

Abstract
Noroviruses are the major cause of human epidemic nonbacterial gastroenteritis. Viral replication requires a 3C cysteine protease that cleaves a 200 kDa viral polyprotein into its constituent functional proteins. Here we describe the X-ray structure of the Southampton norovirus 3C protease (SV3CP) bound to an active site-directed peptide inhibitor (MAPI) which has been refined at 1.7 Å resolution. The inhibitor, acetyl-Glu-Phe-Gln-Leu-Gln-X, which is based on the most rapidly cleaved recognition sequence in the 200 kDa polyprotein substrate, reacts covalently through its propenyl ethyl ester group (X) with the active site nucleophile, Cys 139. The structure permits, for the first time, the identification of substrate recognition and binding groups in a noroviral 3C protease and thus provides important new information for the development of antiviral prophylactics.
Noroviruses are the most common cause of acute viral gastroenteritis in humans with epidemics being particularly common in confined communities such as hospitals, schools, hotels, and ocean liners and in the armed forces (1). The virus is transmitted by the fecal−oral route through contaminated food and water, by direct contact, and possibly by transmission through air-borne viral particles (2). Currently, there is neither vaccine nor antiviral therapy available for the control of the disease, largely because progress in studying human noroviruses has been greatly impeded by the lack of a permissive cell culture system (3).
Comparisons of genome sequence and organization have placed these viruses in the family Caliciviridae: a viral family consisting of five currently recognized genera, one of which is the noroviruses (4,5). Phylogenetic analyses have shown that animal and human noroviruses can be classified into five separate genogroups, with the noroviruses infecting humans most commonly occurring in genogroups 1 and 2. The prototype virus, Norwalk virus, and the Southampton virus, used in this study, both belong to genogroup 1. The Southampton norovirus genome consists of a molecule of single-stranded positive sense RNA of 7708 nucleotides with a polyadenylated 3′ terminus (6,7). The genome is organized into three open reading frames; ORF1 1 is positioned at the 5′ end and encodes a large 200 kDa nonstructural polyprotein, ORF 2 encodes the major capsid protein VP1, and ORF 3 codes for a small basic protein VP2 that is likely to assist in the assembly of newly synthesized viruses (8).
In vitro translation and mutagenesis studies indicated that the 200 kDa ORF 1 polyprotein of Southampton virus is cleaved by the action of a 3C protease to initially generate three separate functional protein products (9). Full processing of the precursor polyprotein generates six mature products (10): an N-terminal protein (p48), an NTPase (p41), a 3A-like protein (p21), a Vpg protein (p16), the 3C protease (p19), and an RNA polymerase (p57), as shown in Figure 1. The protease also inhibits translation of host proteins by cleavage of the poly(A) binding protein, thereby allowing preferential viral protein expression (11).
Figure 1.

Diagramatic representation of Southampton norovirus nonstructural polyprotein showing the protease cleavage sites. The scissile bonds are shown arrowed, and the amino acid residue numbers are shown for the cleavage sites. The functions of the mature viral proteins are indicated below the bars.
The scissile bonds cleaved by the Southampton virus protease within its 200 kDa polyprotein substrate are within the dipeptide recognition sequences Glu-Gly, Gln-Gly, or Gln-Ala, where the “-” indicates the cleavage site (10). The protease has a preference for cleavage at LQ-GP and LQ-GK, but it can also cleave at ME-GK, FE-AP, and LE-GG. It appears that the enzyme preferentially accommodates a glutamine residue at the active site S1 position but will also accept a glutamate residue. Since the proteolytic processing of the 200 kDa precursor polyprotein is essential to yield functional viral proteins, the viral protease presents itself as a particularly viable target for antiviral strategies.
Enzymes in this family are cysteine proteases that display a trypsin- or chymotrypsin-like serine protease fold, a property which distinguishes this family from other viral proteases (12). It has been shown for the human rhinovirus enzyme that the preferred amino acids at the P3, P2, and P1 positions are Leu, Phe, and Gln, respectively (13). Modified peptide inhibitors that include the preferred amino acid recognition sequence but possess a C-terminal chemical moiety capable of reacting with the active site cysteine residue have been developed, and in vitro studies show that these completely inhibit the 3C protease activity (14,15). One class of inhibitor includes a Michael-acceptor group at its C-terminus (Figure 2), which undergoes nucleophilic attack by the active site thiol, resulting in the inhibitor becoming covalently and irreversibly bound to the catalytic cysteine (14). Development of these peptide Michael-acceptor inhibitors has led to compounds that not only inhibit human rhinovirus 3C protease activity in vitro but also display antiviral properties in vivo(16).
Figure 2.

Structure of the Michael acceptor peptidyl inhibitor (MAPI) designed to be specific for the Southampton virus 3C protease (SV3CP). The site of nucleophilic attack by the active site thiol is indicated.
From a comparison between norovirus protease amino acid sequences and those of polio virus (PV), human rhinovirus (HRV), hepatitis A virus (HAV), Chiba virus (CV), and foot and mouth disease virus (FMDV), it is apparent that the norovirus protease belongs to the same small family of (chymo)trypsin-like cysteine proteases, as distinct from the more distant papain-like cysteine proteases. This has been confirmed by the determination of two X-ray crystal structures of the protease from group 1 norovirus (17,18). Cysteine proteases typically possess an active site dyad comprising cysteine and histidine. Interestingly, the three-dimensional structures of the PV, HRV, HAV, FMDV, and CV 3C proteases indicate that a third amino acid, either aspartate or glutamate, is present at the active site, as in all serine proteases. However, site-directed mutagenesis of CV 3C protease has suggested that the third acidic amino acid may not be essential for activity (19).
This paper describes, for the first time, the X-ray structure of the Southampton norovirus protease bound to a polypeptide irreversible inhibitor (MAPI), acetyl-Glu-Phe-Gln-Leu-Gln-X, in which the natural polyprotein substrate recognition sequence is linked to a propenyl ethyl ester moiety (X). The catalytic cysteine at position 139 of the enzyme is rapidly modified by the inhibitor. The high-resolution structure of the complex provides the first detailed information on how a noroviral protease recognizes its peptide substrate and provides a foundation for the knowledge-based design of inhibitors as antiviral prophylactics.
Materials and Methods
Protein Expression and Purification
cDNA specifying the 3C proteinase was obtained as described in ref (10). Expression of the protease was achieved by generating a segment of DNA using PCR primers designed to form extensions of 38 bp upstream of the 5′ end of the protease coding region and 1022 bp downstream of the 3′ end. The 5′ primer was designed to include a NdeI site encoding the ATG start codon followed by commonly used Escherichia coli codons specifying the C-terminal amino acids of the VPg protein which precedes the protease in ORF 1. The 3′ primer contained a BamHI site and bases specifying the N-terminal part of the 3D RNA polymerase gene which follows the protease in ORF 1. The amplified DNA was introduced into the expression vector pRSET (UCB) at the NdeI and BamHI restriction sites by standard methods, and the resulting plasmid was transformed into E. coli BL21(DE3) pLysS. For overexpression, these cells which were grown in Luria−Bertani medium with 50 μg/mL ampicillin in shaken flasks at 37 °C and induced using isopropyl β-d-thiogalactopyranoside (IPTG) which was added to a final concentration of 1 mM for the last 3 h of cell growth. The harvested cells were sonicated, and following ultracentrifugation, the supernatant was applied to a column of SP-Sepharose cation-exchange matrix (GE Healthcare) in 10 mM phosphate buffer at pH 7.65 containing 5 mM 2-mercaptoethanol, followed by elution with a gradient up to 1 M NaCl. After desalting with a Sephadex G25 column, the enzyme was applied to a Source 15S column (GE Healthcare) in the same buffer, followed by elution with a gradient up to 1 M NaCl. After a final desalting step with Sephadex G25, the protein was obtained in a yield of approximately 20 mg/L of culture and was concentrated to 10 mg/mL and stored in 50% glycerol. The Mr of the purified enzyme was determined by electrospray mass spectrometry as 19258, which is consistent with the predicted amino acid sequence of 181 residues and confirmed that the protease had self-excised from the flanking sequences encoded by the expression construct.
