

Abstract
Lhs1p is an Hsp70-related chaperone localized in the endoplasmic reticulum (ER) lumen. Δlhs1 mutant cells are viable but are constitutively induced for the unfolded protein response (UPR). Here, we demonstrate a severe growth defect in Δire1Δlhs1 double mutant cells in which the UPR can no longer be induced. In addition, we have identified a UPR- regulated gene, SIL1, whose overexpression is sufficient to suppress the Δire1Δlhs1 growth defect. SIL1 encodes an ER-localized protein that interacts directly with the ATPase domain of Kar2p (BiP), suggesting some role in modulating the activity of this vital chaperone. SIL1 is a non-essential gene but the Δlhs1Δsil1 double mutation is lethal and correlates with a complete block of protein translocation into the ER. We conclude that the IRE1-dependent induction of SIL1 is a vital adaptation in Δlhs1 cells, and that the activities associated with the Lhs1 and Sil1 proteins constitute an essential function required for protein translocation into the ER. The Sil1 protein appears widespread amongst eukaryotes, with homologues in Yarrowia lipolytica (Sls1p), Drosophila and mammals.
Keywords: endoplasmic reticulum/IRE1/LHS1/SIL1,PER100,YOL031c/translocation/UPR
Introduction
Protein translocation across the membrane of the yeast endoplasmic reticulum (ER) can occur by two distinct pathways. The co-translational pathway targets nascent polypeptides to the ER membrane via signal recognition particle (SRP), whereas the post-translational pathway translocates full-length precursor polypeptides that are targeted to the ER independently of SRP (for a review see Stirling, 1999). Recent evidence suggests a specific role for BiP, an ER lumenal Hsp70, in gating the translocon prior to the initiation of co-translational translocation (Hamman et al., 1998). Such an activity might be expected to be crucial in living cells, and indeed Kar2p (yeast BiP; Normington et al., 1989; Rose et al., 1989) has recently been shown to be essential for the translocation of SRP-dependent precursors in vivo (B.P.Young, R.A.Craven, P.J.Reid and C.J.Stirling, submitted). Kar2p is also essential for the post-translational translocation pathway in yeast where it is required at two distinct stages: (i) for initiation of translocation prior to the stable interaction of precursor with the translocon and (ii) to promote the vectorial transport of polypeptide chains through the translocon into the ER lumen (Sanders et al., 1992; Lyman and Schekman, 1995, 1997). This latter role involves the Hsp70 ATP-dependent reaction cycle, which drives multiple rounds of Kar2p binding to the translocating polypeptide as it enters the lumen. Such binding of the incoming precursor by a lumenal factor prevents ‘backslip’ and is sufficient to drive import (Matlack et al., 1999).
A second ER-resident Hsp70-related protein, encoded by the LHS1 gene, has been characterized in yeast (Baxter et al., 1996; Craven et al., 1996; Hamilton and Flynn, 1996). The Lhs1 protein (Lhs1p) represents a novel branch of the Hsp70 superfamily (Craven et al., 1997) and appears ubiquitous amongst eukaryotes, with homologues including mammalian Grp170/Orp150 (Chen et al., 1996; Kuwabara et al., 1996; Craven et al., 1997). Unlike KAR2, the LHS1 gene is not essential for viability, but lhs1 null mutant cells display a partial defect in post-translational translocation (Baxter et al., 1996; Craven et al., 1996; Hamilton and Flynn, 1996) and are also defective in the repair of misfolded proteins in the ER (Saris et al., 1997). However, the interpretation of the Δlhs1 mutant phenotype is complicated by the fact that these cells exhibit a constitutive induction of the unfolded protein response (UPR) (Baxter et al., 1996; Craven et al., 1996). The UPR is triggered by a reduction in the levels of free Kar2p/BiP, presumably resulting from its sequestration onto misfolded polypeptides (Kohno et al., 1993; Bertolotti et al., 2000). In yeast, this leads to activation of the transmembrane kinase/nuclease Ire1p (Cox et al., 1993; Mori et al., 1993; Shamu and Walter, 1996) which initiates a novel splicing mechanism that removes an intron from the HAC1 mRNA (Cox and Walter, 1996; Mori et al., 1996; Sidrauski et al., 1996; Sidrauski and Walter, 1997). Translation of the spliced mRNA produces a functional Hac1p transcription factor which then mediates the transcriptional induction of at least 381 genes in the yeast genome (Travers et al., 2000). Many of these induced genes are known to encode ER-resident chaperones, hence this response is presumed to maximize the cell’s capacity to process misfolded molecules and thus to tolerate a variety of stresses affecting folding pathways (Cox et al., 1993; Mori et al., 1993). Interestingly, the LHS1 gene is itself UPR regulated, suggesting that Lhs1p plays a role in the normal cellular response to folding stress (Baxter et al., 1996; Craven et al., 1996). However, the fact that the UPR is induced in Δlhs1 cells also raises the possibility that one or more UPR-regulated chaperones might be required to compensate for the loss of Lhs1p. Here we report that a functional UPR is required for the near normal growth rate observed in Δlhs1 cells when compared with the parental strain. Moreover, we have identified a single UPR-regulated gene, which we have named SIL1, that is essential for survival of Δlhs1 cells and whose overexpression is sufficient to suppress the phenotypes associated with the lhs1 deletion. Furthermore, we show that the lethal Δsil1Δlhs1 double mutation results in a complete defect in protein translocation into the ER.
Results
The role of the UPR in cellular adaptation to the Δlhs1 mutation
Yeast cells carrying a null mutation in lhs1 (Δlhs1) grow well but exhibit a constitutive induction of the UPR (Craven et al., 1996). As discussed above, the IRE1 gene encodes a key effector of the yeast UPR that is essential for the induction of UPR-regulated genes. Cells deleted for IRE1 (Δire1) are viable but are highly sensitive to tunicamycin, dithiothreitol (DTT) and a range of other treatments that perturb protein folding in the ER (Cox et al., 1993; Mori et al., 1993). We have reported previously that sporulation of a heterozygous diploid strain (Δire1/IRE1, Δlhs1/LHS1) gave rise to no viable Δire1Δlhs1 haploids (Craven et al., 1996). However, this requirement for Ire1p may be specific to germination after sporulation and may not be reflected in vegetative cells. In order to test the latter, we first transformed a heterozygous diploid (JTY21; Δire1/IRE1, Δlhs1/LHS1) with a URA3-based plasmid containing the functional LHS1 gene (pRC43). Following sporulation, viable Δire1Δlhs1 haploids were recovered which, in every case, were found to be Ura+, indicating the presence of the pRC43 plasmid. These haploids strains were then inoculated onto medium containing 5-fluoro-orotic acid (5-FOA) to counter- select the URA3-based pRC43 as described previously (Wilkinson et al., 1997). Incubation at 30°C gave rise to a low frequency of very slow growing colonies that are barely visible in Figure 1. This contrasted sharply with the numerous rapidly growing colonies observed when pRC43 was lost from either single mutant cells or from the wild-type parent (Figure 1). As expected, our results confirm that both the Δlhs1 and Δire1 single mutants are viable and grow vigorously. However, our findings clearly demonstrate a severe synthetic effect when the two mutations are combined. From these results, we conclude that the Ire1p-dependent induction of the UPR represents a vital adaptation in Δlhs1 cells.
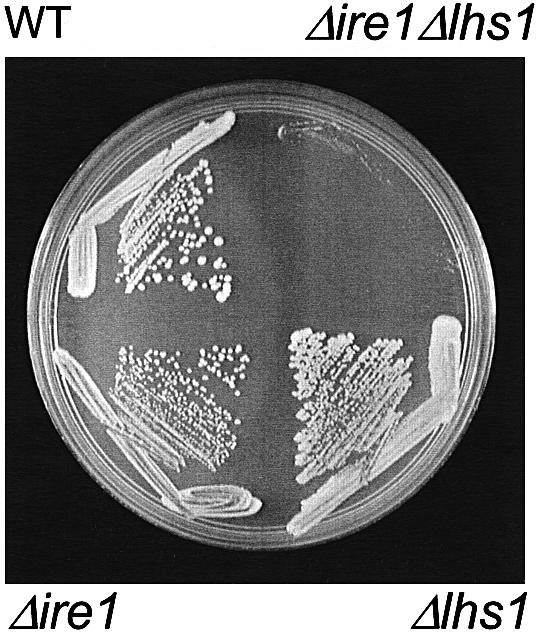
Fig. 1. Δlhs1Δire1 double mutant cells exhibit a severe growth defect. JTY21 (IRE1/ire1::KanMX4, LHS1/lhs1::TRP1) diploids carrying pRC43 (2µ, URA3, LHS1) were sporulated and tetrads dissected. Four plasmid-containing haploids from a single tetrad, with the genotypes as indicated, were then streaked onto minimal medium containing 5-FOA and incubated at 30°C to select for those cells that have lost pRC43 by spontaneous mis-segregation.
The requirement for IRE1 is not strain dependent
The phenotypes of the lhs1 deletion have been reported to be less severe in W303 strain backgrounds when compared with those used above (Baxter et al., 1996; Craven et al., 1996). We therefore constructed a Δlhs1 mutation in W303 (strain JTY33) and found no significant differences in either the extent of induction of the UPR or the accumulation of the untranslocated form of prepro-α-factor (data not shown). We further tested whether there was a synthetic interaction between Δire1 and Δlhs1 in W303 similar to that described for TR (above). As before, the Δire1Δlhs1 double mutant was lethal upon sporulation and in vegetative cells (data not shown). From these results, we conclude that the IRE1-dependent adaptation to the loss of Lhs1p is required in both strain backgrounds. From this point onwards, we report only results relating to the W303-derived strains.
