

Abstract
Mutations in HERG and KCNQ1 (or KVLQT1) genes cause the life-threatening Long QT syndrome. These genes encode K+ channel pore-forming subunits that associate with ancillary subunits from the KCNE family to underlie the two components, IKr and IKs, of the human cardiac delayed rectifier current IK. The KCNE family comprises at least three members. KCNE1 (IsK or MinK) recapitulates IKs when associated with KCNQ1, whereas it augments the amplitude of an IKr-like current when co-expressed with HERG. KCNE3 markedly changes KCNQ1 as well as HERG current properties. So far, KCNE2 (MirP1) has only been shown to modulate HERG current. Here we demonstrate the interaction of KCNE2 with the KCNQ1 subunit, which results in a drastic change of KCNQ1 current amplitude and gating properties. Furthermore, KCNE2 mutations also reveal their specific functional consequences on KCNQ1 currents. KCNQ1 and HERG appear to share unique interactions with KCNE1, 2 and 3 subunits. With the exception of KCNE3, mutations in all these partner subunits have been found to lead to an increased propensity for cardiac arrhythmias.
Keywords: cardiac arrhythmias/IKs current/Long QT syndrome/MirP1
Introduction
KCNE proteins form a family of K+-channel regulatory subunits (Attali, 1996; Barhanin et al., 1998; Sanguinetti, 2000). To date, four related KCNE genes have been identified, but functional studies have been reported for only three of them (Takumi et al., 1988; Abbott et al., 1999; Schroeder et al., 2000). It is probable that other KCNE gene family members will be recognized after a careful analysis of the human genome sequence. They encode small proteins with a single transmembrane domain that do not produce any current by themselves. However, these proteins associate with a complex typically consisting of six transmembrane domain pore-forming subunits, thus modifying the electrophysiological properties of the completed channel. The first channel complex identified was the association KCNQ1–KCNE1, which is responsible for the slow delayed rectifier cardiac current, IKs (Sanguinetti and Jurkiewicz, 1990). KCNQ1 generates a K+-selective current that activates relatively rapidly and deactivates slowly. KCNE1 considerably reduces the speed of activation of the current and shifts its voltage dependency towards more positive values (Barhanin et al., 1996; Sanguinetti et al., 1996). Mutations in either KCNQ1 or KCNE1 result in a loss of IKs function that causes the Long QT1 syndrome (LQTS) (Wang et al., 1996; Splawski et al., 1997) (for review see Priori et al., 1999), a cardiac disorder that is characterized by a propensity for polymorphic ventricular tachycardia and ventricular fibrillation, which may causes sudden death. In the case of homozygous mutations (Neyroud et al., 1997; Schulze-Bahr et al., 1997; Tyson et al., 1997; Duggal et al., 1998), a complete deafness is associated with the cardiac abnormalities to produce the Jervell and Lange-Nielsen syndrome (Jervell and Lange-Nielsen, 1957).
The KCNE3 subunit presents an overall 35% amino acid sequence similarity with KCNE1 and was recently shown to form heteromultimers with KCNQ1 (Schroeder et al., 2000). Surprisingly, the channel remains constitutively open at all membrane voltages as a functional consequence. Therefore, the association of KCNE1 or KCNE3 with the same complex of KCNQ1 pore-forming subunits can yield K+ currents with dramatically different gating properties.
KCNE2, a closer relative to KCNE1 than KCNE3 (overall amino acid sequence similarity of 51%), was only shown to interact with HERG and no association with KCNQ1 could be detected upon co-expression experiments in Xenopus oocytes (Abbott et al., 1999). In our study, we demonstrate that KCNE2 can interact with the KCNQ1 channel when co-expressed in mammalian COS cells. This association results in a KCNE3-like effect, that is a transformation of the voltage-dependent KCNQ1 current to a voltage-independent current. Furthermore, mutations in either KCNE2 or KCNQ1 that have been described as being responsible for human genetic forms of cardiac arrhythmias, markedly alter the functional properties of the KCNQ1–KCNE2 channel complex.
