

Abstract
Among the pathological factors known to be associated with Alzheimer disease (AD), oxidative stress induced by the amyloid-β peptide (Aβ) has been demonstrated to play a key role in human brain and animal models of AD. Recently, we reported elevated levels of oxidative damage in the brain of a transgenic (Tg) AD mouse model with Swedish and Indiana familial AD mutations in human amyloid precursor protein (APP) [PDAPP mice, line J20], as evidenced by increased levels of protein carbonyls, 3-nitrotyrosine, and protein-bound 4-hydroxy-2-nonenal. This oxidative damage was dependent on the methionine 35 residue within the Aβ peptide. Further insight into the molecular pathways affected in this Tg model of AD may be gained with discovery-based proteomics studies; therefore, two-dimensional gel-based expression proteomics was performed to compare differences in brain protein levels of J20 Tg mice with non-transgenic (NTg) littermate controls. Based on our studies, we identified six proteins that had significantly increased levels in J20 Tg relative to NTg mice: calcineurin subunit B type 1, ρ GDP-dissociation inhibitor 1, T-complex protein 1 subunit α A, α-enolase, peptidyl-prolyl cis-trans isomerase (Pin-1), and ATP synthase subunit α mitochondrial. Several of these proteins have previously been implicated in in vitro and in vivo models and subjects with AD. Additionally, using redox proteomics analyses we identified two oxidatively-modified proteins: phosphatidylethanolamine-binding protein 1 and Pin-1 with decreased levels of protein 3-nitrotyrosine in J20 Tg mice relative to NTg. Western blotting and immunoprecipitation analyses were used to validate proteomics results. Overall, these studies provide information about changes in the brain proteome as a result of Aβ deposition and clues with which to further direct studies on elucidating AD pathogenesis.
Keywords: Alzheimer disease, amyloid β-peptide, expression proteomics, transgenic mouse model of Alzheimer disease, redox proteomics
Alzheimer disease (AD) is an age-related neurodegenerative disorder that is clinically manifested by progressive signs of cognitive decline and memory impairment (Price et al., 1998). Familial cases of AD have been linked primarily to mutations in genes such as amyloid precursor protein [APP] (Goate et al., 1991), presenilin-1 (PS-1) and presenilin-2 [PS-2] (Cruts et al., 1998); however, other genes such as apolipoprotein E, allele 4 (Levy-Lahad et al., 1995, Slooter et al., 1998), endothelial nitric oxide synthase-3 (Dahiyat et al., 1999), and α2-macroglobulin (Blacker et al., 1998) have been found in other AD cases. Histopathological hallmarks of AD include senile plaques (SP), neurofibrillary tangles (NFT), and synapse loss. Additionally, oxidative stress has been implicated in the pathogenesis of AD (Smith et al., 1994, Good et al., 1996, Markesbery, 1997, Smith et al., 1997, Butterfield and Lauderback, 2002, Butterfield et al., 2006b).
SPs are largely composed of amyloid-β (Aβ) peptides, which are generated by β- and γ-secretase cleavage of the APP protein. The most common forms of Aβ associated with human AD are Aβ(1-40) and Aβ(1-42) (Selkoe, 1996), and the latter has been shown to be more toxic than Aβ(1-40) in model systems of AD (Butterfield and Boyd-Kimball, 2004, Mohmmad Abdul et al., 2004, Boyd-Kimball et al., 2005a, 2005b, 2005c, Mohmmad Abdul et al., 2006). Overproduction of APP (caused by duplication of chromosome 21 genetic mutations) subsequently leads to increased levels of Aβ (Selkoe, 2001). In familial AD, mutations close to the β- and γ-secretase cleavage sites are thought to lead to increased levels of Aβ. While Aβ peptides can exist as monomers, oligomers, fibrils, or aggregates, the soluble Aβ(1-42) oligomers are believed to be the most toxic species in AD brain (Drake et al., 2003, Demuro et al., 2005, Selkoe, 2008, Viola et al., 2008). The major consequences of this toxicity are the observed cognitive losses in learning and memory (Walsh et al., 2002, Selkoe, 2008). Our laboratory and others have provided considerable evidence that Aβ(1-42) induces oxidative stress in AD and various AD model systems (Pappolla et al., 1999, Drake et al., 2003, Boyd-Kimball et al., 2004, 2005c, Sultana et al., 2005, Ansari et al., 2006, Mohmmad Abdul et al., 2006, Abdul et al., 2008, Resende et al., 2008, Zhu et al., 2008).
Recently, we published reports of increased levels of oxidative stress in a PDAPP transgenic mouse model of AD as measured by protein oxidation [i.e., protein carbonyls and 3-nitrotyrosine (3-NT)] and lipid peroxidation [i.e., 4-hydroxy-2-nonenal (HNE)] (Butterfield et al., 2009). This PDAPP model (hereafter, referred to as J20 Tg) has mutations in human APP corresponding to the Swedish (670/671KM→ML) and Indiana (717V→F) familial forms of AD (Mucke et al., 2000, Galvan et al., 2006). J20 Tg mice have aspects of AD pathology, such as Aβ accumulation, neuritic plaque formation, and memory deficits (Mucke et al., 2000, Galvan et al., 2006). Oxidative stress in J20 Tg mice can be directly correlated with these pathological hallmarks (Butterfield et al., 2009).
In this study, we sought to gain insight into the specific proteins that are altered in this AD model using expression and redox proteomics techniques in a discovery-based approach. Alterations of brain proteins from J20 Tg mice in comparison to non-transgenic (NTg) littermate controls were identified following two-dimensional polyacrylamide gel electrophoresis (2D-PAGE) separation employing mass spectrometry (MS) and database searching techniques. Redox proteomic experiments also were performed to identify brain proteins with significant differences in oxidative modification in J20 Tg mice as measured by 3-NT. PD-Quest results were validated using Western blotting and immunoprecipitation analyses of peptidyl-prolyl cis-trans isomerase (Pin-1). Overall, findings from these proteomics studies enhance the understanding of altered pathways in AD and provide potential targets for AD prevention and treatment that warrant further investigation.
2.0 Materials & Methods
All chemicals, proteases, and antibodies used in these studies were purchased from Sigma-Aldrich (St. Louis, MO) with exceptions noted. Criterion precast polyacrylamide gels, TGS and XT MES electrophoresis running buffers, ReadyStrip™ IPG strips, mineral oil, Precision Plus Protein™ All Blue Standards, Sypro Ruby® Protein Stain, nitrocellulose membranes, dithiothreitol (DTT), iodoacetamide (IA), Biolytes, and urea were purchased from Bio-RAD (Hercules, CA). 2,4-Dinitrophenylhydrazine (DNPH) and the primary antibody used for the protein-bound 2,4-dinitrophenylhydrazone (DNP) products were purchased from Chemicon International (Temecula, CA). Rabbit polyclonal anti-Pin-1 [H-123] primary antibody was purchased from Santa Cruz Biotechnologies, Inc. (Santa Cruz, CA). Amersham ECL rabbit IgG horseradish peroxidase (HRP)-linked secondary antibody and ECL-Plus Western blotting detection reagents were purchased from GE Healthcare (Pittsburgh, PA).
2.1 Animals
The IACUC of the Buck Institute for Age Research approved all animal studies, which were carried out at the Buck Institute's AAALAC-accredited vivarium. The generation of platelet-derived growth factor (PDGF) β-chain promoter-driven human APP minigene (hAPP) carrying the Swedish (670/671KM→ML) and Indiana (717V→F) mutations (hAPPSw,In) has been described previously (Hsia et al., 1999, Mucke et al., 2000). All male PDAPP (J20 line) Tg mice were kept in their original C57BL/J6 background and were originally provided by Professor Lennart Mucke (Gladstone Institute and UC-San Francisco). J20 Tg expresser lines were maintained by heterozygous crosses with C57BL/J6 breeders (The Jackson Laboratory, Bar Harbor, ME), and all Tg animals were heterozygous with respect to the transgene. Male non-transgenic (NTg) littermates were used as controls in all studies. Experimental groups of animals were: NTg, N=5; J20 Tg, N=10.
2.2 Sample Preparation
A one-half portion of brain was homogenized using a Wheaton glass homogenizer (∼100 passes) in Media I buffer [0.32 M sucrose, 0.10 mM Tris HCl (pH 8.0), 0.10 mM MgCl2, 0.08 mM EDTA, 10 μg/ml leupeptin, 0.5 μg/ml pepstatin, 11.5 μg/ml aprotinin; pH 8.0]. Homogenates were vortexed and sonicated for 10 s at 20% power with a Fisher 550 Sonic Dismembrator (Pittsburgh, PA). Protein concentrations were determined according to the Pierce BCA method (Rockford, IL).
2.3 Isoelectric Focusing (IEF)
Proteins from brain homogenates (200 μg) were precipitated by addition of ice-cold 100% trichloroacetic acid (TCA) to obtain a final concentration of 15% (v/v) TCA in solution and incubated on ice for 10 min. For protein carbonyl analysis, samples were first derivatized in 4× the sample volume of 10 mM DNPH in 2 N HCl for 30 min at room temperature (RT), followed by protein precipitation with TCA as described above. For 3-NT analysis, samples were acetone precipitated according to the manufacturer's instructions (Pierce, Rockford, IL), except 100% acetone was chilled to -20 °C and added at 4× the sample volume and incubated for 60 min at -80 °C.
