

Abstract
Microsomal glutathione transferase 1 (MGST1) is representative of a superfamily of membrane proteins where different members display distinct or overlapping physiological functions, including detoxication of reactive electrophiles (glutathione transferase), reduction of lipid hydroperoxides (glutathione peroxidase), and production of leukotrienes and prostaglandin E. It follows that members of this superfamily constitute important drug targets regarding asthma, inflammation and the febrile response. Here we propose that this superfamily consists of a new class of membrane proteins built on a common left-handed four-helix bundle motif within the membrane, as determined by electron crystallography of MGST1 at 6 Å resolution. Based on the 3D map and biochemical data we discuss a model for the membrane topology. The 3D structure differs significantly from that of soluble glutathione transferases, which display overlapping substrate specificity with MGST1.
Keywords: electron crystallography/membrane protein/microsomal glutathione transferase/3D structure
Introduction
Lipids are key components of cell signalling but are also vulnerable to oxidative stress. Membrane proteins belonging to a recently characterized superfamily named MAPEG (membrane-associated proteins in eicosanoid and glutathione metabolism) (Jakobsson et al., 1999a) have evolved divergent functions to protect against reactive lipid intermediates (lipid hydroperoxides and hydroxyalkenals) or indeed harness these molecules for key physiological functions (leukotrienes and prostaglandins). The fundamental nature of this superfamily is underscored by its wide distribution from prokaryotes to humans, and the fact that a recently identified prostaglandin E synthase (Jakobsson et al., 1999b) is one of the genes most strongly upregulated during p53-mediated apoptosis in a colon cancer cell line, referred to as PIG12 (Polyak et al., 1997). Furthermore, prostaglandin E synthase mRNA expression is upregulated in β-amyloid-treated rat astrocytes as well, suggesting a possible role in Alzheimer’s disease (Satoh et al., 2000). The most thoroughly studied member of this superfamily is MGST1, an abundant detoxication enzyme displaying both glutathione S-transferase (GST) and peroxidase activities. The structure now obtained provides a basis for interpreting previous results and indeed changes our view of the topology and structure of the MAPEG proteins.
Microsomal glutathione transferase 1 (MGST1) is a homotrimeric protein (Mr = 51 kDa) that is present at high concentrations both in the endoplasmic reticulum and the mitochondrial outer membrane. A reason for the two different locations is perhaps the need for protection of these membranes against oxidative stress. Abundant presence at two different locations is an unusual feature for a membrane protein. It was shown earlier that the homotrimer binds one glutathione (GSH) molecule and that the hydrophobic xenobiotic binding site faces the cytosol (Andersson et al., 1994; Sun and Morgenstern, 1997). The active site of MGST1 might thus benefit from membrane proximity and/or a xenobiotic substrate gradient extending into the cytosol (as hydrophobic molecules are concentrated in the membrane). It is also possible that specific substrates, such as phospholipid hydroperoxides, can interact while still partly embedded in the membrane.
Depending on the prediction programme applied, three or four transmembrane regions can be seen in hydropathy plots. One transmembrane stretch was shown by trypsin cleavage of the trimer to be situated between Lys4 and Lys41, with the N-terminus on the lumenal side of the membrane (Andersson et al., 1994).
In the projection maps of two different 2D crystal forms (Hebert et al., 1997; Schmidt-Krey et al., 1999), three densities per monomer were identified with a diameter and spacing typical of perpendicular α-helices (Henderson and Unwin, 1975; Henderson et al., 1990; Kühlbrandt et al., 1994; Cheng et al., 1997; Li et al., 1997; Walz et al., 1997; Auer et al., 1998; Rhee et al., 1998; Unger et al., 1999; Williams, 2000). An additional, longer density on the periphery of the monomer within the trimer could not be identified, but was thought to arise from secondary transmembrane structure as well, due to the mostly hydrophobic nature of the protein (Morgenstern et al., 1985).
We have now calculated a 3D structure of p6 2D crystals of MGST1 from images as well as electron diffraction patterns of specimens tilted between 0 and 60°.