Chromogenic Substrate Synthesis, Kinetic Assay, and Inhibitor Synthesis
For kinetic studies of the protease specificity, a series of peptides was synthesized, each with p-nitroaniline (pNA) at the C-terminus to provide a convenient spectrophotometric assay of the SV3CP activity. The following chromogenic peptides, Ac-QLQ-pNA, Ac-FQLQ-pNA, Ac-EFQLQ-pNA, and Ac-DEFQLQ-pNA, were synthesized using a combination of standard Fmoc solid-phase chemistry and synthetic techniques (20). MALDI-Q-TOF-MS was used to confirm correct synthesis following reverse-phase purification of each product in DMSO. Each peptide mimics the SV3CP recognition sequence within the 200 kDa ORF 1 polyprotein which experiences the greatest rate of cleavage (10). All peptides were synthesized with an acetylated N-terminus and the C-terminus linked to a chromogenic pNA group (21,22). Whereas the uncleaved chromogenic pNA peptide is colorless, cleavage of the C-terminal p-nitroanilide group yields free p-nitroaniline which is strongly yellow in color, and its production can be followed spectrophotometrically at 405 nm. The C-terminal glutamine-pNA presents an issue during peptide chain extension since the C-terminal residue of a peptide would normally be attached to the support resin via its main-chain carboxyl. In this instance, pNA has to be attached at the glutamine carboxyl, and therefore this residue must be linked to the support resin by its side chain. Hence, the pNA derivative of glutamate, rather than glutamine, was synthesized and linked via its side chain to a Rink amide MBHA resin (23). Upon cleavage from the resin, the side-chain carboxyl is aminated to yield a glutamine residue.
The rate of cleavage of each pNA substrate by the protease was monitored at 405 nm using a Nanodrop ND1000 spectrophotometer. The assay involved dissolving the substrates in DMSO and diluting them into a solution containing 100 mM Tris, pH 8.5, and 5 mM β-mercaptoethanol to give final substrate concentrations in the 0.1−3.0 mM range and a final enzyme concentration of 0.1 mg/mL SV3CP. The absorbance at 405 nm of 2 μL samples taken from the reaction mixture was measured at 1 min intervals over a 10 min period.
Since it was found that Ac-EFQLQ-pNA had the highest kcat/KM ratio (see Results), a Michael-acceptor polypeptide inhibitor (MAPI) with the same sequence as the optimal substrate was synthesized, essentially by minor adaptation of the methods described by Dragovich et al. (14), and purified by reverse-phase chromatography (full details are given in the Supporting Information). The inhibitor (shown in Figure 2) has the sequence Ac-EFQLQ-X, where X is the propenyl ethyl ester extension to the C-terminal residue which behaves as a Michael acceptor, undergoing nucleophilic attack by the active site thiol and generating a stable thioether product (24). The success of each step in the synthesis and the purity of the final compound was confirmed by MALDI-Q-TOF-MS.
Crystallization and Preliminary X-ray Analysis
For the initial crystallization studies, samples of protease that were stored in glycerol were exchanged using a Sephadex G25 column into 10 mM phosphate buffer, pH 7.45, containing 5 mM β-mercaptoethanol and then concentrated to 3 mg/mL. Crystals of the native enzyme were obtained by the vapor diffusion method in several conditions at room temperature using the Jena Biosciences classic screens, and following further screening the optimum conditions were found to be 7% PEG 8000 and 0.1 M HEPES, pH 7.5, with 8% (v/v) ethylene glycol. Preliminary data collection using station ID14-1 at the ESRF (Grenoble, France) with crystals frozen in 30% glycerol (added stepwise) revealed that they belonged to a hexagonal point group and produced diffraction data to medium resolution.
Crystals of vastly improved diffraction quality were obtained by forming a complex of the protease with the irreversible inhibitor MAPI. To obtain these cocrystals, NaCl was added to the protein sample to a final concentration of 300 mM which allowed the protease to be concentrated to 17 mg/mL using a 10 kDa cutoff Centricon concentration vessel. To provide suitable conditions to form a complex of the protease with the essentially insoluble inhibitor, it was necessary to include 10% DMSO in the buffer. An amount of inhibitor giving a 3-fold molar excess over the protein was dissolved in a volume of DMSO that would, once added to the protein sample, result in the final buffer containing 10% DMSO. The inhibitor in DMSO was added to the protein sample in 10 equal volumes at 10 min intervals. The sample was then passed through a Sephadex G25 minispin column to rid the complex of any excess unbound inhibitor and DMSO. Incubation of a small sample of the complex with the chromogenic substrate Ac-EFQLQ-pNA demonstrated that 100% inhibition had been achieved. Further confirmation and accurate assessment of MAPI binding were accomplished by mass spectrometry which revealed a single major protein peak at 20045 Da corresponding to one molecule of SV3CP plus one molecule of the inhibitor. With the complex at a concentration of 15 mg/mL, large crystals were obtained in 25% (w/v) PEG 5000 MME, 100 mM Tris-HCl, pH 8.5, and 200 mM lithium sulfate (Jena Bioscience screen condition JB4 A1). Data collection at station ID23-1 (ESRF, Grenoble) established that the crystals diffracted to high resolution. Data processing in MOSFLM (25), SCALA (26), and other programs in the CCP4 suite (27) revealed that the crystals belonged to the space group P212121 with unit cell dimensions a = 49.5 Å, b = 84.1 Å, and c = 121.5 Å; the data set had an overall Rmerge of 5.3% to 1.7 Å resolution.
Preparation of selenomethioninyl protease for experimental phasing involved the E. coli cells harboring the expression construct being grown at 37 °C to midlog phase in LB medium with 50 μg/mL ampicillin. The cells were then pelleted by centrifugation prior to resuspension in M9 minimal medium supplemented with 0.4% (w/v) glucose and ampicillin at the above concentration. They were then grown at 37 °C for 30 min to use up remaining amino acids prior to the addition of each of the 20 standard amino acids to final concentrations of 40 mg/L with the exception of methionine, which was replaced by l-selenomethionine at the same final concentration. The cultures were supplemented with the vitamins riboflavin, niacinamide, pyridoxine and thiamin, each at a final concentration of 1 mg/L, and were shaken at 37 °C for another 20 min. The cultures were then induced by addition of IPTG to a final concentration of 1 mM and grown for a further 3 h at 37 °C. Purification of the selenomethioninyl protease was performed as for the native enzyme and yielded approximately 12 mg/L of culture. Analysis of the protein by electrospray mass spectrometry showed that the molecular mass of the Se-Met enzyme was 19518 Da, indicating 100% occupancy of the five Se-Met residues per molecule. Crystals of the selenomethionyl protease complexed with the inhibitor were then grown and frozen as described above.
