

Abstract
Signaling through the receptor/transcriptional regulator Notch pathway plays an important role in tumor cell survival. Recent studies have demonstrated that pharmacological inhibition of the Notch pathway with γ-secretase inhibitor (GSI) induces apoptosis of multiple myeloma (MM) cells via up-regulation of the proapoptotic protein Noxa. ABT-737, a novel BH3 mimetic, was shown to block Bcl-2 and Bcl-xL and induce MM cell apoptosis. Here we investigated whether the inhibition of Notch signaling could enhance the proapoptotic effect of ABT-737. The anti-myeloma effect of ABT-737 on MM cell lines or primary cells was substantially increased by the addition of Notch inhibitor. The synergistic effect of the GSI+ABT-737 combination was mediated by activation of Bak and Bax and release of cytochrome c. While toxic for MM cells, the combination of GSI and ABT-737 did not affect survival of peripheral blood mononuclear cells isolated from healthy donors. In vivo experiments using xenograft and SCID-hu models of MM demonstrated a significant antitumor effect of the GSI/ABT-737 combination as compared to the effect of Notch or Bcl-2/Bcl-xL inhibitors alone. Thus, this drug combination may be therapeutically beneficial for patients with MM.
Keywords: multiple myeloma, Notch, Bcl-2/Bcl-xL inhibitor, GSI
Introduction
Multiple myeloma (MM) is characterized by the uncontrolled proliferation of malignant plasma cells in bone marrow. Despite significant improvements in the treatment of this disease with novel therapeutic agents including bortezomib and lenalidomide, many patients ultimately become resistant to therapy. Therefore, searching for new molecular targets and drug combinations is critical for the development of effective therapeutic options.
Signaling through the receptor/transcriptional regulator Notch plays an important role in the survival of tumor cells and their protection from apoptosis induced by chemotherapeutic agents. The Notch pathway has been shown to be activated in the majority of solid and hematological tumors studied, including MM (1-3). According to the conventional model, Notch signaling is initiated upon binding of the Notch receptors expressed by MM cells to the Notch ligands, Jagged or Delta, that are expressed by both MM cells and the surrounding cellular components of bone marrow microenvironment. This binding initiates two subsequent proteolytic cleavages within the transmembrane domain resulting in the liberation and translocation of the intracellular domain of Notch (ICN) into the nucleus followed by its binding to the transcriptional repressor CSL (CBF-1). Binding of ICN displaces corepressor complexes and recruits coactivators thereby turning CBF-1 into a transcriptional activator (4, 5). A second proteolytic cleavage leading to Notch activation is regulated by a protease complex possessing γ-secretase activity. Pharmacological compounds able to inhibit γ-secretase (γ-secretase inhibitors, GSIs), and therefore Notch signaling, have shown impressive pre-clinical activity and are currently being tested in clinical trials (6, 7). Our previous data demonstrated that inhibition of Notch signaling with GSI resulted in significant cytotoxicity of MM cells. This effect was mediated through a dramatic up-regulation of the proapoptotic bcl-2 family protein Noxa (8).
Noxa belongs to the BH3-only “sensitizer” protein family which act by displacing the BH3-only “activators” like Bid and Bim from the antiapoptotic proteins, allowing the activators to bind Bax and Bak (9). Alternatively, antiapoptotic proteins could directly inhibit Bax and Bak activation (10). It is well known that Noxa has a very high affinity for the antiapoptotic protein Mcl-1, but not Bcl-2, Bcl-xL, or Bcl-w. While activation of Bax is controlled by Bcl-2, Mcl-1, and Bcl-xL, Mcl-1 also cooperates with Bcl-xL to sequester Bak and prevent its activation (11, 12). Recent studies have indicated that targeting of both Mcl-1 and Bcl-xL is necessary in order to release Bak (13).
ABT-737, a small molecule BH3-only mimetic developed by Abbott Laboratories, specifically targets the antiapoptotic bcl-2 members Bcl-2, Bcl-xL and Bcl-w (14). This compound prevents sequestration of Bax and Bak by Bcl-2 and Bcl-xL and induces apoptosis of tumor cells including multiple myeloma cells (14-17). However, ABT-737 does not have a strong affinity for Mcl-1 and therefore does not effectively trigger apoptosis of tumor cells expressing high Mcl-1 level (13, 18, 19). Recently, several groups have reported that the down-regulation of Mcl-1 sensitized tumor cells to apoptosis induced by ABT-737 (18, 20-22). Binding of Noxa to Mcl-1 reduces Mcl-1 levels by promoting proteosomal degradation. Therefore, strategies to increase Noxa could be potentially advantageous by overcoming Mcl-1 related resistance to ABT-737.
Using MM cell lines and primary cells as well as an in vivo xenograft and a SCID-hu model of MM, we demonstrate that the combination of these two agents has a significant synergistic anti-myeloma effect.
Materials and Methods
Cell cultures and reagents
Human MM NCI-H929, U266 and RPMI-8226 cell lines were obtained from the American Type Culture Collection (Manassas, VA) and were kept in culture no longer than 6 weeks. MM1S cell line was a gift from Dr Steven Rosen (Northwestern University, Chicago, IL); no authentication was done by the authors. Cells were cultured as described previously (2). GSI (γ-secretase inhibitor XII) was purchased from Calbiochem (San Diego, CA) and dissolved in DMSO as a 5 mM stock solution. Bcl-2/bcl-xL inhibitor ABT-737 was provided by Abbott Laboratories (Abbott Park, IL) and dissolved in DMSO as a 20 mM stock solution for in vitro studies and in 30% propylene glycol, 5% Tween 80 and 65% D5W (5% dextrose in water) for in vivo studies. Chemical structures of GSI XII and ABT-737 are shown in Figure 1. Pan-caspase inhibitor z-VAD-FMK was obtained from Bachem (Bachem Americas, Torrance, CA) and dissolved in DMSO.