Samples were centrifuged at 14,000 rpm (23,700 × g) for 5 min at 4 °C. Pellets were resuspended and rinsed in a Wash buffer [1:1 (v/v) ethanol:ethyl acetate] a total of four times to remove excess salts. Following the final wash, pellets were dried at RT for ∼10 min and rehydrated for 2 h at RT in 200 μl of a Rehydration buffer [8 M urea, 2 M thiourea, 50 mM DTT, 2.0% (w/v) CHAPS, 0.2% Biolytes, bromophenol blue], and then sonicated for 10 s at 20% power. Samples (200 μl) were applied to 11 cm pH 3-10 ReadyStrip™ IPG strips and after 1 h, 2 ml of mineral oil was added to prevent sample evaporation. Strips were actively rehydrated at 20 °C for 18 h at 50 V, focused at a constant temperature of 20 °C beginning at 300 V for 2 h, 500 V for 2 h, 1,000 V for 2 hr, 8,000 V for 8 hr, and finishing at 8,000 V for 10 hr rapidly. IPG strips were stored at -80 °C until the second dimension of analysis was performed.
2.4 Two-Dimensional Polyacrylamide Gel Electrophoresis (2D-PAGE)
2D-PAGE was performed to separate proteins on IEF strips based on molecular migration rate. IEF strips were thawed and equilibrated for 10 min in equilibration buffer A [50 mM Tris-HCl pH 6.8, 6 M urea, 1% (w/v) SDS, 30% (v/v) glycerol, 0.5% DTT] and then re-equilibrated for 10 min in equilibration buffer B [50 mM Tris-HCl pH 6.8, 6 M urea, 1% (w/v) SDS, 30% (v/v) glycerol, 4.5% IA]. All strips were rinsed in a 1× dilution of TGS running buffer before being placed into Criterion precast linear gradient (8-16%) Tris-HCl polyacrylamide gels. Precision Plus Protein™ All Blue Standards and samples were run at a constant voltage of 200 V for 65 min.
2.5 SYPRO Ruby® Staining
Following 2D-PAGE, gels were incubated in a Fixing solution [7% (v/v) acetic acid, 10% (v/v) methanol] for 20 min at RT. Sypro Ruby® Protein Gel Stain (∼50 ml) was added to gels and allowed to stain overnight at RT on a gently rocking platform. Gels were transferred to ∼50 ml of deionized water at RT until scanning. Gels were scanned into Adobe Photoshop 6.0 with a Molecular Dynamics STORM phosphoimager (λex/λem: 470/618 nm) and stored in deionized water at 4 °C until further use.
2.6 2D-Western Blotting
Following 2D-PAGE, in-gel proteins were transferred to a nitrocellulose membrane (0.45 μm) for immunochemical detection of protein-resident 3-NT. Gels were transferred using a Trans-Blot Semi-Dry Transfer Cell system at 20 V for 2 h (BioRAD, Hercules, CA). Post-transfer, membranes were incubated in a blocking solution of 3% bovine serum albumin (BSA) in Wash Blot [phosphate-buffered saline (PBS) solution containing 0.04% (v/v) Tween 20 and 0.10 M NaCl] at RT for 2 h. Nitrated proteins were detected by incubation with rabbit polyclonal anti-Nitrotyrosine (1:2000) primary antibody in blocking solution at RT on a rocking platform for 2-3 h. Blots were rinsed three times for 5 min each in Wash Blot, followed by a 1 h incubation with rabbit IgG alkaline phosphatase (1:3000) secondary antibody at RT. Blots were rinsed five times for 5, 15, 15, 15, and 5 min each in Wash Blot and developed colorimetrically with a solution of 5-Bromo-4-chloro-3-indolyl phosphate dipotassium combined with Nitrotetrazolium Blue chloride (BCIP/NBT) in ALP buffer [0.1 M Tris, 0.1 M NaCl, 5 mM MgCl2 · 6 H2O (pH 9.5)]. Blots were allowed to dry overnight at RT prior to scanning into Adobe Photoshop 6.0 with a Canon CanoScan 8800F scanner.
2.7 Image Analysis
2.7.1 Differential Expression
Spot intensities from SYPRO Ruby®-stained 2D-gel images of J20 Tg and NTg samples were quantified densitometrically according to the total spot density. Intensities were normalized to total gel densities and/or densities of all valid spots on the gels. Only spots with a 1.5-fold increase or decrease in normalized spot density in J20 Tg samples compared to NTg samples and a statistically significant difference based on a Student's t-test at 95% confidence (i.e., P<0.05) were considered for MS analysis.
2.7.2 Oxidative Modification
Western blot PD-Quest analysis was performed with densitometric quantitation of J20 Tg and NTg spots. Spots on blots were aligned and matched with the corresponding protein spots among differential gels. Blot immunoreactivity (nitration) was normalized to the total protein content as measured by the intensity of SYPRO Ruby®-stained gels. Only spots with statistically significant increases or decreases in protein-resident 3-NT levels, as calculated by a Student's t-test and Mann-Whitney U Statistical test at 95% confidence were selected for in-gel trypsin digestion and subsequent MS analysis.
2.8 In-Gel Trypsin Digestion
In-gel trypsin digestion of selected gel spots was performed as previously described (Thongboonkerd et al., 2002). Briefly, protein spots identified as significantly altered in J20 Tg mice relative to NTg controls were excised from 2D-gels with a clean, sterilized blade and transferred to Eppendorf microcentrifuge tubes. Gel plugs were then washed with 0.1 M ammonium bicarbonate (NH4HCO3) at RT for 15 min, followed by incubation with 100% acetonitrile at RT for 15 min. After solvent removal, gel plugs were dried in their respective tubes under a flow hood at RT. Plugs were incubated for 45 min in 20 μl of 20 mM DTT in 0.1 M NH4HCO3 at 56 °C. The DTT/NH4HCO3 solution was then removed and replaced with 20 μl of 55 mM IA in 0.1 M NH4HCO3 and incubated with gentle agitation at RT in the dark for 30 min. Excess IA solution was removed and plugs incubated for 15 min with 200 μl of 50 mM NH4HCO3 at RT. A volume of 200 μl of 100% acetonitrile was added to this solution and incubated for 15 min at RT. Solvent was removed and gel plugs were allowed to dry for 30 min at RT under a flow hood. Plugs were rehydrated with 20 ng/μl of modified trypsin (Promega, Madison, WI) in 50 mM NH4HCO3 in a shaking incubator overnight at 37 °C. Enough trypsin solution was added in order to completely submerge the gel plugs.
2.9 Mass Spectrometry (MS)
Salts and contaminants were removed from tryptic peptide solutions using C18 ZipTips (Sigma-Aldrich, St. Louis, MO), reconstituted to a volume of ∼15 μL in a 50:50 water acetonitrile solution containing 0.1% formic acid. Tryptic peptides were analyzed with an automated Nanomate electrospray ionization (ESI) [Advion Biosciences, Ithaca, NY] Orbitrap XL MS (ThermoScientific, Waltham, MA) platform. The Orbitrap MS was operated in a data-dependent mode whereby the eight most intense parent ions measured in the Fourier Transform (FT) at 60,000 resolution were selected for ion trap fragmentation with the following conditions: injection time 50 ms, 35% collision energy, MS/MS spectra were measured in the FT at 7500 resolution, and dynamic exclusion was set for 120 sec. Each sample was acquired for a total of ∼2.5 min. MS/MS spectra were searched against the International Protein Index (IPI) database using SEQUEST and the following parameters: 2 trypsin miscleavages, fixed carbamidomethyl modification, variable Methionine oxidation, parent tolerance 10 ppm, and fragment tolerance of 25 mmu or 0.01 Da. Results were filtered with the following criteria: Xcorr > 1.5, 2.0, 2.5, 3.0 for +1, +2, +3, and +4 charge states, respectively, Delta CN >0.1, and P-value (protein and peptide) < 0.01. IPI accession numbers were cross-correlated with SwissProt accession numbers for final protein identification. It should be noted that proteins identified with a single peptide were kept for further analyses if multiple spectral counts (SC, number of observed MS/MS spectra) were observed in a single analysis or if the peptide was identified in a separate analysis and workup of the same protein spot.
2.10 Western Blotting Validation
2.10.1 One-Dimensional Polyacrylamide Gel Electrophoresis (1D-PAGE)
Brain homogenates (75 μg) were suspended in sample loading buffer [0.5 M Tris, pH 6.8; 40% glycerol, 8% SDS, 20% β-mercaptoethanol, 0.01% bromophenol blue], heated at 95 °C for 5 min, and cooled on ice prior to gel loading. Samples and Precision Plus Protein™ All Blue Standards were loaded into a Criterion precast (12%) Bis-Tris polyacrylamide gel and ran at RT in a Criterion Cell™ vertical electrophoresis buffer tank filled with a 1× dilution of XT MES running buffer at 80 V for ∼10 min to ensure proper protein stacking. The voltage was then increased to 140 V for ∼110 min at RT for the duration of the electrophoretic run.
2.10.2 1D-Western Blotting
1D-gels were directly transferred to nitrocellulose membranes for Western blot analysis as described for 2D-PAGE above, using rabbit anti-Pin-1 (H-123) or anti-ATP synthase [1:2000] primary antibody, with a rabbit polyclonal anti-actin (1:8000) primary antibody used as the loading control. Pin-1 (or ATP-synthase) and actin primary antibodies were added simultaneously, as each antibody is specific for particular proteins whose MWs do not overlap. Secondary antibody incubation was completed as described above for 2D-PAGE blotting, with exceptions. Pin-1 blots were incubated with an Amersham ECL rabbit IgG HRP-linked (1:8000) secondary antibody, and developed chemifluorescently with a 40:1 dilution of ECL-Plus Western blotting detection reagents A and B, respectively. After developing for 5 min at RT while covered, blots were scanned on a phosphoimager (λex/λem: 470/618 nm) and quantified using the 1D-component of ImageQuant TL software (GE Healthcare, Pittsburgh, PA).
