppressor mutation restored RNA unwinding activity to ∼40% of the wild-type level and to a level at least 10-fold higher than that of the S635A mutant (Figure 5C). These results show that whereas the intragenic suppressor mutation actually reduced the ATPase activity slightly compared with the S635A single mutant, it revived the coupling of ATP hydrolysis and RNA unwinding.

Fig. 10. Purification and ATPase activity of the intragenic suppressor S27. (A) Aliquots (1 µg) of the glycerol gradient preparation of recombinant wild-type Prp22p, S635A and S27 were analyzed by SDS–PAGE. A Coomassie Blue-stained gel is shown. The asterisk marks the position of the Prp22 proteins. The positions and molecular weights (in kDa) of marker proteins are indicated on the left. (B) The ATPase activity of WT Prp22p, S635A and S27 is plotted as a function of input protein.
The recombinant S27 was fully competent in supporting step 2 transesterification during in vitro splicing of actin pre-mRNA by extracts depleted of Prp22p (Figure 6). Reactions reconstituted with S27 did not accumulate the excised lariat-intron as did reactions containing S635A (Figure 6). Remarkably, the S27 suppressor mutation restored to the S635A mutant protein the ability to catalyze ATP-dependent release of the mature mRNA from the spliceosome (Figure 7). Thus, helicase activity is intimately connected to the ability of Prp22p to perform an essential function in pre-mRNA splicing in vitro and in vivo.
Discussion
The ability of Prp22p to hydrolyse ATP is necessary but not sufficient for Prp22’s function in pre-mRNA splicing. We have shown here that the hydroxyamino acids Ser635 and Thr637 in motif III (SAT) are important for coupling ATP hydrolysis to spliceosome disassembly and to RNA unwinding. A gain-of-function mutation in S635A that restores growth at 30°C coordinately restores spliceosome disassembly in vitro and the coupling of ATP hydrolysis to RNA unwinding. These findings provide the clearest evidence to date that the RNA helicase activity of a DExH/D-box protein is relevant to pre-mRNA splicing.
It is worth emphasizing that it is not a foregone conclusion that enzymatic activity is required for DExH-box protein function in vivo. For example, the yeast DExH-box protein Rad3p is essential for cell growth, but the ATPase and DNA helicase activities of Rad3p are not (Sung et al., 1988). The ATPase and DNA helicase activities of the bacterial DExH-box helicase PriA are not necessary for PriA to assemble the DNA replication primosome in vitro or for PriA to function in homologous recombination and double-strand repair in vivo (Zavitz and Marians, 1992; Kogoma et al., 1996; Sandler et al., 1996). To our knowledge, among the DExH-box RNA helicases, only vaccinia NPH-II and now Prp22p have been shown to require the helicase activity for execution of their essential functions in vivo (Gross and Shuman, 1998).
Uncoupling of the ATPase and helicase activities of Prp22p was achieved by eliminating either of the two hydroxyamino acids in motif III (SAT). The crystal structure of the hepatitis C virus (HCV) RNA helicase NS3 shows that motif III is located in a hinge region that connects one globular domain containing the GKT (motif I) and DExH (motif II) elements to a second globular domain containing motif VI (QxxGRxxR) (Yao et al., 1997; Kim et al., 1998). Motifs I, II, III and VI contain conserved amino acids that are essential for Prp22p function in vivo (Figure 1) and which, by analogy to other well studied DExH/D proteins, are likely to comprise the active site of NTP hydrolysis. Kim et al. (1998) surmised from their crystal structure of NS3 that the conserved side chains in motifs I and II of domain 1 coordinate the divalent cation and contact the β phosphate of ATP, whereas the motif VI arginines of domain 2 contact the α and γ phosphates of ATP, thereby bridging the two domains together in the ATP-bound state. They suggested that ATP hydrolysis leads to a conformational change that opens the cleft between the two domains and translocates the protein in a 3′ to 5′ direction along the polynucleotide. This model invokes flexibility of the hinge region connecting the two domains.