Results
Effects of KCNE2 on KCNQ1 currents expressed in COS cells
KCNQ1 and KCNE2 subunits were co-expressed in mammalian COS cells. As expected, KCNQ1 alone elicited a strongly outward rectifying K+ current that was characterized by: (i) an exponential activation time course; (ii) a slow deactivation process, preceded by a transient increase in the current (resulting from a rapid recovery from inactivation); and (iii) a threshold of activation of approximately –40 mV (Figure 1A and B). Co-expression of KCNE2 drastically modified these characteristics, leading to currents with (i) an apparently instantaneous activation; (ii) a rapid deactivation process; (iii) a linear current–voltage relationship (Figure 1C and D). This current presented the properties of a background current and appeared almost identical to the recently described KCNQ1–KCNE3 current (Schroeder et al., 2000). In addition to the gating effects, a decrease in the amplitude of the outward current, from 14.8 ± 1.0 pA/pF at +30 mV (control KCNQ1; n = 31) to 5.3 ± 0.9 pA/pF (KCNQ1–KCNE2; n = 29, p <0.005), was observed. Clearly, KCNE2 diminished the number of cells expressing current densities larger than 10 pA/pF, which were outnumbered by those expressing currents below 5 pA/pF (Figure 1E and F). The reversal potential of the KCNQ1–KCNE2 currents varied with extracellular K+ concentrations, with a slope of 48 ± 2 mV (n = 6; Figure 1D, inset) per decade, a value slightly lower than the slope expected for a strictly K+-selective current. However, similar low values of 49.9 and 51 mV per decade have been previously reported for the KCNQ1–KCNE1 current by Sanguinetti et al. (1996) and Barhanin et al. (1996).

Fig. 1. Comparison of KCNQ1 (A, B and E) and KCNQ1–KCNE2 (C, D and F) currents recorded in COS-7 cells. Currents were normalized for cell surface area and expressed as current density (pA/pF). (A and C) Representative current traces elicited by 2 s voltage steps from a holding potential of –80 mV over the range of –130 to +60 mV in 10 mV increments (A) or to +70 mV in 20 mV increments (C). Tail currents were recorded upon a repolarization step to –40mV. Density values at +30 mV were 17.1 pA/pF and 4.1 pA/pF for KCNQ1 and KCNQ1–KCNE2, respectively. Cells capacities were 77 pF and 85.4 pF, respectively. (B and D) Corresponding current–voltage relationships. (D, inset) Shift of the reversal potential as a function of the extracellular K+ concentration. (E and F) Histograms of the percentage of cells producing indicated current densities. The current density (pA/pF) was calculated for each recorded cell in relation to the capacitance and the peak current amplitude at +30 mV.
Corresponding co-expression experiments were also performed in CHO and HEK transfected cells, yielding results very similar to those described above in COS cells (not shown). Clearly, the KCNE2 effects are not restricted to COS-expressing cells.
Physical association of KCNQ1 and KCNE2 subunits
The formation of hetero-oligomeric complexes of KCNQ1–KCNE2 was assessed by co-precipitation experiments. [35S]methionine-labelled proteins, M2-flag-KCNQ1, KCNE1 and KCNE2, were synthesized with rabbit reticulocyte lysate. An anti-M2-flag antibody was used to precipitate KCNQ1 after co-incubation with either KCNE1 or KCNE2 or a mixture of KCNE1 and KCNE2. Bands with migrations appropriate for KCNE1 (Figure 2, lane 2) and for KCNE2 (Figure 2, lane 3) were clearly present in the KCNQ1 precipitates and were absent when the same experiments were performed in the presence of Kv9.3 in the test mixture (Figure 2, lanes 5 and 6). Competition experiments between KCNE1 and KCNE2 for their association with KCNQ1 were also performed. When equal amounts of all three subunits are mixed together, both auxillary subunits can be co-precipitated with KCNQ1 (Figure 2, lane 4). It thus appears that, in our assay conditions, KCNQ1 does not display a marked preference for either of the two KCNE subunits.