MAD Phasing and Refinement
Multiwavelength anomalous diffraction data were collected on station ID23-1 at the ESRF (Grenoble, France) to 1.8 Å resolution using 1° oscillations and an exposure time of 1 s per image. The data at each wavelength were scaled and processed as above in space group P212121 (see Table 1 for details). The Se sites and phases were determined using SOLVE (28), which gave a mean figure of merit of 0.56, and density modification using DM (29) gave an excellent electron density map from which an initial model of the structure was obtained using MAID (30) and manual building using TURBO-FRODO (BioGraphics, Marseille). The model was refined with the 1.7 Å resolution “native” data set for the inhibitor complex using the programs SHELX (31) and CNS (32). Validation tests were performed with MOLPROBITY (33), and buried solvent-accessible areas were calculated using AreaIMol (27). The final coordinates and structure factors have been deposited in the Protein Data Bank (www.wwpdb.org) with accession code 2IPH.
Table 1. Crystallographic and Refinement Statisticsa.
| space group | P212121 |
| unit cell dimensions | |
| a (Å) | 49.5 |
| b (Å) | 84.1 |
| c (Å) | 121.5 |
| data set | peak | inflection | remote | native |
|---|---|---|---|---|
| λ (Å) | 0.97925 | 0.97955 | 0.97625 | 0.93400 |
| resolution (Å) | 40.5−2.2 (2.3−2.2) | 46.0−2.5 (2.6−2.5) | 49.2−2.5 (2.6−2.5) | 40.5−1.7 (1.8−1.7) |
| Rmerge (%)b | 8.6 (74.1) | 5.7 (13.7) | 6.4 (15.8) | 5.3 (62.3) |
| completeness (%) | 99.5 (98.7) | 98.2 (99.9) | 97.6 (100.0) | 98.8 (92.8) |
| average I/σ(I) | 29.3 (3.5) | 22.8 (10.5) | 20.7 (8.4) | 22.4 (2.4) |
| multiplicity | 19.6 (18.0) | 6.7 (6.4) | 6.6 (6.3) | 6.7 (5.2) |
| refinementc | |
| R-factor (%) | 20.5 |
| Rfree factor (%) | 22.3 |
| no. of reflections | 51760 |
| rmsd bond lengths (Å) | 0.005 |
| rmsd bond angles (deg) | 1.3 |
| mean B-factor for all atoms (Å2) | 27.3 |
Values for the outer resolution shell of each data set are shown in parentheses; note that data to only 3.0 Å were used in the initial phasing.
Rmerge = ∑h∑i|(Ihi − Ih)|/∑h∑i(Ihi), where Ih is the mean intensity of the scaled observations Ihi.
All data to 1.7 Å resolution with no σ(I) cutoff were used in the refinement except for 5% of the data which were reserved for the Rfree set.
Results
Kinetic Evaluation of the Trial Substrates
Measurement of the initial rates of cleavage of the chromogenic pNA peptides established that Ac-EFQLQ-pNA was the best substrate in terms of specificity constant (kcat/KM ratio); see Table 2. In contrast, the shortest peptide, Ac-QLQ-pNA, was not cleaved at all during the 10 min assay period. The longest substrate, Ac-DEFQLQ-pNA, had a kcat which was higher than that of Ac-EFQLQ-pNA although its proportionately higher KM meant that it was a slightly inferior substrate in terms of specificity constant. The remaining substrate, Ac-FQLQ-pNA, was cleaved with a kcat/KM ratio that was significantly lower than that of optimal substrate. These results facilitated the design and synthesis of the inhibitor MAPI (Figure 2) with a sequence which matches that of the most efficiently cleaved substrate, Ac-EFQLQ-pNA. Preincubation of the enzyme with millimolar concentrations of the inhibitor, prior to addition of substrate, was found to inactivate the enzyme irreversibly within the dead time of the experiment (<30 s). Formation of the complex of SV3CP with this inhibitor proved to be critical in obtaining a crystal form of the enzyme that diffracted to high resolution and allowed structure determination to proceed.
Table 2. Kinetic Data for the Hydrolysis of pNA Substrates by SV3CPa.
| substrate | KM (M) | kcat (s−1) | kcat/KM (M−1 s−1) |
|---|---|---|---|
| Ac-QLQ-pNA | |||
| Ac-FQLQ-pNA | 1.5 × 10−3 | 0.08 | 55 |
| Ac-EFQLQ-pNA | 3 × 10−4 | 0.14 | 463 |
| Ac-DEFQLQ-pNA | 8 × 10−4 | 0.33 | 416 |
The smallest substrate Ac-QLQ-pNA was not cleaved detectably, and so no parameters could be determined.
Three-Dimensional Structure Analysis
The structure of the SV3CP inhibitor complex has been refined at 1.7 Å resolution to an R-factor and Rfree of 20.5% and 22.3%, respectively. The electron density is clearly defined for all residues in both molecules in the asymmetric unit except for a few amino acids at the N-terminus of one monomer and a few side chains in the loop regions of both molecules. The active site-directed inhibitor is clearly shown to be covalently linked to Cys 139 as well as making numerous other interactions with the protein. The two molecules of the protease in the asymmetric unit are very closely related structurally with an rms deviation in Cα atom positions of only 0.4 Å, excluding a partly disordered loop region formed by residues 123−130 and a few residues at the N-terminus. Validation by MOLPROBITY (33) placed all residues within allowed regions of the Ramachandran plot (98.3% in favored regions and 1.7% in permissible regions).
Tertiary Structure of the Complex
Commensurate with the crystallographic structures of other viral 3C proteases, e.g., PV (34), HAV (35,36) and HRV (37), the SV3CP structure displays a chymotrypsin-like fold consisting of two domains connected by a large loop (38). The amino acid sequence of SV3CP and its associated secondary structure are shown in Figure 3 along with a sequence alignment with other family members. Domain I consists of a short N-terminal two-turn α-helical region leading directly into the first of five β-strands, βaI to βeI (Figure 3), that comprise a twisted antiparallel β-sheet. Domain I contributes a number of regions that interact with the active site ligand. These are the first β-hairpin loop (residues 12−15) and the loops involving residues 29−47 and 53−54. Domain II is connected to domain I by a 20-residue loop and is substantially larger than domain I, extending from Ile 72 to Glu 181. This domain consists of six β-strands, βaII to βfII, that comprise an antiparallel β-barrel. The catalytic Cys 139 is contained within the large loop linking βcII and βdII, and accordingly, domain 2 forms the bulk of the interactions with active site ligand (Figure 4a,b).
Figure 3.

(a) Sequence alignment of SV3CP with other noroviral proteases. The sequences were obtained from the UniProtKB/SWISSPROT sequence databank with the following accession numbers: Southampton virus Q04544, Norwalk-like virus Q83883, Lordsdale virus P54634, Camberwell virus Q9W183, and Chiba virus Q9DU47. The amino acids are colored according to the following scheme: cyan = basic, red = acidic, green = neutral-polar, pink = bulky hydrophobic, white = Gly, Ala, and Pro, and yellow = Cys. The secondary structure elements present in SV3CP are indicated in the bottom row. (b) A topology diagram of SV3CP with the putative active site catalytic residues Cys 139, His 30, and Glu 54 indicated in red.
Figure 4.