Multicopy KAR2 or SCJ1 fail to suppress the Δire1Δlhs1 double mutant phenotype
The Δire1Δlhs1 double mutant has a very severe growth defect when compared with either single mutation alone. The simplest interpretation of these findings is to propose that the Ire1p-dependent unfolded protein response is required to induce one or more factors which then compensate for the loss of Lhs1p. Perhaps the most obvious candidate for a suppressor of Lhs1p would be Kar2p, primarily because it is a UPR-regulated member of the Hsp70 superfamily, but also due to its functional overlap with Lhs1p (Baxter et al., 1996; Craven et al., 1996; Hamilton and Flynn, 1996). A second obvious candidate is SCJ1, which encodes a UPR-regulated DnaJ homologue in the yeast ER lumen and has previously been reported to suppress the translocation phenotype associated with the lhs1 deletion when present on a multicopy plasmid (Hamilton and Flynn, 1996). We therefore decided to test whether multicopy plasmids containing either KAR2 (pJT41) or SCJ1 (pJT42) might be able to suppress the growth defect in Δire1Δlhs1 mutant cells. First, we transformed either pJT41 or pJT42 into JTY38 {Δire1Δlhs1 [pRC43 (LHS1, URA3)]} and tested for the ability of these strains to lose pRC43 on 5-FOA medium. Our results indicated that neither multicopy KAR2 nor SCJ1 suppressed the growth defect of the Δire1Δlhs1 double mutant (Figure 2A).
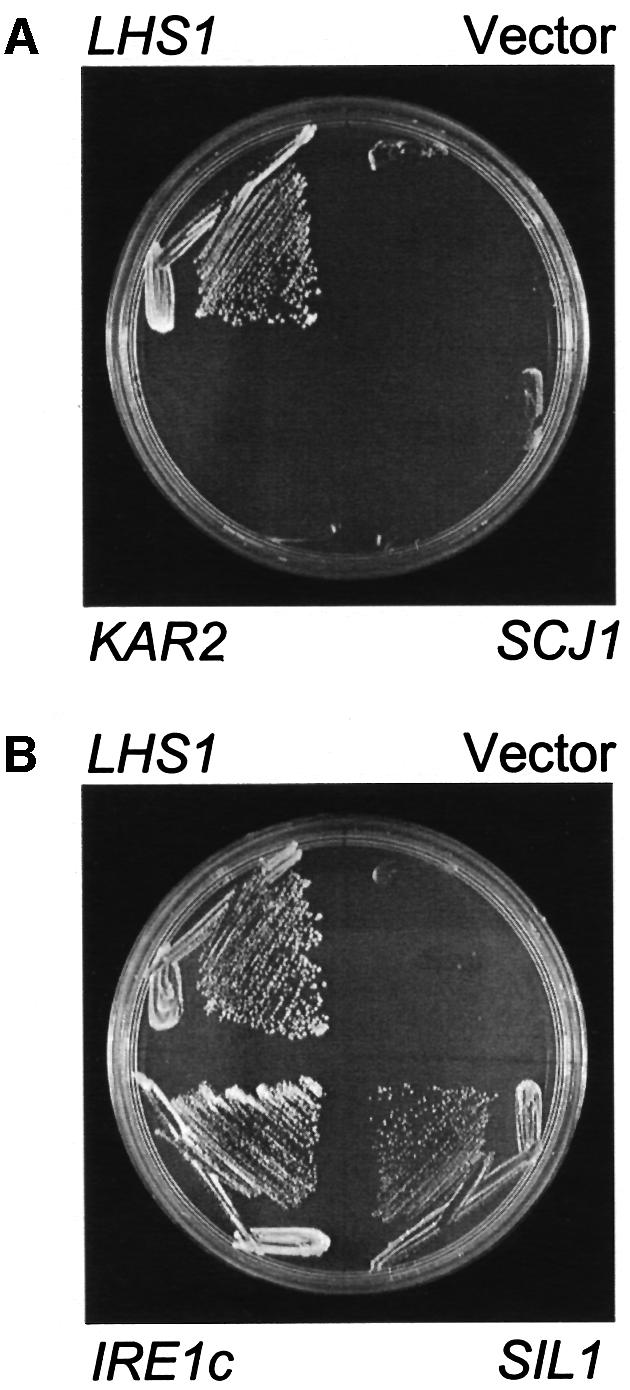
Fig. 2. Plasmid suppressors of the growth defect in Δire1Δlhs1 mutant cells. (A) Multicopy KAR2 or SCJ1 fail to suppress Δire1Δlhs1. JTY38 (Δire1Δlhs1 + pRC43) was transformed with pRS423 (vector control), pRC42 (HIS3, LHS1), pJT41 (2µ, HIS3, KAR2) or pJT42 (2µ, HIS3, SCJ1) and restreaked onto minimal medium containing 5-FOA and incubated at 30°C. (B) Isolation of suppressors from a multicopy library. JTY38 containing pRC43 was transformed with either pRS423 (vector control), pRC42 (HIS3, LHS1), pIRE1c (2µ, LEU2, IRE1 C-terminus) or pJT43 (2µ, HIS3, SIL1). Transformants were then inoculated onto 5-FOA medium and incubated as before.
Isolation of multicopy suppressors of the Δire1Δlhs1 double mutation
We next screened a multicopy plasmid-based library of yeast genomic DNA in order to identify any genes capable of suppressing the Δire1Δlhs1 growth phenotype. Unfortunately, the frequency with which spontaneous suppressors of the double mutant arose in the plasmid-loss assay (on 5-FOA medium) precluded this as a means with which to screen the library. Consequently, it was necessary for us to devise a robust suppression screen based upon a plasmid sectoring assay. First, a strain was constructed in which the Δire1Δlhs1 mutations were combined with mutant alleles of both ade2 and ade3 (JTY62) and whose viability is maintained by the presence of LHS1 on plasmid pJT40 (URA3, ADE3, LHS1). In this strain, pJT40 is stably maintained due to the strong selection for a functional LHS1 gene. Since pJT40 also contains the ADE3 gene, this stability can be visualized easily by virtue of the uniformly red colonies that are characteristic of Ade3+, Ade2– cells (Koshland et al., 1985). However, when this strain was transformed further with a second LHS1-containing plasmid, pRC42 (HIS3, LHS1), then cells could lose pJT40, giving rise to colonies containing Ade3– white sectors (data not shown). Having established a reliable sectoring assay, we first tested the multicopy plasmids containing either KAR2 (pJT41) or SCJ1 (pJT42). As expected, neither plasmid gave rise to sectoring colonies, confirming their inability to suppress the Δire1Δlhs1 growth phenotype (data not shown).
A multicopy LEU2-based library was transformed into JTY62 (Δire1Δlhs1) containing pJT40 (LHS1, URA3, ADE3) and transformants selected on minimal medium with appropriate supplements. A total of 18 000 transformants were examined, from which 29 authentic sectoring clones were identified. Plasmids isolated from these clones not only restored sectoring when re-transformed into JTY62(pJT40), but also rescued the 5-FOA sensitivity of this strain. Results for two suppressor plasmids, named pIRE1c and pSIL1, are shown in Figure 2B. Sequence data were obtained from the insert– vector boundaries of all 29 plasmids. Of these, 27 contained the intact LHS1 gene (with four different overlapping insert fragments represented), one contained a portion of the IRE1 gene and one contained a 13.2 kbp insert from chromosome XV. Clearly, the 27 LHS1-containing clones will complement the Δlhs1 mutation, resulting in cells with a simple Δire1 phenotype. The remaining two suppressors, pIRE1c and pSIL1, were of more interest.
Constitutive induction of the UPR by a C-terminal fragment of Ire1p
The pIRE1c suppressor plasmid contained a 5.54 kbp insert corresponding to residues 254 693–260 233 from yeast chromosome VIII (Johnston et al., 1994). This insert contains only the C-terminal 661 codons of the IRE1 open reading frame (ORF) fused in-frame to an AUG codon located 62 codons upstream in the vector sequence. Initiation at this AUG codon would give rise to a fusion protein lacking the ER-luminal dimerization domain of Ire1p, but which would contain the transmembrane domain and cytosolic kinase/nuclease domains (see Figure 3A) (Cox et al., 1993; Mori et al., 1993). We tested the ability of pIRE1c to induce the UPR by measuring induction of β-galactosidase activity in cells carrying a plasmid in which the lacZ gene is expressed under the control of a UPR-regulated promoter element (UPRE; Wilkinson et al., 2000). Cells carrying the UPRE–lacZ construct were transformed either with pIRE1c or with a vector control. As expected, wild-type cells containing only the vector control exhibited a low level of LacZ activity, with yet lower levels detected in Δire1 cells due to their inactive UPR (Figure 3B). In contrast, vector-transformed Δlhs1 cells exhibited a constitutive UPR induction as reported previously (Figure 3B) (Craven et al., 1996). When transformed with pIRE1c, all three strains now showed substantial expression of UPR-regulated LacZ at levels in excess of those observed in Δlhs1 cells (Figure 3B). The ability of Ire1Cp to induce the UPR was confirmed further by its ability to rescue the tunicamycin sensitivity of Δire1 cells (Figure 3C). These observations demonstrate that the truncated C-terminal fragment of Ire1p, termed Ire1Cp, is a constitutive inducer of the UPR pathway in yeast. The ability to induce the UPR to levels in excess of those normally observed in Δlhs1 cells would therefore appear to be sufficient to suppress the Δire1Δlhs1 double mutant phenotype.


Fig. 3. IRE1c encodes a C-terminal portion of IRE1 that constitutively induces the UPR and suppresses the tunicamycin hypersensitivity of Δire1 mutants. (A) Schematic views of Ire1p and the predicted Ire1Cp domain structures. The black region at the N-terminal end of Ire1Cp indicates potential vector coding sequence in-frame across the junction of the vector and the genomic insert. (B) Constitutive UPR induction in pIRE1c-containing cells. Strains JTY18 (WT), JTY32 (Δire1) and JTY33 (Δlhs1) harbouring the UPRE-lacZ reporter plasmid pJT30 were transformed with the plasmids pIRE1c (IRE1 fragment) or YEplac181 (vector control) and assayed for β-galactosidase activity. Fold induction is relative to the activity measured in JTY18 containing YEplac181. (C) pIRE1c rescues the tunicamycin sensitivity in Δire1 cells. JTY32 (Δire1) was transformed with either pIRE1c (IRE1 fragment) or YEplac181 (vector control), streaked onto YPD containing 1 µg/ml tunicamycin and incubated at 30°C.