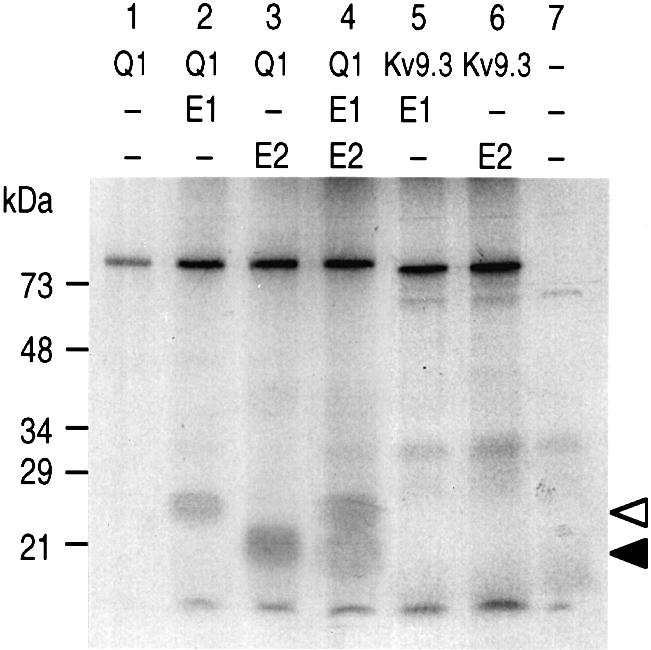
Fig. 2. Physical association of [35S]methionine-labelled KCNQ1, KCNE1 and KCNE2 proteins in the in vitro conditions. Complex formations of M2-flag-KCNQ1 with KCNE subunits. Immunoprecipitations with anti-M2-flag antibodies were carried out by using equal volumes of 35S-labelled in vitro translated products corresponding to M2-flag-KCNQ1 (K1), KCNE1 (E1), KCNE2 (E2) and M2-flag-Kv9.3 (Kv9.3), and mixed as follows: lane 1, K1; lane 2, K1 + E1; lane 3, K1 + E2; lane 4, K1 + E1 + E2; lane 5, Kv9.3 + E1; lane 6, Kv9.3 + E2; lane 7, control reaction performed without adding a cRNA in the in vitro translation mixture. White and black arrows indicate the migration of KCNE1 and KCNE2, respectively.
Effects of the presence of KCNE2 on the chromanol 293B sensitivity
KCNE subunits confer specific pharmacological features to the KCNQ1 current. KCNE1 modifies the sensitivity of the specific blockade exerted on KCNQ1 by the IKs blocker chromanol 293B. This compound inhibits the KCNQ1 current with an IC50 of 50 µM. It is 10 times more potent for the blockade of the KCNQ1–KCNE1 current (Busch et al., 1997). The KCNQ1–KCNE3 current is also much more sensitive to chromanol 293B than the KCNQ1 current (Schroeder et al., 2000). KCNE2 also increases the sensitivity to the KCNQ1 blocker. Application of 10 µM chromanol 293B inhibited KCNQ1 by a mere 23 ± 2% (n = 4), whereas it drastically decreased the intensity of the KCNQ1–KCNE2 current (81 ± 5%; n = 6, p <0.005) (Figure 3).

Fig. 3. Increased sensitivity to chromanol 293B (10 µM) of KCNQ1–KCNE2 (B) as compared with KCNQ1 currents (A). Currents were elicited at +30 mV from a holding potential of –80 mV in the whole-cell configuration.
Effects of arrhythmogenic mutations on the KCNQ1–KCNE2 current
The presence of some gene mutations/polymorphisms of KCNE2 in patients with sporadic, drug-induced or inherited polymorphic ventricular tachycardia supports the hypothesis that KCNE2, like KCNE1, is associated with cardiac arrhythmias (Abbott et al., 1999). Since KCNE2 modulates HERG expression in the Xenopus oocyte (Abbott et al., 1999), their arrhythmogenic effects were attributed to a deleterious effect on IKr. The demonstration of the KCNE2 interaction with KCNQ1 made it important to evaluate the effects of these previously identified KCNE2 mutations on the current produced by the KCNQ1–KCNE2 complexes.
The KCNE2-Q9E mutation, which was previously identified in a patient with a drug-induced arrhythmia (Abbott et al., 1999), exerted a greater inhibition on KCNQ1 current than its wild-type counterpart. As a result, only a small instantaneous current (2.0 ± 0.3 pA/pF; n = 9) was recorded (Figure 4B) upon co-expression of KCNE2-Q9E with KCNQ1. As compared with WT, the KCNE2-Q9E mutation did not significantly change the inhibition produced by 10 µM of chromanol 293B (Figure 4F).