2.11 Immunoprecipitation
Protein samples (250 μg) were first incubated with a Lysing buffer [5 N NaCl, 1 M Tris-HCl, (pH 7.6), 0.5% NP-40, 4 μg/ml leupeptin, 4 μg/ml pepstatin, 5 μg/ml aprotinin] for 30 min at 4 °C with continuous agitation. Samples were precleared with Protein G PLUS/Protein A agarose beads (CalBiochem, La Jolla, CA) for 1 h at 4°C with continuous agitation. Following a 5 min centrifugation at 3000 rpm for 5 min, supernatants were transferred to new Eppendorf microcentrifuge tubes, and incubated overnight in rabbit anti-Pin-1 (H-123) antibody (1:2000) with continuous agitation. Samples were incubated with Protein G PLUS/Protein A agarose beads for 1 h at 4 °C and beads washed five times in RIA buffer [5 N NaCl, 1 M Tris-HCl (pH 7.6), 1% NP-40] at 3000 rpm for 5 min at 4 °C. Following the final wash, beads were prepared for 1D-PAGE separation and 1D-Western blotting, as described above, using rabbit polyclonal anti-Nitrotyrosine (1:2000) primary and Amersham ECL rabbit IgG HRP-linked (1:8000) secondary antibodies. Blots were developed chemifluorescently (λex/λem: 470/618 nm) and quantified using ImageQuant TL software.
2.12 Statistical Analysis
All data are presented as mean ± S.D. or mean ± S.E.M., as noted, and statistical analyses were performed using a two-tailed Student's t-test, wherein P<0.05 was considered significant for Western and Immunoprecipitation analyses. A Mann-Whitney U statistical analysis was performed to determine the significance of differential expression fold-change values and oxidative modification levels, wherein a P<0.05 was considered significant. Significance was also confirmed using a student's t-test. Protein and peptide identifications obtained with the SEQUEST search algorithm with a P-value less than 0.01 were considered as statistically significant. To further validate PD-Quest and SEQUEST identification of significantly different spots, the location of protein spots on the 2D-gels were manually checked to ensure they were near the expected molecular weight (MW) and isoelectric point (pI) values based on SwissProt database information.
3.0 Results
3.1 Proteomics
We performed proteomics analysis on proteins isolated from the brains of J20 Tg and NTg mice using a 2D-PAGE approach. Figure 1 shows examples of 2D-gel images from these analyses. A total of six protein spots were identified as having significant changes in intensity between J20 Tg and NTg mice. Interestingly, all six of these proteins were significantly up-regulated in the brains of J20 Tg mice. Tryptic digestion and MS analysis was carried out on excised protein spots, and Table 1 provides a list of the protein identifications corresponding to these spots. Also given in Table 1 are the number of peptide sequences and SC identified for each protein, MW, pI, fold-change levels, and P-values associated with the identification. Additional information with average spot values and standard deviations for proteins on 2D gels and blots, and peptide identifications corresponding to proteins identified in Tables I and II, are provided in Tables III and IV, respectively. As shown in Table I, half of the proteins were identified with more than a single peptide and several SC (i.e., number of MS/MS events observed that meet SEQUEST filter criteria). For proteins identified with only a single peptide, more than one SC was observed and/or the MS/MS spectra for that protein was manually inspected (see Figures 2-4). We are confident in the identification of these proteins based on the low P-values associated with the SEQUEST identifications (i.e., P≤2.00e-04). In addition, the location of protein spots align with their anticipated MW and pI values on the 2D-gels.
Figure 1.
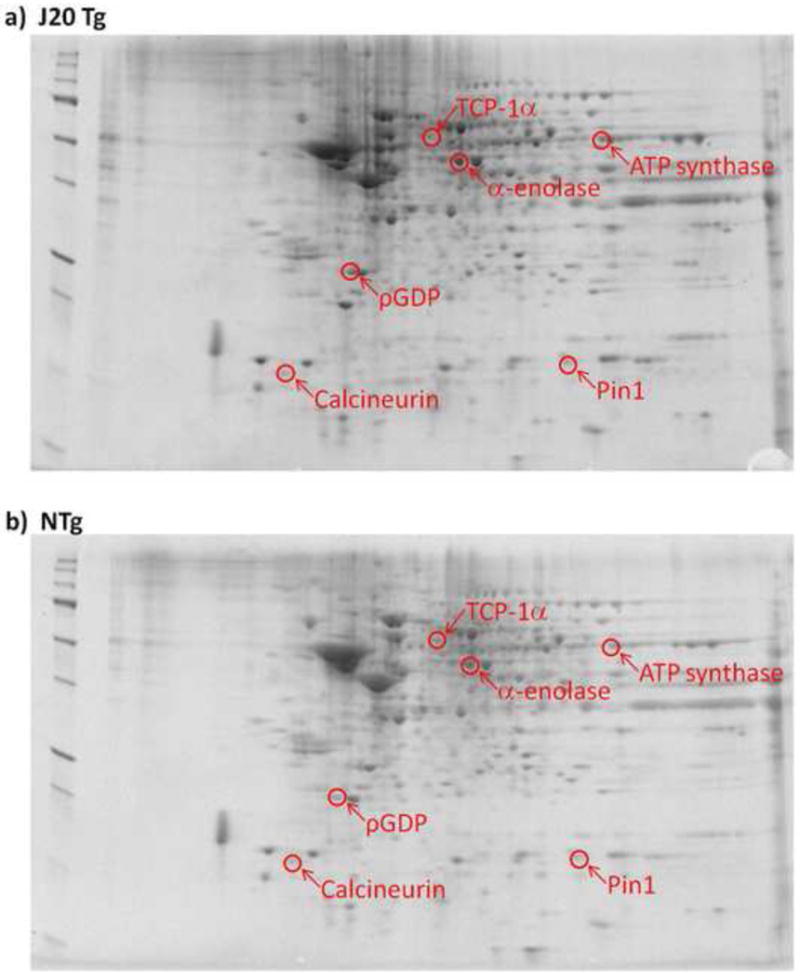
Representative 2D-gel images of proteins isolated from brain in a) J20 Tg and b) NTg mice. Significantly up-regulated spots in J20 Tg mice are labeled with protein identifications obtained from nanospray ESI-MS/MS analyses.
Table 1.
List of proteins with differential levels in J20 Tg mice relative to NTg controls.
| Protein | SwissProt Accession No. | MW (kDa) | pI | Peptides (SC)a | pb | Fold-Changec | P-valued |
|---|---|---|---|---|---|---|---|
| T-complex protein 1, subunit α A (TCP-1αA) | P11984 | 60.30 | 5.70 | 1(2) | 2.00e-07 | 47.9 ↑ | 0.005 |
| ATP synthase, subunit α, mitohondrial | Q03265 | 59.72 | 9.53 | 5(10) | 8.00e-07 | 12.2 ↑ | 0.002 |
| Peptidyl-prolyl cis-trans isomerase 1 (Pin-1) | P17742 | 18.30 | 7.97 | 1(3) | 1.00e-05 | 3.09 ↑ | 0.003 |
| ρ GDP-dissociation inhibitor 1 | Q99PT1 | 23.39 | 4.96 | 3(13) | 5.00e-09 | 2.62 ↑ | 0.027 |
| Calcineurin, subunit B, Type 1 | Q63810 | 19.29 | 4.49 | 3(10) | 3.00e-10 | 2.47 ↑ | 0.020 |
| α-Enolase | P17182 | 47.11 | 6.38 | 5(9) | 8.00e-13 | 1.88 ↑ | 0.027 |
The number of peptide sequences identified by nanospray ESI-MS/MS of tryptic peptides. The total number of MS/MS spectral counts (SC) is indicated in ( ).
The probability of an incorrect identification associated with each protein identification using the SEQUEST search alogorithm.
The fold-change in spot density from J20 Tg mice compared to NTg controls. The arrow indicates the direction of change.
The P-value associated with fold-changec calculated using the Mann-Whitney U statistical test. N=10 for J20 Tg, N=5 for NTg.
Table 2.
List of 3NT-modified proteins in J20 Tg mice relative to NTg controls.
| Protein | SwissProt Accession No. | MW (kDa) | pI | Peptides (SC)a | pb | % Control (NTg)c | P-valued |
|---|---|---|---|---|---|---|---|
| Phosphatidylethanolamine-binding protein 1 (PEBP-1) | P70296 | 20.82 | 5.07 | 2(4) | 3.00e-07 | 27.4 ↓ | 0.007 |
| Peptidyl-prolyl cis-trans isomerase 1 (Pin-1) | P17742 | 18.30 | 7.97 | 1(6) | 3.00e-04 | 19.1 ↓ | 0.003 |
The number of peptide sequences identified by nanospray ESI-MS/MS of tryptic peptides. The total number of MS/MS spectral counts (SC) is indicated in ( ).
The probability of an incorrect identification associated with each protein identification using the SEQUEST search alogorithm.
Percent oxidation in J20 Tg mice compared to NTg controls (oxidative level arbitrarily set to 100%).
The P-value associated with % Control (NTg)c calculated using the Mann-Whitney U statistical test. N=10 for J20 Tg; N=5 for NTg.