Identification of YOL031c as a multicopy suppressor of Δire1Δlhs1
The third class of suppressor identified we termed SIL1 for (suppressor of the Δire1 Δlhs1 double mutant number 1). Unlike pIRE1c, the pSIL1 plasmid did not restore tunicamycin resistance in Δire1 cells, from which it appears unlikely that it functions as an inducer of the UPR (data not shown). The insert in pSIL1 corresponds to a 13.2 kbp portion of chromosome XV (Rad et al., 1997). Subcloning from pSIL1 identified the YOL031c ORF as encoding the SIL1 activity. The SIL1/YOL031c ORF comprises 421 codons and would encode a polypeptide with a predicted mol. wt of 48.26 kDa. The primary sequence contains a potential cleavable N-terminal signal sequence that would direct the protein to the ER lumen, and a C-terminal RDELCOOH likely to function as an ER-retrieval sequence. We therefore predict that the product of the SIL1 gene is an ER-localized protein. The SIL1/YOL031c protein sequence previously was noted to be homologous to that of the Sls1 protein (Sls1p) from Yarrowia lipolytica, which is an ER-resident protein known to interact with Kar2p and which is involved in the biosynthesis and secretion of an alkaline extracellular protease (Boisrame et al., 1996, 1998).
Overexpression of Sil1p suppresses the translocation phenotype associated with Δlhs1
Previous studies have shown that Δlhs1 mutant cells are defective in protein translocation into the ER. In order to characterize the nature of the Sil1p suppressor further, we next sought to determine whether or not it rescued the translocation phenotype of Δlhs1 cells. The Δlhs1 mutant strain JTY33 was transformed with either pJT43 (SIL1) or a vector control and then cell extracts were prepared and examined by immunoblotting. As expected, Δlhs1 cells containing the control vector accumulated cytosolic precursor forms of pre-Kar2, pre-PDI and prepro-α-factor (Figure 4A). The extent of precursor accumulation was significantly reduced in cell extracts prepared from JTY33 cells carrying pJT43 (Figure 4A). These results indicate that pJT43 largely suppresses the translocation phenotype associated with the Δlhs1 mutation. In contrast, neither multicopy KAR2 nor SCJ1 had any effect on the levels of precursor accumulated in Δlhs1 cells (Figure 4A), a result entirely consistent with their failure to suppress the growth defect in the Δlhs1Δire1 double mutant.


Fig. 4. Characterization of the SIL1 suppressor. (A) Multicopy SIL1 suppresses the translocation defect in Δlhs1 cells. Wild-type (JTY18), or JTY33 (Δlhs1) transformed with either a vector control (pRS423), pJT43 (SIL1), pJT42 (SCJ1) or pJT41 (KAR2) were grown to mid-log phase at 30°C in YNB with appropriate supplements. Whole-cell extracts were prepared and separated by SDS–PAGE before immunoblotting with antibodies specific to either Lhs1p, Kar2p, PDI or α-factor serum as indicated. Untranslocated precursor forms of the various proteins are indicated (preKar2p, prePDI and prepro-α-factor). Translocated forms are subject to ER processing; the signal-processed form of Kar2p is indicated, as are the signal-processed and core-glycosylated forms of PDI. Processed forms of α-factor are not evident in this gel system. (B) Δsil1 cells show no obvious defect in protein translocation. Strains JTY33 (Δlhs1) or JTY63 (Δsil1) were grown to mid-log phase in YNB before being harvested and whole-cell extracts prepared and analysed by immunoblotting as indicated. Control extracts were also prepared from wild-type cells (JTY18) treated in either the presence or absence of tunicamycin (10 µg/ml) for 2 h prior to harvesting cells. Protein species are annotated as before but include the unglycosylated, signal-processed forms of both Lhs1p (ugLhs1p) and PDI (ugPDI). (C) Constitutive levels of UPR induction. Wild-type (JTY18), Δsil1 (JTY63) or Δlhs1 (JTY33) cells were transformed with pJT30 (UPRE–lacZ) and grown to mid-log phase before assaying β-galactosidase activity. JTY33 (Δlhs1) containing pJT30 were also assayed after being transformed further with multicopy SIL1 (pJT43). (D) The Δsil1 and Δlhs1 are synthetically lethal in combination. JTY64 (SIL1/sil1::KanMX4, LHS1/lhs1::KanMX4) diploids carrying pRC43 (2µ, URA3, LHS1) were sporulated and tetrads dissected. A representative tetrad (with genotypes as indicated) is shown streaked onto 5-FOA-containing medium to select against pRC43.
Phenotypic analysis of a Δsil1 null mutant
The SIL1 ORF (YOL031c) was disrupted as part of the Eurofan project and shown to be non-essential for viability. We obtained a cloned sil1::kanMX4 disruption cassette (pYORC_YOL031c) from Euroscarf and used this to recreate the Δsil1 mutation by transformation of a haploid W303-derived strain, JTY19, to geneticin resistance. The site of integration in one such transformant, JTY63, was confirmed by PCR and this strain was used for further studies. As expected, our results confirmed that the Δsil1 mutant cells are viable. The Y.lipolytica SLS1 gene is also non-essential but has been implicated in secretory protein biogenesis in this system. We therefore analysed the translocation and processing of various precursor proteins in Δsil1 null mutant cells by immunoblotting. No defects were observed in the biogenesis of either PDI or Kar2, with protein profiles in Δsil1 cell extracts being indistinguishable from those in wild-type extracts (Figure 4B). We next examined UPR status in Δsil1 cells and found a modest induction compared with wild-type cells, suggesting a minor role for Sil1p in protein folding within the lumen (Figure 4C). We also tested for the effect of multicopy SIL1 on the level of UPR induction in Δlhs1 cells and found that this was reduced from 4-fold to 3-fold induction relative to a wild-type control (Figure 4C). While this manuscript was in preparation, a paper appeared identifying SIL1/YOL031c as a UPR-regulated gene which the authors named PER100 (Travers et al., 2000).
SIL1 is essential for the viability of Δlhs1 cells
Thus far, our data indicate that overexpression of Sil1p is sufficient to suppress the Δire1Δlhs1 double mutant. The SIL1 gene is itself UPR inducible (Travers et al., 2000) and so Sil1p may also play a role in the Ire1-dependent adaptation in Δlhs1 cells described earlier. If induction of Sil1p is important in suppressing the Δlhs1 mutation, then one might expect some synthetic effect between Δlhs1 and Δsil1. To test this, we mated JTY33 and JTY63 to generate the doubly heterozygous diploid JTY64 (Δlhs1/LHS1 Δsil1/SIL1). After sporulation, tetrads were dissected onto YPD medium and incubated at 30°C, whereupon no viable haploid Δsil1Δlhs1 progeny were obtained. Genotyping and PCR analysis on viable spores demonstrated the recovery of single mutants and wild-type spores at the expected frequencies. Where a double mutant could be inferred from the genotypes of its siblings, it was examined microscopically and shown to have germinated but to have failed to progress more than three to four cell divisions. We next transformed JTY64 (Δlhs1/LHS1 Δsil1/SIL1) with pRC43 (LHS1, URA3) and repeated the tetrad analysis. In this case, we recovered viable haploid Δsil1Δlhs1 double mutants that were found always to contain pRC43. Incubation of such strains on 5-FOA medium failed to produce any viable colonies, confirming that the double mutant combination was lethal (Figure 4D).
Overexpression of Sil1p does not accelerate ER-associated degradation
It has been reported that deletion of SIL1/PER100 leads to a minor reduction in the rate with which misfolded proteins are exported from the ER lumen for degradation in the cytosol (Travers et al., 2000). If the growth defect in Δire1Δlhs1 cells were due to the accumulation of misfolded proteins in the ER lumen, then an obvious source of suppression might be the induction of a factor that would accelerate ER-associated degradation (ERAD). We therefore tested whether multicopy SIL1 led to accelerated degradation of a mutant form of carboxypeptidase Y (CPY), known as CPY*, which misfolds in the ER lumen and which has been shown to be a substrate for ERAD (Knop et al., 1996). W303-1C cells (expressing CPY* from the prc1-1 allele) were transformed with either pJT43 (SIL1) or a vector control, and the rate of degradation of CPY* was monitored by pulse–chase studies. We found that the half-life of CPY* in fact was marginally extended in cells carrying multicopy SIL1 (pJT43) compared with the wild-type control (data not shown). Clearly, the overexpression of Sil1p does not accelerate CPY* degradation, from which it appears unlikely that observed suppression of Δire1Δlhs1 by pSIL1 is due to accelerated ERAD.