Fig. 4. Modifications of the KCNQ1 current induced by co-expression of mutated KCNE2 subunits. (A, B and C) Traces of currents elicited by 2 s voltage steps from a holding potential of –80 mV over the range of –60 to +60 mV in 10 mV increments for KCNQ1 and KCNQ1–KCNE2-I57T, and over the range –130 to +60 mV in 10 mV increments for KCNQ1–KCNE2-Q9E currents (B). (D) Averaged activation curves for KCNQ1 (filled circle, n = 12) and KCNQ1–KCNE2-I57T (open triangle, n = 9), showing a 40 mV shift towards positive values. (E and F) Influence of the KCNE2-I57T and Q9E mutations on the inhibition by 293B.
The I57T substitution is another mutation found in the KCNE2 gene from patients with inherited LQTS (Abbott et al., 1999). We show that this mutation affects KCNQ1 currents, but in a different manner compared to the Q9E mutation. Figure 4C shows that the KCNE2-I57T subunit (i) decreases the rate of activation of the KCNQ1 current; (ii) abolishes the hump of the tail currents upon repolarization; (iii) accelerates the deactivation process [τdeact (-40mV) 127 ± 21 ms (n = 6) as compared with 522 ± 25 ms (n = 9, p <0.005)]; and (iv) shifts the voltage dependency of the channel activation towards more depolarized potentials (Figure 4D). The slowing down of the activation process and the voltage shift induced by KCNE2-I57T effects on KCNQ1 currents are not seen with the wild-type KCNE2 and resemble those exerted by the KCNE1 subunit. In addition, the KCNE2-I57T mutation totally abolished the increased potency of chromanol 293B that was observed with wild-type KCNE2, since 10 µM chromanol 293B only blocked a mere 20% of the KCNQ1–KCNE2-I57T current instead of ∼80% of the KCNQ1–KCNE2 current (Figure 4E).
Many mutations in KCNQ1 have been reported that account for the majority of congenital LQTS. Most of them cause a total loss of channel function, independently of the presence of KCNE1 (Chouabe et al., 1997; Wollnik et al., 1997; Wang et al., 1999). However, some mutations have been reported that strictly rely on the presence of KCNE1 in the channel complex to alter its voltage-dependent activation (Franqueza et al., 1999; Chouabe et al., 2000). This is the case of the missense mutation KCNQ1-R243H located at the cytoplasmic end of the S4 segment (Chouabe et al., 2000). The power of the KCNE subunit to reveal such an underlying genetic defect was exploited as a putative ‘molecular sensor’ of the KCNQ1-R243H–KCNE2 interaction. Co-transfection of KCNQ1-R243H with WT KCNE2 led to an important decrease in the rate of activation of the KCNQ1 channel [control τact 37 ± 4 ms (n = 9) versus 1807 ± 250 ms for KCNQ1-R243H (n = 5, p <0.005)] and to the typical sigmoidal shape of IKs (Figure 5A and B). In addition, the KCNE2 subunit induced a positive voltage shift (14 mV) of the activation (Figure 5C) and an increase in the deactivation kinetics [control τdeact (-40mV) 611 ± 43 ms (n = 7) versus 157 ± 37 for KCNQ1-R243H–KCNE2 (n = 4, p <0.005; Figure 5D)]. These effects reflect important changes in the intrinsic properties of the KCNQ1 channel that further confirm the formation of KCNQ1–KCNE2 heteromeric channels.

Fig. 5. Slowing down of the activation of the KCNQ1-R243H current (A) by the KCNE2 subunit (B). Currents were recorded from a holding potential of –80 mV over the range of –60 to +50 mV. (C) Averaged activation curves for KCNQ1-R243H in the absence (filled circle; n = 6) or the presence (open circle; n = 9) of KCNE2. (D) Corresponding deactivation time constants fitted to a single exponential function from tail currents elicited from –120 to –40 mV after a test pulse to +30 mV.
Discussion
In a previous study, it was concluded from Xenopus oocyte expression experiments that KCNE2 has no effect on KCNQ1 gating or current amplitude (Abbott et al., 1999). Although we confirmed the absence of evidence for an interaction between the two K+ channel subunits in this expression system, KCNE2 co-expression drastically modified KCNQ1 current characteristics in mammalian cells. In the presence of KCNE2, KCNQ1 is transformed into a permanently open channel. The same KCNE2 effects were observed in COS, CHO and HEK transfected cells. In all three cell types, a net decrease in the mean current density is also observed. Clearly, the mammalian-derived and the Xenopus oocyte expression systems produce different results. Such a difference in the two types of expression systems is not unique and has been reported recently in the case of mutated forms of the human cardiac Na+ channel, for which the co-expression of the regulatory β-subunit plays an important role (Baroudi et al., 2000).
