

Abstract
We report a new pathway of translation regulation that may operate in interferon-treated or virus-infected mammalian cells. This pathway is activated by P56, a protein whose synthesis is strongly induced by interferons or double-stranded RNA. Using a yeast two-hybrid screen, we identified the P48 subunit of the mammalian translation initiation factor eIF-3 as a protein that interacts with P56. The P56–P48 interaction was confirmed in human cells by co-immunoprecipitation assays and confocal microscopy. Gel filtration assays revealed that P56 binds to the large eIF-3 complex that contains P48. Purified recombinant P56 inhibited in vitro translation of reporter mRNAs in a dose-dependent fashion, and that inhibition was reversed by the addition of purified eIF-3. In vivo, expression of transfected P56 or induction of the endogenous P56 by interferon caused an inhibition of overall cellular protein synthesis and the synthesis of a transfected reporter protein. As expected, a P56 mutant that does not interact with P48 and eIF-3 failed to inhibit protein synthesis in vitro and in vivo.
Keywords: double-stranded RNA/eIF-3/interferon/P56/translational regulation
Introduction
Although the antiviral effect of interferon (IFN) is the most well known, IFNs have many other effects on cell physiology (Stark et al., 1998). The multiple effects of IFNs are mediated by the numerous cellular proteins whose synthesis is induced by IFNs (Sen and Ransohoff, 1993). However, the specific biochemical and cellular functions of most of these proteins are unknown. Among the notable exceptions are the enzymes PKR and 2–5(A) synthetases that are known to affect virus replication and cell growth (Stark et al., 1998). Here, we report the function of another IFN-inducible protein, P56.
The cDNA for P56 was cloned in the process of cloning IFN-inducible mRNAs (Chebeth et al., 1983; Kusari and Sen, 1986). We have studied extensively the transcriptional regulation of 561 mRNA that encodes P56 (Bandyopadhyay et al., 1990). The mRNA is undetectable in untreated cells but it is induced rapidly upon IFN treatment (Kusari and Sen, 1986). The 561 mRNA level is quite high in IFN-treated cells and, in a recent gene array analysis, it scored as the most abundant IFN-induced mRNA among >100 such mRNAs (Der et al., 1998). Because the 561 mRNA and the encoded protein, P56, both turn over rapidly, cells are depleted of them quickly after IFN treatment ceases. The 561 gene is transcriptionally induced not only by IFN but also by double-stranded (ds) RNA or virus infection (Tiwari et al., 1987). Our studies using various mutant cells have clearly established that the Jak–STAT pathway used by IFN to induce it is dispensable for its induction by dsRNA or viruses (Bandyopadhyay et al., 1995). Thus, there are alternative ways to induce the 561 gene, and the encoded protein, P56, may have cellular functions beyond the IFN system.
Because the cellular level of P56 is drastically enhanced upon treatment of cells with IFN or dsRNA, we suspected that it has important cellular functions. However, we did not get any clue to such putative functions by examining the primary structure of the protein, which does not contain any functional motifs other than eight tetratricopeptide repeats (TPRs) spaced evenly along the entire protein. Because TPR motifs are known to mediate protein–protein interactions (Das et al., 1998), we speculated that P56 interacts with other human proteins. We conducted a yeast two-hybrid screen using P56 as the bait and identified several proteins that potentially could interact with P56 in mammalian cells.
Here we report the identity of one P56-interacting protein to be Int-6/P48. Human Int-6 is identical in sequence to mouse Int-6, which was originally discovered as the product of a gene whose disruption by the integration of mouse mammary tumor virus genome causes mammary carcinoma in mice (Marchetti et al., 1995; Desbois et al., 1996). Expression of human Int-6 is also affected in many human breast tumors (Miyazaki et al., 1997), suggesting an important cell growth regulatory activity of this protein. An unexpected connection of Int-6 to protein synthesis was made by the observation that the P48 subunit of the translation initiation factor eIF-3 is identical to Int-6 (Asano et al., 1997). Here we show that through its interaction with P48, P56 can bind to eIF-3 and inhibit its functions. As a result, in the presence of P56, protein synthesis is inhibited both in vitro and in vivo. Thus, a new pathway of translational regulation in mammalian cells has been uncovered.
Results
P56 interacts with Int-6/P48
We searched for P56-interacting human proteins using the yeast two-hybrid transcriptional activation assay. A HeLa cell cDNA library was screened using P56 as the bait, and several P56-interacting cDNA clones were isolated. One P56-interacting cDNA clone was clone 6 (Figure 1A, slot 5). The specificity of the observed interaction was confirmed by including various negative controls (slots 1–4) and a positive control (slot 6). The cDNA insert of clone 6 was partially sequenced (Figure 1B) and found to encode the C-terminal 186 residues of the human protein Int-6/P48 fused in-frame with the Gal4 activation domain (AD). Human Int-6 is identical in sequence to mouse Int-6 and it contains 445 residues (Figure 1C). Mouse Int-6 was discovered as the product of a gene whose disruption by the integration of a mouse mammary tumor virus genome causes breast carcinoma in mice (Marchetti et al., 1995). Later, Int-6 was found to be identical to the P48 subunit of the mammalian eIF-3 (Asano et al., 1997).

Fig. 1. The interaction of clone 6 and P56 in yeast. (A) Yeast strain Y190 was co-transfected with the following pairs of expression vectors and plated onto the selection medium without histidine: (1) BD–vector + AD–clone 6; (2) BD–P56 + AD–SV40 large T-antigen; (3) BD–P56 + AD–vector; (4) BD–P53 + AD–clone 6; (5) BD–P56 + AD–clone 6; and (6) BD–P53 + AD–SV40 large T-antigen. (B) Partial cDNA sequence of clone 6. (C) Maps of full-length Int-6/P48 and clone 6.
To examine a possible interaction between P56 and P48 in human cells, an expression vector of an epitope (Flag)-tagged full-length P48 was constructed. As expected of a subunit of a translation initiation factor, P48 was a cytoplasmic protein as revealed by immunofluorescence microscopy using an anti-Flag antibody. IFN-induced P56 was also a cytoplasmic protein as revealed by an anti-P56 antibody, and superimposition of the images of the two proteins indicated that they reside in the same subcellular locations, as shown by the merging of the green and red colors to produce yellow images (Figure 2A). No fluorescence was observed with the Flag antibody in untransfected cells and with the P56 antibody in cells not treated with IFN (data not shown). These results demonstrated that P56 and P48 are localized in the same subcellular compartment and, therefore, they can possibly interact in vivo. That such an interaction indeed occurs was established by co-immunoprecipitation assays shown in Figure 2B. P48 was expressed by transfection (lanes 2 and 4) whereas P56 was either expressed by transfection (lanes 1 and 2) or the endogenous protein was induced by IFN treatment (lanes 3 and 4). In either case, immunoprecipitation of P48 resulted in co-precipitation of P56 (Figure 2B, lanes 2 and 4). These results demonstrated that P56 and P48 interact with each other in mammalian cells just as they do in yeast cells.