A Δlhs1Δsil1 double mutant is severely defective in protein translocation into the ER
The fact that the Δlhs1Δsil1 double mutation is lethal renders phenotypic analysis difficult. In order to overcome this, we created a conditional expression allele of LHS1. Initially, we placed the complete LHS1 ORF under the control of the methionine-repressible MET3 promoter (Cherest et al., 1985). However, this construct was found to complement the Δire1Δlhs1 double mutant even in the presence of methionine (data not shown). Immunoblot analysis indicated a marginal reduction in the levels of Lhs1p antigen when in methionine-containing medium over an 8 h time course (data not shown). We reasoned that the level of Lhs1p antigen observed in cells under repressing conditions might reflect a long half-life of this protein in the ER. We therefore sought to destabilize the protein by deleting its C-terminal RDELCOOH retrieval motif. A plasmid, pJT44, expressing this truncated copy of Lhs1p under control of the MET3 promoter (MET3p-lhs1trun) was found to complement the Δire1Δlhs1 double mutant only on medium lacking methionine (data not shown). The same plasmid was then transformed into Δlhs1Δsil1 cells carrying pRC43 (LHS1, URA3), selecting for the HIS3 marker on pJT44. Transformants were then inoculated onto 5-FOA medium, lacking methionine in order to select for cells cured of pRC43. The resultant strain (JTY66) grew well on medium lacking methionine but not on medium supplemented with 0.2 mM methionine (Figure 5A). Addition of methionine (0.2 mM) to a liquid culture led to a severe growth defect within 3 h, suggesting that Lhs1ptrun activity became limiting for growth at that time (Figure 5B). Immunoblotting analysis verified that JTY66 cells expressed a truncated form of Lhs1p (Lhs1ptrun), which was undetectable 3 h after addition of methionine (Figure 5C, lanes 3 and 4), confirming that the growth defect in these cells correlates with loss of the Lhs1ptrun antigen. We next examined the processing of various secretory precursors in these cells. As before, Δlhs1 mutant cells accumulated precursor forms of pre-Kar2, pre-PDI and prepro-α-factor that were absent in wild-type cells (Figure 5C, lanes 1 and 2). This translocation defect was complemented by MET3p-lhs1trun in the absence of methionine, but slowly re-appeared upon depletion of Lhs1ptrun following addition of methionine to the medium (Figure 5C, compare lane 2 with lanes 6–8). When Δlhs1Δsil1 double mutant cells carrying pJT44 were grown in the absence of methionine, precursors accumulated to a level intermediate between wild-type and Δlhs1, suggesting a partial complementation by MET3p-lhs1trun (Figure 5C, lanes 1–3). However, after 3 h repression in the presence of methionine, these cells exhibited an enormous accumulation of all precursors tested (Figure 5C, lane 4). These results demonstrate a direct correlation between the lethal consequences of the Δlhs1Δsil1 double mutation and a major defect in the translocation of protein precursors into the ER.


Fig. 5. Conditional expression of truncated Lhs1p reveals a severe translocation defect in Δsil1Δlhs1 double mutant cells. (A) JTY65 (Δsil1Δlhs1 + pRC43) was transformed with pJT44 (HIS3, MET3-LHS1trun) and then passaged on 5-FOA medium to remove pRC43 (URA3, LHS1). The resulting strain, JTY66 (Δsil1Δlhs1 + pJT44), was found to be viable on medium lacking methionine but not on medium containing 0.2 mM methionine. (B) Effect of methionine on logarithmically growing cultures. JTY66 (Δsil1Δlhs1 + pJT44) cells were grown in minimal medium lacking methionine to log phase, the culture was split, 0.2 mM methionine was added to one half and the effect on growth examined over time. Sample points for (C) are indicated with arrows and the time after methionine addition to which they correspond. (C) Translocation defects upon Lhs1ptrun depletion. Aliquots of cells from (B) were harvested and whole-cell extracts prepared and analysed by imunoblotting for Lhs1p, Kar2p, PDI, CPY and α-factor as indicated. Protein species are annotated as before but include the untranslocated (preproCPY) and the mature (mCPY) forms of CPY, and the C-terminally truncated form of Lhs1p (Lhs1ptrun).
Previous studies have shown that precursors may be translocated into the ER by two quite different routes, i.e. the SRP-dependent and SRP-independent pathways (Ng et al., 1996). In order to determine which of these pathways might be affected in Δlhs1Δsil1 cells, we next examined the fate of the SRP-dependent precursor, dipeptidylaminopeptidase B (DPAPB; Ng et al., 1996), and the SRP-independent precursor, CPY (Ng et al., 1996), by pulse-labelling during depletion of Lhs1ptrun. Our results show a substantial defect in CPY translocation after 60 min incubation in the presence of methionine, with >50% of newly synthesized protein being accumulated in the precursor form (Figure 6, lanes 1 and 2). This rose to 100% precursor accumulation after 2 h depletion (Figure 6, lane 3). A defect in DPAPB translocation was also observed, but the onset of this defect was delayed. Minor accumulation of pre-DPAPB was evident at 1 h, with a >50% defect after 2 h and a total defect at 3 h (Figure 6). These findings demonstrate that both the SRP-dependent and -independent pathways are blocked in Δsil1Δlhs1 cells but that the SRP-independent pathway is the first to be affected as Lhs1ptrun is depleted.
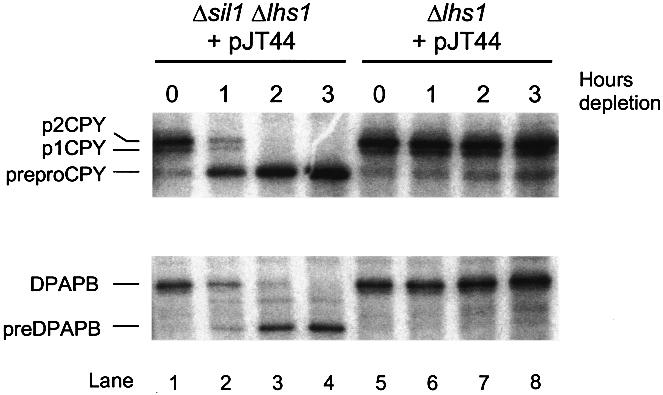
Fig. 6. The translocation defects in Δsil1Δlhs1 occur rapidly and are complete. Strains JTY66 (Δsil1Δlhs1) and JTY33 (Δlhs1) containing pJT44 (MET3p-lhs1trun) were grown to mid-log phase in YNB medium before addition of 0.2 mM methionine to repress the MET3 promoter. Cultures were labelled with a cocktail of [14C]amino acids (as described in Materials and methods) at the time points indicated, then extracts were prepared and subjected to sequential immunoprecipitation with antibodies specific for DPAPB followed by CPY (see Materials and methods). Immunoprecipitates were then resolved by SDS–PAGE, on 8% and 10% gels, respectively, and detected by fluorography. The untranslocated precursor form of CPY is indicated (prepro-CPY), as are the ER-processed (p1 CPY) and Golgi-modified (p2 CPY) forms. The untranslocated and mature forms of DPAPB are also indicated as preDPAPB and DPAPB, respectively.
Sil1p is conserved in mammals and Drosophila
Sil1p is homologous to the Sls1 protein from Y.lipolytica and we have identified homologues in both human and mouse expressed sequence tag (EST) sequence databases, and in the Drosophila genome sequence. A multiple alignment is shown in Figure 7, illustrating significant similarities between all five proteins. Previous studies have shown that Y.lipolytica Sls1p interacts with Kar2p and, intriguingly, we have found that the human and mouse Sil1p sequences also share very significant similarity to the cytosolic Hsp70-binding proteins HspBP1 (human: NP_036399; rat: AAF35834) and HSBp2 (human: AAF35833) (Raynes and Guerriero, 1998, 2000). These may therefore represent a major family of Hsp70 regulators that are present in all compartments of the eukaryotic cell.

Fig. 7. Sequence alignment of Sil1p-like proteins. The Sil1p sequence from S.cerevisiae (Sc) aligned with Sls1p from Y.lipolytica (Yl) and with sequences from Homo sapiens (Hs; DDBJ/EMBL/GenBank accession No. AJ299442), Mus musculus (Mm; accession No. AJ297884) and Drosophila melanogaster (Dm). Alignments were created using the CLUSTAL_X program, with black shading representing 100% conservation of amino acid similarity, dark grey 80% and light grey 60%. The boxed region highlighting the most conserved region motif was used to identify human and mouse EST sequences, allowing the complete sequence to be assembled from overlapping ESTs.
Sil1p interacts with the ATPase domain of Kar2p
In order to test whether yeast Sil1p interacts directly with Kar2p, we next tested for the co-purification of recombinant forms of these two proteins. Sil1p was fused to GST (GST–Sil1p) and purified from Escherichia coli extracts by its binding to glutathione–Sepharose (Figure 8). When a cell extract containing recombinant His-tagged Kar2p was then incubated with GST–Sil1p-bound beads, a substantial quantity of recombinant Kar2p was found to bind in a manner that was dependent upon the presence of GST–Sil1p. As an Hsp70, Kar2p has both an N-terminal ATPase domain and a C-terminal domain that is responsible for binding to unfolded polypeptides (Craven et al., 1997). The binding of Kar2p might therefore indicate merely that the GST–Sil1p fusion protein is partially misfolded. We have excluded this possibility by demonstrating further that a recombinant fragment of Kar2p, corresponding to only the ATPase domain, also interacts specifically with GST–Sil1p (Figure 8). These results demonstrate a biochemical interaction between Sil1p and the ATPase domain of yeast Kar2p.

Fig. 8. Biochemical interaction of GST–Sil1p with His-Kar2p or His-Kar2pATPase. Crude extracts were prepared from bacterial expression of GSTp, GST–Sil1p, His-Kar2p and His-Kar2pATPase, respectively (lanes 1–4). Each of these was then incubated with glutathione–agarose and the bound fraction resolved by SDS–PAGE (lanes 5–8). Lanes 9 and 10 contain the fraction of His-Kar2p and His- Kar2pATPase crude extract that binds to GSTp-bound glutathione–agarose. Lanes 11 and 12 contain the fraction of His-Kar2p and His- Kar2pATPase crude extract that binds to GST–Sil1p-bound glutathione–agarose.
Discussion
Previous characterization of the yeast Δlhs1 null mutant revealed a modest growth phenotype but also identified a substantial induction of UPR-regulated genes. Genetic studies revealed that Δire1Δlhs1 double mutant spores were non-viable, leading us to propose that the Ire1p-dependent UPR represents an important physiological adaptation in Δlhs1 mutant cells (Craven et al., 1996, 1997). This hypothesis is confirmed in this study where we have shown a severe synthetic growth defect in vegetative cells in which the Δlhs1 and Δire1 mutations are combined. Given that Lhs1p and Kar2p are related, we have suggested previously (Craven et al., 1996) that the UPR induction of Kar2p might be required to compensate for the absence of Lhs1p. However, our findings indicate that multicopy KAR2 had no effect upon the growth of the Δire1Δlhs1 double mutant. Interestingly, multicopy KAR2 has been shown to rescue the growth defect associated with accumulation of a misfolded protein in Δire1 cells (Umebayashi et al., 1999). It therefore follows that the growth defect in Δire1Δlhs1 cells is unlikely to be due solely to the accumulation of misfolded proteins. Similarly, overexpression of Kar2p does not rescue the translocation phenotype of a Δlhs1 mutant, from which we conclude that this phenotype is not an indirect consequence of sequestration of Kar2p onto unfolded polypeptides.