Our studies highlight the key role of motif III of Prp22p (SAT) in coupling the energy of hydrolysis of NTP to RNA duplex unwinding. A single Ala mutation of either Ser635 or Thr637 resulted in uncoupling. Single Ala substitution of either Thr in motif III of vaccinia NPH II (TAT) also abrogated RNA unwinding with little effect on NTP hydrolysis (Gross and Shuman, 1995). In the translation initiation factor eIF-4A, a double mutation of motif III (SAT→AAA) also resulted in uncoupling of unwinding from NTP hydrolysis (Pause and Sonenberg, 1992).
There is a notable dichotomy between the effects of motif III mutations in Prp16p versus Prp22p. An Ala mutation of the proximal Ser of motif III (SAT) in Prp16p had no effect on cell growth at any temperature and no effect on pre-mRNA splicing in vitro (Hotz and Schwer, 1998; H.R.Hotz and B.Schwer, unpublished), whereas the equivalent change in Prp22p elicited a severe conditional lethality and a splicing defect in vitro. Replacing the motif III Thr by Ala in Prp16p had no effect on cell growth at 20–37°C, and produced only a slight growth defect at 15°C (Hotz and Schwer, 1998), whereas the same change in Prp22p was conditionally lethal. These differences in mutational response in vivo raise the possibility that the ability of Prp16p to promote step 2 transesterification, though clearly dependent on ATP hydrolysis by Prp16p (Schwer and Guthrie, 1991), may not be tightly coupled to an RNA unwinding event.
The role of motif III in other splicing factors is not well defined. Plumpton et al. (1994) isolated a dominant-negative allele of PRP2 in which the motif III Ser (SAT) was replaced by Leu (LAT). The purified mutant protein was partially active for ATP hydrolysis (∼40% of wild-type activity) and interacted with splicing complexes. Splicing reactions in an extract prepared from a strain overexpressing the mutant Prp2p were blocked at a stage prior to the first cleavage/ligation reaction. Interpretation of this result as indicative of a defect in transmitting the energy of ATP hydrolysis to RNA unwinding is complicated by the following considerations: (i) Prp2p has not been demonstrated to have helicase activity despite vigorous efforts to detect RNA unwinding in vitro (Kim et al., 1992); and (ii) a Ser to Leu change does not engender a clear structure–function relationship, given the larger size of the Leu side chain versus Ser. The value of initially introducing Ala to obtain an instructive mutational effect is underscored by the finding for Prp16p that replacing the motif III Ser by a bulkier Leu or a Pro side chain is lethal and dominant negative, while the Ala substitution has no effect (Hotz and Schwer, 1998; H.R.Hotz and B.Schwer, unpublished).
This and other studies implicate motif III in conformational coupling of ATPase and helicase activities; however, relatively little is known about other residues that may impact on helicase action, e.g. by facilitating conformational changes in the protein. We hypothesized that the identification of intragenic suppressors of the cold sensitivity of S635A might pinpoint structural components that contribute to helicase dynamics. Alternatively, if motif III mutations lead to partial unfolding of the protein, suppressor mutations might stabilize the local domain structure and thus restore growth at 30°C. The intragenic suppressor mutations we identified all mapped to the conserved central and C-terminal region of Prp22p that constitutes the ATPase/helicase functional domain (B.Schwer, unpublished). Previously, Chang et al. (1997) identified an intragenic suppressor of cs growth caused by a motif III TAT to TWT mutation of the DEAD-box splicing factor Prp28p, and they mapped the suppressor just downstream of the DEAD-box. However, the molecular basis for the defect in Prp28-TWT and for suppression was not reported (Chang et al., 1997).
The second-site mutation at Val539 in the S27 suppressor protein is located in motif Ia, the conserved structural element TQPRRVAA in the DEAH-box subfamily (Jankowsky and Jankowsky, 2000). Ala substitution at the Arg immediately flanking the Pro of motif Ia abolishes the ATPase and RNA helicase activities of NPH-II (Gross and Shuman, 1998). Thus, motif Ia can be regarded as a functional component of the active site. Our finding that a V539I change two amino acids downstream of the motif Ia Arg restored coupling of ATP hydrolysis and RNA unwinding in Prp22p attests to the subtlety of internal contacts that contribute to RNA unwinding. In the structure of HCV NS3 protein, motif Ia in the globular domain 1 faces the interdomain cleft opposite motif V in domain 2 (Yao et al., 1997; Kim et al., 1998). We speculate that increasing the size of the Val side chain by one methyl group in the V539I protein facilitates the opening of the interdomain cleft upon ATP hydrolysis (this step being compromised by the S635A mutation) and thereby restores the ratchet-like translocation of Prp22p on RNA. A similar explanation may underlie the suppression of S635A by introduction of a bulky Val in lieu of Ala760 within motif V (TNIAETS in the DEAH-box subfamily).