(a) SV3CP viewed through the β-barrel of domain II. The putative active site catalytic triad of residues Cys 139, His 30, and Glu 54 is indicated in red. Residues delineating the secondary structure elements are indicated numerically. The conformation of the inhibitor MAPI bound to Cys 139 is shown in violet in (b). The β-strands βbII and βcII create an arch that binds with the peptidyl portion of MAPI (corresponding to the natural substrate P residues). (c) Electron density for the inhibitor MAPI. The residues of the inhibitor are shown along with the 2Fo − Fc electron density at 1.7 Å resolution contoured at 1 rms. The contiguous electron density between Q1 and Cys 139 (shown on the far right) is evident; the side-chain sulfur atom is colored green. (d) The β-sheet-like hydrogen-bonding network between the active site cleft residues of SV3CP and the inhibitor MAPI. The SV3CP residues are shown in green and those of MAPI in pink. A number of water-mediated hydrogen bonds are also shown, and donor−acceptor atom distances are given in Å.
The electron density clearly shows that the active site thiol of Cys 139 is bound covalently to the C-terminal Michael-acceptor extension of the inhibitor MAPI (Figure 4c). Two further members of the active site, Glu 54 and His 30, are within feasible bonding distances and complete a putative triad of catalytic residues (see Figure 4b). The involvement of Glu 54 in catalysis was indicated when an E54A mutant was found to exhibit diminished protease activity, although it was not found to be absolutely required for activity (19). This is consistent with other cysteine proteases which, compared to serine proteases, generally exhibit a much reduced requirement for the third member of the catalytic triad, due to the stronger nucleophilicity of the thiol (or thiolate) group.
The β-strands βbII and βcII are of note since they form an “arch” which covers the peptide portion of MAPI (Figure 4b). A structure without the inhibitor bound might have wrongly suggested that substrate would bind through the cleft generated between domains I and II. However, it is clear that the β-barrel motif of domain II will form the bulk of the interactions with peptide substrates. For example, extensive β-sheet-like interactions with the ligand are made by the C-terminal end of β-strand βeII involving residues 158−161 which have the following sequence: -Ala-Ala-Ala-Thr- (Figure 4c,d).
The twisted β-sheet motif of domain I possesses a notable difference in its tertiary structure when compared with the PV, HAV, and HRV 3C proteases which, in contrast, display a complete β-barrel in this domain. The long loop, lpcI, consisting of 19 residues (Thr 29−Ile 47) connecting βcI to βdI, is well-defined by the electron density but lacks any distinguishable secondary structure, and it seems plausible that if it were to possess more β-strand-like character, domain I might adopt a complete β-barrel fold. Further evidence that domain I can be considered an incomplete β-barrel rather than a twisted β-sheet is offered in the form of a marked hydrophobic core, a common feature of β-barrel motifs. The core of this domain is comprised of the following, predominantly aromatic residues: Phe 12, Trp 19, Phe 25, Ile 32, Phe 39, Phe 40, Ile 47, Ile 49, Phe 58, Phe 60, and Ile 64. Interestingly, one end of the hydrophobic core is “capped” by Leu 180 at the C-terminal end of the protein. In contrast, domain II displays a complete β-barrel, the core of which is comprised of the following predominantly aliphatic residues, Val 85, Ile 87, Leu 95, Leu 97, Val 99, Leu 121, Val 152, Val 167, and Val 171, and the somewhat less hydrophobic residues, Tyr 143, Tyr 145, and His 157. This β-barrel motif is important in the orientation of the residues Tyr 143 and His 157 that are intrinsic to substrate binding at the S1 subsite. In addition, this domain contains a number of buried sulfur-containing residues, as will be discussed later.
Gel-filtration studies showed that the SV3CP enzyme is predominantly dimeric in solution. Accordingly, there are two monomers in the crystallographic asymmetric unit which have an interface that is maintained by a network of hydrogen bonds between residues of αaI, βaII, lpaII and βbII and lpcII (Figure 5) resulting in a total buried surface area of 2353 Å2, which is suggestive of a physiologically relevant dimer. However, the SV3CP dimer interface presented here is at odds with that of the 3C protease belonging to the Norwalk noroviral strain (18). This structure suggests that residues on an entirely different face of the molecule are involved in dimer formation, which raises the possibility that the dimer interface of the protease might differ between noroviral strains.
Figure 5.

The dimeric arrangement of SV3CP observed in the crystal structure. Residues involved in the interface are shown in red for molecule 1 and green for molecule 2.
Cysteine Cluster in Domain II
Domain II contains a cluster of cysteine residues (comprising Cys 77, Cys 83, Cys 154, and Cys 169) which is reminiscent of a “cysteine cage” structure that is normally involved in metal coordination. However, the distances between the cysteine sulfurs are in the range of 4−6 Å and are therefore slightly greater than would be expected for a true cysteine cage (approximately 3.5 Å), which probably stems from the absence of a coordinating metal ion. Indeed, the 2Fo − Fc and Fo − Fc density maps revealed a complete lack of density for a coordinating metal ion, and the enzyme showed activity without any exogenous metal ions. As a result it seems more appropriate to call it a “cysteine cluster” rather than a cysteine cage, and its structural role might be quite conventional. Located at the opposite end of the domain II β-barrel to the active site, the cysteines contribute to the hydrophobic core of the barrel. Strengthening the hydrophobic nature of this cluster is a further sulfur-containing hydrophobic residue, Met101. Interestingly, the cysteine/methionine cluster is conserved in a number of norovirus 3C proteases.
Flexible Active Site Loop of Domain II
Domain II possesses a long flexible loop between βcII and βdII which includes residues Thr123 to Asp131 that were refined in two conformations within molecule 1 of the dimer and a third conformation within molecule 2. The position of this loop close to the active site suggests that it may interact with substrate P′ residues, although the details of such interactions cannot be discerned at this stage since the inhibitor does not include residues on the C-terminal side of the scissile bond. Hence, this loop might become more ordered when the P1′ region of the natural substrate is bound. The Chiba and Norwalk 3C protease structures (17,18) lack any bound substrate analogue, and both show similar flexibility in this region, suggesting that the observed disorder is not simply an artifact of crystal packing with SV3CP and that it may have a functional significance. The same loop region is also involved in the dimer interface, implying that it could be involved in quaternary structural changes, should any take place upon substrate binding or during catalysis. Inspection of the dimer interface reveals that three residues in this loop, Thr 123, Ala 125, and Asp 131, make significant intersubunit interactions. Thr 123 interacts directly with Ser 84 that lies within the βaII strand, Ala 125 interacts via a water molecule with Val 82 that also lies within βaII, and Asp 131 interacts directly with Trp 6 and via water molecules with Thr 4 and Leu 5 that all lie within the N-terminal α-helix, αaI.
Binding of the Peptidyl Portion of the Inhibitor
For ease of identification, the residues of the peptide portion of the inhibitor MAPI are named E5, F4, Q3, L2, and Q1, corresponding to residues in the N- to C-terminal direction, Glu, Phe, Gln, Leu, and Gln, respectively. The corresponding enzyme subsites are named S5 to S1 according to the nomenclature of Schechter and Berger (39) in which the substrate residues are labeled Pn−Pn′, where P1−P1′ represents the scissile dipeptide.
As is typical of chymotrypsin-like proteases, the enzyme binds the inhibitor via a hydrogen-bonding network in the manner of an antiparallel β-sheet (Figure 4d). Although such interactions involving the main-chain atoms of the substrate (or inhibitor) are not directly responsible for defining specificity, they are likely to be of great importance in correctly positioning the substrate backbone in relation to the catalytic residues. The key interactions are between Ala 160 and MAPI Q3 and between Ala 158 and MAPI Q1. The MAPI Q3 backbone carbonyl O and NH hydrogen bond to the peptide NH and carbonyl O of Ala 160, respectively, as is typical of an antiparallel β-sheet. A further main-chain to main-chain hydrogen bond exists between the carbonyl O of Ala 158 and the peptide NH of MAPI Q1. Additional hydrogen bonds with the inhibitor include the peptide NH of MAPI L2 and the Oε1 of Gln 110, the carbonyl O of MAPI F4, and the peptide NH of Gln 110 via a water molecule, as well as the peptide NH of MAPI F4 and the carbonyl O of Arg 108, also via a water molecule.