We next attempted to isolate multicopy plasmid suppressors of the Δire1Δlhs1 double mutant growth defect. As expected, we isolated complementing clones of both LHS1 and IRE1, but were struck by the isolation of a clone capable of expressing only the C-terminal portion of Ire1p (Ire1Cp). This clone led to a constitutive induction of the UPR and was of interest as it provides novel insights into the function of Ire1p. Current evidence suggests that the N-terminal ‘dimerization’ domain of Ire1p interacts with Kar2p/BiP in the ER lumen and, upon sequestration of Kar2p/BiP, promotes oligomerization of Ire1p and subsequent activation by phosphorylation (Shamu and Walter, 1996; Welihinda and Kaufman, 1996; Bertolotti et al., 2000). Given that this induction can occur in cells expressing only the C-terminal kinase/nuclease domain, then we must conclude that the dimerization domain of Ire1p is not essential for Ire1p activation. We therefore propose that the lumenal domain functions primarily as a Kar2p-dependent inhibitor of oligomerization/activation of Ire1p.
In addition to complementing clones of LHS1 and IRE1, our screen identified one bona fide suppressor named SIL1. The SIL1 gene corresponds to a reading frame of unknown function, YOL031c, identified in the yeast genome sequence. The SIL1/YOL031c gene has also been characterized recently as PER100, one of 381 UPR-inducible genes in the yeast transcriptome (Travers et al., 2000). The finding that SIL1 is UPR regulated would allow the IRE1-dependent induction of Sil1p in Δlhs1 cells. Such induction of Sil1p would be sufficient to explain the observed requirement for Ire1p in Δlhs1 cells. An absolute requirement for Sil1p in Δlhs1 cells is demonstrated by the synthetically lethal interaction observed between the Δsil1 and Δlhs1 mutations. We therefore conclude that the IRE1-dependent induction of SIL1 is a critical adaptation in Δlhs1 cells.
The SIL1/YOL031c gene encodes a polypeptide previously noted to be 27.5% identical (52% similar) to the Sls1 protein (Sls1p) from Y.lipolytica (Boisrame et al., 1996). The SLS1 gene was identified by virtue of a mutation that is lethal in combination with a defect in the Y.lipolytica SRP, suggesting some role for Sls1p in protein translocation in this system (Boisrame et al., 1996). However, like the Δsil1 mutants in Saccharomyces cerevisiae, the Y.lipolytica Δsls1 mutant is viable and does not accumulate any untranslocated precursor forms of secretory proteins. Nonetheless, the Δsls1 mutant is defective in the rate of synthesis of at least one secretory protein from which a translocation defect has been inferred (Boisrame et al., 1998). Some role in translocation is supported further by the finding that Sls1p can be co-immunoprecipitated with Sec61p (Boisrame et al., 1996), the core component of the translocon complex responsible for protein translocation across the ER membrane (Stirling et al., 1992; Broughton et al., 1997). Moreover, Sls1p has been shown to interact with the ATP-binding domain of Kar2p, leading to the proposal that Sls1p might function as a co-chaperone responsible for modulating the activity of Kar2p during its ATP-dependent reaction cycle (Boisrame et al., 1998; Kabani et al., 2000).
The translocation defects previously reported in Δlhs1 cells would be consistent with a defect that is exclusive to the post-translational SRP-independent protein translocation pathway (Baxter et al., 1996; Craven et al., 1996; Hamilton and Flynn, 1996). However, the lethal depletion of the truncated form of Lhs1p in Δsil1Δlhs1 cells correlates with a severe translocation phenotype for all precursors. This might be due to a gross perturbation of the structural integrity of the ER that might simply eliminate all ER functions. However, we observed substantial ER glycosylation of newly synthesized DPAPB in cells where translocation of prepro-CPY is blocked (Figure 6, lanes 2 and 3). From this, it is evident that the defect in CPY translocation must precede any gross loss of ER function. Our data are therefore entirely consistent with a direct role for Lhs1p in the post-translational translocation reaction. We cannot, at this time, exclude the possibility that a secondary consequence of this defect may then lead to the observed defect in co-translational translocation. Alternatively, the delayed onset of the co-translational translocation defect may reflect a direct requirement for Lhs1p/Sil1p that is satisfied by lower levels of Lhs1ptrun than are needed in the post-translational reaction. Circumstantial evidence in support of such a direct role includes the fact that the Y.lipolytica SLS1 gene was identified via its genetic interactions with SRP (Boisrame et al., 1996), and also that the mammalian homologue of Lhs1p, GRP170, is a major component of an ATP-binding fraction of ER lumenal proteins required for co-translational translocation into microsomes in vitro (Dierks et al., 1996).
The translocation defect observed in Δsil1Δlhs1 double mutant cells clearly demonstrates that Kar2p is not sufficient to drive ER translocation in vivo. We therefore conclude that Sil1p/Lhs1p provide a novel lumenal function(s) that is essential for the translocation process. Clearly, this function might affect either translocon gating, vectorial transport into the lumen, termination of translocation or the recycling of components of the translocon. Current evidence indicates that Kar2p/BiP is sufficient to gate the translocon during initiation of translocation (Hamman et al., 1998). However, the Lhs1 protein (Lhs1p) has been shown to bind to peptides in an ATP-dependent manner similar to that demonstrated for BiP (Hamilton et al., 1999). We therefore propose that Lhs1p binds to translocating polypeptides, thus contributing to the driving force for post-translational translocation in a manner similar to that previously suggested for Kar2p. There is no evidence for Sil1p interacting directly with precursors, but, given the observed interactions between Sil1p and Kar2p, we propose that a complex of Sil1p– Kar2p acts in concert with Lhs1p to drive the import reaction. Lhs1p is sufficient for this activity since the Δsil1 mutant has no translocation defect. Indeed, the fact that Δlhs1 cells exhibit a significant translocation defect, despite UPR induction of Sil1p, suggests that Lhs1p plays a major role in protein translocation in yeast. In the absence of Lhs1p, we propose that the Ire1-dependent induction of Sil1p is required to activate/potentiate Kar2p in order to enhance its contribution to the import reaction. The finding that Sil1p interacts directly with the ATPase domain of Kar2p suggests a direct mechanism by which Sil1p could modulate Kar2p activity. The proposed role of Kar2p in translocon gating would be independent of Sil1p, since the latter is non-essential, and since translocation is not blocked in a Δsil1 mutant. Our model makes a number of experimentally verifiable predictions that currently are under investigation. Finally, the identification of homologues of Sil1p in a variety of eukaryotes suggests that this newly defined role for Sil1p may in fact be widespread.
Materials and methods
Materials
DNA restriction and modification enzymes were purchased from Roche Molecular Biochemicals. [35S]methionine and the [14C]amino acid cocktail were from NEN Life Science Products. Oligonucleotides were purchased from Perkin-Elmer (Warrington, UK). All other reagents were from Roche Molecular Biochemicals, Sigma or Melford Labs (Suffolk, UK) at analytical grade.
Strain and growth conditions
Escherichia coli and yeast strains are listed in Table I. The E.coli cells were grown at 37°C in LB (1% tryptone, 0.5% yeast extract, 1% NaCl). Antibiotics were used when appropriate as follows: ampicillin (100 µg/ml), kanamycin (40 µg/ml) and chloramphenicol (34 µg/ml). Yeast strains were grown routinely at 30°C in YP medium (2% peptone, 1% yeast extract) containing 2% glucose (YPD) or in minimal medium (0.67% yeast nitrogen base; YNB) with 2% glucose plus appropriate supplements for selective growth. When screening for sectoring colonies using the ade2 ade3 mutations, 15 µM adenine was added to the medium; this level of adenine is sufficient to allow growth but also allows the red/white colony phenotype to develop and be observed. Solid media were supplemented with 2% Bacto-agar. All media were from Difco Laboratories. Repression of LHS1trun expressed from the MET3 promoter was achieved by addition of l-methionine to early log-phase cultures at a final concentration of 0.2 mM. Yeast transformations and 5-FOA counter-selection of Ura3+ cells were carried out as described previously (Wilkinson et al., 2000). Diploids were sporulated on 1% KOAc, 0.1% yeast extract, 0.05% glucose, plus appropriate supplements at 24°C. Tetrad dissection was as described previously (Wilkinson et al., 2000). Where appropriate, geneticin was used at a final concentration of 200 µg/ml in YPD. Cell density in liquid culture was monitored by A600 nm using a UVmini1240 spectrophotometer (Shimadzu).