Table III. Gel and Blot Data for Proteins Listed in Tables I and II*.
| Protein Accession No. | SSP | Avg J20_NTg | STD J20_NTg | Avg J20_Tg | STD J20_Tg | Fold change Tg/NTg | MannWhitneya |
|---|---|---|---|---|---|---|---|
| Q63810 | 2 | 490.78 | 386.68 | 1213.51 | 558.47 | 2.4726 | 0.02 |
| Q99PT1 | 2201 | 620.38 | 707.30 | 1626.67 | 756.72 | 2.6221 | 0.027 |
| P17182 | 4608 | 977.78 | 641.86 | 1838.27 | 607.52 | 1.8800 | 0.027 |
| P11984 | 4701 | 9.84 | 2.21 | 472.15 | 289.28 | 47.9827 | 0.005 |
| P17742 | 7002 | 622.58 | 591.92 | 1924.84 | 383.51 | 3.0917 | 0.003 |
| Q03265 | 7710 | 105.2 | 214.50 | 1279.92 | 391.35 | 12.1665 | 0.002 |
| Raw Data for Proteins Listed inTable II. | |||||||
|---|---|---|---|---|---|---|---|
| Protein Accession No. | SSP | Avg J20_NTg | STD J20_NTg | Avg J20_Tg | STD J20_Tg | Actual % J20Tg/J20NTg | MannWhitney |
| P70296 | 2101 | 1.07 | 0.50 | 0.29 | 0.23 | 27.37 | 0.007 |
| P17742 | 6101 | 0.64 | 0.42 | 0.12 | 0.09 | 19.08 | 0.003 |
N=10 J20 Tg, N=5 NTg
P-value associated with a Mann-Whitney U Statistical Test at 95% confidence level.
Table IV. SEQUEST Results for Proteins Identified in Tables I and II.
Database… ipi.MOUSE_090305.fasta; searched against SEQUEST using BioworksBrowser rev. 3.3.1 SP1
Search Criteria: parent 10 ppm, fragment 25 mmu, trypsin 2 MC
Filter(s)… deltacn>=0.100 ; xc (± 1,2,3,4)=1.50,2.00,2.50,3.00 ; peptide probability<=5e-002 ; # distinct peptides>=2
Mods: (M* +15.99492) C=160.03068
| Reference Scan(s) |
Sequence | MH+ | z | P P |
Sf Sf |
Score XC |
Coverage DeltaCn |
Accession Sp |
RSp | Ions |
|---|---|---|---|---|---|---|---|---|---|---|
| Gene_Symbol=Ppp3r1 Isoform 1 of Calcineurin subunit B type 1 (Q63810) | 3e-010 | 2.9 | 30.2 | 0.0 | 0 | |||||
| 2 - 5 | K.DTQLQQIVDK.T | 1187.6266 | 2 | 0.001 | 0.91 | 2.416 | 0.702 | 828.6 | 1 | 12/18 |
| 12 | K.DTQLQQIVDK.T | 1187.6266 | 2 | 0.003 | 0.95 | 2.941 | 0.721 | 1043.8 | 1 | 13/18 |
| 15 - 18 | R.VIDIFDTDGNGEVDFK.E | 1783.8385 | 2 | 5e-010 | 0.98 | 4.484 | 0.781 | 2695.4 | 1 | 24/30 |
| 37 | R.VIDIFDTDGNGEVDFK.E | 1783.8385 | 2 | 3e-010 | 0.99 | 4.590 | 0.713 | 3384.3 | 1 | 26/30 |
| 97 | K.DTQLQQIVDK.T | 1187.6266 | 2 | 3e-005 | 0.95 | 3.050 | 0.697 | 1005.6 | 1 | 13/18 |
| 98 | K.EFIEGVSQFSVK.G | 1369.6998 | 2 | 9e-006 | 0.92 | 2.289 | 0.758 | 792.9 | 1 | 15/22 |
| 101 | K.DTQLQQIVDK.T | 1187.6266 | 2 | 0.002 | 0.92 | 2.869 | 0.671 | 794.8 | 1 | 12/18 |
| 102 | K.EFIEGVSQFSVK.G | 1369.6998 | 2 | 6e-005 | 0.89 | 2.323 | 0.765 | 590.1 | 1 | 13/22 |
| 105 | K.DTQLQQIVDK.T | 1187.6266 | 2 | 0.002 | 0.95 | 3.092 | 0.667 | 1081.4 | 1 | 13/18 |
| 106 | K.EFIEGVSQFSVK.G | 1369.6998 | 2 | 5e-005 | 0.95 | 2.841 | 0.762 | 994.3 | 1 | 16/22 |
| Gene_Symbol=Arhgdia Rho GDP-dissociation inhibitor 1 (Q99PT1) | 5e-009 | 2.6 | 30.2 | 0.0 | 0 | |||||
| 5 | R.VAVSADPNVPNVIVTR.L | 1650.9173 | 2 | 2e-006 | 0.87 | 2.771 | 0.660 | 505.9 | 1 | 15/30 |
| 6 | R.AEEYEFLTPM*EEAPK.G | 1799.8044 | 2 | 1e-005 | 0.76 | 2.207 | 0.668 | 415.6 | 1 | 12/28 |
| 12 | R.VAVSADPNVPNVIVTR.L | 1650.9173 | 2 | 1e-007 | 0.97 | 4.179 | 0.823 | 864.3 | 1 | 19/30 |
| 15 | R.AEEYEFLTPM*EEAPK.G | 1799.8044 | 2 | 4e-006 | 0.89 | 2.478 | 0.723 | 583.9 | 1 | 15/28 |
| 22 | R.VAVSADPNVPNVIVTR.L | 1650.9173 | 2 | 7e-008 | 0.93 | 3.157 | 0.827 | 600.2 | 1 | 16/30 |
| 24 | R.AEEYEFLTPM*EEAPK.G | 1799.8044 | 2 | 3e-005 | 0.88 | 3.036 | 0.698 | 434.4 | 1 | 13/28 |
| 25 | K.IDKTDYM*VGSYGPR.A | 1617.7577 | 3 | 2e-005 | 0.89 | 2.829 | 0.674 | 859.9 | 1 | 21/52 |
| 31 | K.IDKTDYM*VGSYGPR.A | 1617.7577 | 3 | 0.001 | 0.92 | 3.117 | 0.680 | 989.2 | 1 | 22/52 |
| 126 | R.VAVSADPNVPNVIVTR.L | 1650.9173 | 2 | 3e-008 | 0.94 | 3.188 | 0.762 | 750.2 | 1 | 18/30 |
| 133 | R.VAVSADPNVPNVIVTR.L | 1650.9173 | 2 | 2e-007 | 0.96 | 3.722 | 0.735 | 841.3 | 1 | 19/30 |
| 134 | R.AEEYEFLTPM*EEAPK.G | 1799.8044 | 2 | 2e-006 | 0.78 | 2.415 | 0.650 | 395.3 | 1 | 12/28 |
| 139 | R.VAVSADPNVPNVIVTR.L | 1650.9173 | 2 | 5e-009 | 0.96 | 3.960 | 0.851 | 855.9 | 1 | 19/30 |
| 143 | K.IDKTDYM*VGSYGPR.A | 1617.7577 | 3 | 0.004 | 0.90 | 2.955 | 0.685 | 940.9 | 1 | 20/52 |
| Gene_Symbol=LOC100044223;Eno1;EG433182;EG103324 Alpha-enolase (P17182) | 8e-013 | 4.5 | 50.2 | 0.0 | 0 | |||||
| 55 | R.YITPDQLADLYK.S | 1439.7417 | 2 | 5e-006 | 0.84 | 2.254 | 0.662 | 421.5 | 1 | 13/22 |
| 57 | R.GNPTVEVDLYTAK.G | 1406.7162 | 2 | 5e-006 | 0.90 | 2.472 | 0.817 | 380.1 | 1 | 15/24 |
| 60 | R.AAVPSGASTGIYEALELR.D | 1804.944 | 2 | 1e-010 | 0.92 | 3.440 | 0.748 | 304.0 | 1 | 19/34 |
| 61 | K.IDKLM*IEM*DGTENK.S | 1668.7819 | 3 | 0.004 | 0.75 | 2.683 | 0.717 | 305.6 | 1 | 15/52 |
| 65 | R.GNPTVEVDLYTAK.G | 1406.7162 | 2 | 6e-006 | 0.91 | 2.493 | 0.773 | 440.1 | 1 | 16/24 |
| 69 | R.AAVPSGASTGIYEALELR.D | 1804.944 | 2 | 1e-011 | 0.95 | 3.614 | 0.794 | 488.6 | 1 | 22/34 |
| 70 | K.DATNVGDEGGFAPNILENK.E | 1960.9247 | 2 | 1e-010 | 0.97 | 4.575 | 0.854 | 885.0 | 1 | 20/36 |
| 78 | R.AAVPSGASTGIYEALELR.D | 1804.944 | 2 | 8e-013 | 0.89 | 2.870 | 0.793 | 269.4 | 1 | 18/34 |
| 79 | K.IDKLM*IEM*DGTENK.S | 1668.7819 | 3 | 0.0001 | 0.90 | 3.196 | 0.681 | 752.1 | 1 | 21/52 |
| Gene_Symbol=Tcp1 T-complex protein 1 subunit alpha A (P11984) | 2e-007 | 2.5 | 40.1 | 0.0 | 0 | |||||
| 4 | K.FATEAAITILR.I | 1205.6888 | 2 | 3e-006 | 0.94 | 2.268 | 0.714 | 1032 | 1 | 15/20 |
| 85 | K.FATEAAITILR.I | 1205.6888 | 2 | 2e-007 | 0.90 | 2.111 | 0.753 | 742.3 | 1 | 13/20 |
| Gene_Symbol=Ppia Peptidyl-prolyl cis-trans isomerase (P17742) | 1e-005 | 0.9 | 10.1 | 0.0 | 0 | |||||
| 4 | K.FEDENFILK.H | 1154.5728 | 2 | 4e-005 | 0.87 | 2.124 | 0.746 | 385.1 | 1 | 11/16 |
| 18 | K.FEDENFILK.H | 1154.5728 | 2 | 0.0001 | 0.93 | 2.279 | 0.704 | 636.0 | 1 | 14/16 |
| 136 | K.FEDENFILK.H | 1154.5728 | 2 | 1e-005 | 0.91 | 2.318 | 0.787 | 441.9 | 1 | 12/16 |
| Gene_Symbol=Atp5a1 ATP synthase subunit alpha, mitochondrial (Q03265) | 8e-007 | 4.0 | 50.1 | 0.0 | 0 | |||||
| 4 | R.TGAIVDVPVGEELLGR.V | 1624.8905 | 2 | 0.02 | 0.76 | 2.601 | 0.770 | 223.9 | 1 | 11/30 |
Figure 2.