An outstanding question at this point is the nature of the substrate for the helicase activity of Prp22p. The RNA base-pairing interactions posited during the second step of splicing are not very extensive and Prp22p may only need to disrupt a short duplex segment to elicit disassembly of the spliceosome after mature mRNA is formed. Alternatively, the ATP-driven translocation of Prp22p along an RNA strand may serve to disrupt protein–RNA contacts.
We conclude that a multiplicity of single amino acid changes within the core structure of a DExH-box helicase can sufficiently alter the conformation or flexibility of the protein domains to suppress an RNA unwinding defect incurred by altering motif III, which links two of the domains. Further structural and genetic analyses will be necessary to appreciate fully the intra- and intermolecular dynamics of Prp22p, and of the DExH-box helicases in general.
Materials and methods
Targeted mutagenesis of PRP22
Missense mutations were introduced into the PRP22 gene using the two-stage PCR overlap extension method. Plasmid p358-PRP22 (CEN TRP1 PRP22), which contains the PRP22 coding region (3435 bp) plus 232 bp upstream and 255 bp downstream of flanking yeast genomic DNA, was used as the template for the first amplification step. Residues targeted for Ala substitutions were Asp603, Glu604 and His606 in motif II, Ser635 and Thr637 in motif III, and Gln804, Arg805, Gly807, Arg808, Gly810 and Arg811 in motif VI. The mutated DNA products of the second-stage amplification were digested with restriction endonucleases and inserted into p358-PRP22 in lieu of the corresponding wild-type restriction fragments (yielding PRP22-Ala plasmids p358-D603A, p358-E604A, etc.). The presence of the desired mutation was confirmed by DNA sequencing and the segments corresponding to the inserted restriction fragment were then sequenced completely in order to exclude the acquisition of unwanted mutations during amplification or cloning.
Test of mutational effects on PRP22 function in vivo by plasmid shuffle
Viability of yeast strain YBST1 (Mata ura3-52 trp1-63 his3-Δ200 leu2Δ1 ade2-101 lys2-801 prp22::LEU2) depends on p360-Prp22 (URA3 CEN), which complements the chromosomal deletion of PRP22. YBST1 was transformed with TRP1 CEN plasmids carrying the various PRP22-Ala mutant alleles. Trp+ transformants were selected and streaked onto agar medium containing 0.75 mg/ml 5-FOA to select against maintenance of the URA3 gene on p360-Prp22. The ability of the mutant alleles to support growth of YBST1 on 5-FOA was tested at 14, 25, 30, 34 and 37°C.
Dominant-negative effects of PRP22 mutations
The PRP22-Ala genes were inserted into pYX133 (CEN TRP1), where their expression is under the transcriptional control of the GAL1 promoter. The p133-PRP22-Ala plasmids were introduced into a strain carrying the wild-type PRP22 allele and transformants were selected on glucose-containing SC-Trp medium. Liquid cultures were grown in glucose-containing SC-Trp medium at 30°C. The A600 was adjusted to 0.05, and 5 µl of each suspension were spotted to SC-Trp medium with glucose or galactose as carbon source. The plates were incubated at 37, 34, 30, 25 and 14°C.
Expression and purification of recombinant Prp22 proteins
Plasmid pET16b-PRP22 expresses an N-terminal His-tagged version of wild-type Prp22p in bacteria under the control of a T7 promoter (Schwer and Gross, 1998). Here we constructed pET-based plasmids for expression of His-tagged Prp22p missense mutants S635A, T637A and the intragenic suppressor mutant S27. The expression plasmids were transformed into Escherichia coli strain BL21-Codon Plus(DE3) RIL (Stratagene). Cultures were inoculated from single colonies of freshly transformed cells and maintained in logarithmic growth at 37°C in Luria–Bertani medium containing 0.1 mg/ml ampicillin to a final volume of 1 l. When the A600 reached 0.6–0.8, the cultures were chilled on ice for 30 min, and then adjusted to 0.4 mM isopropyl-β-d-thiogalactopyranoside (IPTG) and 2% ethanol. The cultures were then incubated for 16 h at 17°C with constant shaking. Cells were harvested by centrifugation and the pellets were stored at –80°C.