Table I. Strains.
| Strains | Genotype |
|---|---|
| Saccharomyces cerevisiae | |
| CH1305 | MATa ade2 ade3 lys2, leu2, ura3 |
| RCY104 | MATa ade2 his3 lys2 trp1 ura3 Δlhs1::TRP1 |
| TR3 | MATα ade2 his3 lys2 trp1 ura3 |
| W303 | MATα/MATa ade2-1/ade2-1 CAN1-100/CAN1-100 his3-11,15/his3-11,15 leu2-3,112/leu2-3,112 trp1-1/trp1-1 ura3-1/ura3-1 |
| W303-1C | MATα ade2 CAN1 his3 leu2 trp1 ura3 prc1-1 |
| JTY18 | MATα ade2-1 CAN1-100 his3-11,15 leu2-3,112 trp1-1 ura3-1 (derived from W303) |
| JTY19 | MATa ade2-1 CAN1-100 his3-11,15 leu2-3,112 trp1-1 ura3-1 (derived from W303) |
| JTY20 | MATα ade2 his3 lys2 trp1 ura3 Δire1::kanMX4 |
| JTY21 | MATα/MATa ade2/ade2 his3/his3 lys2/lys2 trp1/trp1 ura3/ura3 Δire1::kanMX4/+ Δlhs1::TRP1/+ [pRC43] |
| JTY25 | MATa ade2 his3 lys2 trp1 ura3 Δire1::kanMX4 Δlhs1::TRP1 [pRC43] |
| JTY30 | MATα ade2 his3 lys2 trp1 ura3 Δlhs1::TRP1 |
| JTY32 | MATa ade2-1 CAN1-100 his3-11,15 leu2-3 112 trp1-1 ura3-1 Δire1::kanMX4 |
| JTY33 | MATα ade2-1 CAN1-100 his3-11,15 leu2-3 112 trp1-1 ura3-1 Δlhs1::kanMX4 |
| JTY34 | MATα/MATa ade2-1/ade2-1 CAN1-100/CAN1-100 his3-11,15/his3-11,15 leu2-3 112/leu2-3 112 trp1-1/trp1-1 ura3-1/ura3-1Δire1::kanMX4/+ Δlhs1::kanMX4/+ [pRC43] |
| JTY38 | MATa ade2-1 CAN1-100 his3-11,15 leu2-3 112 trp1-1 ura3-1 Δire1::kanMX4 Δlhs1::kanMX4 |
| JTY62 | MATa ade2 ade3 his3 leu2 trp1 ura3 Δire1::kanMX4 Δlhs1::kanMX4 [pJT40] |
| JTY63 | MATa ade2-1 CAN1-100 his3-11,15 leu2-3 112 trp1-1 ura3-1Δsil1::kanMX4 |
| JTY64 | MATα/MATa ade2-1/ade2-1 CAN1-100/CAN1-100 his3-11,15/his3-11,15 leu2-3 112/leu2-3 112 trp1-1/trp1-1 ura3-1/ura3-1Δsil1::kanMX4/+ Δlhs1::kanMX4/+ |
| JTY65 | MATα ade2-1 CAN1-100 his3-11,15 leu2-3 112 trp1-1 ura3-1Δsil1::kanMX4 Δlhs1::kanMX4 [pRC43] |
| JTY66 | MATα ade2-1 CAN1-100 his3-11,15 leu2-3 112 trp1-1 ura3-1Δsil1::kanMX4 Δlhs1::kanMX4 [pJT44] |
| Escherichia coli | |
| DH5α | supE44 ΔlacU169 (φ80lacZM15) hsdR17 recA1 endA1 gryA96 thi-1 relA1 |
| BL21(DE3)pLysS | F–, ompT, hsdSB (rB–, mB–) dcm, gal, (DE3) pLysS, Cmr |
Yeast strain construction
JTY33 (Δlhs1) was created by transformation of JTY18 with a linear Δlhs1::kanMX4 cassette replacing LHS1 codons 1–881 with kanMX4. The cassette was generated by PCR amplification of a kanMX4 module from pFA6-kanMX4 (Wach et al., 1994) using primers 5′-TGCAGTATTCTGGCATTATTAGTGCAAATAAGTACGCATATTACCCGTACGCTGCAGGTCGAC-3′ and 5′-TGATATCGGAAAATAAATCTA GTGCTATATATTATAAAGATTCTTATCGATGAATTCGAGCTCG-3′ with 5′ homology to the LHS1 ORF-flanking regions and 3′ homology to kanMX4. JTY18 was transformed with 1 µg of the resultant PCR product with selection on YPD containing 200 µg/ml geneticin. Correct integration of the Δlhs1::kanMX4 cassette was confirmed by PCR analysis of the integrated locus, and loss of Lhs1p confirmed by immunoblotting.
Insertion of the Δire1::kanMX4 deletion allele in both the TR (TR3) and W303 (JTY19) backgrounds to produce JTY20 and JTY32, respectively, was performed by a method previously described (Wilkinson et al., 2000). JTY20 was then mated with RCY104 (Craven et al., 1996) to produce a TR-derived heterozygous diploid (IRE/Δire1, LHS1/Δlhs1). Similarly, strains JTY32 (Δire1) and JTY33 (Δlhs1) were mated to produce a W303-based heterozygous diploid (IRE/Δire1, LHS1/Δlhs1). The two heterozygous diploids were then transformed with pRC43, a multicopy yeast vector containing the URA3 and LHS1 genes, to produce W303- or TR-derived heterozygous diploids, JTY21 and JTY34, respectively. Diploids were then sporulated and dissected to produce plasmid-containing haploid derivatives JTY38 and JTY25.
As a first step in creating JTY62 {ade2 ade3 Δire1 Δlhs1 [pJT40 (2µ), URA3, ADE3, LHS1]}, CH1305 (Dr D.Sweet) was mated with JTY33, and upon sporulation and subsequent tetrad dissection a haploid progeny with an ade2 ade3 Δlhs1::kanMX4 genotype was identified due to geneticin resistance and a white colony colour on YPD medium. This strain was then transformed with pJT40 and a heterozygous diploid created by mating to a haploid strain containing the Δire1::kanMX4 allele. This diploid strain was then sporulated and, upon tetrad dissection, JTY62 was isolated.
The SIL1 gene was deleted from JTY19 by replacement with a Δyol031c::kanMX4 allele obtained from the Eurofan project carried on the plasmid pYORC_YOL031c to create JTY63. The pYORC_YOL031c plasmid was digested with NotI and transformed into JTY19 followed by growth on YPD medium containing 2 µg/ml geneticin to select for integration events. Correct replacement was confirmed using diagnostic PCRs. The heterozygous Δlhs1::kanMX4/+ Δsil1::kanMX4/+ diploid, JTY64, was created by mating JTY33 with JTY63. JTY65 (Δlhs1, Δsil1, [pRC43]) was isolated from sporulation and tetrad dissection of JTY64 transformed with pRC43. JTY66 (Δlhs1, Δsil1, [pJT44]) was created by transforming pJT44 (HIS3, MET3p-LHS1trun) into JTY65 (Δlhs1, Δsil1, [pRC43]) and counter-selecting pRC43 on 5-FOA containing minimal medium lacking methionine.
Plasmids and nucleic acid manipulation
The E.coli–S.cerevisiae shuttle vectors pRS423 (2µ, HIS), pRS313 (CEN6, ARS4, HIS), pRS315 (CEN6, ARS4, LEU2) and YEplac181 (2µ, LEU2) have been described (Gietz and Sugino, 1988; Sikorski and Hieter, 1989; Christianson et al., 1992). The plasmid pJT30 (CEN, URA3) contains a UPRE–lacZ reporter cassette used for measuring UPR activity (Wilkinson et al., 2000); pPS177 (2µ, TRP1) encoding wild-type SCJ1 (Blumberg and Silver, 1991); pRC42 (CEN, HIS3) encoding wild-type LHS1; and pRC43 (2µ, URA3) also encoding wild-type LHS1 (Craven et al., 1996) have been described previously. pJT40 (2µ, URA3, ADE3, LHS1) was constructed by ligation of a 3.7 kbp BamHI fragment from pBamADE3 (Dr D.Sweet) containing the ADE3 gene into the single BamHI site of pRC43. pJT41 (2µ, HIS3, KAR2) was constructed by ligation of a 2.8 kbp XhoI fragment carrying KAR2 from pMR109 (Dr J.Leighton) into the single XhoI site of pRS423. pJT42 (2µ, HIS3, SCJ1) was constructed by first cloning a 4 kbp KpnI fragment containing SCJ1 from pPS177 into the single KpnI site of YEplac181, followed by removal of a 4 kbp SacI–BamHI fragment and cloning into pRS423 at the SacI–BamHI sites. pJT43 (2µ, HIS3, SIL1) was constructed by ligation of a 2.17 kbp XhoI fragment from pSIL1 containing SIL1 into the single XhoI site of pRS423. pJT44 (CEN6, ARS4, HIS3, MET3p-LHS1trun) is a single copy yeast vector based on pRS313 containing a MET3 regulatable allele of a truncated LHS1 gene. As a first stage in the construction of pJT44, an ∼700 bp SalI–EcoRV fragment from pHAM8 (Mountain and Korch, 1991) containing the MET3 promoter was ligated into pRS315, which had been cut with HindIII, blunted by Klenow fill-in and then cut with SalI. This was then cut with BamHI, and a 3265 bp LHS1-containing BamHI fragment ligated in. The LHS1-containing 3265 bp BamHI fragment was obtained by first cloning a 3261 bp PstI–SalI LHS1 fragment from pRC43 into PstI–SalI-digested pRS315 and then digesting the resulting vector with BamHI. After this joining of the MET3 promoter and LHS1-containing fragments, they were excised as a single 3823 bp SalI–NotI fragment and ligated into SalI–NotI-digested pRS313. This vector was then digested with SmaI to remove a 12 bp palindromic sequence between the MET3 promoter and the LHS1 gene, a step required to allow methionine-regulatable expression of Lhs1p. To create a truncated allele of LHS1 (amino acids 1–835 plus an alanine residue), pRC44 (Craven et al., 1996) was digested with HindIII followed by Klenow fill-in and re-circularization. A 1239 bp BamHI–Bsu36I fragment containing the truncated allele was excised from this vector, purified and used to replace the 1235 bp BamHI–Bsu36I fragment from the MET3p–LHS1 allele-containing vector, thus generating pJT44. JT45 is a derivative of pGEX-4T (Invitrogen) expressing a Sil1p (30–413) N-terminal GST-tagged protein. The SIL1 fragment was generated by PCR using the primers 5′-CGGGATCCACAATATTGCATTCATCCATAC-3′ and 5′-GCGTCGACGCCAAAGATCAAGTGTCTGC-3′. This was then digested with BamHI and SalI and cloned into the corresponding sites in pGEX-4T. pJT46 is a derivative of pET30a (Novagen) expressing a Kar2p (39–674) His-tagged protein, His-Kar2p. The KAR2 fragment was generated by PCR using the primers 5′-CGGGATCCGTTAGAGGTGCCGATGATG-3′ and 5′-GCGTCGACTTCGTCGTCATAATCAGCG-3′. This was then digested with BamHI and SalI and cloned into the corresponding sites in pET30a. pJT47 is a derivative of pET30a (Novagen) expressing a Kar2p (39–425) His-tagged protein, His-Kar2pATPase. The KAR2 fragment was generated by PCR using the primers 5′-CGGGATCCGTTAGAGGTGCCGATGATG-3′ and 5′-GCGTCGACTAAGACACCAGCTTGAACG-3′. This was then digested with BamHI and SalI and cloned into the corresponding sites in pET30a.