Results
The crystals grown at regular intervals and from several protein purification batches displayed no significant variation in frequency, quality and unit cell parameters (Schmidt-Krey et al., 1998). Amplitude data from electron diffraction patterns of specimens tilted up to 60° were merged at 3.5 Å resolution whereas the image data within the same tilt range were truncated at 6 Å. The vertical resolution was 12 Å as estimated by the point spread function (see Figure 3). The 3D data were sampled from 84 unique lattice lines (Table I; Figure 1). RFriedel and Rmerge of the electron diffraction data merged at 3.5 Å were 26.5 and 34.8%, respectively. The residuals of the experimental phase data from the merged data set calculated in resolution ranges were: 34.1 (100–14.1 Å), 32.2 (14.1–10.0 Å), 46.5 (10.0–8.2 Å), 41.8 (8.2–7.0 Å) and 50.6 (7.0–6.0 Å).

Fig. 3. The MGST1 trimer viewed along the plane of the membrane. The proposed cytoplasmic domain containing the active site is in the upper part of the figure. The anisotropy of the resolution is illustrated by the elongation of the point spread function along the direction perpendicular to the plane of the membrane (inset). In the plane, the resolution is isotropic. Scale bar, 5 Å.
Table I. Crystallographic data.
| Symmetry | p6 |
| Lattice constants | a = b = 81.8 Å, γ = 120° |
| Images | |
| no. of images | 34 (0°, 2; 20°, 4; 45°, 20; 60°, 8) |
| resolution limit for merging | 6 Å |
| phase residual (100–6 Å) | 43.2° (36.1° weighted phase residual, weighting scheme using individual 1/σ2) |
| Electron diffraction | |
| no. of electron diffraction patterns | 64 (0°, 8; 20°, 12; 45°, 24; 60°, 20) |
| resolution limit for merging | 3.5 Å |
| observed reflections | 26 455 |
| sampled reflections | 7495 |
| RFriedel | 26.5% |
| Rmerge | 34.8% |
| completeness | 78.1% |

Fig. 1. Phases from electron micrographs (upper curves) and amplitudes from electron diffraction (lower curves) along the lattice lines (4,0), (5,0), (6,1) and (8,0).
The packing of MGST1 into the hexagonal crystal form shows that the unit cell contains two ordered trimers centred on the 3-fold axes (Figure 2). The density with 6-fold symmetry, indicated by a circle in Figure 2, is due to partially ordered trimers (Schmidt-Krey et al., 1999).

Fig. 2. The crystal packing of MGST1 two-dimensional crystals. Two trimers are located on the crystallographic 3-fold axes of the p6 unit cell (outlined in red). The appearance of the 6-fold symmetrical structure (circled) arises from partially ordered trimers (Schmidt-Krey et al., 1999).
The view of the 3D potential distribution along the plane of the membrane (Figure 3) shows that the MGST1 trimer has a 30–40 Å thick part consisting mainly of rod-like structures that are oriented nearly perpendicular to, or in some cases slightly tilted relative to the plane of the membrane. There are four of these rods per monomeric unit of the protein (Figure 4). Rod A (Figure 5) is the most highly inclined with a tilt of ∼27°, while B and C are barely tilted by only ∼12°, and the last rod, D, has a tilt of ∼18°. Rod C is bent as it approaches one side of the membrane. This side contains relatively little protein mass extending from the rod-like regions (Figure 4), whereas the other face (Figure 5) has several additional domains. This suggested asymmetric mass distribution relative to the layer of rods is also evident from the side view in Figure 3.

Fig. 4. A stereo pair from the lumenal side of the membrane at a slight tilt relative to the normal of the membrane plane of the MGST1 trimer.

Fig. 5. The trimer viewed from the cytoplasmic side of the membrane. The four unique transmembrane helices are indicated by coloured cylinders.
Discussion
The most prominent features of the 3D map of MGST1 are the rod-like domains separated by ∼10 Å and running roughly perpendicular to the plane of the membrane (Figures 3–5). We interpret these as transmembrane helices in accordance with other EM observations of integral membrane proteins (Henderson and Unwin, 1975; Henderson et al., 1990; Kühlbrandt et al., 1994; Cheng et al., 1997; Li et al., 1997; Walz et al., 1997; Auer et al., 1998; Rhee et al., 1998; Unger et al., 1999; Williams, 2000). Thus, the membrane-spanning part of the protein monomer consists of a left-handed α-helical bundle. In the projection structures of both the orthorhombic (Hebert et al., 1997) and the hexagonal crystal form (Schmidt-Krey et al., 1999), the two inner and one of the peripheral helices per subunit in the trimer were identified. The previously unidentified elongated density in the projection structures of both crystal forms can now be interpreted as the most highly tilted transmembrane α-helix. Most hydropathy plot algorithms have predicted three membrane-spanning segments for MGST1 but this must be re-evaluated based on the 3D structure. Thus, the central hydrophobic segment in the sequence should be interpreted as two short stretches with a small intervening loop (Figure 6). A sequence alignment of the currently known members of the MAPEG family has shown comparable hydropathy profiles (Jakobsson et al., 1999a). Therefore, one might expect that all of the MAPEG proteins have a similar membrane topology consisting of a left-handed α-helical bundle (Figure 7).