All subsequent operations were performed at 4°C. The cell pellets were suspended in 100 ml of buffer A (50 mM Tris pH 7.4, 250 mM NaCl, 10% sucrose). Lysozyme was added to 0.2 mg/ml and the suspensions were mixed gently for 30 min, then adjusted to 0.1% Triton X-100. The lysates were sonicated to reduce viscosity and insoluble material was removed by centrifugation for 30 min at 14 000 r.p.m. in a Sorvall SS34 rotor. The soluble lysate was mixed for 1 h with 10 ml of a 50% slurry of Ni-NTA–agarose (Qiagen) that had been equilibrated in buffer A. The resin was recovered by centrifugation, resuspended in 40 ml of buffer A, and collected again by centrifugation. The washed resin was suspended in 40 ml of buffer A and poured into a column. Adsorbed proteins were eluted stepwise with 25, 100 and 500 mM imidazole in buffer E (50 mM Tris pH 7.4, 250 mM NaCl, 10% glycerol). The elution profiles of recombinant Prp22p were monitored by SDS–PAGE of the column fractions. Wild-type and mutant Prp22p were recovered predominantly in the 100 mM imidazole eluate (comprising 6–10 mg of protein). Peak fractions were pooled and aliquots (5 mg) of the Ni–agarose Prp22p preparations were diluted 1:5 with buffer D [50 mM Tris pH 7.4, 2 mM dithiothreitol (DTT), 1 mM EDTA, 10% glycerol] and then mixed for 1 h with 1 ml of phosphocellulose resin that had been equilibrated with 50 mM NaCl in buffer D. The resin was recovered by centrifugation, washed twice with 5 ml of 50 mM NaCl in buffer D, then poured into a column, which was eluted stepwise with 100, 200, 300 and 500 mM NaCl in buffer D. Wild-type and mutant Prp22p were recovered predominantly in the 300 mM NaCl eluate (2–4 mg of protein). Aliquots (160–200 µg) of the phosphocellulose protein preparations were applied to 4.8 ml 15–30% glycerol gradients containing 250 mM NaCl, 50 mM Tris–HCl pH 8.0, 2 mM DTT, 1 mM EDTA, 0.1% Triton X-100. The gradients were centrifuged for 18 h at 47 000 r.p.m. in a Sorvall SW50 rotor. Fractions (0.18 ml) were collected from the tops of the tubes. The Prp22p elution profiles were gauged by SDS–PAGE. Protein concentrations were determined by using the Bio-Rad dye-binding reagent with bovine serum albumin as the standard.
ATPase assay
Reaction mixtures (20 µl) containing 40 mM Tris–HCl pH 8.0, 2 mM DTT, 2 mM MgCl2, 1 mM [γ-32P]ATP, 0.5 µg of poly(A) and Prp22p as specified were incubated for 30 min at 30°C. The reactions were stopped by the addition of 200 µl of a 5% (w/v) suspension of activated charcoal in 20 mM phosphoric acid. The samples were incubated on ice for 10 min and the charcoal was recovered by centrifugation. 32P radioactivity in the supernatant was quantitated by liquid scintillation counting. The results are average values from duplicate reaction mixes with a standard deviation of <5%.