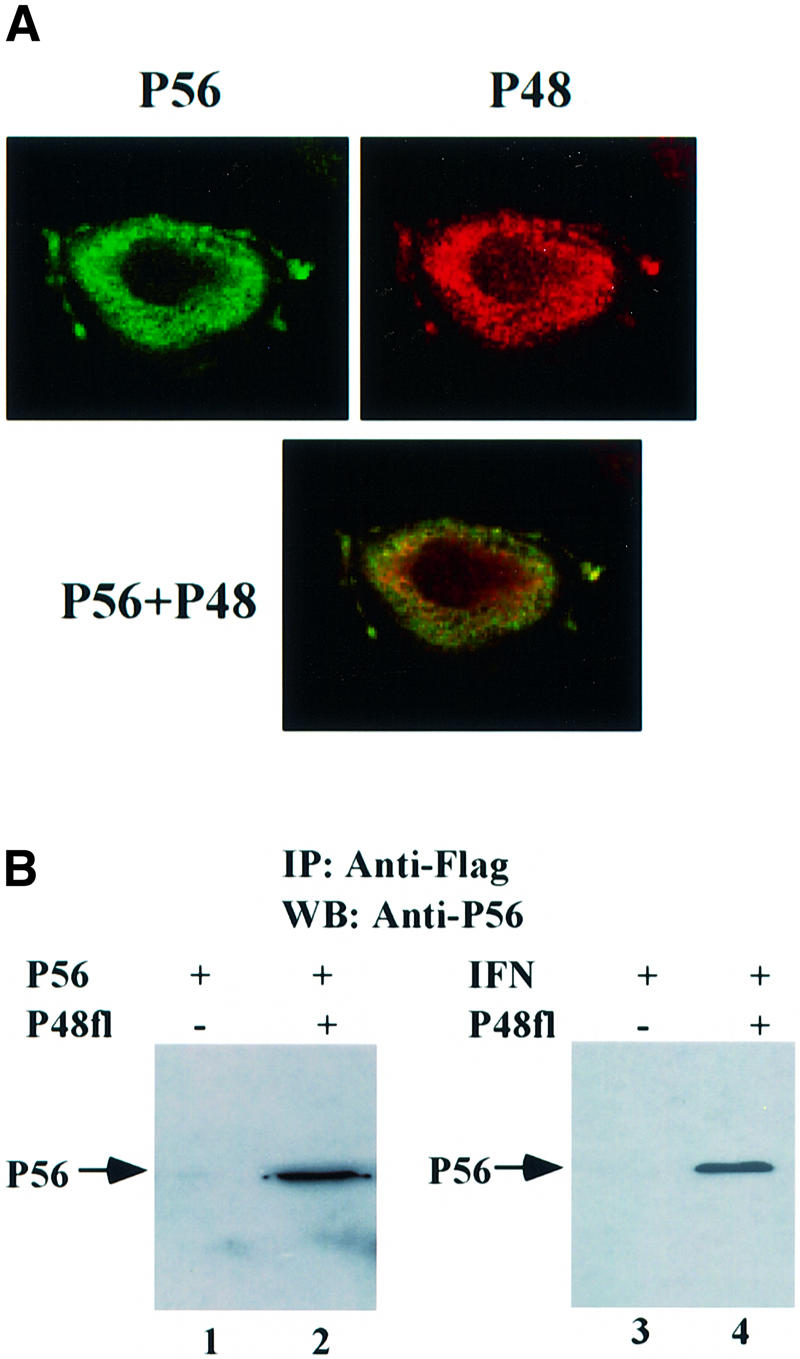
Fig. 2. The interaction of P56 and Int-6/P48 in human cells. (A) Co-localization of P56 and P48 in the cytoplasm. HT1080 cells were transfected with pCMV-P48Fl and, 16 h post-transfection, cells were treated with 1000 U/ml IFN-β for 16 h. A confocal immunofluorescence assay was performed using anti-P56 antibody and anti-Flag antibody. The subcellular locations of P56 (green) and P48 (red) and their co-localization (yellow) are shown. (B) The interaction between both exogenous and endogenous P56 with P48. In lanes 1 and 2, HT1080 cells on a 100 mm plate were transfected with 10 µg of pCMV-P56 alone (lane 1) or co-transfected with 8 µg of pCMV-P56 and 8 µg of pCMV-P48Fl (lane 2). In lanes 3 and 4, cells were transfected with 8 µg of vector alone (lane 3) or pCMV-P48Fl (lane 4) and, 24 h post-transfection, cells were treated with 1000 U/ml IFN-β to induce endogenous P56. After 24 h, cell extracts were made and immunoprecipitation was performed with Flag antibody-conjugated Sepharose beads followed by western blotting with P56 antibody.
P56 binds to eIF-3
Since the only known biochemical function of P48 is as a subunit of eIF-3, we wanted to examine whether P56 can bind to the whole eIF-3 complex, which contains 10 subunits and has a molecular mass of >600 kDa (Pain, 1996). A gel filtration analysis was performed to monitor P56–eIF-3 interactions. For this purpose, recombinant P56 was expressed in Escherichia coli and purified to homogeneity, and eIF-3 was purified to homogeneity from reticulocyte lysate (data not shown). Purified P56 alone (Figure 3A) or P56 mixed with ferritin, a large protein of 440 kDa (Figure 3B), or with purified eIF-3 (Figure 3C and D) was gel filtered. The proteins present in different fractions were analyzed by western blotting with P56 antibody (Figure 3A–C) or by Coomassie Blue staining (Figure 3D). As shown in Figure 3A, P56 itself was entirely in the low molecular weight fractions resolving as mostly dimers and monomers. This pattern did not change when ferritin was mixed with P56 (Figure 3B), demonstrating the absence of an interaction between the two proteins. In contrast, when eIF-3 was mixed with P56, a substantial amount of P56 co-migrated with eIF-3 in the large molecular weight range (Figure 3C). As revealed by staining, eIF-3 was eluted at and around fraction 18, whereas the peak of free P56 was at fractions 40 and 42 (Figure 4D). Western blotting revealed that a peak of P56 was also present in fraction 18, indicating an association with the eIF-3 complex. Lesser amounts of P56 were also present in the intervening fractions (Figure 3C), suggesting partial dissociation of the P56–eIF-3 complex during gel filtration.

Fig. 3. The interaction between P56 and eIF-3 in vitro. Binding of P56 to eIF-3 was monitored by gel filtration chromatography. (A) Recombinant purified P56 protein (65 µg); (B) recombinant purified P56 protein (65 µg; mol. wt 56 kDa) and ferritin (261 µg; mol. wt 440 kDa) mixture (ferritin:P56 molar ratio = 1:2); and (C) recombinant purified P56 protein (65 µg; mol. wt 56 kDa) in the upper panel or recombinant purified MP56 protein (92 µg; mol. wt 40 kDa) in the lower panel was mixed with purified rabbit eIF-3 (350 µg; mol. wt 600 kDa) at a 1:2 molar ratio of eIF-3:P56 and a 1:4 molar ratio of eIF-3:MP56. (D) The same as the upper panel of (C). The different mixtures of proteins were analyzed by gel filtration chromatography on a Superdex 200 packed XK 16/70 column. In (A–C), 25 µl of each even fraction were used for western blotting analysis with P56 antibody. In (D), 500 µl of each even fraction were acetone precipitated and the precipitates were subjected to gel electrophoresis on a 10% SDS–polyacrylamide gel. The gel was run and stained with Coomassie Blue. Fraction numbers are indicated on the top. The positions of the different molecular weight markers are noted by arrows on the top. The positions of P56 detected by western blotting in (A–C) are indicated by arrows on the left. The fractions that contain eIF-3 or P56 are indicated at the bottom. M: pre-stained molecular weight marker.