Immunoblotting
Whole yeast extracts were prepared by glass bead lysis in SDS sample buffer from cultures grown to mid-log phase, resolved by SDS–PAGE, transferred to a nitrocellulose membrane (HybondC, Amersham) and probed with reagent antisera essentially as described previously (Stirling et al., 1992).
Radiolabelling and immunoprecipitation
Strains were grown at 30°C in YNB medium containing 1 mM (NH4)2SO4 and the required supplements. For pulse labelling of strains with [35S]methionine, 5 OD600 equivalents of cells were pelleted, resuspended in 500 µl of culture supernatant, and 100 µCi of [35S]methionine added prior to a 5 min incubation at 30°C. For pulse labelling of strains in methionine gene repression experiments, 5 OD600 equivalents of cells were pelleted, resuspended in 150 µl of 2× YNB, 2% glucose and 150 µl of a 0.1 µCi/µl [14C]l-amino acid mix (NEN) added prior to a 5 min incubation at 30°C. Labelling reactions were stopped by addition of an equal volume of ice-cold 20 mM NaN3, and incubation on ice for 5 min. Cells were pelleted and resuspended in 1 ml of sphaeroplast buffer (1.4 M sorbitol, 50 mM Tris–HCl pH 7.4, 2 mM MgCl2, 10 mM NaN3), followed by addition of yeast lytic enzyme (ICN) to a concentration of 1.5 U/OD600 and incubation at 30°C for 30 min. Sphaeroplasts were pelleted and lysed by resuspension in 200 µl of IP lysis buffer (50 mM Tris–HCl pH 7.4, 1 mM EDTA, 1% SDS) and heating to 95°C for 5 min. After a 2 min cooling step on ice, 800 µl of IP buffer (187.7 mM NaCl, 62.5 mM Tris–HCl pH 8.0, 6.25 mM EDTA, 1.25% Triton X-100) was added to each sample followed by 50 µl of insoluble protein A suspension (Sigma). Samples were cleared by rotation at 4°C for 30 min, and then the insoluble protein A was pelleted by a 5 min microcentrifugation. Supernatants were transferred to a clean tube and 1 µl of the appropriate antibody added per OD600 of cells used followed by incubation at room temperature with rotation for 1 h. After this, 50 µl of protein A–Sepharose beads (20% w/v suspension in IP buffer) was added and samples were rotated at room temperature for a further hour. The beads were then pelleted in a microfuge and washed three times with 1 ml of IP buffer followed by dissociation of antigen by addition of 50 µl of 2× SDS–PAGE sample buffer and heating to 95°C for 5 min. For sequential immunoprecipitation, the bead-free supernatant was taken and cleared with insoluble protein A for 15 min before pelleting, transfer to a clean tube and addition of second antibody.
GST pull-down experiments
GST (GSTp or GST–SIL1p) and His-tagged (His-Kar2p or His-Kar2pATPase) proteins were expressed in BL21(DE3) pLysS (Novagen) by induction with 0.4 mM isopropyl-β-d-thiogalactopyranoside (IPTG) for 4 h. Crude extracts were then made by two freeze–thaw cycles in GST binding buffer [20 mM HEPES pH 7.4, 100 mM KCl, 5 mM MgCl2, 0.1% NP-40, 2% glycerol, 1 mM DTT, 1 mM EDTA, 1 µg/ml aminoethylbenzene sulfonyl fluoride (AEBSF), 2 µg/ml E64, 1 µg/ml aprotinin, 1 µg/ml pepstatin A and 1 µg/ml chemostatin] and cleared by centrifugation at 17 000 g for 30 min. Bindings to glutathione–agarose were performed by adding 100 µl of crude extract to 40 µl of glutathione–agarose (50% slurry) (Sigma) followed by incubation at 4°C for 1 h. The agarose beads were then washed three times with 100 µl of GST binding buffer before incubation with 100 µl of crude extract from strains expressing either His-Kar2p or His-Kar2pATPase for 1 h at 4°C. Following three further washes with GST binding buffer, the bound samples were eluted in SDS sample buffer at 95°C. Samples were then analysed by SDS–PAGE using 10% polyacrylamide gels and Coomassie Blue staining.
Antibodies
Lhs1p and Kar2p antiserum was raised using His6-tagged C-terminal portions of Lhs1p (residues 504–834) and Kar2p (residues 494–652). The fusion proteins for both these protein fragments were expressed using pET16b- (Novagen) derived expression vectors as described previously for His-tagged proteins (Wilkinson et al., 1996). These antigens were then used to inoculate sheep at Diagnostics Scotland (Lanark, Scotland). Antibodies to CPY (residues 155–525), α-factor (residues 10–120) and DPAPB (residues 200–642) were also raised in this laboratory using the same His-tagging approach. The following antibodies were used at the dilutions indicated in parentheses for immunoblotting: α-factor (sheep, 1:10 000; Stirling Lab), (CPY, 1:10 000; Stirling Lab), Lhs1p (sheep, 1:30 000; this study), Kar2p (sheep, 1:30 000; this study), PDI (rabbit, 1:30 000; Dr Jakob Winther, Carlsberg Reasearch Centre), peroxidase-conjugated goat anti-rabbit IgG (1:10 000; Sigma) and peroxidase-conjugated rabbit anti-sheep IgG (1:10 000; Daco). For immunoprecipitations, CPY and DPAPB antibodies were used at 1 µl per OD600 of radiolabelled cells.
β-galactosidase assays
β-galactosidase assays were performed on strains transformed with pJT30 (CEN, URA3, UPRE-lacZ) as previously described (Wilkinson et al., 2000).
Acknowledgments
Acknowledgements
We thank P.A.Silver (Harvard), J.Leighton (Dubendorf), J.Winther (Carlsberg Research Centre) and D.Sweet (MRC Cambridge) for providing various plasmids, strains and antibodies. This work was supported by The Wellcome Trust.
References
- Baxter B.K., James,P., Evans,T. and Craig,E.A. (1996) SSI1 encodes a novel Hsp70 of the Saccharomyces cerevisiae endoplasmic reticulum. Mol. Cell. Biol., 16, 6444–6456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertolotti A., Zhang,Y., Hendershot,L.M., Harding,H.P. and Ron,D. (2000) Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nature Cell Biol., 2, 326–332. [DOI] [PubMed] [Google Scholar]
- Blumberg H. and Silver,P.A. (1991) A homologue of the bacterial heat-shock gene DnaJ that alters protein sorting in yeast. Nature, 349, 627–630. [DOI] [PubMed] [Google Scholar]
- Boisrame A., Beckerich,J.M. and Gaillardin,C. (1996) Sls1p, an endoplasmic reticulum component, is involved in the protein translocation process in the yeast Yarrowia lipolytica. J. Biol. Chem., 271, 11668–11675. [DOI] [PubMed] [Google Scholar]
- Boisrame A., Kabani,M., Beckerich,J.M., Hartmann,E. and Gaillardin,C. (1998) Interaction of Kar2p and Sls1p is required for efficient co-translational translocation of secreted proteins in the yeast Yarrowia lipolytica. J. Biol. Chem., 273, 30903–30908. [DOI] [PubMed] [Google Scholar]
- Broughton J., Swennen,D., Wilkinson,B.M., Joyet,P., Gaillardin,C. and Stirling,C.J. (1997) Cloning of SEC61 homologues from Schizosaccharomyces pombe and Yarrowia lipolytica reveals the extent of functional conservation within this core component of the ER translocation machinery. J. Cell Sci., 110, 2715–2727. [DOI] [PubMed] [Google Scholar]
- Chen X., Easton,D., Oh,H.J., Lee-Yoon,D.S., Liu,X. and Subjeck,J. (1996) The 170 kDa glucose regulated stress protein is a large HSP70-, HSP110-like protein of the endoplasmic reticulum. FEBS Lett., 380, 68–72. [DOI] [PubMed] [Google Scholar]
- Cherest H., Nguyen,N.T. and Surdin-Kerjan,Y. (1985) Transcriptional regulation of the MET3 gene of Saccharomyces cerevisiae. Gene, 34, 269–281. [DOI] [PubMed] [Google Scholar]
- Christianson T.W., Sikorski,R.S., Dante,M., Shero,J.H. and Hieter,P. (1992) Multifunctional yeast high-copy-number shuttle vectors. Gene, 110, 119–122. [DOI] [PubMed] [Google Scholar]
- Cox J.S. and Walter,P. (1996) A novel mechanism for regulating activity of a transcription factor that controls the unfolded protein response. Cell, 87, 391–404. [DOI] [PubMed] [Google Scholar]
- Cox J.S., Shamu,C.E. and Walter,P. (1993) Transcriptional induction of genes encoding endoplasmic reticulum resident proteins requires a transmembrane protein kinase. Cell, 73, 1197–1206. [DOI] [PubMed] [Google Scholar]
- Craven R.A., Egerton,M. and Stirling,C.J. (1996) A novel Hsp70 of the yeast ER lumen is required for the efficient translocation of a number of protein precursors. EMBO J., 15, 2640–2650. [PMC free article] [PubMed] [Google Scholar]
- Craven R.A., Tyson,J.R. and Stirling,C.J. (1997) A novel subfamily of Hsp70s in the endoplasmic reticulum. Trends Cell Biol., 7, 277–282. [DOI] [PubMed] [Google Scholar]
- Dierks T. et al. (1996) A microsomal ATP-binding protein involved in efficient protein transport into the mammalian endoplasmic reticulum. EMBO J., 15, 6931–6942. [PMC free article] [PubMed] [Google Scholar]
