

Abstract
The COX pathway has been a target for pharmaceutical intervention in diseases with a high inflammatory component ranging from asthma and Alzheimer's to arthritis and cancer. A major transcriptional promoter of the malignant phenotype, HIF-1α, has been observed to be regulated by the COX-2 product PGE2. Here we show that HIF-1α protein significantly accumulated in human breast cancer MDA-MB-231 cells in response to the pro-inflammatory cytokine IL-1β, but not in COX-2-silenced MDA-MB-231 cells. In contrast HIF-α expression could be detected in COX-2-silenced cells in response to the hypoxia-mimetic agent CoCl2 and hypoxia. Gene expression profiling in COX-2-containing and COX-2-silenced cells showed that the hypoxia-induced transcriptional response is largely unaffected by COX-2 silencing. These data suggest that the profound effects of COX-2 silencing on inhibiting invasion, tumor growth and metastasis from MDA-MB-231 cells are dependent on the induction of IL-1β-dependent COX-2 and HIF-1α but are independent of hypoxia.
Keywords: breast cancer, inflammation, COX-2, COX-2-silencing, hypoxia, HIF-1α
Introduction
To grow larger than a few millimeters solid tumors have to adapt to decreasing oxygen availability.1,2 The hypoxia-induced transcriptional response is mediated by the stabilization of hypoxia-inducible factor (HIF) and involves the induction of genes associated, among others, with altered metabolism, angiogenesis and increased invasion and metastasis.3–6 Another important physiological environment encountered by growing tumor cells, circulating cancer cells and metastasizing cells involves the inflammatory response. Immune cells orchestrating the response to pathogens as well as transformed cells secrete pro-inflammatory cytokines in the local microenvironment, among them IL-1β.7,8 IL-1 signaling is responsible for the induction of cyclooxygenase 2 (COX-2) an enzyme that catalyzes the conversion of arachidonic acid to secondary lipid mediators such as prostanoids.9 The role of prostanoids in cancer is under intense scrutiny particularly in situations of chronic inflammation or in cancerous lesions overexpressing COX-2. Data from several studies have suggested a co-operation between COX-2 and HIF-1α.10–13
We previously reported that COX-2-silenced cells fail to metastasize to the lung in an experimental model of metastasis and displayed reduced tumor onset when grafted in the mammary fat pad of SCID mice.14 Since hypoxia is known to mediate tumor angiogenesis through vascular endothelial growth factor (VEGF)-A as well as to increase metastasis,5 we examined differences in the hypoxia and inflammation-driven functional activation of HIF-1α in COX-2-containing and COX-2-silenced cells. Previous studies with pharmacologic agents selective for COX-2 inhibition have given conflicting results regarding their effect on HIF-1α levels.10–13 The nonselective COX inhibitors ibuprofen, diclofenac and ketorolac have been shown to reduce HIF-1α and −2α levels in response to hypoxia; however, the reduction of HIF levels was also seen in prostate cancer DU145 cells that did not express COX-2.15 Additionally, in the same study, the selective COX-2 inhibitor NS-398 did not significantly inhibit HIF-1α accumulation in response to hypoxia in neither “COX-2-positive” PC-3 cells, nor “COX-2-negative” DU145 cells. In contrast, another study reported that a similar concentration of NS-398 significantly reduced HIF-1α levels11 in the COX-2-containing PC-3 prostate carcinoma cells and the COX-2-negative HCT116 colon carcinoma cells. Interestingly, a ten-fold lower dose of NS-398 inhibited HIF-1α transcriptional activity, as measured by hypoxia response element activity and VEGF production, in the HT-29 colorectal cancer cell line in a separate study.13 HIF-1α was also shown to directly bind to the COX-2 promoter thereby regulating the expression of COX-2 protein in two colorectal carcinoma cell lines, HCT116 and HT29, although HCT116 cells are generally referred to as COX-2 negative11,13 even in response to hypoxia.13 Since IL-1β signaling also induces HIF-1α stabilization in a pathway dependent on functional NFκB, COX-2 and VHL10 even under normoxic conditions, here, for the first time, we have examined the effect of siRNA-mediated silencing of COX-2 on the expression of HIF-1α in response to inflammation and hypoxia.