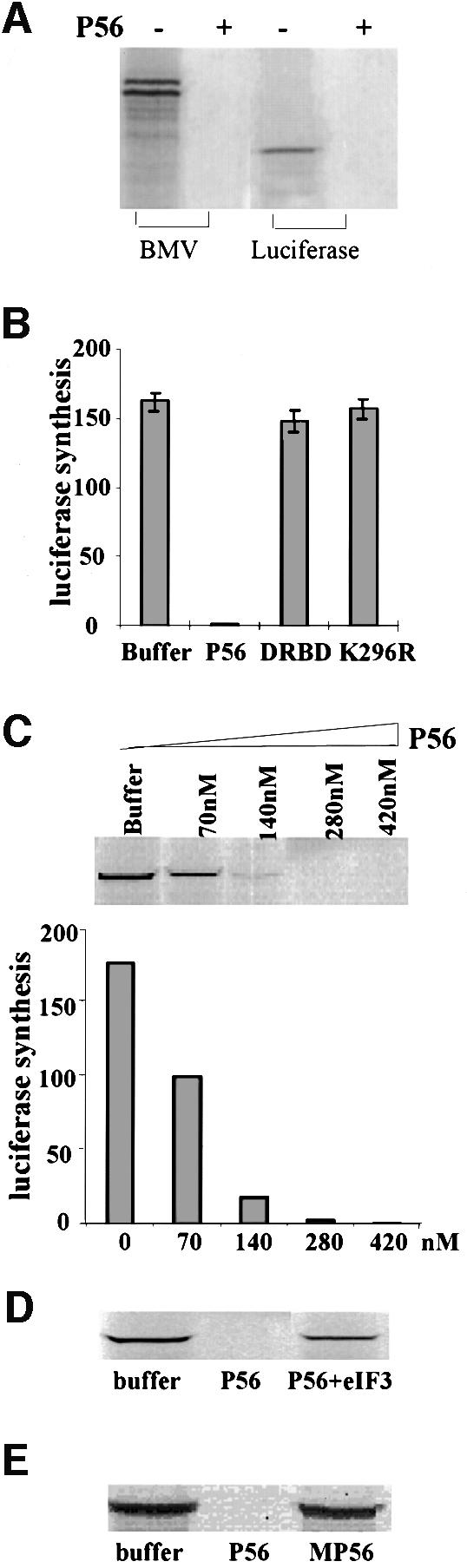
Fig. 4. Inhibition of translation by P56 and rescuing the P56-mediated translation inhibition by eIF-3 in vitro. (A) Luciferase or BMV mRNA was translated in an in vitro translation system with or without 280 nM purified recombinant P56 protein. Synthesized BMV or luciferase was monitored by gel electrophoresis followed by fluorography. (B) Luciferase mRNA was translated in the presence of 280 nM recombinant purified P56, DRBD or K296R protein, or an equivalent volume of the dialysis buffer. The amounts of luciferase synthesized were quantified by phosphorimager analysis after gel electrophoresis. The averages of results from three experiments are shown. (C) Luciferase mRNA was translated in the presence of 70, 140, 280 or 420 nM recombinant purified P56 protein or an equivalent volume of the dialysis buffer. The synthesized luciferase was analyzed by gel electrophoresis and quantified by phosphorimager. (D) Purified rabbit eIF-3 (500 nM) and/or P56 (280 nM) was added to the translation system as indicated. Luciferase synthesis was analyzed by gel electrophoresis. (E) P56 (280 nM) or MP56 (280 nM) was added to the translation system as indicated and luciferase synthesis was analyzed by gel electrophoresis.
To confirm that the observed interaction of P56 with eIF-3 was mediated by P48, we took advantage of a mutant of P56 that does not interact with P48 (Guo et al., 2000). This mutant protein, MP56, was expressed in bacteria as a histidine-tagged protein and purified in the same way as wild-type P56. Its possible interaction with eIF-3 was measured by gel filtration analysis of a mixture of the two proteins. As shown in the lower panel of Figure 3C, MP56 was entirely in the free form, eluting as a monomer. Thus, MP56, a mutant that does not bind to P48, also failed to interact with eIF-3.
P56 inhibits in vitro translation
Once we established that P56 could bind to eIF-3, we wanted to examine the functional consequences of that interaction. Because eIF-3 supports several steps in peptide chain initiation (Pain, 1996), we monitored the effects of P56 on protein synthesis in vitro. An mRNA-dependent in vitro translation system was used for this purpose, and bromomosaic virus (BMV) RNA and luciferase mRNA were translated in the presence or absence of added P56 (Figure 4A). P56 completely inhibited the synthesis of the two BMV proteins and luciferase. The specificity of this observed inhibition was explored further in the experiment shown in Figure 4B. For this purpose, two other proteins, DRBD and K296R (Patel et al., 1995), were expressed in E.coli and purified using the same protocol as that used for P56 purification. DRBD is the dsRNA-binding domain and K296R is an inactive point mutant of PKR, an IFN-induced protein kinase. All three proteins were dialyzed against the same buffer in the same containers, and the buffer after dialysis was used as a negative control. P56 completely inhibited luciferase synthesis, whereas equimolar amounts of DRBD or K296R did not have any effect on translation, demonstrating that the observed effect of P56 is not due to any contaminant co-purifying with the protein. The phenomenon was characterized further by performing a dose–response analysis of P56. Increasing amounts of P56 inhibited translation increasingly, with a complete inhibition occurring between P56 concentrations of 140 and 280 nM (Figure 4C). As anticipated, addition of exogenous eIF-3 substantially relieved the P56-mediated inhibition, although a complete restoration was not achieved at the eIF-3 concentration tested (Figure 4D). Under the conditions of these experiments, P56 did not cause an enhanced degradation of the luciferase mRNA as judged by analysis of a radiolabeled mRNA after the translation incubation (data not shown). The above experiments demonstrated that P56 causes a specific and dose-dependent inhibition of translation by interacting with eIF-3. That interaction with eIF-3 was essential for inhibiting translation was confirmed by testing the effects of MP56, the mutant that does not interact with P48 and eIF-3. As anticipated, MP56 did not inhibit the synthesis of luciferase (Figure 4E).
P56 inhibits protein synthesis in vivo
The physiological relevance of the observed P56-mediated inhibition of translation was established by experiments carried out with whole cells. In human cells, both wild-type P56 (Figure 5A, lane 1) and the mutant MP56 (Figure 5A, lane 2) were expressed equally after transfection but, as expected, only the wild-type protein co-immunoprecipitated with P48 (Figure 5A, lanes 3 and 4).