The S5 pocket is rather poorly defined due to the limited number of interactions at this subsite, although kinetic measurements demonstrate that occupation of the S5 site affects the rate of substrate cleavage significantly, since substrate lacking P5 (Ac-FQLQ-pNA) has a much lower kcat/KM than Ac-EFQLQ-pNA (Table 2). The main-chain carbonyl O of E5 hydrogen bonds via a surface water molecule to the main-chain NH of Lys162, and a second interaction, mediated by a water molecule, occurs between the side-chain carboxylate of E5 and the main-chain NH of Gln 110. Comparison of residues at the P5 position of the five points of cleavage within the natural substrate, Asp, Glu (×2), Ser, and Lys, reveals a diversity of residue types (10). The ability of the enzyme to accommodate a range of residues at the P5 position is presumably due to the lack of specific side-chain interactions at this subsite, although the presence of a P5 residue significantly enhances the rate of cleavage. The observation that this subsite is at the extremity of the active site cleft is consistent with the kinetic data which show no enhancement in kcat/KM when the peptide is extended to P6.
In spite of its surface-exposed location, the S4 binding pocket is comprised predominantly of hydrophobic residues Met 107, Ile 109, and Val 168 along with the aliphatic side-chain moieties of Thr 161 and Thr 166, all of which interact with the Phe side chain of the inhibitor. Sequence comparison of the five ORF 1 polyprotein cleavage sites shows that the following predominantly hydrophobic residues are present at the P4 position: Phe (×2), Ala, Ile, and Thr (10). The P4 main-chain NH and CO groups interact by hydrogen bonding via ordered water molecules to the carbonyl O of Arg 108 and main-chain NH of Gln 110, respectively.
For the Chiba virus 3C protease structure (17), it has been suggested that the side chains of Gln 110 and Lys 162 act to clamp substrate at the P4 position by forming a bridge under which the main chain of the substrate lies, and this feature appears to be present in the SV3CP structure (Figure 6) although the two residues are only interacting in one of the monomers, and they appear to have a greater role in binding at the adjacent P3 site. In the monomer where Gln 110 and Lys 162 interact, the side chain of Gln P3 is oriented toward the side chain of Gln P1 so that they form a hydrogen bond. In the other monomer, where Gln 110 and Lys 162 do not interact, the P3 side chain is oriented away from the catalytic center (Figure 6).
Figure 6.

Coordination of the inhibitor side chain at S3 in both monomers. The disorder of the MAPI Q3 side chain is indicated, as is that of the Gln 110/Lys 162 clamp. For molecule 1 of the SV3CP dimer, the Gln 110 and Lys 162 side chains are shown in blue, and MAPI is shown in light blue. For molecule 2 of the SV3CP dimer, the Gln 110/Lys 162 clamp and the corresponding rendered surface are shown in green, with MAPI in pink. Movement of the Lys 162 side chain from the open “skewed” position to the closed “bridged” position appears to be coupled to rotation of the χ1 torsion angle of the MAPI Q3 side chain. The resultant conformation of Q3 is stabilized by hydrogen bonding between side-chain O of MAPI Q3 and side-chain N of MAPI Q1.
The P3 main chain is held by an antiparallel β-sheet-like hydrogen-bonding pattern involving its main-chain carbonyl and amide which interact with the main-chain NH and CO groups of Ala 160, respectively, as shown in Figure 4d. This antiparallel β-sheet-like hydrogen-bonding pattern is typical of other chymotrypsin-like proteases. Comparison of the residues at the P3 position of the five cleavage sites of the natural substrate, His, Gln, Thr (×2), and Ser, reveals a diversity of amino acid types, suggesting that the nature of the side chain at P3 is less important for substrate binding than the main-chain interactions (10). Accordingly, the structure clearly shows the side-chain group of Gln P3 is not positioned within a defined binding pocket and the side chain can adopt more than one conformation, as shown in Figure 6.
The buried, hydrophobic, and relatively large S2 binding pocket is comprised of the following amino acids: His 30 (one of the catalytic residues), Ile 109, and Val 114, as well as the aliphatic moieties of Gln 110 and Arg 112. The generally hydrophobic nature of the pocket is underlined by the absence of any solvent molecules and a relative dearth of hydrogen bond interactions between the enzyme and the inhibitor, although the main-chain NH of P2 interacts via hydrogen bonding to the side-chain Oε1 of Gln 110. Comparison of the five points of cleavage in the ORF 1 polyprotein reveals that the residues at the P2 position always possess a hydrophobic side chain: Leu (×3), Met, and Phe (10). Accordingly, the large size of the S2 pocket allows the P2 Leu side chain to be in the gauche positive (g+) orientation in molecule 1 and in the trans (t) conformation in molecule 2 of the dimer (Figure 6). This bifurcation is consistent with the known ability of the S2 pocket to accommodate larger hydrophobic residues such as Met and Phe.
The S1 and S1′ binding pockets arguably play the most important role in substrate sequence recognition and orientation. Comparison of the five dipeptide sequences that occupy the S1 and S1′ sites, QG (×2), EG (×2), and EA, suggests that the enzyme will accept only a Gln or Glu at the S1 site and only a Gly or Ala at the S1′ site (10). Inspection of the structure shows the S1 site is relatively large and able to accommodate the Gln side chain of the inhibitor in a fully extended conformation. It is clear from the biochemical evidence alone that the S1 site differs quite dramatically from the S1′ binding site which is only able to accommodate the small side chains of glycine and alanine.
Structurally, the S1 binding site is less distinctive than the S2 and S4 sites, although the P1 Gln side chain can clearly be seen to lie parallel to and sandwiched between loop lpcII and the C-terminal region of the β-strand βeII (Figure 7). The residues Thr 134 of lpcII and His 157 of βeII are located at the end of the S1 binding pocket and hydrogen bond with the P1 Gln side chain (presumably Glu at this position can make the same interactions). Thr 134 and His 157 are therefore likely to be of great importance in substrate recognition and in defining substrate specificity at the S1 site. The important role played by His 157 has been demonstrated by mutational studies (19) which showed that a His157Ala mutant displays a reduced rate of substrate turnover, presumably due to a reduction in the affinity for substrate. It also appears that Tyr 143 may play an important role in substrate interactions at the S1 site, though not directly. The structure suggests that, for correct orientation of the His 157 side chain, the OH group of Tyr 143 must hydrogen bond with the Nδ1 of His 157 so that the imidazole is oriented to hydrogen bond with the substrate side chain occupying the S1 site.
Figure 7.

The S1−S1′ binding sites of SV3CP. The residues involved in coordination of MAPI at the S1 binding site including Gly 137 and His 157 which hydrogen bond with the Q1 side chain. Gly 137 is also a member of the putative oxyanion hole. The loop lpcII and the β-strand βeII form a cleft that accommodates the side chain of P1.