Figure 1. Chemical structures of compounds.
Chemical structures of (A) γ-secretase inhibitor XII and (B) ABT-737.
Isolation of primary myeloma cells and mononuclear cells
Collection of BM was in accordance with University of South Florida Institutional Review Board approved protocol. CD138+ myeloma cells were isolated from BM samples using CD138 MicroBeads and the MidiMACS magnetic cell separator according to the manufacturer's protocol (Miltenyi Biotec, Auburn, CA). Cells were cultured in α-minimal essential medium supplemented with 10% FBS with or without GSI, ABT-737, or their combination for 48 hrs. Blood samples were obtained from healthy volunteers, and mononuclear cells were isolated by Ficoll-Paque gradient centrifugation, and then cultured in α-minimal essential medium supplemented with 10% FBS.
MTT assay
Cells were treated with various concentrations of GSI and/or ABT-737 for 48 hrs. 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide (MTT) dye was added for the last four hours of incubation. Cells were then pelleted and the medium aspirated. Insoluble formazan complexes were solubilized with DMSO and the absorbance was measured at 540 nm using a Benchmark Plus microplate spectrophotometer (Bio-Rad, Hercules, CA). MM cell survival and the combination index (CI) were calculated using CalcuSyn software (Biosoft, Cambridge, UK). Each experimental condition was done in triplicate and performed at least twice.
Flow cytometry
Apoptosis of myeloma cells was detected by the Annexin V binding assay as described earlier (2). Ten thousand events were acquired and analyzed using a FACSCalibur or LSR II flow cytometers (BD) and FlowJo software (Tree Star, Ashland, OR).
Activation of Bak and Bax were determined by flow cytometry as described previously using specific antibodies that recognize only conformationally active forms of these proteins (anti-Bax (clone 3, BD Biosciences) or anti-Bak (Ab-1, Calbiochem, San Diego, CA))(23).
In order to detect cytochrome c release, cells were collected and permeabilized with 100 μl of digitonin (50 μg/ml) for 5 min on ice. Cells were then fixed with 4% paraformaldehyde followed by blocking (3% bovine serum albumin in PBS with 0.05% saponin) for 1 hr. Cells were incubated with an anti-cytochrome c antibody (BD Bioscience, San Jose, CA) overnight at 4°C followed by labeling with a secondary FITC-conjugated anti-mouse IgG antibody. Ten thousand events were acquired using a FACSCalibur flow cytometer (BD).
Western blotting
Western blotting was performed as described previously (2, 24). The antibodies against the following proteins were used: mcl-1, β-actin (Santa Cruz, Santa Cruz, CA); bcl-xL (BD Bioscience, San Jose, CA); bcl-2, PARP, and cleaved caspase 3 (Cell Signaling Technology, Danvers, MA); and Noxa (clone 114C307, Calbiochem).
Cell transfection
Control non-targeting pool siRNA or specific Noxa, Bak or Bax siRNA were obtained from Ambion (Austin, TX). Myeloma RPMI 8226 cells were transfected with siRNA using Amaxa nucleofection technology (Lonza, Allendale, NJ). Briefly, 5×106 cells were re-suspended in buffer V and electroporated using program A20. Cells were cultured for overnight and then treated with GSI, ABT-737, or combination thereof.
Immunohistochemistry
Tumor tissues were fixed in formalin, embedded in paraffin and slides were prepared. Immunohistochemical staining was performed using Ventana Discovery XT automated system (Ventana Medical Systems, Tucson, AZ). Heat-induced antigen retrieval method was used for cleaved caspase 3 staining and enzymatic retrieval method (4 minutes in Protease 1(Ventana) was used for Noxa staining. Slides were then incubated with primary antibodies against cleaved caspase 3 (#9661, Cell Signaling, 1:400 dilution) or Noxa (#3665-100, Biovision, Mountain View, CA, 1:25 dilution) for 1 hr followed by incubation with secondary anti-rabbit antibody (Ventana) for 20 min. Ventana OmniMap kit was used as a detection system. Slides were then counterstained with Hematoxylin.
In vivo studies
SCID-beige (C.B-Igh-1b/GbmsTac-Prkdcscid-Lystbg N7) mice were purchased from Taconic (Germantown, NY) and kept in pathogen-free conditions in the animal facility of the H. Lee Moffitt Cancer Center. All experimental procedures were performed in accordance with the guidelines of the University of South Florida Institutional Animal Care and Use Committee.
Xenograft model was established by subcutaneous inoculation of 1×107 MM 8226 cells in the right flank of 6-8 week old SCID/beige mice. In approximately 3 weeks when tumor became measurable, mice were assigned to one of the following four groups: control group, GSI or ABT-737 treated groups, or a group treated with the combination of GSI and ABT-737. GSI and ABT-737 were administered daily for 14 days i.p. at a dose of 5 mg/kg/day and 75 mg/kg/day, respectively. Tumor size was measured twice a week during and after treatment. Mice were followed up for approximately two weeks after finishing treatment.