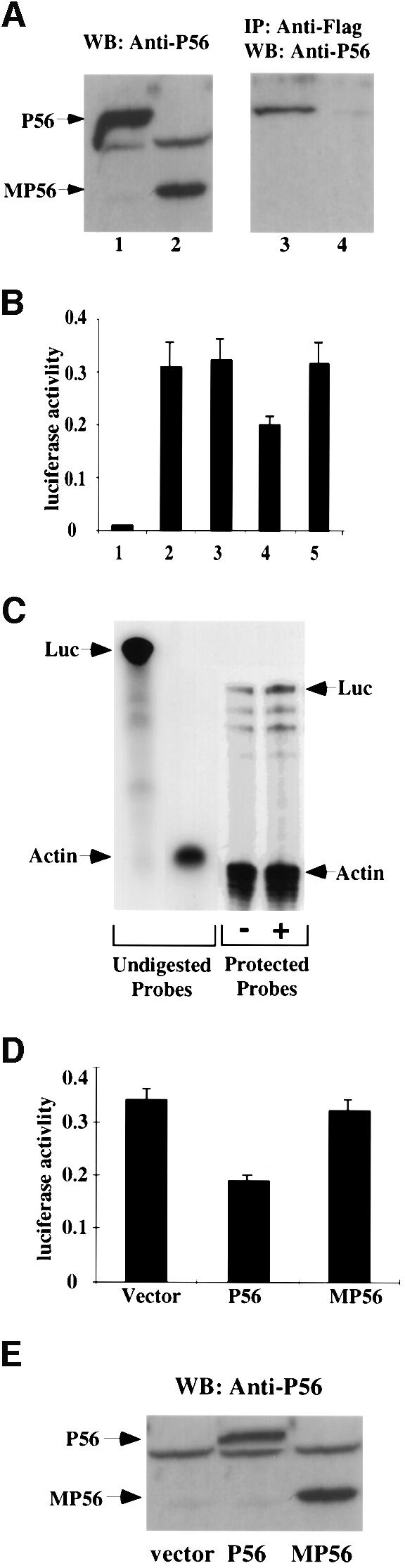
Fig. 5. Inhibition of synthesis of the luciferase reporter gene by P56 in vivo. (A) Interaction of P48/Int-6 with P56 but not MP56. pCMV-P56 (lanes 1 and 3) or pCMV-MP56 (lanes 2 and 4) was co-transfected with pCMV-P48Fl into cells. At 48 h post-transfection, cells were harvested and whole-cell extracts were prepared. A 50 µg aliquot of total cell protein was subjected to gel electrophoresis followed by western blotting with P56 antibody (lanes 1 and 2). A 1 mg aliquot of cell protein was subjected to immunoprecipitation with anti-Flag-conjugated Sepharose beads followed by western blot analysis with P56 antibody (lanes 3 and 4). (B) Cells were co-transfected with E-selectin-Luc and pCMV-P56 (bar 4), pCMV-MP56 (bar 5), pCMV-DRBP76 (bar 3) or the empty expression vector (bars 1 and 2). After 48 h, cells were treated with TNF-α (bars 2–5) for 4 h. Cell extracts were made and luciferase activity was measured. The averages of results from three experiments are shown. (C) Cells were co-transfected with E-selectin-Luc and pCMV-P56 (+) or vector (–). At 48 h post-transfection, cells were treated with TNF-α for 4 h. Cells were harvested and total RNA was isolated for RNase protection assay. A 40 µg aliquot of total RNA was hybridized with 32P-labeled Luc (370 bases) and γ-actin (140 bases) antisense RNA probes shown on the left as undigested probes. Following RNase digestion, the protected RNA probes were resolved in a 6% polyacrylamide, 8 M urea gel. Luciferase mRNA levels, shown on the right as protected probes, were quantified by phosphorimager and, after normalizing against the γ-actin mRNA levels, they were comparable in the two samples. (D) Cells were co-transfected with E-selectin-Luc and vector, pCMV-P56 or pCMV-MP56, as indicated. The experimental protocol was the same as in (B). (E) The same three cell extracts from (D) were western blotted with P56 antibody.
To test the effects of P56 in vivo, we attempted to establish cell lines constitutively expressing P56. These attempts failed, however, probably because P56 expression caused an inhibition of cell growth. We therefore resorted to testing the effect of P56 in a transient transfection assay at the level of expression of a co-transfected reporter gene. To ensure that a major portion of the high cellular level of eIF-3 had a chance to interact with the newly expressed P56, before the reporter mRNA was translated, the reporter gene was driven by an inducible promoter. A cytomegalovirus (CMV) promoter-driven expression vector for P56 or other proteins and a tumor necrosis factor-α (TNF-α)-inducible promoter-driven luciferase reporter gene were co-transfected. P56 was allowed to accumulate in the cells for 48 h and then the reporter gene was activated by TNF-α treatment for 4 h, at which time cell extracts were made and luciferase assays were performed. In vector-transfected cells, luciferase was only expressed after TNF-α treatment (Figure 5B, bars 1 and 2). Expression of wild-type P56 inhibited luciferase expression substantially (Figure 5B, bar 4), whereas MP56 (bar 5) or DRBP76 (Patel et al., 1999), an unrelated protein (bar 3), did not cause any inhibition. That the observed inhibition of luciferase synthesis was at the level of translation was established by quantitating the levels of luciferase mRNA in the vector-transfected (Figure 5B, bar 2) and P56-transfected (Figure 5B, bar 4) cells. RNase protection assays were used for this purpose (Figure 5C). Quantitation of the protected luciferase bands and their normalization against the corresponding γ-actin bands showed that the vector-transfected and the P56-transfected cells contained almost identical levels of luciferase mRNA. Another control for this series of experiments is shown in Figure 5D and E. A part of the experiment shown in Figure 5B was repeated in Figure 5D. A portion of the cell extracts was used for luciferase activity assay (Figure 5D) and another portion containing equal amounts of total protein was used for western blot analysis (Figure 5E). Although similar levels of P56 and MP56 were expressed in the transfected cell (Figure 5E), only P56 inhibited luciferase synthesis (Figure 5D).
The effect of P56 on the overall rate of cellular protein synthesis was investigated further in the experiment shown in Figure 6. For these experiments, we needed to generate a population of cells, all of which were expressing P56. This was achieved by co-expressing, along with P56, the cell surface marker CD20, followed by a fluorescence-activated cell sorting (FACS) selection of the transfected cells using an antibody to CD20. The selected cells were plated out and used for measuring their rates of protein synthesis (Figure 6A). To provide us with a physiological perspective, we compared the level of P56 expression in these cells with that in IFN-treated vector-transfected, and CD20-selected cells. The P56-expressing cells had a level of P56 comparable to that in cells treated with 200 U/ml IFN-β (Figure 6B). The rate of overall protein synthesis as measured by pulse labeling in vivo was inhibited equally strongly in the P56-expressing and IFN-β-treated cells (Figure 6C). There was, however, no inhibition in cells expressing MP56. These results demonstrated that P56 expression by either transfection or IFN treatment leads to a partial but substantial inhibition of the overall rate of cellular protein synthesis.