In addition to electrophysiological evidence, biochemical studies have been performed to demonstrate the association of KCNE2 subunit with KCNQ1. In these experiments it was also demonstrated that the presence of KCNE1 does not prevent the KCNQ1–KCNE2 complex formation. These in vitro data are in agreement with the hypothesis that the KCNQ1–KCNE2 association can take place in vivo, even in cells also expressing KCNE1. However, they do not allow clarification of the mechanisms governing the formation of the different hetero-complexes in native cells.
The KCNE2 effects are very different from those of KCNE1, which induces a considerable decrease in the speed of activation of KCNQ1 together with an increase in the current amplitude. However, the KCNE2-I57T and KCNQ1-R243H mutations modify the effects of KCNE2, making them qualitatively similar to those of KCNE1, i.e. the reduction in the speed of activation, the abolition of the hook on the tail currents and the induction of a positive voltage shift of the activation curve. These findings suggest that the two KCNE subunits, 1 and 2, may share a similar mode of action on KCNQ1, even though they produce dramatically different macroscopic effects.
In conclusion, it is likely that KCNE2, which is present in the heart together with KCNE1, also determines the expression pattern of repolarizing currents through an interaction with the KCNQ1 subunit. Thus, it appears that the KCNQ1 current is highly regulated through an array of interactions with different subunits. The interaction with KCNE1 leads to a slow channel, thought necessary to prevent extensive action-potential prolongation at low heart rates (Drici et al., 1998). The interaction with KCNE2 will produce a background current that may have a role in the control of the resting potential, therefore influencing the length of the refractory periods and action-potential firing frequency. An N-terminal truncated isoform of the KCNQ1 gene product has also been reported in human heart that acts as dominant-negative regulator of the IKs current (Demolombe et al., 1998). Such a complex regulation of KCNQ1 activity reinforces the idea that this K+ current-generating subunit has a crucial role in controlling cardiac action-potential shaping and in regulating heart rate.
It is thought that KCNE2 mutations predispose to arrhythmia because they cause a reduction in the IKr current (Abbott et al., 1999). In this paper, we show that the KCNE2-I57T and Q9E mutations also reduce the K+ current carried by the KCNQ1 channel, by reducing the instantaneous KCNQ1–KCNE2 current. Added to the decrease in IKr, this effect is likely to participate in the reduction of the cardiac ‘repolarization reserve’ (Roden, 1998), thereby predisposing mutation carriers to torsade de pointes and ventricular fibrillation.
Materials and methods
Functional expression in COS-7 cells
Human KCNE2 cDNA (DDBJ/EMBL/GenBank accession No. AP 000052) was obtained by PCR from human brain cDNAs (Clontech) and cloned in the pIRES-CD8 mammalian expression vector (Fink et al., 1998). The KCNE2-Q9E and I57T mutants were constructed using a classical PCR technique. Cells were transiently transfected by the DEAE-Dextran precipitate method, with 0.5 µg or 1 µg (in the co-expression experiments) of pIRES-CD8-KCNE2 and pCI-KCNQ1 per dish. Whole-cell recordings on transfected cells, as visualized by CD8 expression, were performed as described (Chouabe et al., 2000). Current densities, voltage dependence of activation, and activation and deactivation kinetics values are presented as the mean ± SEM.
Co-immunoprecipitation
M2-flagged KCNQ1 and M2-flagged Kv9.3 were obtained by PCR and subcloned into the pCI vector (Promega). cRNAs for M2-flagged KCNQ1, KCNE1 WT, KCNE2 WT and M2-flagged Kv9.3 were synthesized using the RNA transcription kit (Stratagene) and then in vitro translated with the Speedread Lysate 2 (Novagen). Proteins were [35S]methionine labelled. Products were mixed in buffer containing 150 mM NaCl, 50 mM Tris–Cl pH 7.4, 1 mM iodoacetamide, 1× mixture of protease inhibitors (Roche), 1.5% Tween 20, and incubated for 4 h on ice before immunoprecipitation with an anti-M2-flag monoclonal antibody (2EL-1B11, Euromedex). A control experiment was performed without adding cRNA. Proteins were resolved on 12% SDS–PAGE.