Results
COX-2-silenced cells accumulate HIF-1α in response to the hypoxia-mimetic agent CoCl2 and hypoxia
To examine the role of COX-2 in the hypoxic response we initially probed HIF-1α accumulation in COX-2-containing and COX-2-silenced cells in a dose-dependent experiment using CoCl2. CoCl2 is a hypoxia-mimetic agent known to inhibit the oxygen-dependent degradation of HIF-1α. HIF-1α accumulated in COX-2-silenced cells (Clone 2, Pooled) although in smaller amounts than in COX-2-containing cells (MDA-MB-231) in a dose-dependent manner (Fig. 1A). We then probed the response of COX-2-silenced Clone 2 and Pooled cells, to hypoxia (pO2~0.7%—Fig. 1B). A robust and comparable HIF-1α accumulation was observed in both COX-2-containing and COX-2-silenced cells in response to hypoxia (Fig. 1B).
Figure 1.


COX-2-silenced cells accumulate functional HIF-1α and maintain an intact hypoxic response. (A) Immunoblotting for HIF-1α in COX-2-containing parental MDAMB-231 (231) cells, and COX-2-silenced Clone 2 and Pooled cells subjected to the indicated amounts of CoCl2 for 24 h. Total cell lysate from each cell line was subjected to immunoblotting for HIF-1α, COX-2 and GAPDH. (B) Immunoblotting for HIF-1α in COX-2-containing parental MDA-MB-231 (231) cells, and COX-2-silenced Clone 2 and Pooled cells subjected to 0.7% hypoxia for the indicated times. Total cell lysate from each cell line was subjected to immunoblotting for HIF-1α, COX-2 and GAPDH. (C) Fold induction of HIF-1α-induced transcripts SLC2A1, VEGF and LDHA in COX-2-containing (MDA-MB-231) and COX-2-silenced cells (Clone 2 and Pooled) in response to 24 h of hypoxia. Fold induction was calculated using the 2ΔΔCt method using 18S Ct values as control. (D) Microarray analysis of total mRNA in COX-2-containing (MDA-MB-231) and COX-2-silenced cells (Clone 2 and Pooled) in response to hypoxia for 24 h. Fold-induction of known HIF-1α target genes is shown. (E) Fold induction of transcripts significantly upregulated by hypoxia in MDA-MB-231 cells and comparison with the fold induction of transcripts that were similarly regulated between Clone 2 and Pooled cells.
COX-2 silencing does not affect the hypoxia-dependent HIF-1α response
Quantitative PCR and microarray analysis were performed to examine differences among hypoxia-inducible transcripts between COX-2-containing and COX-2-silenced cells. We probed the induction of three known hypoxia-responsive genes VEGF, LDHA and SLC2A1 in response to hypoxia. HIF-1α-responsive transcripts VEGF and LDHA were regulated in a similar manner in the presence (MDA-MB-231) or absence of COX-2 (Clone 2 and Pooled) following 24 h of hypoxia (pO2~0.7%) as shown by qPCR in two independent experiments. A modest increase in the induction of SLC2A1 was observed in both Clone 2 and Pooled COX-2 silenced cells (Fig. 1C). To further address the role of COX-2 in the hypoxia-induced response we performed microarray analysis on total RNA isolated from cells subjected to hypoxia and normoxia for 24 h and examined the expression of 9 genes known to be HIF targets (Fig. 1D). Expression of HIF-induced transcripts was not affected by the absence of COX-2. To further understand the response of hypoxia-induced transcription in COX-2-containing and COX-2-silenced cells we selected transcripts that have been significantly induced by hypoxia in parental MDA-MB-231 cells (posterior probability >0.5) and compared their expression with that of the same probesets in Clone 2 and Pooled cells. Of the 21 Affymetrix probeset IDs that fulfilled the criteria outlined in the materials and methods section, and have been assigned a gene title, only lysyl oxidase (LOX) was differentially regulated by hypoxia in COX-2-containing and COX-2-silenced cells to a significant extent (higher than 3-fold in both Clone 2 and Pooled cells, Fig. 1E). Higher levels of LOX mRNA were observed in COX-2-silenced cells relative to parental cells following hypoxia, although normoxic COX-2-silenced cells contained lower levels of LOX mRNA than parental cells (data not shown). The remaining 20 genes were similarly regulated by hypoxia regardless of the presence or absence of COX-2.