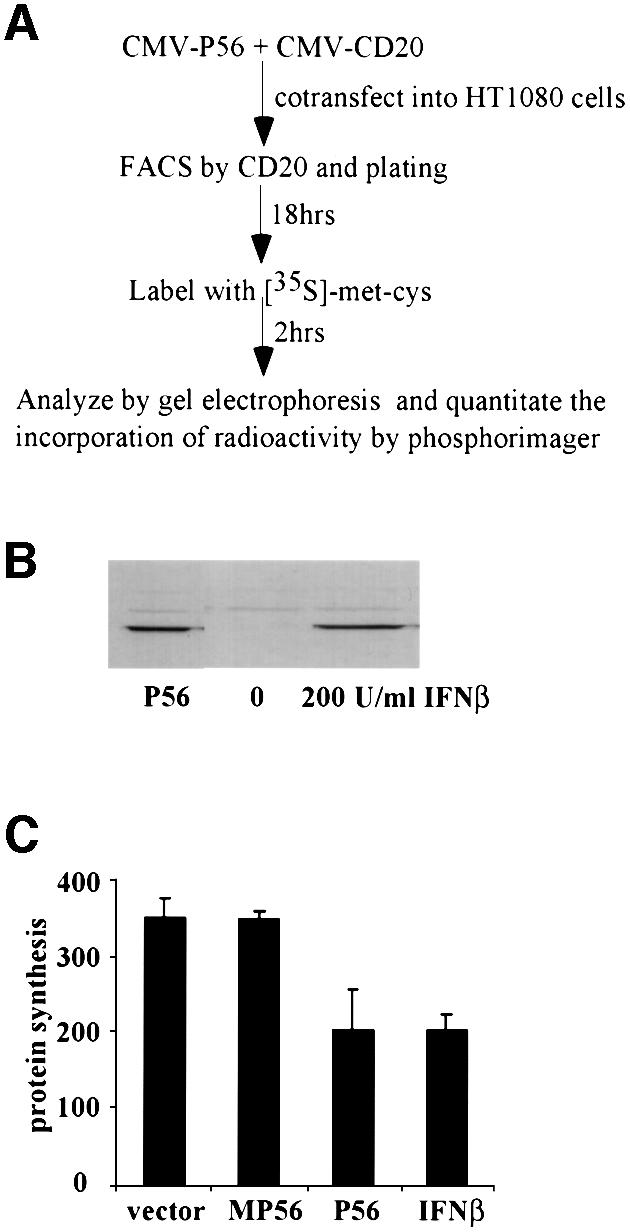
Fig. 6. Inhibition of overall protein synthesis by P56 in vivo. (A) The outline of the in vivo protein synthesis inhibition assay. (B and C) Cells were co-transfected with CMV-CD20 and pCMV-P56, pCMV-MP56 or vector. Transfected cells expressing CD20 were selected by FACS and plated. If IFN treatment was required, 4 h after plating, the sorted vector-transfected cells were treated with 200 U/ml IFN-β. (B) At 18 h after plating, cells were harvested and cell lysate was prepared. A 50 µg aliquot of total cell protein was subjected to gel electrophoresis followed by western blot analysis with P56 antibody. (C) At 18 h after plating, cells were labeled with [35S]methionine and cysteine labeling mix for 2 h. Cell extracts were made and equal amounts of protein were analyzed by gel electrophoresis. The level of radioactivity incorporated in all proteins was quantified by phosphorimager. The averages of results from three experiments are shown.
Discussion
Our study revealed a new mode of regulation of mammalian protein synthesis by modulating the function of eIF-3. The function of translation initiation factor eIF-2 has long been known to be regulated by phosphorylation of its α-subunit (Samuel, 1993). It is also known that the function of another translation initiation factor, eIF-4E, is regulated by its phosphorylation or by phosphorylation of the eIF-4E-binding protein (Sonenberg, 1996). In contrast, the results presented here showed that eIF-3 function was modulated by the binding of one of its subunits, P48, to the IFN-induced cellular protein P56. It remains to be seen whether P48 can be the target of regulation by additional cellular stimuli. A potential regulatory function of P48 in mammalian cells has been suggested before (Asano et al., 1997). This was based on the observation that yeast eIF-3 lacks a counterpart of mammalian P48, suggesting that, although P48 is not required for eIF-3 function, it is used in mammalian cells to regulate eIF-3 function. Our results provide the first experimental support for this hypothesis. If P48 is not required for eIF-3 function, one would expect that lowering the cellular level of functional P48 by engaging free P48 in a complex with P56 will not affect eIF-3 functions because a P48-less eIF-3 should be active. Moreover, extrapolating from the yeast situation, P48 is probably not essential for the other nine subunits of eIF-3 to assemble properly and form a functional complex. Thus, the more plausible scenario for P56 to act as an inhibitor of eIF-3 action is by joining the complex through its interaction with P48 (Figure 7). Results shown in Figure 3 support this model; there was specific binding of P56 to the eIF-3 complex. More detailed biochemical analysis will be needed to establish the stoichiometry of this interaction and to ascertain whether functional P56 is dimeric or monomeric. The P56–eIF-3 interaction demonstrated in vitro is quite likely to occur in vivo because when an extract of IFN-treated cell was subjected to the same gel filtration procedure, P56 was present not only in the low molecular weight fractions, but also in high molecular weight fractions containing eIF-3 (data not shown). Biochemical analysis after cell fractionation and confocal microscopy at a higher resolution will be required to confirm this point further. A difficult technical hurdle in pursuing a more rigorous analysis of P56–eIF-3 interactions is the lack of availability of appropriate eIF-3 antisera. Those that are available do not recognize many subunits of eIF-3, including P48, and, more importantly, they do not immunoprecipitate the complex effectively. Without the immunological reagents, neither microscopy nor biochemical analysis of protein–protein interaction can be done in a quantitative fashion.

Fig. 7. Regulation of cellular protein synthesis by P56. Transcription of 561 mRNA is induced by IFN or virus/dsRNA using two different signaling pathways. P56, produced upon translation of 561 mRNA, binds to the P48 subunit of eIF-3 and blocks its function in peptide chain initiation. As a result, synthesis of cellular proteins, including that of P56 itself, is inhibited.
Inhibition of eIF-3 function by P56 provides a new pathway for blocking protein synthesis in IFN-treated cells (Figure 7). Two such IFN-induced pathways have been studied extensively in the past. The IFN-induced protein kinase PKR phosphorylates eIF-2α and blocks translation initiation, whereas IFN-induced 2–5(A) synthetases synthesize 2′–5′ linked oligoadenylates that activate RNase L and degrade mRNAs (Stark et al., 1998). Both of these pathways, however, require dsRNA for their function. The two key enzymes in the two pathways, PKR and 2–5(A) synthetase, are inactive as such until they are activated by dsRNA. Physiologically, the activating dsRNA is provided by virus infection and therefore these pathways remain latent in uninfected cells. The P56–eIF-3 pathway, in contrast, does not require an activator; induction of P56 synthesis is sufficient to trigger this inhibitory pathway. Thus it should operate in IFN-treated cells both before and after virus infection. Experiments shown in Figures 5 and 6 were designed to mimic the situation in IFN-treated uninfected cells. We chose levels of P56 expression in transfected cells that are comparable to those in cells treated with relatively low doses of IFN because we wanted to examine the cellular effects of P56 at a physiological concentration but in the absence of other IFN-induced proteins. Under these conditions, we observed a significant inhibition of protein synthesis in both transfected and IFN-treated cells. Data with the mutant MP56 strongly suggest that the observed inhibition is mediated by the interaction of P56 with P48 and eIF-3. The degree of inhibition is probably dictated by the relative abundance of eIF-3 and P56 in the cells. An autoregulatory loop may prevent a stronger inhibition of protein synthesis by preventing accumulation of higher levels of P56. This autoregulation of P56 synthesis may be achieved partly by accumulated P56 inhibiting its own synthesis and partly by its rapid turnover (Figure 7).