SCID-hu model was established as described previously (8). Human fetal tissues from 20 to 23 weeks of gestation were obtained from Advance Bioscience Resource (Alameda, CA). Fragments of human fetal bones (humerus, femur, or tibia) were implanted subcutaneously in 6 week old female SCID-beige mice. Six weeks after implantation, 5 × 104 8226 myeloma cells in 50 μL PBS were injected directly into each implanted bone. Treatment with GSI, ABT-737, or the combination began approximately 4 weeks following tumor inoculation. GSI was administered at a dose of 5 mg/kg/day and ABT-737 at a dose of 100 mg/kg/day i.p. daily for 14 days. Mice in the control group did not receive injections. Blood (50-100 γL) was drawn from a submandibular vein and the level of free lambda light chain of human immunoglobulin was measured by an enzyme-linked immunosorbent assay (Bethyl, Montgomery, TX) before and after treatment.
Statistical analysis
Statistical significance of differences observed in cells treated with single drug compared to cells treated with drug combination was determined using the Student t test. The p values less than 0.05 were considered significant. In order to assess for a possible synergistic effect of combined therapies CalcuSyn software (Cambridge, UK) was used. A combination index (CI) less than 0.9 indicates synergism, while CI between 0.9 - 1.0 indicates an additive effect (25). Two-way ANOVA and Mann-Whitney nonparametric test were used for statistical analysis of the differences between in vivo treatment groups and was calculated using GraphPad Prism software (La Jolla, CA).
Results
Combined effect of a Notch inhibitor and Bcl-2/Bcl-xL inhibitor on the viability of myeloma cells
To investigate the combined effect of the Notch inhibitor GSI and ABT-737 on MM cells, we initially measured cellular viability using the MTT assay in different myeloma cell lines. Treatment with either GSI or ABT-737 alone resulted in decreased survival of myeloma cells, while the combination of these two compounds had a significantly stronger anti-myeloma effect (Fig. 2A-C). The effect of the drug combination was evaluated by calculating the combination index (CI) and considered to be synergistic when the CI value was less than 0.9. The combination of GSI and ABT-737 had a significant synergistic cytotoxic effect on all MM cell lines studied. The lowest CI values were 0.745 for 8226 cells, 0.63 for U266 and 0.35 for MM1S cells. The effect of the GSI combination with ABT-737 on primary CD138+ MM cells was similar to its effect on MM cell lines with CI between 0.4 and 0.7 (Fig. 2D).
Figure 2. Combined effect of GSI and ABT-737 on the viability of MM cells.

Fifty thousand human MM (A) 8226, (B) U266, or (C) MM1S cells as well as (D) 4x105 primary CD138+ myeloma cells isolated from BM of one patient with MM were plated in quadruplicates per well of 96-well plates and treated with various concentrations of GSI (dotted line), ABT-737 (thin line), or their combination (thick line) for 48 hrs followed by the MTT assay. Cell viability curves are shown. Combination indices (CI) were calculated using CalcuSyn software. For cell lines, at least three independent experiments with the same results were performed.
Our previous data have demonstrated that treatment with GSI resulted in apoptosis of MM cells (8). ABT-737 was also shown to induce MM cell death (17). Therefore to confirm the effect of the drug combination we next evaluated MM cell apoptosis using the Annexin V binding assay. A significant (p<0.05) increase in apoptosis of cells treated with combination of GSI and ABT-737 was observed as compared to cells treated with either GSI or ABT-737 alone (Fig. 3A). To confirm the validity of the data obtained with cell lines we used primary CD138+ MM cells isolated from the bone marrow of four different patients with MM. Addition of ABT-737 increased the proapoptotic effect of GSI in primary CD138+ cells (Fig 3B). In contrast, the cytotoxic effect of the GSI combination with ABT-737 was not observed in peripheral blood mononuclear cells (MNC) isolated from healthy donors (Fig. 3C). Thus, taken together these results demonstrate that ABT-737 synergizes with GSI in induction of apoptosis in MM cells and does not affect normal MNC cells.
Figure 3. Synergistic effect of combination of GSI with ABT-737 on apoptosis of MM cells.
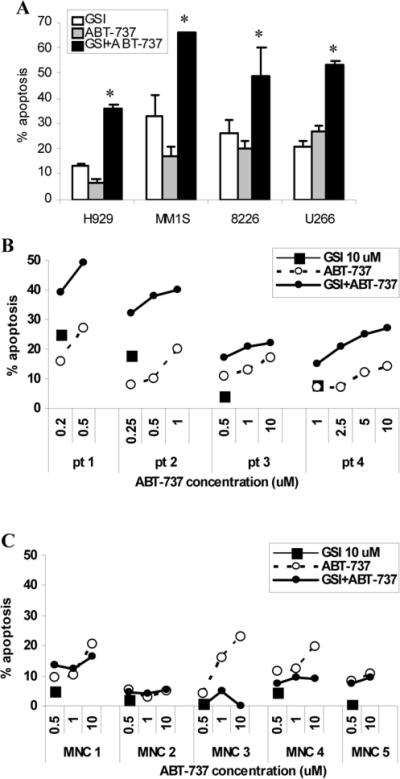
(A) MM cell lines were treated with ABT-737 at a dose either 5 µM (H929 and U266 cells), 0.5 μM (MM1S cells), or 0.2 μM (8226 cells), GSI, or their combination for 48 hrs. * - statistically significant (p<0.05) difference between combination group and single agent groups. (B) primary CD138+ myeloma cells isolated from BM of four patients with MM or (C) peripheral blood mononuclear cells isolated from five healthy donors were treated with indicated concentrations of GSI, ABT-737, or their combination for 48 hrs. The proportion of apoptotic cells was analyzed using Annexin V binding assay by flow cytometry. Presented are the values of specific drug-induced apoptosis that were determined by subtraction of the background value of apoptosis (cells treated with vehicle control) from apoptosis induced by compounds. Experiments with each cell line were performed 3-4 times and mean±SD is shown.