COX-2-silencing inhibits IL-1β-dependent accumulation of functional HIF-1α
We next examined the role of COX-2 and HIF-1α in response to the pro-inflammatory cytokine IL-1β. COX-2-containing, MDA-MB-231 but not COX-2-silenced (Clone 2 and Pooled) cells accumulated HIF-1α in response to IL-1β (Fig. 2A). The accumulating HIF-1α protein was functional as it induced the expression of the known HIF-1α target genes CXCR4 and VEGFA in COX-2 containing cells (Fig. 2B). The fold-induction of VEGF in COX-2-silenced cells suggests that IL-1β may induce VEGF independently of COX-2 and HIF-1α.
Figure 2.
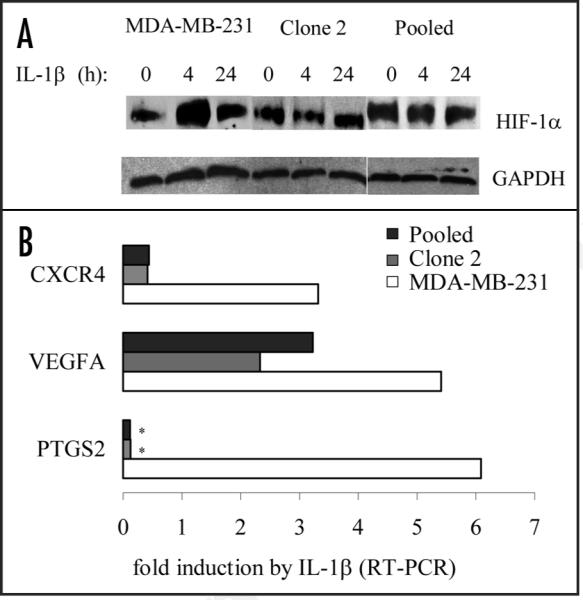
COX-2 silencing inhibits functional HIF-1α activation by IL-1β. (A) Immunoblotting for HIF-1α in COX-2-containing parental MDA-MB-231 (231) cells, and COX-2-silenced Clone 2 and Pooled cells in response to the pro-inflammatory cytokine IL-1β (10 ng/ml). (B) Fold induction of HIF-1α-related transcripts by RT-PCR. * Below detection limits.
Discussion
We have previously shown that silencing of COX-2 expression in MDA-MB-231 cells reduced orthotopic tumor growth in SCID mice and abolished extrapulmonary metastasis in an experimental model of metastasis.14 Given the importance of hypoxia in tumor growth and metastasis we sought to examine whether COX-2 silencing compromised the hypoxia-dependent response thus accounting for reduced tumor onset and loss of metastatic potential observed in these cells. COX-2-silenced cells maintained the capacity to induce HIF-1α to a significant, although slightly reduced, extent in response to the hypoxia-mimetic agent CoCl2 or hypoxia. Real-time RT-PCR and microarray analysis of total RNA demonstrated that COX-2 silencing did not significantly affect hypoxia-driven gene expression. One notable exception was the higher levels of LOX mRNA in COX-2-silenced cells relative to parental cells following hypoxia. LOX was recently shown to be necessary for metastasis of MDA-MB-231 cells in lungs and livers of nude mice17 an observation attributed to the reduction of hypoxia-stimulated cell invasion. More recently the accumulation of LOX was found to inversely correlate with LOX activity in response to hypoxia in poorly invasive MCF-7 and T47D breast cancer cells18 due to the decreased oxygen availability necessary for enzyme function. It was proposed that poorly invasive breast cancer cells subjected to hypoxia overexpress LOX but that its enzymatic function is reduced. This observation is consistent with our finding that poorly invasive COX-2-silenced MDA-MB-231 cell14 express more LOX in response to hypoxia. Our data suggest that COX-2 modulates the expression of LOX, but the interplay between inflammation, hypoxia and the effect of these environments on LOX-dependent metastasis remains to be more precisely defined.
In contrast to the intact response to hypoxia in COX-2 silenced cells, we found that IL-1β-dependent induction of functional HIF-1α depended on COX-2 expression in poorly differentiated breast cancer MDA-MB-231 cells consistent with an earlier study.10 Our results indicate that COX-2 is important for IL-1β, but not hypoxia, driven HIF-1α activation. Our data also suggest that the previously observed inability of COX-2-silenced cells to metastasize and their significantly reduced ability to establish tumor growth in the mammary fat pad cannot be attributed to differential responses to hypoxia. Instead it is likely, that the decreased tumor onset we previously reported14 is mediated, at least in part, as a result of silencing COX-2-dependent HIF-1α activation. Some of the research on the influence of inflammation on hypoxia has been conducted using pharmacologic inhibitors that are now known to have a wide array of functions independent of the COX-2 pathway.19 Our results support the conclusion that COX-2-dependent HIF-1α activation is an important factor in the cellular response to IL-1β but not hypoxia.12 Research results presented here and elsewhere14 indicate that the signaling mediated through the IL-1β-COX-2-HIF-1α axis plays an important role in the tumorigenicity and metastatic potential of poorly differentiated breast tumors.