What is the overall effect of P56-mediated inhibition of protein synthesis on cellular health? We speculate that this pathway may be a major contributor to the often-observed inhibitory effect of IFN on cell growth. Indeed, when the P56-expressing sorted cells were cultured for 3 days, we observed a 30–40% reduction in the rate of their growth as compared with the vector-transfected cells that have been sorted similarly (data not shown). Since, unlike the PKR and 2–5(A) synthetase pathways, functioning of the P56 pathway does not require dsRNA, it could be instrumental in mediating IFN’s cell growth regulatory effects. However, the cellular effects of P56 may not be restricted to the IFN-treated cells. Because virus infection can directly induce P56, it may also play a role in modulating protein synthesis in virus-infected cells. A partial inhibition of protein synthesis may provide a relative advantage to viral mRNA translation over cellular mRNA translation. Similar selective discrimination has been observed in cells containing phosphorylated eIF-2 (Chou et al., 1995).
P56 belongs to a family of IFN-induced proteins. Human P54, P58 and P60 are structurally related to P56, as are mouse P54, P56 and P60 (Wathelet et al., 1988; Bluyssen et al., 1994; Niikura et al., 1997; Yu et al., 1997; de Veer et al., 1998). The sequence identities of these proteins range from 40 to 55%. All of them, however, contain multiple TPR motifs, although the exact sequences of these motifs are quite diverse. It remains an open question whether the other members of this family can bind to P48 and inhibit protein synthesis. Results shown in Figure 6 suggest that it may not be the case because the degrees of inhibition of protein synthesis were very similar in cells expressing P56 and the IFN-treated cells that presumably contained the other related proteins as well. Because different TPR units can mediate interactions with different proteins (Das et al., 1998), it is conceivable that the other members of the P56 family of proteins can bind to cellular proteins other than P48 through their unique TPR motifs. The identities of their putative partners and the cellular consequences of these interactions remain to be investigated in the future.
Materials and methods
Cell culture, IFN and poly(I)⋅poly(C)
HT1080 human fibrosarcoma cells (Leonard and Sen, 1997) were maintained in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 100 U/ml penicillin and 100 µg/ml streptomycin (Gibco-BRL). Human IFN-β was from Hoffman-LaRoche (Nutley, NJ), and poly(I)⋅poly (C) was purchased from Pharmacia Biotech.
Antibody
The rabbit polyclonal antibody that recognizes P56 was raised by injection of rabbit with purified bacterially expressed P56 (our unpublished results).
Construction of P56 clones and MP56
The complete coding sequence of P56 is in the DDBJ/EMBL/GenBank database (accession No. NM001548). The full-length P56 cDNA was constructed by PCR using an existing partial clone (our unpublished data). The cDNA sequence was inserted into pBluescript KS (II) (Strategene). pCMV-P56 was constructed by excising full-length P56 cDNA from pBluescript KS (II) and inserting into pCB6+, a eukaryotic expression vector (Patel et al., 1996). BD–P56, a yeast expression vector that expresses a fusion protein of the Gal4 DNA-binding domain and P56, was constructed by inserting full-length cDNA of P56 in-frame into pGBT9 (Clontech). pET-P56 was constructed by inserting full-length P56 cDNA into pET15b to generate an in-frame fusion of P56 sequence to the histidine tag-coding sequence from pET15b (Novagen). MP56 contained 1–339 amino acids of P56. The cDNA was generated by PCR and subcloned into pcDNA3 (Invitrogen) to produce pCMV-MP56.
Construction of the P48 clone
pCMV-P48Fl contains the full-length Int-6/P48-coding sequence with a Flag tag at the C-terminus. Full-length P48Fl was generated by PCR using pSGF-Int-6 (Desbois et al., 1996) (a gift of Dr P.Jalinot) as a template. The 5′ PCR primer encoded 14 amino acids including residues 2–9 of Int-6, which were missing in pSGF-Int-6, and eliminated the Flag sequence and 13 extraneous residues present at the N-terminus of pSGF-Int-6. The 3′ PCR primer included the Flag tag sequence. The cDNA was inserted into pCB6+.
Yeast two-hybrid assay
The full-length P56 expressed as BD–P56 was used as the bait. A total of 1 × 107 transformants from a human HeLa cell matchmaker library (Clontech) were screened in the yeast strain Y190 (Clontech) and 150 colonies were recovered as His+, out of which eight were positive for β-galactosidase. On further analysis, two of these were dependent on P56 to give a positive β-galactosidase reaction. These clones were subjected to a second screen to ensure that they gave a positive β-galactosidase reaction in a manner specific for co-expression of P56. Analysis of one of these clones (clone 6) is reported herein. Sequence analysis of the cDNA clone revealed that it encoded a protein that is identical to the C-terminal 260–445 amino acids of the P48 protein, a subunit of translation initiation factor eIF-3. For known–known protein yeast two-hybrid assay, 0.1 µg of each plasmid in combination, as described in Figure 1A, were co-transfected into yeast strain Y190 using the lithium acetate transfection method. Growth selection and scoring for growth were performed as described (Patel and Sen, 1998).
Transfection and confocal immunofluorescence
Transfection was performed using the Fugene 6 transfection method (Boehringer Mannheim Co.). Confocal immunofluorescence was performed as described previously (Leonard and Sen, 1996) with the following modifications. A 1.6 µg aliquot of pCMV-P48Fl was transfected into HT1080 cells on coverslips in a 6-well plate. At 12 h post-transfection, cells were treated with 1000 U/ml IFN-β to induce endogenous P56. At 16 h after IFN treatment, cells were fixed and incubated with 1:2000 anti-P56 antibody and 1:2000 anti-Flag M2 antibody (Kodak Scientific Imaging Systems) to detect P56 and P48Fl, respectively. Antibody binding was detected with fluorescein isothiocyanate (FITC)-conjugated goat anti-rabbit antibody (Gibco) and Texas red-conjugated goat anti-mouse antibody (Molecular Probe). Finally, the coverslips were washed, mounted and examined with a Leica confocal laser scanning microscope. Digitized images were acquired with software provided with the microscope. Double-stained images were acquired by a split image of both fluorochromes filtered by FITC and Texas red filters, and subsequent overlay of the two-color images.