Fig. 6. The hydropathy plot of MGST1. Possible locations for helices 1–4 are marked with white arrows and the vertical arrow points at the connection, possibly a short loop, between helices 2 and 3.
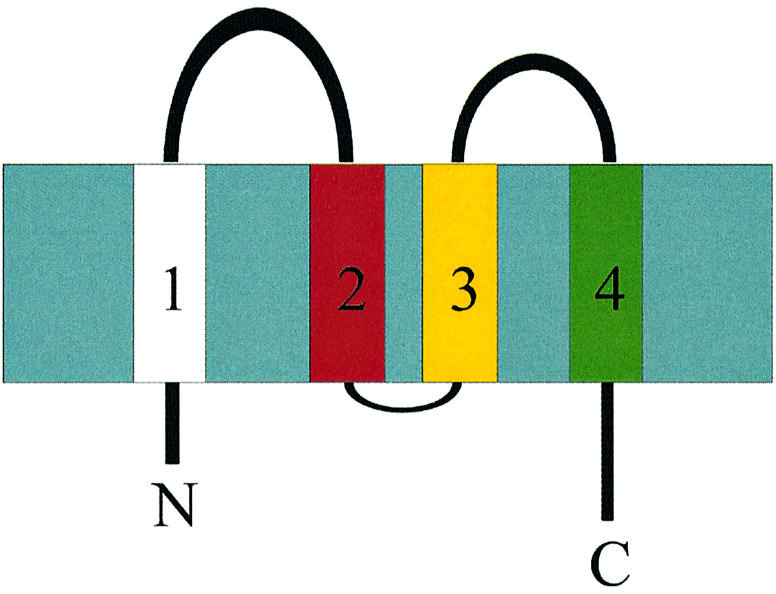
Fig. 7. Membrane topology of MGST1 with both the N- and C-terminus facing the lumen of the ER.
Since the topology of helix 1 is known (Andersson et al., 1994) and four helices can be observed in the 3D map, both the N- and the C-terminus must face the lumen (Figure 7). Furthermore, the N-terminus including the first hydrophobic segment can be cleaved off by trypsin and separated from the trimer, leaving the remainder of the protein functional (Andersson et al., 1994). Thus, it is likely that one of the peripheral α-helices, A, C or D, corresponds to the N-terminal hydrophobic segment, since helix B is located in the central cavity of the trimer where it is protected from the outside by the rest of the structure (Figure 5).
The deduced topology with four transmembrane helices and the N- and C-terminus on the lumenal side of the membrane places two segments connecting α-helix 1 to 2 and 3 to 4, respectively, on the cytoplasmic side of the membrane. These hydrophilic domains most likely contain the active site since it has been shown to face the cytoplasm (Andersson et al., 1994). The present structure displays more mass on one side of the membrane. As the N- and C-termini usually display a relatively high degree of flexibility, and comprise only one third of the amino acid content compared with the cytoplasmic domains, we suggest that the most prominent hydrophilic density observed in the 3D map faces the cytosolic side of the membrane.