The role of IL-1β and pro-inflammatory signaling is currently being investigated for the induction promotion, or exacerbation of cancer,8 diabetes20 premature labor,21 auto-immune,22 neurodegenerative23 and cardiovascular diseases.24 Our results show that treatment of illnesses with exacerbated inflammatory components should also account for the implications of functional HIF-1α activation even under normoxic conditions; targeting COX-2 could minimize these effects. Trauma from cancer treatments such as surgery and ionizing radiation are also likely to activate the inflammatory response. Our data support the administration of anti-inflammatory agents immediately following surgery and ionizing radiation treatment of patients to minimize activation of the IL-1β-COX-2-HIF-1α axis of oncogenic signaling.
Materials and Methods
Cell culture and hypoxia studies
All cells were cultured with RPMI-1640 medium (Sigma-Aldrich, St. Louis, MO) supplemented with 8.25% FBS (Sigma-Aldrich, St. Louis, MO) without penicillin or streptomycin. Cells used in the microarray experiment were seeded (1 × 106) in 100 mm dishes and allowed to attach to the plate overnight. The following day cells were placed in a hypoxic chamber at 0.7% oxygen or a normoxic incubator for 24 h at 37°C. Oxygen tension in the hypoxia chamber was monitored in real-time using the ProOXc system (Model 110, Biospherix, Ltd.,).
Immunoblotting
Cells were lysed using M-PER (Pierce, Rockford, IL) and a protease inhibitor cocktail (Sigma-Aldrich, St. Louis, MO) was added at a 1:750 dilution. Crude cell extracts were quantified using Dc protein assay (Bio-Rad, Hercules, CA). Fifty micrograms of protein were subjected to SDS-PAGE and immunobloting following standard protocols. Membranes were probed overnight with antibodies against COX-2 (CaymanChem, Ann Arbor, MI), HIF-1α (BD Biosciences, Franklin Lakes, NJ) and GAPDH (Santa Cruz, Santa Cruz, CA) and washed and probed with appropriate HRP-labeled secondary antibodies (Jackson ImmunoResearch, West Grove, PA) following standard protocols. Substrate (SuperSignal West Pico-Pierce, Rockford, IL) was added to the probed membranes and films were developed using automatic film processors (Kodak, Rochester, NY—Agfa, Ridgefield Park, NJ).
Real-time RT-PCR
One-step real-time RT-PCR was performed as previously described14 using 10 ng of total RNA isolated following the Qiagen RNeasy purification protocol as described by the manufacturer (Qiagen, Valencia, CA). Primer sequences for 18S transcript used as a control were: 5'-GGTTGATCCTGCCAGTAGC-3' and 5'-GCGACCAAAGGAACCATAAC-3'. QuantiTect real-time primers for CXCR4, VEGF, SLC2A1 and LDHA were also purchased from Qiagen's geneglobe collection and they are catalogue numbers: QT00223188, QT01036861, QT00068957 and QT00001687 respectively. Fold differences were calculated using the 2ΔΔCt method.
Microarray analysis
RNA from COX-2-containing (MDA-MB-231) and COX-2-silenced (Clone 2 and Pooled) cells was isolated as described above. Samples were subjected to agarose formaldehyde electrophoresis to determine the quality of the RNA by comparing the ratios of 18S and 28S RNA species. All samples were run in commercial arrays from Affymetrix (Santa Clara, CA), using Human Genome U133Plus 2.0 GeneChip arrays as described in the Affymetrix web site (http://www.affymetrix.com). Probe hybridization and analysis was run as suggested by the manufacturer in the aforementioned website. These descriptions include all information currently considered under the MIAME supportive guidelines, with which the JHMI Microarray Core Facility abides in all its procedures. Expression signals were obtained by Robust Multiarray Analysis.16 The posterior probabilities of the differential expression of genes between the treatments were estimated with an empirical Bayses method using Gamma-Gamma modeling by the bioconductor R package, EBarrays. The methodology for selecting transcripts shown in Figure 2C is as follows: the criterion of the posterior probability >0.5 was used to produce a list of genes upregulated in hypoxic over normoxic conditions in parental MDA-MB-231 cells. Affymetrix probeset IDs corresponding to transcripts upregulated in hypoxic over normoxic conditions from MDA-MB-231 cells were matched and compared with those from Clone 2 and Pooled cells. Differences in the hypoxic response of MDA-MB-231 cells compared to Clone 2 and Pooled cells higher than 3-fold was considered significant.