pre-mRNA splicing in vitro
Yeast whole-cell extract from strain BJ2168 was prepared using the liquid nitrogen method (Umen and Guthrie, 1995; Ansari and Schwer, 1995). The extract was immunodepleted of Prp22p using Prp22p affinity-purified polyclonal antibodies. Briefly, 120 µl of extract were mixed with 30 µl of anti-Prp22 for 45 min on ice, then adsorbed to 75 µl of protein A– Sepharose resin that had been equilibrated with phosphate-buffered saline (10 mM sodium phosphate pH 7.2, 150 mM KCl) for 45 min. The Sepharose was recovered by centrifugation and the Prp22p-depleted supernatant (Δ22 extract) was stored at –80°C. Splicing reaction mixtures (200 µl) contained 50% Prp22p-depleted extract, 2 × 107 c.p.m. (∼400 fmol) of 32P-GMP-labeled actin precursor RNA, 60 mM potassium phosphate, 2.5 mM MgCl2 and 2 mM ATP (Lin et al., 1985). The reaction mixtures were incubated for 10 min at 23°C, then 0.6–0.8 µg of wild-type or mutant Prp22p (phosphocellulose fractions) were added and incubation continued for another 15 min at 23 or 30°C. The reaction mixtures were halted by transfer to ice. An aliquot (10 µl) was removed from each mixture and added directly to 200 µl STOP solution (50 mM sodium acetate pH 5.2, 1 mM EDTA, 0.1% SDS, 30 µg/ml tRNA). The remaining aliquots (190 µl) were layered onto 15–40% glycerol gradients containing 20 mM HEPES pH 6.5, 100 mM KCl, 2 mM MgCl2, and then centrifuged at 4°C for 12 h at 35 000 r.p.m. in a Sorvall TH641 rotor. Fractions (400 µl) were collected from the tops of the tubes. RNA was recovered by phenol extraction and ethanol precipitation. RNA from alternate gradient fractions was analyzed by electrophoresis through a 6% polyacrylamide gel containing 7 M urea in TBE. Radiolabeled RNA was visualized by autoradiographic exposure of the dried gel.
RNA unwinding assay
Radiolabeled dsRNA helicase substrate was prepared as described previously (Gross and Shuman, 1995). Briefly, the component RNA strands were transcribed in vitro from linear plasmid DNA templates using SP6 and T7 RNA polymerase. The 103-nucleotide lower strand (5′-GGGAGACCGGAAUUAGCUUGUAUUCUAUAGUGUCUCCUA AAUCGUAUGUGUAUGAUACAUAAGGUUAUGUAUUAAUUGU AGCCGCGUUCUAACGACAAUAUGU-3′) was labeled to high specific activity with [α-32P]GTP. The partially complementary 107mer strand (5′-GAAUACAAGCUAAUUCCGGUCUCCCUAUAGUGAGUCGUAUUAAUUUCGAUAAGCCAGCUGCAUUAAUGAAUCGGCCAACGCGCGGGGAGAGGCGGUUUGCGUAUUGU-3′) was labeled at 300-fold lower specific activity. The 25-bp duplex is underlined. The transcription reaction products were gel purified and annealed at a 2:1 molar ratio of the 107mer to 103mer. The tailed RNA duplexes were then purified by native gel electrophoresis (Lee and Hurwitz, 1992). The helicase assays were performed as described previously (Schwer and Gross, 1998) and the extent of unwinding (displaced RNA/total RNA) after 25 min of incubation at 37°C was quantitated by scanning the gel using a STORM phosphoimager.
Acknowledgments
Acknowledgements
We are grateful to Christian Gross and Susanne Schneider for their contributions and Stewart Shuman for comments on the manuscript. This work was supported by NIH grant GM50288.