The structure of the MGST1 trimer shows a striking similarity to subunit 1 of both bacterial and bovine cytochrome c oxidase (Iwata et al., 1995; Tsukihara et al., 1996), in spite of the fact that there is neither any shared functional property nor any sequence similarity between MGST1 and subunit 1 of cytochrome c oxidase. The three proteins all have 12 transmembrane α-helices and similar molecular masses, 53 kDa for MGST1 and 57 and 60 kDa for the bovine and paracoccal subunits, respectively. The helices of the cytochrome c oxidase subunits are slightly more tilted than those of MGST1 but their positions fall on arcs in a spiral arrangement similar to MGST1, as displayed in Figure 5. The handedness of the spiral of, for instance, the Paracoccus subunit is the same as for MGST1, provided that viewing directions are from the periplasmic and the cytoplasmic sides, respectively. In fact, this seems to be the correct way of comparing the two proteins since the N- and C-termini of the Paracoccus cytochrome c oxidase subunit 1 are on the cytoplasmic side with the largest hydrophilic domains in the periplasm. In the MGST1 monomer the N- and C-termini face the lumenal side of the membrane while the largest hydrophilic structures, forming the connections from helix 1 to 2 and from 3 to 4, are located in the cytoplasm. The cytochrome c oxidase subunit 1 is a continous polypeptide chain with pseudo 3-fold symmetry. When the helices are numbered according to their positions in the sequence the first four helices have the same positions as the MGST1 helices labelled in Figure 5, indicating that this may be the correct assignment of a monomer of MGST1. An alternative interpretation with the helices in one arc forming the monomer is less likely since this would lead to a large separation of the residues of importance for the active site, which are located in the hydrophilic domains connecting helix 1 to 2 and 3 to 4, respectively.
Soluble GSTs share a wide range of similar substrates with MGST1. In fact, we have speculated earlier that the enzymes might actually share structural elements (Hebert et al., 1997). However, the 3D model of MGST1 now shows a decidedly different tertiary structure compared with the cytosolic GSTs (Sinning et al., 1993). Further more, the apparent formation of one active site in the MGST1 homotrimer distinguishes the protein even further from cytosolic GSTs, where each 25 kDa subunit contributes an independent active site (Mannervik and Danielsson, 1988). Thus, it can be concluded that integral membrane GSTs form a structurally independent superfamily of proteins. To what degree then is MGST1 a good model for the rest of the MAPEG superfamily? As the four-helix bundle is now experimentally verified in mammalian MGST1 and is in agreement with topology predictions for prokaryotes (Cserzo et al., 1997), this structural motif forms an attractive candidate for the tertiary structure of MAPEG proteins. Another MAPEG member (leukotriene C4 synthase) has been proposed to form dimers based on gel filtration experiments in the presence of detergent (Nicholson et al., 1993). Con sequently, generalization of the oligomeric structure to other MAPEG family members must await further experiments.
Materials and methods
Crystallization
Two-dimensional crystals of MGST1 were prepared according to Schmidt-Krey et al. (1998).
Electron microscopy and data processing
Trehalose was added as an embedding medium (3%) to the suspension of crystalline membranes. The specimen was placed on molybdenum grids using the back-injection method (Wang and Kühlbrandt, 1991). After an incubation period of 30–60 s, the grids were blotted with filter paper and plunged into liquid ethane. The grids were observed at a specimen temperature of 4 K and an accelerating voltage of 300 kV in a JEOL JEM-3000SFF electron microscope (Fujiyoshi et al., 1991; Fujiyoshi, 1998). Electron diffraction patterns were recorded at a camera length of 2.5 m with a 2048 × 2048 Gatan slow scan CCD camera. The electron diffraction patterns were processed according to Henderson et al. (1986). Images were collected on Kodak SO-163 film and developed with full-strength Kodak D19 developer for 14 min. The best images selected by optical diffraction were scanned either with a Zeiss SCAI scanner (step size 7 µm, scanning area ∼7000 × 8000 pixels) or a Scitex LeafScan 45 (step size 5 µm, scanning area 6000 × 8000 pixels) and processed on a DEC/Alpha workstation using the MRC image processing programs (Crowther et al., 1996). The image phases were obtained after several cycles of lattice unbending, and determination and refinement of the contrast transfer function, lattice parameters, tilt angle and tilt axis, and merging to 6 Å of phase data from 34 images. The image phases were merged with the electron diffraction amplitudes, and a 3D map was calculated with the CCP4 program suite (CCP4, 1994). The map was displayed with the program O (Jones et al., 1991).
The hydropathy plot was calculated according to Kyte and Doolittle (1982), with an 11-residue window.
Acknowledgments
Acknowledgements
We thank Gerd Lundqvist for technical assistance. This project was supported by the Swedish Medical Research Council, the Swedish Natural Science Research Council, the Swedish Cancer Society and Magnus Bergvalls Stiftelse.