Mechanisms of apoptosis induced by simultaneous inhibition of Notch and bcl-2/bcl-xL
Initially we investigated whether the inhibition of Notch signaling with GSI affected the levels of the antiapoptotic members of the bcl-2 family. MM cells were treated with different concentrations of GSI and the levels of Bcl-2, Bcl-xL and Mcl-1 were evaluated by Western blotting. GSI only slightly down-regulated the level of Bcl-xL and did not affect the level of Bcl-2 (Fig. 4A). Consistent with our previous data, inhibition of Notch resulted in dramatic up-regulation of the pro-apoptotic BH3-only protein Noxa (Fig 4A) (8). All MM cells studied expressed Mcl-1 protein and there was no substantial difference in Mcl-1 level between cell lines. We did not observe GSI-induced Mcl-1 degradation in MM cells (Fig. 4A).
Figure 4. Effect of GSI/ABT-737 combination on apoptosis of MM cell is mediated through activation of Bak and Bax.

(A) MM H929, RPMI 8226, or U266 cells were treated with GSI for 48 hrs. After that time cells were collected and the level of indicated proteins was determined by Western blotting. Vertical lines separate part of the membranes that were developed using maximum strength enhanced chemiluminescence. (B, D) MM 8226 cells were transfected with siRNA specific for Noxa, bak, bax, or control non-targeting siRNA. After overnight incubation cells were treated with GSI, ABT-737 or drug combination. Western blotting with antibodies specific for Noxa (B) or Bak and Bax (D) was performed. Apoptosis in different treatment groups of cells was detected by Annexin V/DAPI staining using LSR II flow cytometer (BD). Shown are combined results of three independent experiments performed in duplicates. (B) * - statistically significant difference (p<0.05) between indicated group and GSI+ABT-737 group; # - statistically significant difference (p<0.05) between cells transfected with control or Noxa siRNA and treated with combination of GSI and ABT-737. (D) * - statistically significant difference (p<0.05) between cells transfected with control or Bak and Bax siRNA. (C) MM U266, H929, or RPMI 8226 cells were cultured for 48 hrs with or without GSI, ABT-737, or combination thereof. Cells were then collected and the level of Bax and Bak activation was evaluated by flow cytometry as described in the Methods section. Mean fluorescence intensities were determined and compared between cells treated with ABT-737, GSI, of combination thereof.
To clarify the role of Noxa in apoptosis induced by drug combination we down-regulate its level in MM cells using specific siRNA (Fig. 4B). Cells were then treated with GSI, ABT-737, or combination of these compounds and apoptosis was detected by Annexin V/DAPI staining by flow cytometry. Consistent with our previous data (8), inhibition of Noxa abrogated the effect of GSI (Fig. 4B). Decrease in Noxa level also resulted in reduced ABT-737 cytotoxicity and significantly reduced cell death induced by combination of GSI with ABT-737 (Fig. 4B).
Inhibition of Bcl-2 and Bcl-xL with ABT-737 has been shown to induce caspase-dependent apoptosis (14, 21). Here we investigated whether ABT-737 synergizes with GSI in Bak and Bax activation followed by release of cytochrome c and cleavage of caspase 3 and PARP. Substantially increased proapoptotic conformational changes of Bak and Bax were observed in MM cells treated with the combination of GSI and ABT-737 as compared to cells treated with either GSI or ABT-737 alone (Fig. 4C). The combination of these two drugs was able to activate Bax in 1.8±0.53 fold (range from 1.3 to 2.5 fold) and Bak in 1.9×0.6 fold (range 1.4 to 2.8 fold) in MM cells when compared to the level of activation in cells treated with either GSI or ABT-737 alone. To clarify the role of Bak and Bax in apoptosis induced by GSI in combination with ABT-737 we down-regulate expression of these proteins in the cells using siRNA. MM 8226 cells were transfected with either control non-targeting pool siRNA or specific Bak and Bax siRNA. This resulted in a substantial decrease of Bak and/or Bax protein levels (Fig. 4D). Cells were then treated with GSI, ABT-737 or drug combination and apoptosis was evaluated by Annexin V/DAPI staining. Depletion of Bak/Bax level significantly decreased cell death induced by either GSI or ABT-737 alone or in combination (Fig. 4D).