Acknowledgements
The authors thank Dr. Balaji Krishnamachary and Dr. Kristine Glunde for useful discussions and advice.
This work was supported by NIH research grants 2R01CA82337 and P50 CA103175.
Abbreviations
- COX
cyclooxygenase
- HIF
hypoxia inducible factor
- PGE
prostaglandin E
- VEGF
vascular endothelial growth factor
- LDHA
lactate dehydrogenase A
- SLC
solute carrier
- SCID
severe combined immunodeficiency
- LOX
lysyl oxidase
References
- 1.Fukumura D, Jain RK. Tumor microvasculature and microenvironment: targets for anti-angiogenesis and normalization. Microvasc Res. 2007;74:72–84. doi: 10.1016/j.mvr.2007.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Naumov GN, Akslen LA, Folkman J. Role of angiogenesis in human tumor dormancy: animal models of the angiogenic switch. Cell Cycle. 2006;5:1779–87. doi: 10.4161/cc.5.16.3018. [DOI] [PubMed] [Google Scholar]
- 3.Wang GL, Jiang BH, Rue EA, Semenza GL. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc Natl Acad Sci USA. 1995;92:5510–4. doi: 10.1073/pnas.92.12.5510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Huang LE, Gu J, Schau M, Bunn HF. Regulation of hypoxia-inducible factor 1alpha is mediated by an O2-dependent degradation domain via the ubiquitin-proteasome pathway. Proc Natl Acad Sci USA. 1998;95:7987–92. doi: 10.1073/pnas.95.14.7987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liao D, Johnson RS. Hypoxia: a key regulator of angiogenesis in cancer. Cancer Metastasis Rev. 2007;26:281–90. doi: 10.1007/s10555-007-9066-y. [DOI] [PubMed] [Google Scholar]
- 6.Zhang H, Gao P, Fukuda R, Kumar G, Krishnamachary B, Zeller KI, Dang CV, Semenza GL. HIF-1 inhibits mitochondrial biogenesis and cellular respiration in VHL-deficient renal cell carcinoma by repression of C-MYC activity. Cancer Cell. 2007;11:407–20. doi: 10.1016/j.ccr.2007.04.001. [DOI] [PubMed] [Google Scholar]
- 7.Allavena P, Sica A, Solinas G, Porta C, Mantovani A. The inflammatory micro-environment in tumor progression: the role of tumor-associated macrophages. Crit Rev Oncol Hematol. 2008;66:1–9. doi: 10.1016/j.critrevonc.2007.07.004. [DOI] [PubMed] [Google Scholar]
- 8.Apte RN, Dotan S, Elkabets M, White MR, Reich E, Carmi Y, Song X, Dvozkin T, Krelin Y, Voronov E. The involvement of IL-1 in tumorigenesis, tumor invasiveness, metastasis and tumor-host interactions. Cancer Metastasis Rev. 2006;25:387–408. doi: 10.1007/s10555-006-9004-4. [DOI] [PubMed] [Google Scholar]
- 9.Smith WL, DeWitt DL, Garavito RM. Cyclooxygenases: structural, cellular and molecular biology. Annu Rev Biochem. 2000;69:145–82. doi: 10.1146/annurev.biochem.69.1.145. [DOI] [PubMed] [Google Scholar]
- 10.Jung YJ, Isaacs JS, Lee S, Trepel J, Neckers L. IL-1beta-mediated upregulation of HIF-1alpha via an NFkappaB/COX-2 pathway identifies HIF-1 as a critical link between inflammation and oncogenesis. Faseb J. 2003;17:2115–7. doi: 10.1096/fj.03-0329fje. [DOI] [PubMed] [Google Scholar]
- 11.Zhong H, Willard M, Simons J. NS398 reduces hypoxia-inducible factor (HIF)-1alpha and HIF-1 activity: multiple-level effects involving cyclooxygenase-2 dependent and independent mechanisms. Int J Cancer. 2004;112:585–95. doi: 10.1002/ijc.20438. [DOI] [PubMed] [Google Scholar]