References
- Andersson C., Weinander,R., Lundqvist,G., DePierre,J.W. and Morgenstern,R. (1994) Functional and structural membrane topology of rat liver microsomal glutathione transferase. Biochim. Biophys. Acta, 1204, 298–304. [DOI] [PubMed] [Google Scholar]
- Auer M., Scarborough,G.A. and Kühlbrandt,W. (1998) Three-dimensional map of the plasma membrane H+-ATPase in the open conformation. Nature, 392, 840–843. [DOI] [PubMed] [Google Scholar]
- Cheng A., van Hoek,A.N., Yeager,M., Verkman,A.S. and Mitra,A. (1997) Three-dimensional organisation of a human water channel. Nature, 387, 627–630. [DOI] [PubMed] [Google Scholar]
- Collaborative Computational Project No. 4 (1994) The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D, 50, 760–763. [DOI] [PubMed] [Google Scholar]
- Crowther R.A., Henderson,R. and Smith,J.M. (1996) MRC image processing programs. J. Struct. Biol., 116, 9–16. [DOI] [PubMed] [Google Scholar]
- Cserzo M., Wallin,E., Simon,I., von Heijne,G. and Elofsson,A. (1997) Prediction of transmembrane α-helices in prokaryotic membrane proteins: the dense alignment surface method. Protein Eng., 10, 673–676. [DOI] [PubMed] [Google Scholar]
- Fujiyoshi Y. (1998) The structural study of membrane proteins by electron crystallography. Adv. Biophys., 35, 25–80. [DOI] [PubMed] [Google Scholar]
- Fujiyoshi Y., Mizusaki,T., Morikawa,K., Yamagishi,H., Aoki,Y., Kihara,H. and Harada,Y. (1991) Development of a superfluid helium stage for high-resolution electron microscopy. Ultramicroscopy, 38, 241–251. [Google Scholar]
- Hebert H., Schmidt-Krey,I., Morgenstern,R., Murata,K., Hirai,T., Mitsuoka,K. and Fujiyoshi,Y. (1997) The 3.0 Å projection structure of microsomal glutathione transferase as determined by electron crystallography of p21212 two-dimensional crystals. J. Mol. Biol., 271, 751–758. [DOI] [PubMed] [Google Scholar]
- Henderson R. and Unwin,P.N.T. (1975) Three-dimensional model of purple membrane obtained by electron microscopy. Nature, 257, 28–32. [DOI] [PubMed] [Google Scholar]
- Henderson R., Baldwin,J.M., Downing,K.H., Lepault,J., and Zemlin,F. (1986) Structure of purple membrane from Halobacterium halobium: recording, measurement and evaluation of electron micrographs at 3.5 Å resolution. Ultramicroscopy, 19, 147–178. [Google Scholar]
- Henderson R., Baldwin,J.M., Ceska,T.A., Zemlin,F., Beckman,E. and Downing,K.H. (1990) A model for the structure of bacteriorhodopsin based on high resolution electron crystallography. J. Mol. Biol., 213, 899–929. [DOI] [PubMed] [Google Scholar]
- Iwata S., Ostermeier,C., Ludwig,B. and Michel,H. (1995) Structure at 2.8 Å resolution of cytochrome c oxidase from Paracoccus denitrificans. Nature, 376, 660–669. [DOI] [PubMed] [Google Scholar]
- Jakobsson P.-J., Morgenstern,R., Mancini,J., Ford-Hutchinson,A. and Persson,B. (1999a) Common structural features of MAPEG—a widespread superfamily of membrane associated proteins with highly divergent functions in eicosanoid and glutathione metabolism. Protein Sci., 8, 1–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakobsson P.-J., Thorén,S., Morgenstern,R. and Samuelsson,B. (1999b) Identification of human prostaglandin E synthase: a microsomal, glutathione dependent, inducible enzyme, constituting a potential novel drug target. Proc. Natl Acad. Sci. USA, 96, 7220–7225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones T.A., Zou,J.Y., Cowan,S.W. and Kjeldgaard,M. (1991) Improved methods for building protein models in electron density maps. Acta Crystallogr. A, 47, 110–119. [DOI] [PubMed] [Google Scholar]
- Kühlbrandt W., Wang,D.N. and Fujiyoshi,Y. (1994) Atomic model of the plant light harvesting complex by electron crystallography. Nature, 367, 614–621. [DOI] [PubMed] [Google Scholar]