When activated, Bak and Bax can permeabilize the outer mitochondrial membrane and release proapoptogenic factors including cytochrome c needed to activate the caspases and induce DNA fragmentation. To confirm the downstream effect of Bak and Bax activation, we evaluated the release of cytochrome c in MM cells using flow cytometry. Treatment of three myeloma cell lines with the combination of GSI and ABT-737 resulted in a dramatic release of cytochrome c as compared to cells exposed to either GSI or ABT-737 alone (Fig. 5A). Release of cytochrome c was followed by the cleavage of caspase 3 and PARP (Fig. 5B). Thus, taken together these data demonstrate that the combination of GSI with ABT-737 is synergistic in inducing mitochondria-dependent apoptosis. Pre-treatment of MM cells with the pan-caspase inhibitor z-VAD-FMK abrogated the effect of the GSI/ABT-737 drug combination as was evaluated by measuring MM cell apoptosis using the Annexin V binding assay (Fig. 5C) and release of cytochrome c by flow cytometry (Fig. 5D).
Figure 5. Treatment with z-VAD abrogates the effect of GSI/ABT-737 combination.

(A) MM U266, H929, or RPMI 8226 cells were cultured for 48 hrs with or without GSI, ABT-737, or combination threof. Release of cytochrome c was evaluated by flow cytometry as described in the Methods section. The fraction of cells that retained their mitochondrial cytochrome c appears to be highly fluorescent while cells that lost their cytochrome c emerged as a population of low fluorescent cells. (B) MM cell lines were treated with ABT-737 at either 5 μM (H929 and U266 cells), 0.5 μM (MM1S cells), or 0.2 μM (8226 cells), GSI, or their combination for 48 hrs. Cells were collected and Western blot with antibodies against cleaved caspase 3 and PARP was performed. Equal loading was confirmed by re-probing of the membrane with a beta-actin antibody. (C-D) MM U266 (top panels) or H929 (bottom panels) cells were pre-treated with 100 μM z-VAD-FMK for 2 hrs followed by treatment with 5 μM GSI, 10 μM ABT-737, or their combination. (C) Apoptosis of myeloma cells was detected using an Annexin V binding assay by flow cytometry. (D) Cytochrome c release was detected by flow cytometry using a specific antibody as described in Methods. Indicated are the cells undergoing apoptosis. Experiments were performed twice with similar results.
ABT-737 enhances the effect of GSI on MM cells in vivo
To investigate whether the addition of ABT-737 would enhance the anti-tumor effect of GSI in vivo, we used two different MM mouse models, the xenograft and SCID-hu model. Initial experiments were performed using the xenograft model, where MM 8226 cells were injected s.c. into a flank of SCID-beige mice. When the tumor mass became palpable, mice were split into four groups and treated with GSI, ABT-737, or a combination thereof for 14 days. No apparent signs of toxicity such as behavioral changes or weight loss were observed in treated animals. At selected doses the anti-tumor effect of GSI or ABT-737 alone was rather moderate. The combination of these two drugs had significantly stronger anti-tumor effect (Fig. 6A). After treatment stops, a rapid tumor growth was observed in GSI and ABT-737 groups while tumor growth in combination group was significantly delayed (Fig. 6A). Immunohistochemical analysis of tumor tissues using antibodies against cleaved caspase 3 and Noxa indicated that GSI and ABT-737 induced substantially stronger apoptosis of tumor cells when used in combination than separately (Fig. 6B). The disadvantage of the xenograft model is that MM cells grow as a solid tumor, which is artificial for MM. Therefore, to evaluate the effect of the drug combination in the context of human BM, we also used a SCID-hu model. In this model 8226 MM cells were injected directly into previously engrafted fragments of human bone and tumor growth was monitored by measuring the level of a specific human paraprotein in mouse serum. Mice were treated with GSI, ABT-737, or a combination of these two drugs. Serum level of free human lambda chain was used to evaluate tumor growth. Administration of each of these drugs alone decreased the level of free human lambda chain in serum when compared to a control group (Fig. 6C,D). The combination of GSI and ABT-737 had significantly more potent anti-myeloma effect than each of the drug alone (Fig. 6D).
Figure 6. In vivo anti-myeloma effect of combination of GSI with ABT-737.

(A) RPMI-8226 tumors were established s.c. in SCID-beige mice. Mice were split into 4 groups (4 mice per group) with equal size tumors and treated with GSI, ABT-737, or a combination thereof for 14 days. Tumor growth was monitored during and after treatment. Statistical analysis was performed using two-way ANOVA. Shown are statistical differences between treatment groups and vehicle control-treated group. (B) Tumor tissues were collected at the end of the treatment and immunohistochemical staining for cleaved caspase 3 (top row) and Noxa (bottom row) was performed. Representative photomicrographs (×200 magnification) are shown. Scale bars, 50 μM. (C, D) The SCID-hu model was established as described in Methods. Tumor growth was monitored by measuring the level of human paraprotein in mouse sera. Approximately 4 weeks after tumor injection, mice were split into four groups (4-5 mice per group) with an equal average level of free lambda light chain in the sera. Mice were treated with GSI, ABT-737, or a combination of GSI with ABT-737 for 14 days. Please note the different scale for non-treated control group (C) and treated groups of mice (D). Two-way ANOVA and nonparametric Mann-Whitney test were used to determine differences between treatment groups. * - statistically significant (p<0.05) difference between indicated group and GSI+ABT-737 group. # - statistically significant (p<0.05) difference between indicated group and ABT-737 group.
Discussion
In this study, we examined the effect of simultaneous inhibition of Notch signaling and the bcl-2 family antiapoptotic members Bcl-2/Bcl-xL on MM cell growth and survival. We found that the combination of the Notch inhibitor GSI and the Bcl-2/Bcl-xL inhibitor ABT-737 had a synergistic cytotoxic effect on MM cells in vitro and in vivo.