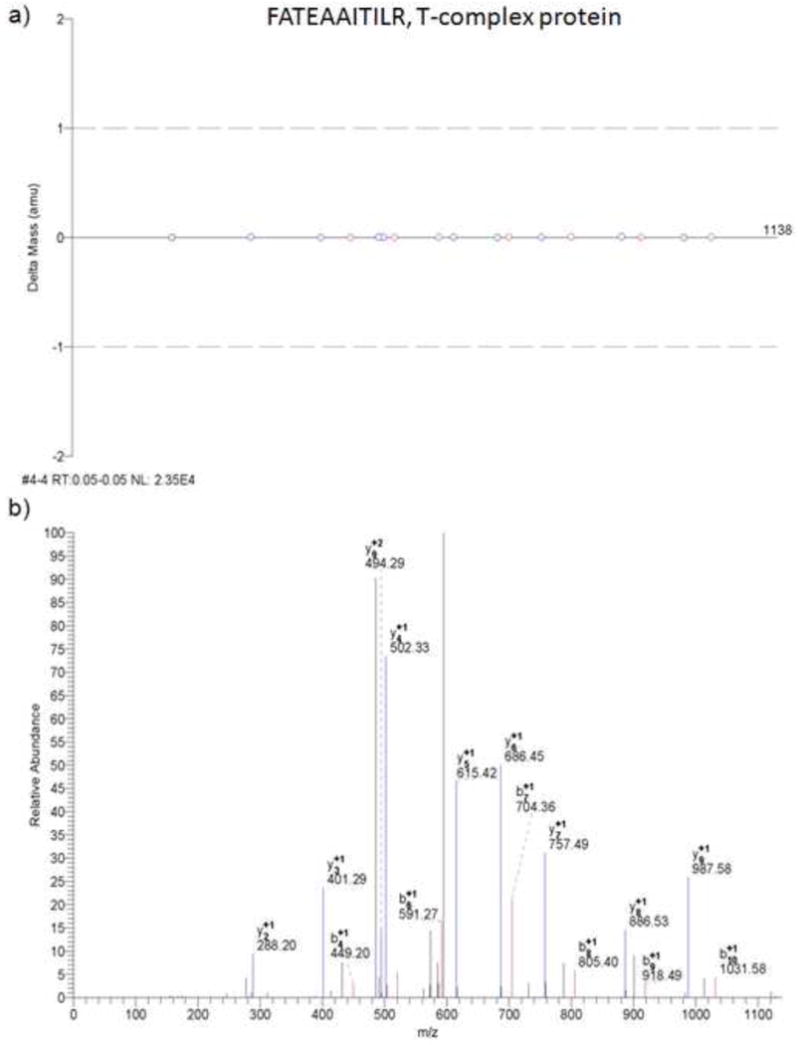
MS/MS results from Bioworks Browser. a) Mass error for b- and y-type fragment ions detected for the doubly-charged peptide FATEAAITILR. b) Example MS/MS spectra for the peptide FATEAAITILR of the T-complex protein.
Figure 4.
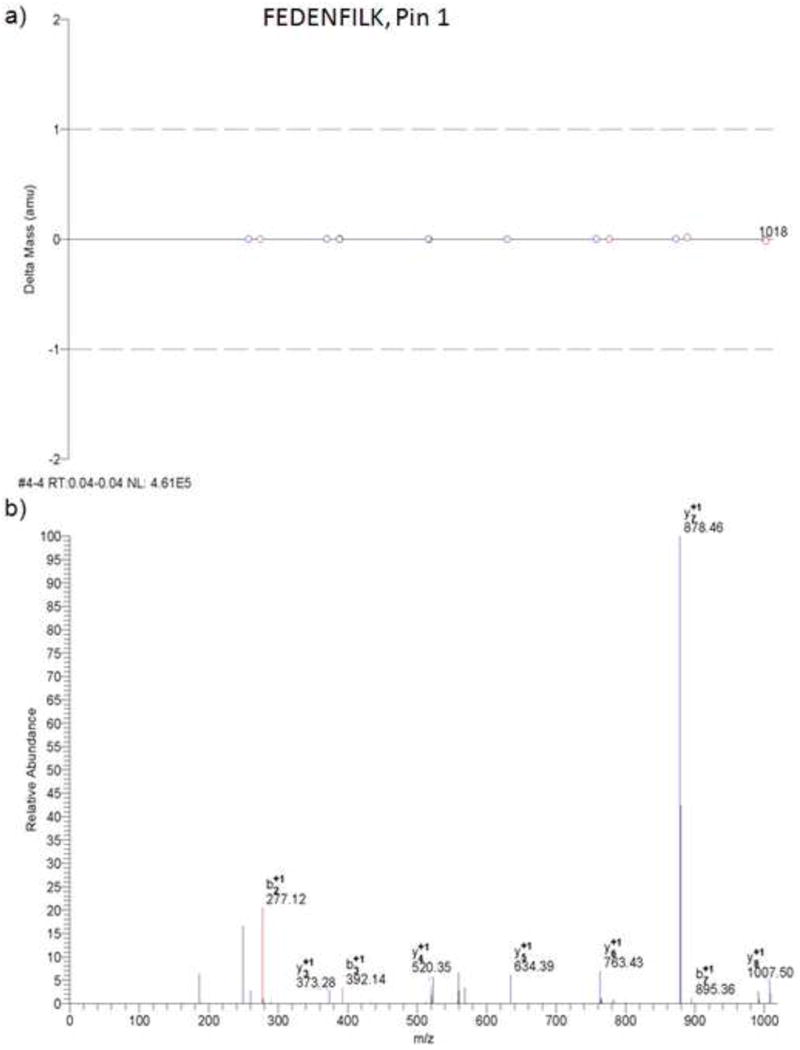
MS/MS results from Bioworks Browser. a) Mass error for b- and y-type fragment ions detected for the peptide FEDENFILK. b) Example MS/MS spectra for the peptide FEDENFILK of the Pin-1 protein. These spectra were generated from a separate experiment other than that shown in Figure 3.
Two of the brain proteins detected as significantly up-regulated in J20 Tg mice had fold-change levels greater than 10-fold compared to NTg mice. These proteins are T-complex protein 1 subunit α A (TCP-1;↑48-fold, P<0.00069), and ATP synthase subunit α, mitochondrial (↑12.2-fold, P<5.09e-06). The other significantly up-regulated proteins in J20 Tg mice are Pin-1 (↑3.09-fold, P<0.0047), ρ GDP-dissociation inhibitor 1 (↑2.62-fold, P<0.032), calcineurin subunit B type 1 (↑2.47-fold, P<0.013), and α-enolase (↑1.88-fold, P<0.038).
3.2 Western Validation Experiments
Figure 5a shows Western blot lanes corresponding to Pin-1 (18 kDa) and the loading control actin (42 kDa) levels. The figure shows that the location of Pin-1 agrees with its expected position based on MW. The histogram plot in Figure 5b shows a ∼15% significant increase (P<0.04) in the brain level of Pin-1 of J20 Tg relative to NTg mice. These results are consistent with the proteomics analyses described above, wherein Pin-1 had a ↑3.09 fold-increase in J20 Tg mouse brain relative to NTg mice. We note that quantitative differences in the % increase in Pin-1 levels from Western and 2D PAGE experiments may be attributed to Pin-1 existing in multiple forms. For example, post-translational modifications of Pin-1 (e.g., phosphorylation) would exist as different spots on the 2D gel in which case certain spots may not change in expression. The analysis carried out in these studies focused on individual protein spots which exhibited significant changes in expression. The contribution of other potential post-translationally modified forms of Pin-1 in the 1D Western band, would cause differences in the quantitative levels detected in J20 Tg and NTg mice. Herein, both the 2D PAGE and 1D Western results detect a significant increase in Pin-1 brain levels in J20 Tg mice.
Figure 5.