Acknowledgments
Acknowledgements
We thank M.D.Drici for helpful discussions, M.Jodar for expert technical assistance, A.Patel for reading the manuscript and V.Lopez for secretarial assistance. This work was supported by the Centre National de la Recherche Scientifique (CNRS), the Association Française contre les Myopathies (AFM Grant and AFM Fellowship for N.T.), the Institut National de la Santé et de la Recherche Médicale (PROGRES N°4P009D) and the Conseil Regional PACA.
References
- Abbott G.W., Sesti,F., Splawski,I., Buck,M.E., Lehmann,M.H., Timothy,K.W., Keating,M.T. and Goldstein,S.A. (1999) MiRP1 forms IKr potassium channels with HERG and is associated with cardiac arrhythmia. Cell, 97, 175–187. [DOI] [PubMed] [Google Scholar]
- Attali B. (1996) Ion channels—a new wave for heart rhythms. Nature, 384, 24–25. [DOI] [PubMed] [Google Scholar]
- Barhanin J., Lesage,F., Guillemare,E., Fink,M., Lazdunski,M. and Romey,G. (1996) K(v)LQT1 and IsK (minK) proteins associate to form the IKs cardiac potassium current. Nature, 384, 78–80. [DOI] [PubMed] [Google Scholar]
- Barhanin J., Attali,B. and Lazdunski,L. (1998) IKs, a slow and intriguing cardiac K+ channel and its associated long QT diseases. Trends Cardiovasc. Med., 8, 207–214. [DOI] [PubMed] [Google Scholar]
- Baroudi G., Carbonneau,E., Pouliot,V. and Chahine,M. (2000) SCN5A mutation (T1620M) causing Brugada syndrome exhibits different phenotypes when expressed in Xenopus oocytes and mammalian cells. FEBS Lett., 467, 12–16. [DOI] [PubMed] [Google Scholar]
- Busch A.E., Busch,G.L., Ford,E., Suessbrich,H., Lang,H.J., Greger,R., Kunzelmann,K., Attali,B. and Stuhmer,W. (1997) The role of the I-sK protein in the specific pharmacological properties of the IKs channel complex. Br. J. Pharmacol., 122, 187–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chouabe C., Neyroud,N., Guicheney,P., Lazdunski,M., Romey,G. and Barhanin,J. (1997) Properties of KvLQT1 K+ channel mutations in Romano-Ward and Jervell and Lange-Nielsen inherited cardiac arrhythmias. EMBO J., 16, 5472–5479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chouabe C., Neyroud,N., Richard,P., Denjoy,I., Hainque,B., Romey,G., Drici,M.D., Guicheney,P. and Barhanin,J. (2000) Novel mutations in KvLQT1 that affect IKs activation through interactions with Isk. Cardiovasc. Res., 45, 971–980. [DOI] [PubMed] [Google Scholar]
- Demolombe S. et al.(1998) A dominant negative isoform of the long QT syndrome 1 gene product. J. Biol. Chem., 273, 6837–6843. [DOI] [PubMed] [Google Scholar]
- Drici M.D., Arrighi,I., Chouabe,C., Mann,J.R., Lazdunski,M., Romey,G. and Barhanin,J. (1998) Involvement of IsK-associated K+ channel in heart rate control of repolarization in a murine engineered model of Jervell and Lange-Nielsen syndrome. Circ. Res., 83, 95–102. [DOI] [PubMed] [Google Scholar]
- Duggal P., Vesely,M.R., Wattanasirichaigoon,D., Villafane,J., Kaushik,V. and Beggs,A.H. (1998) Mutation of the gene for IsK associated with both Jervell and Lange-Nielsen and Romano-Ward forms of Long-QT syndrome. Circulation, 97, 142–146. [DOI] [PubMed] [Google Scholar]
- Fink M., Lesage,F., Duprat,F., Heurteaux,C., Reyes,R., Fosset,M. and Lazdunski,M. (1998) A neuronal two P domain K+ channel stimulated by arachidonic acid and polyunsaturated fatty acids. EMBO J., 17, 3297–3308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franqueza L., Lin,M., Splawski,I., Keating,M.T. and Sanguinetti,M.C. (1999) Long QT syndrome-associated mutations in the S4-S5 linker of KvLQT1 potassium channels modify gating and interaction with minK subunits. J. Biol. Chem., 274, 21063–21070. [DOI] [PubMed] [Google Scholar]