Antiapoptotic members of bcl-2 family including Bcl-2, Bcl-xL, and Mcl-1 are highly overexpressed by myeloma cells and are considered to be among the major contributing factors to survival, growth, and chemoresistance of these cells (26-30). Therefore targeting these proteins represents an attractive therapeutic strategy. ABT-737, a potent small molecule BH3 mimetic was recently developed by Abbott (14). As a single agent it has been shown to induce apoptosis in various solid and hematopoietic tumor cells including multiple myeloma (16, 17). ABT-737 binds with high affinity to Bcl-2, Bcl-xL, and Bclw of prosurvival bcl-2 family members (14). This binding results in activation (oligomerization) of Bax and Bak that subsequently leads to apoptosis (31). While activation of Bax is controlled by Bcl-2, in order to liberate Bak, both Bcl-xL and Mcl-1 also need to be neutralized (11, 12). While ABT-737 disrupts the interaction of Bcl-xL with Bak this compound has a low affinity for Mcl-1. As a result, tumor cells overexpressing Mcl-1 are resistant to this agent, and neutralization of Mcl-1 has been shown to be effective in enhancing the anti-tumor effect of ABT-737 (18, 31). According to our results RPMI-8226 cells were significantly more sensitive to ABT-737 as compared to H929 and U266 cells while the level of Mcl-1 in these cell lines was similar. The higher sensitivity of RPMI-8226 cells to ABT-737 could be explained by the fact that these cells have substantially lower level of Bcl-2 and higher level of Noxa expression.
One of the potent regulators of Mcl-1 is the proapoptotic BH3-only protein Noxa (12, 32). When overexpressed, Noxa specifically and tightly binds to Mcl-1 targeting it to proteasomal degradation, and displaces Bak. While GSI was able to significantly induce Noxa, we did not observe Mcl-1 degradation in myeloma cells following GSI treatment. Recent study indicated that it is rather Mcl-1 functional inactivation than degradation and elimination that is critical to activation of Bak (33). Although Mcl-1 level was not affected in our study, GSI was able to induce apoptosis and synergize with ABT-737 in anti-myeloma effect confirming these data. Why Mcl-1 was not undergoing proteasomal degradation after dramatic induction of Noxa by GSI is not clear. However it is possible that beside inhibition of Notch GSI could potentially have off-target effects. Recently it was suggested that selected gamma-secretase inhibitors could also block proteasome activity (34). Therefore, one could speculate that degradation of Mcl-1 is abrogated in myeloma cells due to off-target effect of GSI on proteasome activity while the cytotoxic effect of the compound is preserved due to induction of Noxa and neutralization of Mcl-1.
We have previously demonstrated that by blocking Notch signaling with GSI, Noxa is dramatically up-regulated and induces activation of Bak in MM cell lines and primary patient myeloma cells (8). These data suggest that the anti-tumor effect of Notch inhibition could be substantially improved by pharmacological targeting of Bcl-2 and Bcl-xL. Indeed, our results demonstrated that simultaneous inhibition of Notch and Bcl-2/Bcl-xL by using GSI and ABT-737 resulted in significant induction of Bak and Bax activation, followed by cytochrome c release, cleavage of caspase 3, and cell apoptosis. This is a first report showing synergism between inhibitions of Notch signaling and Bcl-2/Bcl-xL in vitro and in vivo using xenograft and SCID-hu model. Consistent with previous observations GSI or ABT-737 alone moderately decreased myeloma tumor growth in mice. When treatment has stopped GSI group did not retain significant anti-tumor effect that we observed previously using the same concentrations of the compound (8). This is most likely attributed to the fact that different mouse strains were used for xenograft model in these experiments. Our data indicated that MM 8226 tumor grew substantially slower in SCID/NOD mice as compared to SCID-beige mice (Fig. 6A and (8). Although GSI had anti-tumor effect in both models, due to kinetics of tumor growth the effect of GSI was less prominent in SCID-beige mice. Although at selected doses GSI and ABT-737 alone has only moderate effect on MM growth, the combination of these two drugs has statistically significant anti-tumor effect that retained after the end of the treatment.
Our finding of synergism between Notch and Bcl-2/bcl-xL inhibitors is consistent with the studies where the induction of Noxa with topoisomerase I inhibitor CPT-11 in colorectal cancer cells and with proteasome inhibitors in melanoma and lymphoid malignancies sensitized these cells to ABT-737 (35-37).
In summary, we show for the first time that the inhibition of Notch synergistically cooperates with ABT-737 to enhance apoptosis in myeloma cells. Our findings indicate that this therapeutic strategy could be potentially beneficial for the treatment of patients with multiple myeloma.
Grant Support
National Institute of Health grant CA130923 and Multiple Myeloma Research Foundation Senior Research Award (Y.N.). This work was supported in part by the Flow Cytometry Core Facility at the H. Lee Moffitt Cancer Center.
Abbreviations list
- MM
multiple myeloma
- BM
bone marrow
- GSI
γ-secretase inhibitor
- MNC
mononuclear cells
- ICN
intracellular domain of Notch
- CI
combination index
Footnotes
Conflict of Interest: The authors declare no competing financial interests.