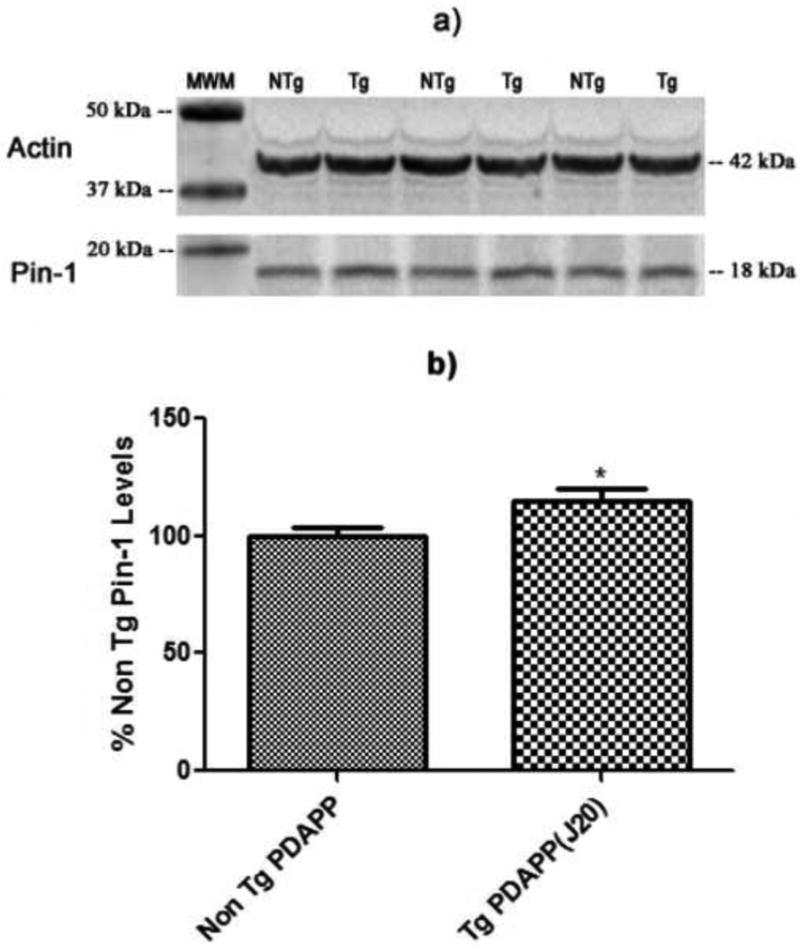
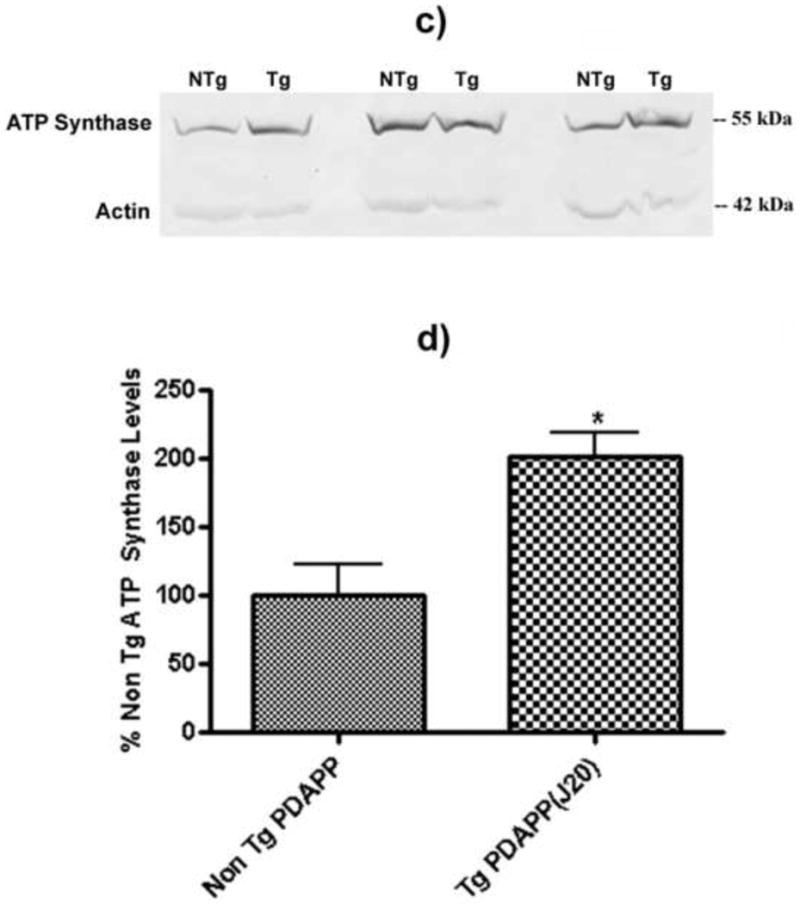
Pin-1 levels in the brains of J20 and NTg mice. a) Western blot image showing lanes corresponding to Pin-1 and actin (loading control) in J20 Tg and NTg mice. b) Histogram representation of data shown in a). N = 5 for J20 Tg, N = 5 for NTg; *P<0.04. c) Western blot image showing lanes corresponding to ATP-synthase and actin (loading control) in J20 Tg and NTg mice. d) Histogram representation of data shown in a). N = 3 for J20 Tg, N = 3 for NTg; *P<0.03. Values shown represent normalized (Pin-1/actin or ATP-synthase/actin) percentages relative to NTg and are plotted as average±SEM.
Figure 5c shows Western blot lanes corresponding to ATP-synthase (55 kDa) and the loading control actin (42 kDa) levels. The histogram plot in Figure 5d shows that the level of ATP-synthase is a factor of 2× greater in J20 Tg mouse brain relative to NTg (P<0.03). Again, the direction of change in the Western is similar to that measured in the 2D gel, however the quantitative levels differ. In both cases shown here, it appears that the Western levels are lower than those obtained in the 2D gel indicating that the dynamic range and sensitivity of the detection schemes in the two techniques (i.e., colorimetric and fluorescence detection, respectively) varies substantially, thus causing differences in the final expression levels between J20 Tg and NTg mice.
3.3 Redox Proteomics
Utilizing previously established redox proteomics approaches for the detection of protein carbonyls and 3-NT (Sultana et al., 2006b, Butterfield and Sultana, 2008), we identified two oxidatively-modified proteins, listed in Table 2, that were significantly different in J20 Tg mice relative to NTg. Phosphatidylethanolamine-binding protein 1 (PEBP-1; P<0.022), and Pin-1 (P<0.049) were detected with lower levels of 3-NT modification in J20 Tg relative to NTg mice (see Figure 6). These results and the above described proteomic results will be discussed in detail below with respect to protein functions and how the detected changes may be implicated in the etiology of AD.
Figure 6.
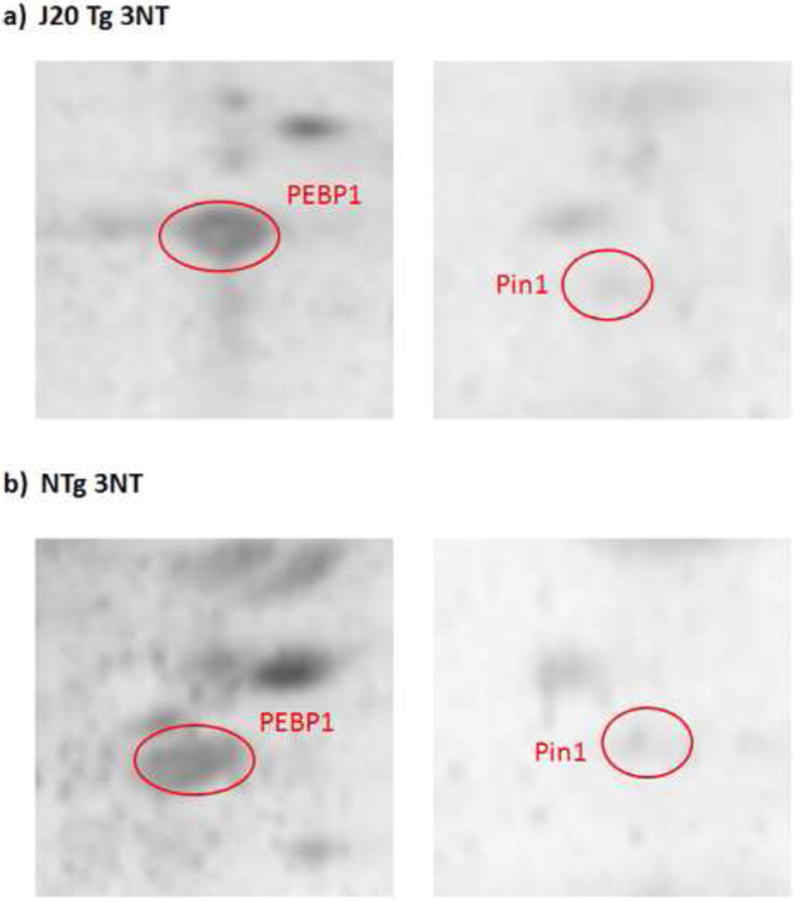
Representative 2D-Western blot images of nitrated PEBP-1 and Pin-1 proteins (images have been zoomed in to focus on these spots) in a) J20 Tg and b) NTg mice. Proteins were probed with anti-3NT antibody.
3.4 Pin-1 Immunoprecipitation
In order to confirm our findings of oxidatively modified proteins with redox proteomics analysis, we performed immunoprecipitation of Pin-1. The Western blot image shown in Figure 7a represents nitrated Pin-1 in J20 Tg mice and NTg controls. Figure 7b is a histogram representation showing a significant decrease (P<0.02) in the level of nitrated Pin-1 in J20 Tg mice compared to NTg. This result supports our finding with 2D-redox proteomics, in which Pin-1 nitration is significantly decreased in J20 Tg mice relative to NTg (i.e., 19.1%, see Table 2).
Figure 7.