- Kyte J. and Doolittle,R.F. (1982) A simple method for displaying the hydrophobic character of a protein. J. Mol. Biol., 157, 105–132. [DOI] [PubMed] [Google Scholar]
- Li H., Lee,S. and Jap,B. (1997) Molecular design of aquaporin-1 water channel as revealed by electron crystallography. Nature Struct. Biol., 4, 263–265. [DOI] [PubMed] [Google Scholar]
- Mannervik B. and Danielsson,U.H. (1988) Glutathione transferases—structure and catalytic activity. CRC Crit. Rev. Biochem., 23, 283–337. [DOI] [PubMed] [Google Scholar]
- Morgenstern R., DePierre,J.W. and Jörnvall,H. (1985) Microsomal glutathione transferase: primary structure. J. Biol. Chem., 260, 13976–13983. [PubMed] [Google Scholar]
- Nicholson D.W., Ali,A., Vaillancourt,J.P., Calaycay,J.R., Mumford,R.A., Zamboni,R.J. and Fordhutchinson,A.W. (1993) Purification to homogeneity and the N-terminal sequence of human leukotriene C4 synthase—a homodimeric glutathione S-transferase composed of 18 kDa subunits. Proc. Natl Acad. Sci. USA, 90, 2015–2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polyak K., Xia,Y., Zweier,J.L., Kinzler,K.W. and Vogelstein,B. (1997) A model for p53-induced apoptosis. Nature, 389, 300–305. [DOI] [PubMed] [Google Scholar]
- Rhee K.H., Morris,E.P., Barber,J. and Kühlbrandt,W. (1998) Three-dimensional structure of the plant photosystem II reaction centre at 8 Å resolution. Nature, 396, 283–286. [DOI] [PubMed] [Google Scholar]
- Satoh K., Nagano,Y., Shimomura,C., Suzuki,N., Saeki,Y. and Yokota,H. (2000) Expression of prostaglandin E synthase mRNA is induced in β-amyloid treated rat astrocytes. Neurosci. Lett., 283, 221–223. [DOI] [PubMed] [Google Scholar]
- Schmidt-Krey I., Lundqvist,G., Morgenstern,R. and Hebert,H. (1998) Parameters for the two-dimensional crystallization of the membrane protein microsomal glutathione transferase. J. Struct. Biol., 123, 87–96. [DOI] [PubMed] [Google Scholar]
- Schmidt-Krey I., Murata,K., Hirai,T., Mitsuoka,K., Cheng,Y., Fujiyoshi,Y. and Hebert,H. (1999) The projection structure of the membrane protein microsomal glutathione transferase at 3 Å resolution as determined from two-dimensional hexagonal crystals. J. Mol. Biol., 288, 243–253. [DOI] [PubMed] [Google Scholar]
- Sinning I. et al. (1993) Structure determination and refinement of human α class glutathione transferase A1-1 and a comparison with the µ and π class enzymes. J. Mol. Biol., 232, 192–212. [DOI] [PubMed] [Google Scholar]
- Sun T.-H. and Morgenstern,R. (1997) Binding of glutathione and an inhibitor to microsomal glutathione transferase. Biochem. J., 326, 193–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukihara T., Aoyama,H., Yamashita,E., Tomizaki,T., Yamaguchi,H., Shinzawa-Itoh,K., Nakashima,R., Yaono,R. and Yoshikawa,S. (1996) The whole structure of the 13-subunit oxidized cytochrome c oxidase at 2.8 Å. Science, 272, 1136–1144. [DOI] [PubMed] [Google Scholar]
- Unger V.M., Kumar,N.M., Gilula,N.B. and Yeager,M. (1999) Three-dimensional structure of a recombinant gap junction membrane channel. Science, 283, 1176–1180. [DOI] [PubMed] [Google Scholar]
- Walz T., Hirai,T., Murata,K., Heymann,J.B., Mitsuoka,K., Fujiyoshi,Y., Smith,B.L., Agre,P. and Engel,A. (1997) The three-dimensional map of aquaporin-1. Nature, 387, 624–627. [DOI] [PubMed] [Google Scholar]
- Wang D.N. and Kühlbrandt,W. (1991) High-resolution electron crystallography of light-harvesting chlorophyll a/b protein complex in different media. J. Mol. Biol., 217, 691–699. [DOI] [PubMed] [Google Scholar]
- Williams K.A. (2000) Three-dimensional structure of the ion-coupled transport protein NhaA. Nature, 403, 112–115. [DOI] [PubMed] [Google Scholar]