References
- 1.Nickoloff BJ, Osborne BA, Miele L. Notch signaling as a therapeutic target in cancer: a new approach to the development of cell fate modifying agents. Oncogene. 2003;22:6598–608. doi: 10.1038/sj.onc.1206758. [DOI] [PubMed] [Google Scholar]
- 2.Nefedova Y, Cheng P, Alsina M, Dalton W, Gabrilovich D. Involvement of Notch-1 signaling in bone marrow stroma-mediated de novo drug resistance of myeloma and other malignant lymphoid cell lines. Blood. 2004;103:3503–10. doi: 10.1182/blood-2003-07-2340. [DOI] [PubMed] [Google Scholar]
- 3.Jundt F, Probsting KS, Anagnostopoulos I, et al. Jagged1-induced Notch signaling drives proliferation of multiple myeloma cells. Blood. 2004;103:3511–5. doi: 10.1182/blood-2003-07-2254. [DOI] [PubMed] [Google Scholar]
- 4.Allman D, Punt J, Izon DJ, Aster JC, Pear WS. An invitation to T and more: Notch signaling in lymphopoiesis. Cell. 2002;109:S1–S11. doi: 10.1016/s0092-8674(02)00689-x. [DOI] [PubMed] [Google Scholar]
- 5.Artavanis-Tsakonas S, Rand M, Lake R. Notch signaling: cell fate control and signal integration in development. Science. 1999;284:770–6. doi: 10.1126/science.284.5415.770. [DOI] [PubMed] [Google Scholar]
- 6.Rizzo P, Osipo C, Foreman K, Golde T, Osborne B, Miele L. Rational targeting of Notch signaling in cancer. Oncogene. 2008;27:5124–31. doi: 10.1038/onc.2008.226. [DOI] [PubMed] [Google Scholar]
- 7.Nefedova Y, Gabrilovich D. Mechanisms and clinical prospects of Notch inhibitors in the therapy of hematological malignancies. Drug Resist Updat. 2008;11:210–8. doi: 10.1016/j.drup.2008.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nefedova Y, Sullivan D, Bolick S, Dalton W, Gabrilovich D. Inhibition of Notch signaling induces apoptosis of myeloma cells and enhances sensitivity to chemotherapy. Blood. 2008;111:2220–9. doi: 10.1182/blood-2007-07-102632. [DOI] [PubMed] [Google Scholar]
- 9.Letai A, Bassik M, Walensky L, Sorcinelli M, Weiler S, Korsmeyer S. Distinct BH3 domains either sensitize or activate mitochondrial apoptosis, serving as prototype cancer therapeutics. Cancer Cell. 2002;2:183–92. doi: 10.1016/s1535-6108(02)00127-7. [DOI] [PubMed] [Google Scholar]
- 10.Willis S, Fletcher J, Kaufmann T, et al. Apoptosis initiated when BH3 ligands engage multiple Bcl-2 homologs, not Bax or Bak. Science. 2007;315:856–9. doi: 10.1126/science.1133289. [DOI] [PubMed] [Google Scholar]
- 11.Zha H, Reed J. Heterodimerization-independent functions of cell death regulatory proteins Bax and Bcl-2 in yeast and mammalian cells. J Biol Chem. 1997;272:31482–288. doi: 10.1074/jbc.272.50.31482. [DOI] [PubMed] [Google Scholar]
- 12.Willis SN, Chen L, Dewson G, et al. Proapoptotic Bak is sequestered by Mcl-1 and Bcl-xL, but not Bcl-2, until displaced by BH3-only proteins. Genes Dev. 2005;19:1294–305. doi: 10.1101/gad.1304105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dai Y, Grant S. Targeting Multiple Arms of the Apoptotic Regulatory Machinery. Cancer Res. 2007;67:2908–11. doi: 10.1158/0008-5472.CAN-07-0082. [DOI] [PubMed] [Google Scholar]
- 14.Oltersdorf T, Elmore S, Shoemaker A, et al. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature. 2005;435:677–81. doi: 10.1038/nature03579. [DOI] [PubMed] [Google Scholar]
- 15.Cory S, Adams J. Killing cancer cells by flipping the Bcl-2/Bax switch. Cancer Cell. 2005;8:5–6. doi: 10.1016/j.ccr.2005.06.012. [DOI] [PubMed] [Google Scholar]
- 16.Trudel S, Stewart A, Li Z, et al. The Bcl-2 family protein inhibitor, ABT-737, has substantial antimyeloma activity and shows synergistic effect with dexamethasone and melphalan. Clin Cancer Res. 2007;13:621–9. doi: 10.1158/1078-0432.CCR-06-1526. [DOI] [PubMed] [Google Scholar]
- 17.Chauhan D, Velankar M, Brahmandam M, et al. A novel Bcl-2/Bcl-X(L)/Bcl-w inhibitor ABT-737 as therapy in multiple myeloma. Oncogene. 2007;26:2374–80. doi: 10.1038/sj.onc.1210028. [DOI] [PubMed] [Google Scholar]
- 18.Konopleva M, Contractor R, Tsao T, et al. Mechanisms of apoptosis sensitivity and resistance to the BH3 mimetic ABT-737 in acute myeloid leukemia. Cancer Cell. 2006;10:375–88. doi: 10.1016/j.ccr.2006.10.006. [DOI] [PubMed] [Google Scholar]
- 19.Certo M, Del Gaizo Moore V, Nishino M, et al. Mitochondria primed by death signals determine cellular addiction to antiapoptotic BCL-2 family members. Cancer Cell. 2006;9:351. doi: 10.1016/j.ccr.2006.03.027. [DOI] [PubMed] [Google Scholar]