Inspection of the map reveals a paucity of electron density for the ethyl ester extension of the inhibitor, suggesting that it has been hydrolyzed to yield a carboxylate group which appears to interact with the catalytic His 30 and Gly 137. The Nε2 of His 30 hydrogen bonds to the carboxylate via water molecules whereas the main-chain amide nitrogen of Gly 137 makes a direct interaction. The latter residue is likely to constitute part of the oxyanion-binding hole of the enzyme. Hence it appears that fortuitous hydrolysis of the ethyl ester moiety to generate a C-terminal carboxylate provides a unique structural insight into the nature of the oxyanion hole. The pseudo-first-order rate constant for base-catalyzed hydrolysis of n-alkyl carboxylic acid esters at the pH of crystallization (pH 8.5) is approximately 0.03 day−140,41, which suggests that, following formation of the enzyme−inhibitor complex, the ethyl ester group will be substantially hydrolyzed during the 3 week crystallization period at ambient temperature by nonenzymic processes. However, there is weak electron density for the ethyl group in the structure, suggesting that although the extent of hydrolysis is significant, it is not complete.
In both molecules of the SV3CP−MAPI complex, the side chain of His 30 is separated by too great a distance from the side chains of Glu 54 and Cys 139 to allow the necessary hydrogen bonding required to form the putative catalytic triad. With MAPI covalently bound to the Sγ of Cys 139, it appears that the His 30 side chain adopts a conformation pointing away from Cys 139 and Glu 54. Indeed, the His 30 side chain appears to interact electrostatically with the C-terminal carboxylate of the hydrolyzed inhibitor. In the absence of the inhibitor, the His 30 side chain would most likely adopt a conformation in which the imidazole is within hydrogen-bonding distance of both Cys 139 and Glu 54. This agrees with the biochemical and structural studies of a related enzyme (17,19) which showed that it is indeed His 30 and not His 157 (which is also close to Cys 139) that participates in catalysis.
Prediction of the S′ Substrate Binding Sites of SV3CP
Visual inspection of a surface-rendered model of the SV3CP-MAPI structure suggests that the substrate P′ residues may be bound in a rather deep surface cleft that follows on from the S5−S1 sites occupied by MAPI. Hence, the S1′ binding site is likely to consist of residues Ser 14, Val 31, and Gly 137 and the catalytic residue Cys 139. Biochemical evidence suggests the P1′ residue of the substrate always possesses a small side-chain group, therefore requiring only a small S1′ pocket in which to be accommodated. The relatively close proximity of Ser 14, Val 31, Gly 137, and Cys 139 would provide such a small-sized S1′ binding pocket.
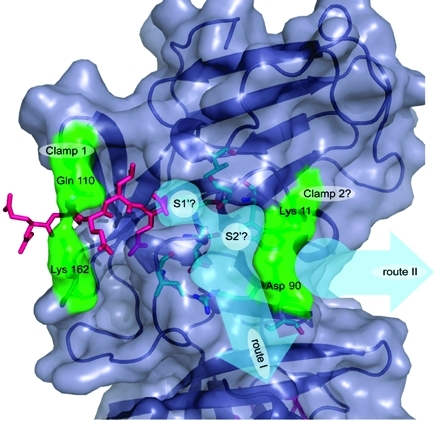
The subsequent S′ sites are less easy to define although it is possible to suggest two putative routes along which they may lie. As discussed above, the flexible loop region comprised of residues Thr 123 to Asp 131 may contribute to substrate binding at the S′ sites. The most obvious route for the P′ residues directly follows the deep surface cleft mentioned in the previous paragraph; this is shown as “route I” in Figure 8. If substrate were to lie along this route, the residues that would interact with it are Lys 11, Ser 14, Trp 16, Arg 89, Asp 90, Ser 91, Ile 135, Gly 137, and Asp 138 as well as residues in the disordered loop involving 123−131. Alternatively, substrate could lie along the route named “route II” in Figure 8, although this is dependent upon the conformations of Lys 11 and Asp 90 when ligand is bound. From visual inspection of the structure it seems possible that these residues may operate as a clamp in a similar manner to the Gln 110/Lys 162 clamp. If so, the putative Lys 11/Asp 90 clamp may bind the substrate P3′ or P4′ residues. However, in the absence of a structure for a ligand occupying the S′ sites of any noroviral protease, this hypothesis remains speculative. For the substrate to follow the alternative route (route I) it would have to kink appreciably at P1′ and P2′ (see Figure 8), which would be entirely consistent with the preference of the enzyme for the conformationally disruptive residues Gly and Pro at these positions of the substrate.
Figure 8.

The predicted S′ binding sites of SV3CP. The S1′ site is likely to be comprised of the residues Ser 14, Val 31, Gly 137, and Cys 139. The subsequent P′ residues may lie along one of two routes labeled “route I” and “route II” (shown as thick pale blue arrows). The residues predicted to form the S′ sites of “route I” are colored in cyan. The alternative route (II) would involve the substrate being held at P3′ or P4′ by the putative clamp formed of Lys 11 and Asp 90. MAPI is shown in pink.
Discussion
The use of a peptide irreversible inhibitor with a sequence based on that of the most rapidly cleaved site of the viral polyprotein (EFQLQ-GKMYD) has permitted the detailed nature of substrate binding by a noroviral 3C protease to be elucidated structurally for the first time. This work establishes that the N-terminal Glu of the peptide is largely outside the active site cleft, and relatively few direct contacts with the enzyme are evident. In contrast, the P4 residue, Phe, makes many contacts with the enzyme, and the observation that the chromogenic peptide Ac-FQLQ-pNA is cleaved but that Ac-QLQ-pNA is not suggests that the S4 binding site plays a very important role in productive substrate binding. The main chain of the substrate at P3 interacts extensively with the enzyme, although the side chain has some flexibility. The known cleavage sites for the SV3C protease possess a hydrophobic residue in the S2 subsite, and accordingly the structure confirms this is a region of extensive contacts between the enzyme and substrate, which is typical of other cysteine proteases. Within the viral polyprotein this residue may be Leu (three occurrences), Met (one occurrence), or Phe (one occurrence) (10). In the current study the reason for the preference for a hydrophobic residue is clear since there are several nonpolar residues making up the S2 site, including the hydrophobic side chains of Ile 109, Val 114, and Ala 159. In addition, the methylene chains of Gln 110 and Arg 112 form opposite sides of the S2 site.
SV3C protease is relatively specific for the residues on either side of the scissile bond and can only cleave significantly when the P1 residue is Gln or Glu and the P1′ residue is very small such as Gly or Ala, the optimal combination being Gln-Gly. The most important residues at the far extremity of the S1 site appear to be Thr 134 and His 157 which make hydrogen bonds with the amide group of Gln.
The relative lack of specificity of the enzyme at several subsites close to the scissile bond can be rationalized by the structural studies, which demonstrate that these pockets allow the ligand side chains to adopt more than one conformation. Similar conformational heterogeneity is observed for residues of the enzyme that form these pockets.
One further feature of the enzyme which, although not absolutely conserved in noroviral proteases, raises a number of intriguing questions is the buried cysteine cluster in domain II. Perhaps the sensitivity of the enzyme to oxidizing environments can be partly attributed to disulfide formation. In addition, it may be that these residues have a physiological role in the viral life cycle as a “redox switch”, possibly preventing excessive proteolysis following maturation of the virus particle.