References
- Ansari A. and Schwer,B. (1995) SLU7 and a novel activity, SSF1, act during the PRP16-dependent step of yeast pre-mRNA splicing. EMBO J., 14, 4001–4009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arenas J.E. and Abelson,J.N. (1997) Prp43: an RNA helicase-like factor involved in spliceosome disassembly. Proc. Natl Acad. Sci. USA, 94, 11798–11802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burge C.B., Tuschl,T. and Sharp,P.A. (1999) Splicing of precursors to mRNA by the spliceosome. In Gesteland,R.F. and Atkins,J.F. (eds), The RNA World. 2nd edn. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, pp. 525–560. [Google Scholar]
- Burgess S., Couto,J.R. and Guthrie,C. (1990) A putative ATP binding protein influences the fidelity of branchpoint recognition in yeast splicing. Cell, 60, 705–717. [DOI] [PubMed] [Google Scholar]
- Chang T.-H., Latus,L.J., Liu,Z. and Abbott,J.M. (1997) Genetic interactions of conserved regions in the DEAD-box protein Prp28p. Nucleic Acids Res., 25, 5033–5040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J.H. and Lin,R.J. (1990) The yeast PRP2 protein, a putative RNA-dependent ATPase, shares extensive sequence homology with two other pre-mRNA splicing factors. Nucleic Acids Res., 18, 6447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Company M., Arenas,J. and Abelson,J. (1991) Requirement of the RNA helicase-like protein PRP22 for release of messenger RNA from spliceosomes. Nature, 349, 487–493. [DOI] [PubMed] [Google Scholar]
- de la Cruz J., Kressler,D. and Linder,P. (1999) Unwinding RNA in Saccharomyces cerevisiae: DEAD-box proteins and related families. Trends Biochem. Sci., 24, 192–198. [DOI] [PubMed] [Google Scholar]
- Edwalds-Gilbert G., Kim,D.-H., Kim,S.-H., Tseng,Y.-H., Yu,Y. and Lin,R.-J. (2000) Dominant negative mutants of the yeast splicing factor Prp2 map to a putative cleft region in the helicase domain of DExD/H-box proteins. RNA, 6, 1106–1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorbalenya A.E., Koonin,E.V., Donchenko A.P. and Blinov,V.M. (1989) Two related superfamilies of putative helicases involved in replication, recombination, repair and expression of DNA and RNA genomes. Nucleic Acids Res., 17, 4713–4730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross C.H. and Shuman,S. (1995) Mutational analysis of vaccinia virus nucleoside triphosphate phosphohydrolase II, a DExH box RNA helicase. J. Virol., 69, 4727–4736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross C.H. and Shuman,S. (1996) The QRxGRxGRxxxG motif of the vaccinia virus DExH box RNA helicase NPH-II is required for ATP hydrolysis and RNA unwinding but not for RNA binding. J. Virol., 70, 1706–1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross C.H. and Shuman,S. (1998) The nucleoside triphosphatase and helicase activities of vaccinia virus NPH-II are essential for virus replication. J. Virol., 72, 4729–4736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guthrie C. (1991) Messenger RNA splicing in yeast: clues to why the spliceosome is a ribonucleoprotein particle. Science, 253, 157–163. [DOI] [PubMed] [Google Scholar]
- Heilek G.M. and Peterson,M.G. (1997) A point mutation abolishes the helicase but not the nucleoside triphosphatase activity of hepatitis C virus NS3 protein. J. Virol., 71, 6264–6266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotz H.R. and Schwer,B. (1998) Mutational analysis of the yeast DEAH-box splicing factor Prp16. Genetics, 149, 807–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jankowsky J. and Jankowsky,A. (2000) The DExH/D protein family database. Nucleic Acids Res., 28, 333–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D.W., Kim,J., Gwack,Y., Han,J.H. and Choe,J. (1997) Mutational analysis of hepatitis C virus RNA helicase. J. Virol., 71, 9400–9409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J.L., Morgenstern,K.A., Griffith,J.P., Dwyer,M.D., Thomson,J.A., Murcko,M.A., Lin,C. and Caron,P.R. (1998) Hepatitis C virus NS3 RNA helicase domain with a bound oligonucleotide: the crystal structure provides insights into the mode of unwinding. Structure, 6, 89–100. [DOI] [PubMed] [Google Scholar]