- 20.Tahir S, Yang X, Anderson M, et al. Influence of Bcl-2 family members on the cellular response of small-cell lung cancer cell lines to ABT-737. Cancer Res. 2007;67:1176–83. doi: 10.1158/0008-5472.CAN-06-2203. [DOI] [PubMed] [Google Scholar]
- 21.Chen S, Dai Y, Harada H, Dent P, Grant S. Mcl-1 down-regulation potentiates ABT-737 lethality by cooperatively inducing Bak activation and Bax translocation. Cancer Res. 2007;67:782–91. doi: 10.1158/0008-5472.CAN-06-3964. [DOI] [PubMed] [Google Scholar]
- 22.Lin X, Morgan-Lappe S, Huang X, et al. ‘Seed’ analysis of off-target siRNAs reveals an essential role of Mcl-1 in resistance to the small-molecule Bcl-2/Bcl-XL inhibitor ABT-737. Oncogene. 2007;26:3972–9. doi: 10.1038/sj.onc.1210166. [DOI] [PubMed] [Google Scholar]
- 23.Gómez-Benito M, Marzo I, Anel A, Naval J. Farnesyltransferase inhibitor BMS-214662 induces apoptosis in myeloma cells through PUMA up-regulation, Bax and Bak activation, and Mcl-1 elimination. Mol Pharmacol. 2005;67:1991–8. doi: 10.1124/mol.104.007021. [DOI] [PubMed] [Google Scholar]
- 24.Nefedova Y, Cheng P, Gilkes D, et al. Activation of dendritic cells via inhibition of Jak2/STAT3 signaling. J Immunol. 2005;175:4338–46. doi: 10.4049/jimmunol.175.7.4338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Reynolds C, Maurer B. Evaluating response to antineoplastic drug combinations in tissue culture models. Methods Mol Med. 2005;110:173–83. doi: 10.1385/1-59259-869-2:173. [DOI] [PubMed] [Google Scholar]
- 26.Ong F, van Nieuwkoop J, de Groot-Swings G, et al. Bcl-2 protein expression is not related to short survival in multiple myeloma. Leukemia. 1995;9:1282–4. [PubMed] [Google Scholar]
- 27.Wuillème-Toumi S, Robillard N, Gomez P, et al. Mcl-1 is overexpressed in multiple myeloma and associated with relapse and shorter survival. Leukemia. 2005;19:1248–52. doi: 10.1038/sj.leu.2403784. [DOI] [PubMed] [Google Scholar]
- 28.Le Gouill S, Podar K, Harousseau J, Anderson K. Mcl-1 regulation and its role in multiple myeloma. Cell Cycle. 2004;3:1259–62. doi: 10.4161/cc.3.10.1196. [DOI] [PubMed] [Google Scholar]
- 29.Puthier D, Pellat-Deceunynck C, Barillé S, et al. Differential expression of Bcl-2 in human plasma cell disorders according to proliferation status and malignancy. Leukemia. 1999;13:289–94. doi: 10.1038/sj.leu.2401302. [DOI] [PubMed] [Google Scholar]
- 30.Pettersson M, Jernberg-Wiklund H, Larsson L, et al. Expression of the bcl-2 gene in human multiple myeloma cell lines and normal plasma cells. Blood. 1992;79:495–502. [PubMed] [Google Scholar]
- 31.van Delft M, Wei A, Mason K, et al. The BH3 mimetic ABT-737 targets selective Bcl-2 proteins and efficiently induces apoptosis via Bak/Bax if Mcl-1 is neutralized. Cancer Cell. 2006;10:389–99. doi: 10.1016/j.ccr.2006.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen L, Willis SN, Wei A, et al. Differential targeting of prosurvival Bcl-2 proteins by their BH3-only ligands allows complementary apoptotic function. Mol Cell. 2005;17:393–403. doi: 10.1016/j.molcel.2004.12.030. [DOI] [PubMed] [Google Scholar]
- 33.Lee E, Czabotar P, van Delft MF, et al. A novel BH3 ligand that selectively targets Mcl-1 reveals that apoptosis can proceed without Mcl-1 degradation. J Cell Biol. 2008;180:341–55. doi: 10.1083/jcb.200708096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Clementz A, Osipo C. Notch versus the proteasome: what is the target of gamma-secretase inhibitor-I? Breast Cancer Res. 2009;11:110–2. doi: 10.1186/bcr2407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Okumura K, Huang S, Sinicrope F. Induction of Noxa sensitizes human colorectal cancer cells expressing Mcl-1 to the small-molecule Bcl-2/Bcl-xL inhibitor, ABT-737. Clin Cancer Res. 2008;14:8132–42. doi: 10.1158/1078-0432.CCR-08-1665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Paoluzzi L, Gonen M, Bhagat G, et al. The BH3-only mimetic ABT-737 synergizes the antineoplastic activity of proteasome inhibitors in lymphoid malignancies. Blood. 2008;112:2906–16. doi: 10.1182/blood-2007-12-130781. [DOI] [PubMed] [Google Scholar]
- 37.Miller L, Goldstein N, Johannes W, et al. BH3 mimetic ABT-737 and a proteasome inhibitor synergistically kill melanomas through Noxa-dependent apoptosis. J Invest Dermatol. 2009;129:964–71. doi: 10.1038/jid.2008.327. [DOI] [PubMed] [Google Scholar]