Immunoprecipitation and western blotting
Immunoprecipitation of Flag-tagged protein was performed in low salt buffer [20 mM Tris pH 7.5, 50 mM KCl, 200 mM NaCl, 1 mM EDTA, 20% glycerol, 0.05% Triton 100, 0.2 mM phenylmethylsulfonyl fluoride (PMSF)]. The M2 anti-Flag Sepharose beads (Kodak Scientific Imaging System) were pre-soaked with 3 µg of bovine serum albumin for 15 min. The cell lysate was prepared as described (Leonard and Sen, 1996), and 1 mg of whole-cell extracts was mixed with 500 µl of low salt buffer and 20 µl of pre-incubated anti-Flag Sepharose beads at 4°C for 2 h. The immune complexes were washed four times with the low salt buffer and subjected to denaturing gel electrophoresis through a 10% polyacrylamide gel. Western blotting was performed with a 1:2000 dilution of P56 antibody.
Purification of recombinant protein from E.coli
P56, MP56, K296R (PKR inactive mutant) and DRBD (dsRNA-binding domain of PKR) (Patel et al., 1995) were subcloned in pET15b vector (Novagen). The proteins were expressed in bacteria and purified using Ni-chromatography as described in the pET system manual (Novagen). The proteins were purified in the same way and dialyzed in the same container against 2 l of dialysis buffer (20 mM Tris pH 7.5 and 20% glycerol) with three changes every 4 h followed by a dialysis overnight. The dialyzed proteins were concentrated using Centricon (Amicon Inc.) to an appropriate concentration and stored as aliquots at –70°C.
Gel filtration chromatography
Gel filtration was performed on an XK 16/70 column (16 mm diameter, 70 cm long, column volume ∼100 ml, V0 ∼35 ml; Amersham Pharmacia Biotech) packed with Superdex 200. The column was equilibrated with gel filtration equilibration buffer [20 mM Tris, pH 7.9, 150 mM KCl, 1 mM dithiothreitol (DTT), 0.1 mM EDTA and 5% glycerol]. Recombinant purified P56 protein with or without purified rabbit eIF-3 or ferritin (Sigma) was diluted with gel filtration equilibration buffer to a final volume of 500 µl and incubated at 30°C for 15 min. The mixture was applied to the column and separation was performed at 4°C with a flow rate of 1 ml/min using an FPLC system (Amersham Pharmacia Biotech). The void volume of the column is ∼35 ml, and 1 ml fractions were collected after the first 30 ml had passed. A 25 µl aliquot of each even fraction was subjected to gel electrophoresis followed by western blot analysis to detect the P56 protein. A 500 µl aliquot of each even fraction was acetone precipitated, and the precipitates were dissolved in gel electrophoresis sample buffer and subjected to gel electrophoresis on 10% SDS–PAGE. The gel was stained with Coomassie Blue.
In vitro translation inhibition assay
A 1 µg aliquot of luciferase mRNA or BMV mRNA (Promega) was added to 50 µl of a rabbit reticulocyte lysate in vitro translation reaction (Promega) in the presence of recombinant purified P56, MP56, DRBD, K296R or purified rabbit eIF-3 (Asano et al., 1997). The reaction mixture was incubated at 30°C for 2 h. Newly synthesized 35S-labeled proteins were analyzed by subjecting an equal volume of reaction mixture to 10% SDS–PAGE and were quantified by a phosphorimager.
In vivo reporter synthesis inhibition assay
A 500 ng aliquot of vector pCB6+, pCMV-P56, pCMV-MP56 or pCMV-DRBP76 (CMV promoter-driven dsRNA-binding protein P76) (Patel et al., 1999) was co-transfected with 500 ng of E-selectin-Luc (E-selectin promoter-driven luciferase) (Askew et al., 1993) into HT1080 cells in 6-well plates in triplicate. After 48 h, cells were induced with 20 ng/ml TNF-α for 4 h. Cell extracts were prepared in 1× reporter lysis buffer (Promega) and luciferase activity was measured using the luciferase reporter gene assay kit (Promega).
RNase protection assay
A 500 ng aliquot of vector pCB6+ or pCMV-P56 was co-transfected with 500 ng of E-selectin-Luc (E-selectin promoter-driven luciferase) (Askew et al., 1993) into HT1080 cells in 6-well plates. At 48 h post-transfection, cells were induced with 20 ng/ml TNF-α for 4 h. Cells were harvested and total RNA was isolated from the cells using the RNAzol B reagent according to the manufacturer’s protocol (Teltest, Friendswood, TX). The antisense probe to Luc was transcribed with T7 RNA polymerase from the pGEM-luc plasmid (Promega) cut with EcoRV to generate a 370 nucleotide probe. The γ-actin probe was synthesized as described (Patel and Sen, 1998). The RNase protection assay was performed as described before (Patel and Sen, 1998).
In vivo protein synthesis inhibition assay
pCB6+, pCMV-P56 or pCMV-MP56 was co-transfected with CMV-CD20 at a ratio of 8:1 into HT1080 cells on a 100 mm plate. At 18 h post-transfection, cells were trypsinized and incubated for 30 min with 20 µl/100 mm plate of FITC-conjugated anti-CD20 antibody (Becton Dickinson Immunocytometry Systems). Cells were washed twice in phosphate-buffered saline (PBS) and resuspended in DMEM. Cell sorting was performed to detect FITC-conjugated anti-CD20 antibody for CD20 expression by FACS. A total of 2 × 105 sorted cells were plated into each 6-well plate. At 18 h after plating, the sorted cells were washed twice with labeling medium (DMEM minus methionine and cysteine and plus dialyzed serum) and pulse labeled with 100 µCi of [35S]methionine and cysteine labeling mix in 0.5 ml of labeling medium for 2 h. Cell extracts were made and protein concentration was measured by the Bradford method; an equal amount of protein was analyzed by 10% SDS–PAGE. The gel was dried and total radioactivity incorporated was quantified by phosphorimager. The final quantitation was the average of triplicate samples.
Acknowledgments
Acknowledgements
We thank Dr P.Jalinot for pSGF-Int-6 construct, Drs Xiaoxia Li and George Stark for the E-selectin-Luc construct, Dr Judy Drazba for help with confocal immunofluorescence microscopy, Mr Rune Hartman and Dr Nancy Richter for help with gel filtration chromatography, and Ms Theresa Rowe for help with the RNase protection assay. The FACS sorting was dedicated through a gift from the W.M.Keck Foundation. We also thank Drs Bryan Williams, Robert Silverman and George Stark for helpful discussion. This work was supported in part by National Institutes of Health grants CA-68782 and CA-62220.