- Gietz R.D. and Sugino,A. (1988) New yeast–Escherichia coli shuttle vectors constructed with in vitro mutagenized yeast genes lacking six-base pair restriction sites. Gene, 74, 527–534. [DOI] [PubMed] [Google Scholar]
- Hamilton T.G. and Flynn,G.C. (1996) Cer1p, a novel Hsp70-related protein required for posttranslational endoplasmic reticulum translocation in yeast. J. Biol. Chem., 271, 30610–30613. [DOI] [PubMed] [Google Scholar]
- Hamilton T.G., Norris,T.B., Tsuruda,P.R. and Flynn,G.C. (1999) Cer1p functions as a molecular chaperone in the endoplasmic reticulum of Saccharomyces cerevisiae. Mol. Cell. Biol., 19, 5298–5307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamman B.D., Hendershot,L.M. and Johnson,A.E. (1998) BiP maintains the permeability barrier of the ER membrane by sealing the lumenal end of the translocon pore before and early in translocation. Cell, 92, 747–758. [DOI] [PubMed] [Google Scholar]
- Johnston M. et al. (1994) Complete nucleotide sequence of Saccharo myces cerevisiae chromosome VIII. Science, 265, 2077–2082. [DOI] [PubMed] [Google Scholar]
- Kabani M., Boisrame,A., Beckerich,J.M. and Gaillardin,C. (2000) A highly representative two-hybrid genomic library for the yeast Yarrowia lipolytica. Gene, 241, 309–315. [DOI] [PubMed] [Google Scholar]
- Knop M., Finger,A., Braun,T., Hellmuth,K. and Wolf,D.H. (1996) Der1, a novel protein specifically required for endoplasmic reticulum degradation in yeast. EMBO J., 15, 753–763. [PMC free article] [PubMed] [Google Scholar]
- Kohno K., Normington,K., Sambrook,J., Gething,M.J. and Mori,K. (1993) The promoter region of the yeast KAR2 (BiP) gene contains a regulatory domain that responds to the presence of unfolded proteins in the endoplasmic reticulum. Mol. Cell. Biol., 13, 877–890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koshland D., Kent,J.C. and Hartwell,L.H. (1985) Genetic analysis of the mitotic transmission of minichromosomes. Cell, 40, 393–403. [DOI] [PubMed] [Google Scholar]
- Kuwabara K. et al. (1996) Purification and characterization of a novel stress protein, the 150-kDa oxygen-regulated protein (ORP150), from cultured rat astrocytes and its expression in ischemic mouse brain. J. Biol. Chem., 271, 5025–5032. [DOI] [PubMed] [Google Scholar]
- Lyman S.K. and Schekman,R. (1995) Interaction between BiP and Sec63p is required for the completion of protein translocation into the ER of Saccharomyces cerevisiae. J. Cell Biol., 131, 1163–1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyman S.K. and Schekman,R. (1997) Binding of secretory precursor polypeptides to a translocon subcomplex is regulated by BiP. Cell, 88, 85–96. [DOI] [PubMed] [Google Scholar]
- Matlack K.E., Misselwitz,B., Plath,K. and Rapoport,T.A. (1999) BiP acts as a molecular ratchet during posttranslational transport of prepro-α factor across the ER membrane. Cell, 97, 553–564. [DOI] [PubMed] [Google Scholar]
- Mori K., Ma,W., Gething,M.J. and Sambrook,J. (1993) A trans membrane protein with a cdc2+/CDC28-related kinase activity is required for signaling from the ER to the nucleus. Cell, 74, 743–756. [DOI] [PubMed] [Google Scholar]
- Mori K., Kawahara,T., Yoshida,H., Yanagi,H. and Yura,T. (1996) Signalling from endoplasmic reticulum to nucleus: transcription factor with a basic-leucine zipper motif is required for the unfolded protein-response pathway. Genes to Cells, 1, 803–817. [DOI] [PubMed] [Google Scholar]
- Mountain H.A. and Korch,C. (1991) TDH2 is linked to MET3 on chromosome X of Saccharomyces cerevisiae. Yeast, 7, 873–880. [DOI] [PubMed] [Google Scholar]
- Ng D.T., Brown,J.D. and Walter,P. (1996) Signal sequences specify the targeting route to the endoplasmic reticulum membrane. J. Cell Biol., 134, 269–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Normington K., Kohno,K., Kozutsumi,Y., Gething,M.J. and Sambrook,J. (1989) S.cerevisiae encodes an essential protein homologous in sequence and function to mammalian Bip. Cell, 57, 1223–1236. [DOI] [PubMed] [Google Scholar]
- Rad M.R., Habbig,B., Jansen,G., Hattenhorst,U., Kroll,M. and Hollenberg,C.P. (1997) Analysis of the DNA sequence of a 34,038 bp region on the left arm of yeast chromosome XV. Yeast, 13, 281–286. [DOI] [PubMed] [Google Scholar]
- Raynes D.A. and Guerriero,V.,Jr (1998) Inhibition of Hsp70 ATPase activity and protein renaturation by a novel Hsp70-binding protein. J. Biol. Chem., 273, 32883–32888. [DOI] [PubMed] [Google Scholar]
- Raynes D.A. and Guerriero,V. (2000) Isolation and characterization of isoforms of HspBP1, inhibitors of Hsp70. Biochim. Biophys. Acta, 1490, 203–207. [DOI] [PubMed] [Google Scholar]
- Rose M.D., Misra,L.M. and Vogel,J.P. (1989) KAR2, a karyogamy gene, is the yeast homolog of the mammalian Bip/GRP78 gene. Cell, 57, 1211–1221. [DOI] [PubMed] [Google Scholar]
- Sanders S.L., Whitfield,K.M., Vogel,J.P., Rose,M.D. and Schekman,R.W. (1992) Sec61p and BiP directly facilitate polypeptide translocation into the ER. Cell, 69, 353–365. [DOI] [PubMed] [Google Scholar]
- Saris N., Holkeri,H., Craven,R.A., Stirling,C.J. and Makarow,M. (1997) The Hsp70 homologue Lhs1p is involved in a novel function of the yeast endoplasmic reticulum, refolding and stabilization of heat-denatured protein aggregates. J. Cell Biol., 137, 813–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shamu C.E. and Walter,P. (1996) Oligomerization and phosphorylation of the Ire1p kinase during intracellular signaling from the endoplasmic reticulum to the nucleus. EMBO J., 15, 3028–3039. [PMC free article] [PubMed] [Google Scholar]
- Sidrauski C. and Walter,P. (1997) The transmembrane kinase Ire1p is a site-specific endonuclease that initiates mRNA splicing in the unfolded protein response. Cell, 90, 1031–1039. [DOI] [PubMed] [Google Scholar]
- Sidrauski C., Cox,J.S. and Walter,P. (1996) tRNA ligase is required for regulated mRNA splicing in the unfolded protein response. Cell, 87, 405–413. [DOI] [PubMed] [Google Scholar]
- Sikorski R.S. and Hieter,P. (1989) A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics, 122, 19–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stirling C.J. (1999) Protein targeting to the endoplasmic reticulum in yeast. 1997 Fleming Lecture. Microbiology, 145, 991–998. [DOI] [PubMed] [Google Scholar]
- Stirling C.J., Rothblatt,J., Hosobuchi,M., Deshaies,R. and Schekman,R. (1992) Protein translocation mutants defective in the insertion of integral membrane proteins into the endoplasmic reticulum. Mol. Biol. Cell, 3, 129–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Travers K.J., Patil,C.K., Wodicka,L., Lockhart,D.J., Weissman,J.S. and Walter,P. (2000) Functional and genomic analyses reveal an essential coordination between the unfolded protein response and ER-associated degradation. Cell, 101, 249–258. [DOI] [PubMed] [Google Scholar]
- Umebayashi K., Hirata,A., Horiuchi,H., Ohta,A. and Takagi,M. (1999) Unfolded protein response-induced BiP/Kar2p production protects cell growth against accumulation of misfolded protein aggregates in the yeast endoplasmic reticulum. Eur. J. Cell Biol., 78, 726–738. [DOI] [PubMed] [Google Scholar]
- Wach A., Brachat,A., Pohlmann,R. and Philippsen,P. (1994) New heterologous modules for classical or PCR-based gene disruptions in Saccharomyces cerevisiae. Yeast, 10, 1793–1808. [DOI] [PubMed] [Google Scholar]
- Welihinda A.A. and Kaufman,R.J. (1996) The unfolded protein response pathway in Saccharomyces cerevisiae. Oligomerization and trans-phosphorylation of Ire1p (Ern1p) are required for kinase activation. J. Biol. Chem., 271, 18181–18187. [DOI] [PubMed] [Google Scholar]
- Wilkinson B.M., Critchley,A.J. and Stirling,C.J. (1996) Determination of the transmembrane topology of yeast Sec61p, an essential component of the endoplasmic reticulum translocation complex. J. Biol. Chem., 271, 25590–25597. [DOI] [PubMed] [Google Scholar]
- Wilkinson B.M., Esnault,Y., Craven,R.A., Skiba,F., Fieschi,J., Kepes,F. and Stirling,C.J. (1997) Molecular architecture of the ER translocase probed by chemical crosslinking of Sss1p to complementary fragments of Sec61p. EMBO J., 16, 4549–4559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkinson B.M., Tyson,J.R., Reid,P.J. and Stirling,C.J. (2000) Distinct domains within yeast sec61p involved in post-translational translocation and protein dislocation. J. Biol. Chem., 275, 521–529. [DOI] [PubMed] [Google Scholar]