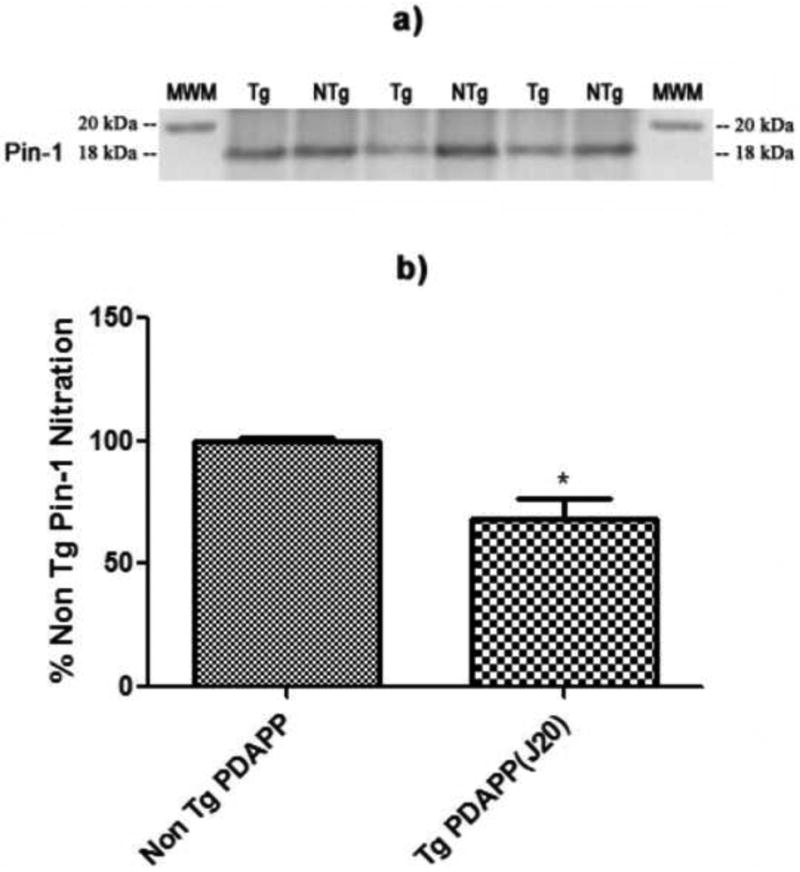
Levels of Pin-1 nitration in the brains of J20 Tg and NTg mice. a) Western blot image showing lanes corresponding to nitrated Pin-1 in J20 Tg and NTg mice. b) Histogram representation of data shown in a). N = 3 for J20 Tg, N = 3 for NTg; *P<0.02. Values are plotted as average±SEM.
4.0 Discussion
The J20 line used in these studies was 9 months of age and has pathological and behavioral characteristics that mimic those encountered in subjects with AD. For example, J20 Tg mice have age-related progressive accumulation of Aβ peptide, SP formation, and cognitive decline that is associated with disease progression (Mucke et al., 2000, Galvan et al., 2006). In the current research, we measured changes in the brain proteomes of J20 Tg mice in comparison to littermate NTg controls using 2D-PAGE and MS/MS. The goal of these studies was to carry-out a discovery-based series of experiments in order to identify specific proteins that vary in expression and oxidative modification in J20 Tg mice as a result of AD-associated pathology. We note that the results we obtained only provide information about expression levels of proteins in the J20Tg mouse model and that other studies are needed in order to assess how the function or dysfunction of the identified differentially-expressed proteins may influence pathology in this AD mouse model. We identified six proteins with significantly increased levels in J20 Tg mouse brain relative to NTg controls. These proteins are calcineurin subunit B type 1, ρ GDP-dissociation inhibitor 1, TCP-1 subunit α A, α-enolase, Pin-1, and ATP synthase subunit α mitochondrial. Recent studies from our laboratory indicate increased oxidative stress in the brains of J20 Tg mice as measured by the levels of protein carbonyls and 3NT (Butterfield et al., 2009). This oxidative stress is directly correlated with levels of Aβ deposition in the brains of these mice (Butterfield et al., 2009), and with cognitive decline as measured by Morris water-maze (Mucke et al., 2000, Butterfield et al., 2009). To gain further insight into oxidative modification of proteins in J20 Tg mice, we also performed a redox proteomics analysis of brain proteins from J20 Tg mice and NTg controls. The biochemical processes associated with altered proteins include energy dysfunction, metabolism alterations, calcium signaling, antioxidant defense, neuritic/structural abnormalities, cell-cycle/signaling, lipid abnormalities, and cholinergic dysfunction. The relevance of increased expression of these proteins in J20 Tg mice is discussed below with regards to biochemical pathways and their significance in AD.
4.1 Energy Dysfunction & Metabolism Alterations
Energy metabolism, particularly cerebral glucose uptake, is known to be altered in the brains of AD patients based on positron emission tomography (PET) scanning (Watson and Craft, 2004). We observed a significant increase in the levels of α-enolase and ATP synthase subunit α mitochondrial proteins in J20 Tg mice. α-enolase is involved in the glycolytic pathway, as an enzyme responsible for the catalysis of 2-phosphoglycerate to 2-phosphoenolpyruvate in the last step of the glycolytic cycle. Increased levels of α-enolase in the brains of J20 Tg mice suggests that there may be an increased need for ATP production in which one response of the glycolysis pathway is to complete the conversion of glucose to pyruvate, which is only possible with α-enolase catalysis. Thus, the brain in J20 Tg mice may recognize early the need for maintaining levels of ATP that eventually become lowered under conditions of AD pathology (Watson and Craft, 2004). Increased levels of α-enolase have also been reported in proteomic studies of brain from subjects with AD and mild cognitive impairment (MCI), arguably the earliest form of AD (Castegna et al., 2002b, Sultana et al., 2007). Also, increased oxidative modification of α-enolase, as indexed by protein carbonyls, 3-NT, and HNE, has been reported in the brains of subjects with AD and AD model systems (Castegna et al., 2002a, 2002b, 2003). Oxidative modification of proteins leads to a loss of enzyme function (Butterfield and Stadtman, 1997), which applied to α-enolase, may result in impaired ATP production. These results are consistent with the PET studies in AD brains that show altered glucose metabolism (Watson and Craft, 2004). As noted, the age of J20 Tg mice used in these studies was 9 months, an age at which significant Aβ deposition is present in the brains (Mucke et al., 2000, Galvan et al., 2006, Shankar et al., 2009). Thus, increased levels of α-enolase may represent an early attempt to prevent lowered levels of ATP production that will inevitably occur with increased Aβ deposition and SP formation, signatures of advanced stages of AD. In addition, α-enolase has been reported to have many functions and roles in AD as well as other neurodegenerative disorders (Butterfield and Lange, 2009).
ATP synthase subunit α is a part of the ATP synthase mitochondrial enzyme involved in the production of ATP. Mitochondrial dysfunction has been linked to AD pathogenesis (Blass et al., 2002, Bubber et al., 2005). In advanced stages of AD, ATP synthase activity is reportedly decreased in AD brain (Schagger and Ohm, 1995) and the protein also is oxidatively modified (Sultana et al., 2006c). J20 Tg mice used in these studies show increased levels of ATP synthase subunit α, which could be related to early cellular stress responses by the brain to maintain sufficient levels of energy production.
4.2 Calcium Signaling
Dysregulation of Ca2+ homeostasis has been linked to brain aging (Foster et al., 2001) and to AD (Mattson and Chan, 2001). Calcineurin is a Ca2+/calmodulin-dependent serine/threonine protein phosphatase (also known as PP2B) that is highly expressed in brain (Goto et al., 1986) and is important in Ca2+ signaling and cellular responses (Klee et al., 1998). Calcineurin is a heterodimeric enzyme that consists of two subunits: a 61 kDa calmodulin binding subunit, calcineurin A, and a 19 kDa Ca2+ binding subunit, calcineurin B (Aitken et al., 1984, Merat and Cheung, 1987, Hemenway and Heitman, 1999). Reports of calcineurin levels and activity are somewhat conflicting in AD brain and various AD models. Calcineurin reportedly has reduced basal activity in AD frontal cortex (Lian et al., 2001), although studies of cortical cells treated with Aβ(25-35) and Aβ(1-40) suggest increased calcineurin activity with levels being unaffected (Agostinho et al., 2008). Hata et al. observed calcineurin subunit B to be the most up-regulated gene in the hippocampus of AD brains (Hata et al., 2001). However, in primary neuronal cultures treated with Aβ peptide, expression of calcineurin was reduced (Celsi et al., 2007). We observed an increased level of calcineurin subunit B type 1 in the brains of J20 Tg mice. Consistent with this observation, Norris et al. report significant overexpression of calcineurin in hypertrophic astrocytes that surround SP in the brains of APP/PS-1 double transgenic mice (Norris et al., 2005) and in the hippocampus of patients with MCI and AD (Abdul et al., 2009). Overall, increased levels of calcineurin in this AD model are consistent with alterations in glutamate release, neuroinflammation, synaptic plasticity, cell survival, and cognitively related changes in behavior.