Based partly on the accepted mechanism of catalysis by cysteine proteases, the SV3CP structure presented here sheds light on a number of specific details of the enzyme-catalyzed reaction. As described above, the structure establishes that the catalytically important members of the active site are Cys 139, His 30, and potentially Glu 54. The latter residue is likely to facilitate catalysis by immobilizing the His 30 side chain via a hydrogen bond such that the imidazole can act as a Brønsted−Lowry base to deprotonate Cys 139. With the correct positioning of the substrate within the active site cleft, the scissile bond is brought into sufficiently close proximity with Cys 139 to facilitate nucleophilic attack by the thiolate on the carbonyl C of the scissile peptide and yield an unstable, covalently bound tetrahedral intermediate. During nucleophilic attack on the substrate carbonyl group, the shift of negative charge onto the carbonyl O would probably be favored by hydrogen bonds involving the backbone amide nitrogens of Cys 139 and Gly 137 that are likely to form the oxyanion hole. We propose that Asp 138 does not contribute to the oxyanion hole, as suggested by others (18), although we suggest that it is vital in orientation of the oxyanion hole members through indirect means. The peptide NH of Asp 138 hydrogen bonds to the main-chain carbonyl O of Ile 135, thereby stabilizing the type II reverse turn that correctly orients the peptide N−H group of Gly 137 toward the main-chain nitrogen of Cys 139 to form the oxyanion hole. Collapse of the tetrahedral intermediate complex is promoted by His 30, now acting as a Brønsted−Lowry acid, protonating the substrate amide N. This results in formation of an acyl-enzyme intermediate and the amine product (i.e., the C-terminal portion of the substrate) which dissociates from the active site cleft. Diffusion of a water molecule into the catalytic site followed by nucleophilic attack on the substrate carbonyl carbon, promoted by His 30 and potentially Glu 54, results in hydrolysis of the acyl-enzyme and release of the N-terminal portion of the substrate.
Further work on this enzyme will structurally define the binding pockets forming the “prime” side of the active site cleft, will allow the relevance of the putative dimeric states observed in the crystal structures to be assessed, and will aid the development of further inhibitory compounds with therapeutic potential.
Supporting Information Available
Full details of the synthesis of the Michael-acceptor polypeptide inhibitor (MAPI). This material is available free of charge via the Internet at http://pubs.acs.org.
We acknowledge the School of Biological Sciences, University of Southampton, for a studentship to R.J.H., Hope (Southampton General Hospital) for a grant to P.M.S.-J., the Wellcome Trust for a program grant to I.N.C. and P.R.L. (No. 086112), and the ESRF (Grenoble, France) for provision of synchrotron beamtime and travel support.
Footnotes
Abbreviations: Å, 10−10 m; Ac, acetyl; Da, dalton; ESRF, European Synchrotron Radiation Facility; LB, Luria−Bertani; MALDI-Q-TOF-MS, matrix-assisted laser desorption ionization quadrapole time-of-flight mass spectrometry; MBHA, 4-methylbenzhydrylamine hydrochloride; ORF, open reading frame; pNA, p-nitroaniline; VP, viral protein.
Supplementary Material
References
- Clarke I. N., and Lambden P. R. (2005) Human enteric RNA viruses: noroviruses and sapoviruses, in Virology (Mahy B. W. J., and ter Meulen V., Eds.) pp 911−931, Hodder Arnold, London. [Google Scholar]
- Green K. Y. (2007) Caliciviridae: the noroviruses, in Fields Virology (Knipe D. M., and Howley P. M., Eds.) pp 949−980, Kluwer, Philadelphia, PA. [Google Scholar]
- Duizer E.; Schwab K. J.; Neill F. H.; Atmar R. L.; Koopmans M. P. G.; Estes M. K. (2004) Laboratory efforts to cultivate noroviruses. J. Gen. Virol. 85, 79–87. [DOI] [PubMed] [Google Scholar]
- Oliver S. L.; Asobayire E.; Dastjerdi A. M.; Bridger J. C. (2006) Genomic characterization of the unclassified bovine enteric virus Newbury agent-1 (Newbury1) endorses a new genus in the family Caliciviridae. Virology 350, 240–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smiley J. R.; Chang K. O.; Hayes J.; Vinje J.; Saif L. J. (2002) Characterization of an enteropathogenic bovine calicivirus representing a potentially new calicivirus genus. J. Virol. 76, 10089–10098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambden P. R.; Caul E. O.; Ashley C. R.; Clarke I. N. (1993) Sequence and genome organization of a human small round-structured (Norwalk-like) virus. Science 259, 516–519. [DOI] [PubMed] [Google Scholar]
- Lambden P. R.; Liu B. L.; Clarke I. N. (1995) A conserved sequence motif at the 5′ terminus of the Southampton virus genome is characteristic of the Caliciviridae. Virus Genes 10, 149–152. [DOI] [PubMed] [Google Scholar]
- Bertolotti-Ciarlet A.; Crawford S. E.; Hutson A. M.; Estes M. K. (2003) The 3′ end of Norwalk virus mRNA contains determinants that regulate the expression and stability of the viral capsid protein VP1: a novel function for the VP2 protein. J. Virol. 77, 11603–11615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B. L.; Clarke I. N.; Lambden P. R. (1996) Polyprotein processing in Southampton virus: identification of 3C-like protease cleavage sites by in vitro mutagenesis. J. Virol. 70, 2605–2610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B. L.; Viljoen G. J.; Clarke I. N.; Lambden P. R. (1999) Identification of further proteolytic cleavage sites in the Southampton calicivirus polyprotein by expression of the viral protease in E. coli. J. Gen. Virol. 80, 291–296. [DOI] [PubMed] [Google Scholar]
- Kuyumcu-Martinez M.; Belliot G.; Sosnovtsev S. V.; Chang K. O.; Green K. Y.; Lloyd R. E. (2004) Calicivirus 3C-like proteinase inhibits cellular translation by cleavage of poly(A)-binding protein. J. Virol. 78, 8172–8182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews D. A.; Smith W. W.; Ferre R. A.; Condon B.; Budahazi G.; Sisson W.; Villafranca J. E.; Janson C. A.; McElroy H. E.; Gribskov C. L.; Worland S. (1994) Structure of human rhinovirus 3C protease reveals a trypsin-like polypeptide fold, RNA-binding site, and means for cleaving precursor polyprotein. Cell 77, 761–771. [DOI] [PubMed] [Google Scholar]
- Cordingley M. G.; Callahan P. L.; Sardana V. V.; Garsky V. M.; Colonno R. J. (1990) Substrate requirements of human rhinovirus 3C protease for peptide cleavage in vitro. J. Biol. Chem. 265, 9062–9065. [PubMed] [Google Scholar]
- Dragovich P. S.; Webber S. E.; Babine R. E.; Fuhrman S. A.; Patick A. K.; Matthews D. A.; Lee C. A.; Reich S. H.; Prins T. J.; Marakovits J. T.; Littlefield E. S.; Zhou R.; Tikhe J.; Ford C. E.; Wallace M. B.; Meador J. W.; Ferre R. A.; Brown E. L.; Binford S. L.; Harr J. E. V.; DeLisle D. M.; Worland S. T. (1998) Structure-based design, synthesis, and biological evaluation of irreversible human rhinovirus 3C protease inhibitors. 1. Michael acceptor structure-activity studies. J. Med. Chem. 41, 2806–2818. [DOI] [PubMed] [Google Scholar]