References
- Asano K., Merrick,W.C. and Hershey,J.W. (1997) The translation initiation factor eIF3-p48 subunit is encoded by int-6, a site of frequent integration by the mouse mammary tumor virus genome. J. Biol. Chem., 272, 23477–23480. [DOI] [PubMed] [Google Scholar]
- Askew G.R., Doetschman,T. and Lingrel,J.B. (1993) Site-directed point mutations in embryonic stem cells: a gene-targeting tag-and-exchange strategy. Mol. Cell. Biol., 13, 4115–4124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bandyopadhyay S.K., Kalvakolanu,D.V. and Sen,G.C. (1990) Gene induction by interferons: functional complementation between trans-acting factors induced by α interferon and γ interferon. Mol. Cell. Biol., 10, 5055–5063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bandyopadhyay S.K., Leonard,G.T.,Jr, Bandyopadhyay,T., Stark,G.R. and Sen,G.C. (1995) Transcriptional induction by double-stranded RNA is mediated by interferon-stimulated response elements without activation of interferon-stimulated gene factor 3. J. Biol. Chem., 270, 19624–19629. [DOI] [PubMed] [Google Scholar]
- Bluyssen H.A., Vlietstra,R.J., Faber,P.W., Smit,E.M., Hagemeijer,A. and Trapman,J. (1994) Structure, chromosome localization, and regulation of expression of the interferon-regulated mouse IFi54/IFi56 gene family. Genomics, 24, 137–148. [DOI] [PubMed] [Google Scholar]
- Chebeth J., Merlin,G., Metz,R., Benech,P. and Revel,M. (1983) Interferon-induced 56,000 Mr protein and its mRNA in human cells: molecular cloning and partial sequence of the cDNA. Nucleic Acids Res., 11, 1213–1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou J., Chen,J.J., Gross,M. and Roizman,B. (1995) Association of a M(r) 90 000 phosphoprotein with protein kinase PKR in cells exhibiting enhanced phosphorylation of translation initiation factor eIF-2α and premature shutoff of protein synthesis after infection with γ134.5-mutants of herpes simplex virus 1. Proc. Natl Acad. Sci. USA, 92, 10516–10520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das A.K., Cohen,P.W. and Barford,D. (1998) The structure of the tetratricopeptide repeats of protein phosphatase 5: implications for TPR-mediated protein–protein interactions. EMBO J., 17, 1192–1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Der S.D., Zhou,A., Williams,B.R. and Silverman,R.H. (1998) Identifi cation of genes differentially regulated by interferon α, β, or γ using oligonucleotide arrays. Proc. Natl Acad. Sci. USA, 95, 15623–15628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desbois C., Rousset,R., Bantignies,F. and Jalinot,P. (1996) Exclusion of Int-6 from PML nuclear bodies by binding to the HTLV-I Tax oncoprotein. Science, 273, 951–953. [DOI] [PubMed] [Google Scholar]
- de Veer M.J., Sim,H., Whisstock,J.C., Devenish,R.J. and Ralph,S.J. (1998) IFI60/ISG60/IFIT4, a new member of the human IFI54/IFIT2 family of interferon-stimulated genes. Genomics, 54, 267–277. [DOI] [PubMed] [Google Scholar]
- Guo J. and Sen,G.C. (2000) Characterization of the interaction between the interferon-induced protein P56 and the Int6 protein encoded by a locus of insertion of the mouse mammary tumor virus. J. Virol., 74, 1892–1899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kusari J. and Sen,G.C. (1986) Regulation of synthesis and turnover of an interferon-inducible mRNA. Mol. Cell. Biol., 6, 2062–2067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonard G.T. and Sen,G.C. (1996) Effects of adenovirus E1A protein on interferon-signaling. Virology, 224, 25–33. [DOI] [PubMed] [Google Scholar]
- Leonard G.T. and Sen,G.C. (1997) Restoration of interferon responses of adenovirus E1A-expressing HT1080 cell lines by overexpression of p48 protein. J. Virol., 71, 5095–5101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchetti A., Buttitta,F., Miyazaki,S., Gallahan,D., Smith,G.H. and Callahan,R. (1995) Int-6, a highly conserved, widely expressed gene, is mutated by mouse mammary tumor virus in mammary preneoplasia. J. Virol., 69, 1932–1938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyazaki S., Imatani,A., Ballard,L., Marchetti,A., Buttitta,F., Albertsen,H., Nevanlinna,H.A., Gallahan,D. and Callahan,R. (1997) The chromosome location of the human homolog of the mouse mammary tumor-associated gene INT6 and its status in human breast carcinomas. Genomics, 46, 155–158. [DOI] [PubMed] [Google Scholar]
- Niikura T., Hirata,R. and Weil,S.C. (1997) A novel interferon-inducible gene expressed during myeloid differentiation. Blood Cells Mol. Dis., 23, 337–349. [DOI] [PubMed] [Google Scholar]
- Pain V.M. (1996) Initiation of protein synthesis in eukaryotic cells. Eur. J. Biochem., 236, 747–771. [DOI] [PubMed] [Google Scholar]
- Patel R.C. and Sen,G.C. (1998) PACT, a protein activator of the interferon-induced protein kinase, PKR. EMBO J., 17, 4379–4390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel R.C., Stanton,P., McMillan,N.M., Williams,B.R. and Sen,G.C. (1995) The interferon-inducible double-stranded RNA-activated protein kinase self-associates in vitro and in vivo. Proc. Natl Acad. Sci. USA, 92, 8283–8287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel R.C., Stanton,P. and Sen,G.C. (1996) Specific mutations near the amino terminus of double-stranded RNA-dependent protein kinase (PKR) differentially affect its double-stranded RNA binding and dimerization properties. J. Biol. Chem., 271, 25657–25663. [DOI] [PubMed] [Google Scholar]
- Patel R.C., Vestal,J.D., Xu,Z., Bandyopadhyay,S., Guo,W., Erme,S.M., Williams,B.R.G. and Sen,G.C. (1999) DRBP76, a double-stranded RNA-binding nuclear protein, is phosphorylated by the interferon-induced protein kinase, PKR. J. Biol. Chem., 274, 20432–20437. [DOI] [PubMed] [Google Scholar]
- Samuel C.E. (1993) The eIF-2α protein kinases, regulators of translation in eukaryotes from yeasts to humans. J. Biol. Chem., 268, 7603–7606. [PubMed] [Google Scholar]
- Sen G.C. and Ransohoff,R.M. (1993) Interferon-induced antiviral actions and their regulation. Adv. Virus Res., 42, 57–102. [DOI] [PubMed] [Google Scholar]
- Sonenberg N. (1996) mRNA 5′ cap-binding protein eIF4E and control of cell growth. In Hershey,J.W.D., Matthews,M.B. and Sonenberg,N. (eds), Translational Control. Cold Spring Harbor Laboratory Press, Plainview, NY, pp. 245–269. [Google Scholar]
- Stark G.R., Kerr,I.M., Williams,B.R.G., Silverman,R.H. and Schreiber,R.D. (1998) How cells respond to interferons. Annu. Rev. Biochem., 67, 227–264. [DOI] [PubMed] [Google Scholar]
- Tiwari R.K., Kusari,J. and Sen,G.C. (1987) Functional equivalents of interferon-mediated signals needed for induction of an mRNA can be generated by double-stranded RNA and growth factors. EMBO J., 6, 3373–3378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wathelet M.G., Clauss,I.M., Content,J. and Huez,G.A. (1988) The IFI-56K and IFI-54K interferon-inducible human genes belong to the same gene family. FEBS Lett., 231, 164–171. [DOI] [PubMed] [Google Scholar]
- Yu M. et al. (1997) Cloning of a gene (RIG-G) associated with retinoic acid-induced differentiation of acute promyelocytic leukemia cells and representing a new member of a family of interferon-stimulated genes. Proc. Natl Acad. Sci. USA, 94, 7406–7411. [DOI] [PMC free article] [PubMed] [Google Scholar]