- Jervell A. and Lange-Nielsen,F. (1957) Congenital deaf-mutism, functional heart disease with prolongation of Q-T interval and sudden death. Am. Heart J., 54, 59–68. [DOI] [PubMed] [Google Scholar]
- Neyroud N. et al. (1997) A novel mutation in the potassium channel gene KVLQT1 causes the Jervell and Lange-Nielsen cardioauditory syndrome. Nature Genet., 15, 186–189. [DOI] [PubMed] [Google Scholar]
- Priori S.G. et al. (1999) Genetic and molecular basis of cardiac arrhythmias: impact on clinical management parts I and II. Circulation, 99, 518–528. [DOI] [PubMed] [Google Scholar]
- Roden D.M. (1998) Taking the “idio” out of “idiosyncratic”: predicting torsades de pointes. Pacing Clin. Electrophysiol., 21, 1029–1034. [DOI] [PubMed] [Google Scholar]
- Sanguinetti M.C. (2000) Maximal function of minimal K+ channel subunits. Trends Pharmacol. Sci., 21, 199–201. [DOI] [PubMed] [Google Scholar]
- Sanguinetti M.C. and Jurkiewicz,N.K. (1990) Two components of cardiac delayed rectifier K+ current. Differential sensitivity to block by class-III antiarrhythmic agents. J. Gen. Physiol., 96, 195–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanguinetti M.C., Curran,M.E., Zou,A., Shen,J., Spector,P.S., Atkinson,D.L. and Keating,M.T. (1996) Coassembly of K(v)LQT1 and MinK (IsK) proteins to form cardiac IKs potassium channel. Nature, 384, 80–83. [DOI] [PubMed] [Google Scholar]
- Schroeder B.C., Waldegger,S., Fehr,S., Bleich,M., Warth,R., Greger,R. and Jentsch,T.J. (2000) A constitutively open potassium channel formed by KCNQ1 and KCNE3. Nature, 403, 196–199. [DOI] [PubMed] [Google Scholar]
- Schulze-Bahr E. et al. (1997) KCNE1 mutations cause Jervell and Lange-Nielsen syndrome. Nature Genet., 17, 267–268. [DOI] [PubMed] [Google Scholar]
- Splawski I., Tristani-Firouzi,M., Lehmannn,M.H., Sanguinetti,M.C. and Keating,M.T. (1997) Mutations in hminK gene cause long-QT syndrome and suppress IKs function. Nature Genet., 17, 338–340. [DOI] [PubMed] [Google Scholar]
- Takumi T., Ohkubo,H. and Nakanishi,S. (1988) Cloning of a membrane protein that induces a slow voltage-gated potassium current. Science, 242, 1042–1045. [DOI] [PubMed] [Google Scholar]
- Tyson J. et al.(1997) IsK and KvLQT1: mutation in either of the two subunits of the slow component of the delayed rectifier potassium channel can cause Jervell and Lange-Nielsen syndrome. Hum. Mol. Genet., 6, 2179–2185. [DOI] [PubMed] [Google Scholar]
- Wang Q. et al.(1996) Positional cloning of a novel potassium channel gene: KVLQT1 mutations cause cardiac arrhythmias. Nature Genet., 12, 17–23. [DOI] [PubMed] [Google Scholar]
- Wang Z., Tristani-Firouzi,M., Xu,Q., Lin,M., Keating,M.T. and Sanguinetti,M.C. (1999) Functional effects of mutations in KvLQT1 that cause long QT syndrome. J. Cardiovasc. Electrophysiol., 10, 817–826. [DOI] [PubMed] [Google Scholar]
- Wollnik B., Schroeder,B.C., Kubisch,C., Esperer,H.D., Wieacker,P. and Jentsch,T.J. (1997) Pathophysiological mechanisms of dominant and recessive KVLQT1 K+ channel mutations found in inherited cardiac arrhythmias. Hum. Mol. Genet., 6, 1943–1949. [DOI] [PubMed] [Google Scholar]