- Dragovich P. S.; Webber S. E.; Babine R. E.; Fuhrman S. A.; Patick A. K.; Matthews D. A.; Reich S. H.; Marakovits J. T.; Prins T. J.; Zhou R.; Tikhe J.; Littlefield E. S.; Bleckman T. M.; Wallace M. B.; Little T. L.; Ford C. E.; Meador J. W.; Ferre R. A.; Brown E. L.; Binford S. L.; DeLisle D. M.; Worland S. T. (1998) Structure-based design, synthesis, and biological evaluation of irreversible human rhinovirus 3C protease inhibitors. 2. Peptide structure-activity studies. J. Med. Chem. 41, 2819–2834. [DOI] [PubMed] [Google Scholar]
- Dragovich P. S.; Prins T. J.; Zhou R.; Johnson T. O.; Hua Y.; Luu H. T.; Sakata S. K.; Brown E. L.; Maldonado F. C.; Tuntland T.; Lee C. A.; Fuhrman S. A.; Zalman L. S.; Patick A. K.; Matthews D. A.; Wu E. Y.; Guo M.; Borer B. C.; Nayyar N. K.; Moran T.; Chen L. J.; Rejto P. A.; Rose P. W.; Guzman M. C.; Dovalsantos E. Z.; Lee S.; McGee K.; Mohajeri M.; Liese A.; Tao J. H.; Kosa M. B.; Liu B.; Batugo M. R.; Gleeson J. P. R.; Wu Z. P.; Liu J.; Meador J. W.; Ferre R. A. (2003) Structure-based design, synthesis, and biological evaluation of irreversible human rhinovirus 3C protease inhibitors. 8. Pharmacological optimization of orally bioavailable 2-pyridone-containing peptidomimetics. J. Med. Chem. 46, 4572–4585. [DOI] [PubMed] [Google Scholar]
- Nakamura K.; Someya Y.; Kumasaka T.; Ueno G.; Yamamoto M.; Sato T.; Takeda N.; Miyamura T.; Tanaka N. (2005) A norovirus protease structure provides insights into active and substrate binding site integrity. J. Virol. 79, 13685–13693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeitler C. E.; Estes M. K.; Prasad B. V. V. (2006) X-ray crystallographic structure of the Norwalk virus protease at 1.5 Å resolution. J. Virol. 80, 5050–5058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Someya Y.; Takeda N.; Miyamura T. (2002) Identification of active-site amino acid residues in the Chiba virus 3C-like protease. J. Virol. 76, 5949–5958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merrifield R. B. (2007) Solid phase peptide synthesis. I. The synthesis of a tetrapeptide. J. Am. Chem. Soc. 85, 2149–2154. [Google Scholar]
- Whitmore A. J.; Daniel R. M.; Petach H. H. (1995) A general method for the synthesis of peptidyl substrates for proteolytic enzymes. Tetrahedron Lett. 36, 475–476. [Google Scholar]
- Kaspari A.; Schierhorn A.; Schutkowski M. (1996) Solid-phase synthesis of peptide-4-nitroanilides. Int. J. Pept. Protein Res. 48, 486–494. [DOI] [PubMed] [Google Scholar]
- Rink H. (1987) Solid-phase synthesis of protected peptide fragments using a trialkoxy-diphenyl-methylester resin. Tetrahedron Lett. 28, 3787–3790. [Google Scholar]
- Govardhan C. P.; Abeles R. H. (1996) Inactivation of cysteine proteases. Arch. Biochem. Biophys. 330, 110–114. [DOI] [PubMed] [Google Scholar]
- Leslie A. G. W. (2006) The integration of macromolecular diffraction data. Acta Crystallogr. D62, 48–57. [DOI] [PubMed] [Google Scholar]
- Evans P. (2006) Scaling and assessment of data quality. Acta Crystallogr. D62, 72–82. [DOI] [PubMed] [Google Scholar]
- (1994) The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D50, 760–763. [DOI] [PubMed] [Google Scholar]
- Terwilliger T. C.; Berendzen J. (1999) Automated MAD and MIR structure solution. Acta Crystallogr. D55, 849–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowtan K. (1994) DM: an automated procedure for phase improvement by density modification, Joint CCP4 ESF-EACBM Newsletter on Protein Crystallography, Vol. 31, pp 34−38, Daresbury Laboratory, Warrington, U.K. [Google Scholar]
- Levitt D. G. (2001) A new software routine that automates the fitting of protein X-ray crystallographic electron-density maps. Acta Crystallogr. D57, 1013–1019. [DOI] [PubMed] [Google Scholar]
- Sheldrick G. M.; Schneider T. R. (1997) SHELXL: high-resolution refinement. Methods Enzymol. 277, 319–343. [PubMed] [Google Scholar]
- Brünger A. T.; Adams P. D.; Clore G. M.; DeLano W. L.; Gros P.; Grosse-Kunstleve R. W.; Jiang J.-S.; Kuszewski J.; Nilges N.; Pannu N. S.; Read R. J.; Rice L. M.; Simonson T.; Warren G. L. (1998) Crystallography and NMR system (CNS): a new software system for macromolecular structure determination. Acta Crystallogr. D54, 905–921. [DOI] [PubMed] [Google Scholar]
- Chen V. B.; Arendall W. B.; Headd J. J.; Keedy D. A.; Immormino R. M.; Kapral G. J.; Murray L. W.; Richardson J. S.; Richardson D. C. (2010) MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D66, 12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosimann S. C.; Cherney M. M.; Sia S.; Plotch S.; James M. N. G. (1997) Refined X-ray crystallographic structure of the poliovirus 3C gene product. J. Mol. Biol. 273, 1032–1047. [DOI] [PubMed] [Google Scholar]
- Bergmann E. M.; Mosimann S. C.; Chernaia M. M.; Malcolm B. A.; James M. N. G. (1997) The refined crystal structure of the 3C gene product from hepatitis A virus: specific proteinase activity and RNA recognition. J. Virol. 71, 2436–2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergmann E. M.; Cherney M. M.; McKendrick J.; Frormann S.; Luo C.; Malcolm B. A.; Vederas J. C.; James M. N. G. (1999) Crystal structure of an inhibitor complex of the 3C proteinase from hepatitis A virus (HAV) and implications for the polyprotein processing in HAV. Virology 265, 153–163. [DOI] [PubMed] [Google Scholar]
- Matthews D. A.; Dragovich P. S.; Webber S. E.; Fuhrman S. A.; Patick A. K.; Zalman L. S.; Hendrickson T. F.; Love R. A.; Prins T. J.; Marakovits J. T.; Zhou R.; Tikhe J.; Ford C. E.; Meador J. W.; Ferre R. A.; Brown E. L.; Binford S. L.; Brothers M. A.; DeLisle D. M.; Worland S. T. (1999) Structure-assisted design of mechanism-based irreversible inhibitors of human rhinovirus 3C protease with potent antiviral activity against multiple rhinovirus serotypes. Proc. Natl. Acad. Sci. U.S.A. 96, 11000–11007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allaire M.; Chernaia M. M.; Malcolm B. A.; James M. N. G. (1994) Picornaviral 3C cysteine proteinases have a fold similar to chymotrypsin-like serine proteinases. Nature 369, 72–76. [DOI] [PubMed] [Google Scholar]
- Schechter I.; Berger A. (1967) On the size of the active site in proteases. I. Papain. Biochem. Biophys. Res. Commun. 27, 157–162. [DOI] [PubMed] [Google Scholar]
- Larson R. A., and Weber E. J. (1994) Reaction mechanisms in environmental organic chemistry, Lewis Publishers, Boca Raton, FL. [Google Scholar]
- Schwarzenbach R. P., Gschwend P. M., and Imboden D. M. (1993) Environmental organic chemistry, Wiley-Interscience, Hoboken, NJ. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.