- Kim S.H., Smith,J., Claude,A. and Lin,R.J. (1992) The purified yeast pre-mRNA splicing factor PRP2 is an RNA-dependent NTPase. EMBO J., 11, 2319–2326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kogoma T., Cadwell,G.W., Barnard,K.G. and Asai,T. (1996) The DNA replication priming protein, PriA, is required for homologous recombination and double-strand break repair. J. Bacteriol., 178, 1258–1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C.G. and Hurwitz,J. (1992) A new RNA helicase isolated from HeLa cells that catalytically translocates in the 3′ to 5′ direction. J. Biol. Chem., 267, 4398–4407. [PubMed] [Google Scholar]
- Lin R.J., Newman,A.J., Cheng,S.C. and Abelson,J. (1985) Yeast mRNA splicing in vitro. J. Biol. Chem., 260, 14780–14792. [PubMed] [Google Scholar]
- Madhani H.D. and Guthrie,C. (1994) Dynamic RNA–RNA interactions in the spliceosome. Annu. Rev. Genet., 28, 1–26. [DOI] [PubMed] [Google Scholar]
- Martins A., Gross,C.H. and Shuman,S. (1999) Mutational analysis of vaccinia virus nucleoside triphosphate phosphohydrolase I, a DNA-dependent ATPase of the DExH box family. J. Virol., 73, 1302–1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore M.J., Query,C.C. and Sharp,P.A. (1993) Splicing of precursors to mRNA by the spliceosome. In Gesteland,R.F. and Atkins,J.F. (eds), Splicing of Precursors to mRNA by the Spliceosome. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, pp. 303–357. [Google Scholar]
- Nilsen T.W. (1994) RNA–RNA interactions in the spliceosome: unraveling the ties that bind. Cell, 78, 1–4. [DOI] [PubMed] [Google Scholar]
- Pause A. and Sonenberg,N. (1992) Mutational analysis of a DEAD box RNA helicase: the mammalian translation initiation factor eIF-4A. EMBO J., 11, 2643–2654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plumpton M., McGarvey,M. and Beggs,J.D. (1994) A dominant negative mutation in the conserved RNA helicase motif ‘SAT’ causes splicing factor PRP2 to stall in spliceosomes. EMBO J., 13, 879–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandler S.J., Samra,H.S. and Clark,A.J. (1996) Differential suppression of priA::kan phenotypes in Escherichia coli K-12 by mutations in priA, lexA and dnaC. Genetics, 143, 5–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwer B. and Gross,C.H. (1998) Prp22, a DExH-box RNA helicase, plays two distinct roles in yeast pre-mRNA splicing. EMBO J., 17, 2086–2094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwer B. and Guthrie,C. (1991) PRP16 is an RNA-dependent ATPase that interacts transiently with the spliceosome. Nature, 349, 494–499. [DOI] [PubMed] [Google Scholar]
- Staley J.P. and Guthrie,C. (1998) Mechanical devices of the spliceosome: motors, clocks, springs and things. Cell, 92, 315–326. [DOI] [PubMed] [Google Scholar]
- Sung P., Higgins,D., Prakash,L. and Prakash,S. (1988) Mutation of lysine-48 to arginine in the yeast RAD3 protein abolishes its ATPase and DNA helicase activities but not the ability to bind ATP. EMBO J., 7, 3263–3269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umen J.G. and Guthrie,C. (1995) A novel role for a U5 snRNP protein in 3′ splice site selection. Genes Dev., 9, 855–868. [DOI] [PubMed] [Google Scholar]
- Velankar S.S., Soultanas,P., Dillingham,M.S., Subramanya,H.S. and Wigley,D.B. (1999) Crystal structures of complexes of PcrA DNA helicase with a DNA substrate indicate an inchworm mechanism. Cell, 97, 75–84. [DOI] [PubMed] [Google Scholar]
- Wagner J.D.O., Jankowsky,E., Company,M., Pyle,A.M. and Abelson,J.N. (1998) The DEAH-box protein PRP22 is an ATPase that mediates ATP-dependent mRNA release from spliceosomes and unwinds RNA duplexes. EMBO J., 17, 2926–2937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y. and Guthrie,C. (1998) The DEAH-box splicing factor Prp16 unwinds RNA duplexes in vitro. Curr. Biol., 8, 441–451. [DOI] [PubMed] [Google Scholar]
- Will C.L. and Lührmann,R. (1997) Protein functions in pre-mRNA splicing. Curr. Opin. Cell Biol., 9, 320–328. [DOI] [PubMed] [Google Scholar]
- Yao N., Hesson,T., Cable,M., Hong,Z., Kwong,A.D., Le,H.V. and Weber,P.C. (1997) Structure of the hepatitis C virus RNA helicase domain. Nature Struct. Biol., 4, 463–467. [DOI] [PubMed] [Google Scholar]
- Zavitz K.H. and Marians,K.J. (1992) ATPase-deficient mutants of the Escherichia coli DNA replication protein PriA are capable of catalyzing the assembly of active primosomes. J. Biol. Chem., 267, 6933–6940. [PubMed] [Google Scholar]