- 12.Kaidi A, Qualtrough D, Williams AC, Paraskeva C. Direct transcriptional upregulation of cyclooxygenase-2 by hypoxia-inducible factor (HIF)-1 promotes colorectal tumor cell survival and enhances HIF-1 transcriptional activity during hypoxia. Cancer Res. 2006;66:6683–91. doi: 10.1158/0008-5472.CAN-06-0425. [DOI] [PubMed] [Google Scholar]
- 13.Agarwal B, Swaroop P, Protiva P, Raj SV, Shirin H, Holt PR. Cox-2 is needed but not sufficient for apoptosis induced by Cox-2 selective inhibitors in colon cancer cells. Apoptosis. 2003;8:649–54. doi: 10.1023/A:1026199929747. [DOI] [PubMed] [Google Scholar]
- 14.Stasinopoulos I, O'Brien DR, Wildes F, Glunde K, Bhujwalla ZM. Silencing of cyclooxygenase-2 inhibits metastasis and delays tumor onset of poorly differentiated metastatic breast cancer cells. Mol Cancer Res. 2007;5:435–42. doi: 10.1158/1541-7786.MCR-07-0010. [DOI] [PubMed] [Google Scholar]
- 15.Palayoor ST, Tofilon PJ, Coleman CN. Ibuprofen-mediated reduction of hypoxia-inducible factors HIF-1alpha and HIF-2alpha in prostate cancer cells. Clin Cancer Res. 2003;9:3150–7. [PubMed] [Google Scholar]
- 16.Irizarry RA, Hobbs B, Collin F, Beazer-Barclay YD, Antonellis KJ, Scherf U, Speed TP. Exploration, normalization and summaries of high density oligonucleotide array probe level data. Biostatistics. 2003;4:249–64. doi: 10.1093/biostatistics/4.2.249. [DOI] [PubMed] [Google Scholar]
- 17.Erler JT, Bennewith KL, Nicolau M, Dornhofer N, Kong C, Le QT, Chi JT, Jeffrey SS, Giaccia AJ. Lysyl oxidase is essential for hypoxia-induced metastasis. Nature. 2006;440:1222–6. doi: 10.1038/nature04695. [DOI] [PubMed] [Google Scholar]
- 18.Postovit LM, Abbott DE, Payne SL, Wheaton WW, Margaryan NV, Sullivan R, Jansen MK, Csiszar K, Hendrix MJ, Kirschmann DA. Hypoxia/reoxygenation: a dynamic regulator of lysyl oxidase-facilitated breast cancer migration. J Cell Biochem. 2008;103:1369–78. doi: 10.1002/jcb.21517. [DOI] [PubMed] [Google Scholar]
- 19.Grosch S, Maier TJ, Schiffmann S, Geisslinger G. Cyclooxygenase-2 (COX-2)-independent anticarcinogenic effects of selective COX-2 inhibitors. J Natl Cancer Inst. 2006;98:736–47. doi: 10.1093/jnci/djj206. [DOI] [PubMed] [Google Scholar]
- 20.Tilg H, Moschen AR. Inflammatory mechanisms in the regulation of insulin resistance. Mol Med. 2008;14:222–31. doi: 10.2119/2007-00119.Tilg. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Romero R, Gotsch F, Pineles B, Kusanovic JP. Inflammation in pregnancy: its roles in reproductive physiology, obstetrical complications and fetal injury. Nutr Rev. 2007;65:194–202. doi: 10.1111/j.1753-4887.2007.tb00362.x. [DOI] [PubMed] [Google Scholar]
- 22.Schett G. Review: Immune cells and mediators of inflammatory arthritis. Autoimmunity. 2008;41:224–9. doi: 10.1080/08916930701694717. [DOI] [PubMed] [Google Scholar]
- 23.Shaftel SS, Griffin WS, O'Banion MK. The role of interleukin-1 in neuroinflammation and Alzheimer disease: an evolving perspective. J Neuroinflammation. 2008;5:7. doi: 10.1186/1742-2094-5-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Apostolakis S, Vogiatzi K, Krambovitis E, Spandidos DA. IL-1 cytokines in cardiovascular disease: diagnostic, prognostic and therapeutic implications. Cardiovasc Hematol Agents Med Chem. 2008;6:150–8. doi: 10.2174/187152508783955006. [DOI] [PubMed] [Google Scholar]