4.3 Neuritic Abnormalities & Structural Integrity
Cellular structural integrity is known to be altered in AD (Butterfield et al., 2006b). TCP-1 subunit α A is a part of the TCP-1 family, which has significant structure and sequence homology to heat shock protein 60 (Gupta, 1995). While TCP-1 is involved in molecular chaperoning, it is also necessary for maintaining normal actin and tubulin function (Chen et al., 1994, Ursic et al., 1994) and is an initiator in microtubule growth (Brown et al., 1996). Because actin and tubulin are major substrates of TCP-1, we intuit that an increase in TCP-1 would require an increase in actin and actin-related proteins such as F-acting-capping protein. TCP-1 ε unit was reported as having decreased levels in the parietal cortex (Yoo et al., 2001) and brains of subjects with AD (Schuller et al., 2001). Our results of a substantial increase in TCP-1 subunit α A levels in J20 Tg mice further supports the notion that in the presence of Aβ deposits and oxidative stress at 9 months of age, cytoskeletal structural integrity is not yet compromised in these transgenic mice.
4.4 Lipid Abnormalities & Cholinergic Dysfunction
PEBP-1 is a precursor of the hippocampal cholinergic neurostimulating peptide (HCNP), a signaling transduction peptide that helps to regulate choline acetyltransferase (ChAT). Alterations to ChAT lead to reduced levels of acetyl choline, a neurotransmitter that is necessary for regulating normal neurotransmission (Ojika, 1998). We observed decreased nitration of PEBP-1 in brains of J20 Tg mice relative to NTg controls. This result is somewhat striking as PEBP-1, also known as neuropolypeptide h3 and Raf-kinase inhibitor protein, has been reported as having increased nitration (Castegna et al., 2003) and decreased mRNA expression (Maki et al., 2002) in AD brain. In addition, PEBP-1 had 20% greater expression in the hippocampus of subjects with AD (Chen et al., 2006) and is believed to be a potential calpain substrate leading to proteasome dysfunction. Moreover, PEBP-1, is known as phosphatidylethanolamine binding protein-1. Hence, results of increased nitration in AD brain could contribute to the known disruption to lipid asymmetry in AD, which leads to apoptosis and poor neurotransmission due to reduced levels of acetyl choline. On the other hand, our observation of lower levels of nitrated PEBP-1 in J20 Tg mice could be an indication that acetyl choline levels and lipid asymmetry are not yet disturbed at this stage of AD pathology in these animals. Thus, under this scenario, altered acetyl choline levels and lipid abnormalities could be a result of more advanced plaque formation that occurs in later stages of AD. Finally, our current results suggest that J20 Tg mice have not yet experienced proteasome dysfunction. Taken together, it appears that specific biochemical changes that are observed in AD may occur later in disease progression in J20 Tg mice despite the presence of significant Aβ(1-42) deposition and oxidative stress.
4.5 Cell Signaling, Cell-Cycle, Tau Phosphorylation, & Aβ Production
Pin-1 belongs to the family of PPIases and is unique in that it acts as a regulatory protein by binding to and isomerizing a proline on the C-terminal side of a pSer/pThr moiety to the cis-trans conformation, thereby regulating the activity of the target protein (Butterfield et al., 2006a). The functions of Pin-1 are diverse and include regulation of pathways such as the cell-cycle, transcription, acting as a cytokine, apoptosis, and DNA damage response (Butterfield et al., 2006a). Pin-1 has been heavily implicated in AD, through its regulation of phosphorylation/dephosphorylation of tau and production of Aβ (Butterfield et al., 2006a). Pin-1 is oxidatively modified and has lowered expression and activity in the hippocampus of AD and MCI brain (Sultana et al., 2006a). In the current study, Pin-1 expression was increased and nitration was decreased in the brains of J20 Tg mice compared to NTg controls. It should be noted that in this study, one-half of the brain was homogenized and thus regional comparison to AD was not possible. As noted above, AD-specific roles of Pin-1 include regulation of Aβ production through APP binding (Pastorino et al., 2006) and phosphorylation/dephosphorylation of tau (Lee and Tsai, 2003, Lu et al., 2003). Increased Pin-1 expression levels could possibly indicate early response strategies of the brain to maintain low levels of Aβ peptides and subsequent SP formation. Furthermore, the current increase in Pin-1 levels may keep postmitotic neurons from entering the cell-cycle, maintain normal transcriptional regulation of key proteins, and allow the brain to respond to DNA damage that may be occurring in J20 Tg mouse brain as a result of Aβ deposition and oxidative stress.
Additionally, we observed the cell signaling protein, ρ GDP-dissociation inhibitor 1, to be increased in expression in J20 Tg mice. This inhibitor belongs to the ρ GTPase family that regulates membrane trafficking and recycling of GTPases (Wu et al., 1996) and APP processing (Maillet et al., 2003). In the brain, ρ GDP proteins are involved in neurotransmitter release (Maillet et al., 2003). Aβ(1-42) exposure to the SN56 cell line leads to reduced phosphorylation levels of ρ GDP-dissociation inhibitor (Joerchel et al., 2008). In studies of concanavalin A-associated proteins, ρ GDP-dissociation inhibitor is reported as decreased in the hippocampus of subjects with AD (Owen et al., 2009). Increased levels of ρ GDP-dissociation inhibitor 1 and Pin-1 in J20 Tg mice suggests that normal neurotransmission may still be occurring at this disease stage in this AD model, despite significant levels of Aβ deposition.
4.6 Conclusions
The changes observed in this differential expression and redox proteomics-based investigation of the J20 Tg mouse brain proteome supports the notion that biochemical pathways are altered in AD. Alterations in brain proteins in this AD mouse model, consistent with previous reports of AD, involve altered proteins associated with calcium signaling and neuritic abnormalities. Interestingly, some of the changes associated with advanced stages of AD, in which significant Aβ deposition, SP, and NFT are present, do not appear to occur at 9 months of age in J20 Tg mice. In fact, it appears that the central nervous system of J20 Tg mice may be responding to increased Aβ deposition to try to maintain normal levels of ATP, cytoskeletal structural integrity, and cellular signaling. In the presence of oxidative stress, there is also an antioxidant defense response against ROS and RNS. While these analyses focused on entire brain homogenate, proteomic studies of specific brain regions may provide more information regarding localization of altered proteins in relation to their proximity of Aβ deposits (e.g., hippocampus, cortex). The results presented also suggest that further insight can be gained through biochemical analyses that explore specific activity and functional consequences of altered proteins identified in these studies. In addition, it appears that some biological responses to Aβ deposition and Aβ(1-42)-induced oxidative stress may appear at later ages in J20 Tg mice, where pathologically the animals mimic advanced stages of AD. We are continuing to investigate the downstream effects of Aβ(1-42)-induced oxidative stress and deposition in J20 Tg mice, and future studies will involve gaining more insight through functional investigations of altered proteins and examination of changes localized to specific brain regions or age-related changes in protein expression. More interestingly, are studies that probe Aβ(1-42)-associated mechanisms of methionine 35-mediated oxidation via genetic modification of the PDAPP J20 Tg in vivo model of AD.
Figure 3.
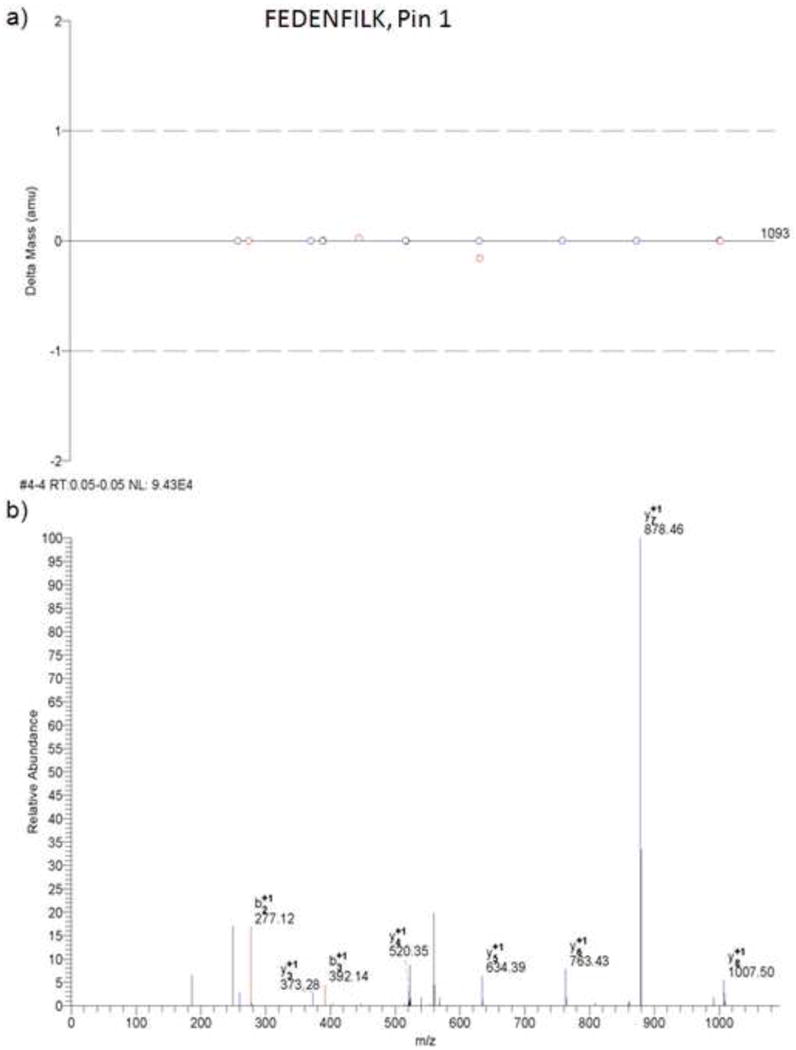
MS/MS results from Bioworks Browser. a) Mass error for b- and y-type fragment ions detected for the peptide FEDENFILK. b) Example MS/MS spectra for the peptide FEDENFILK of the Pin-1 protein.
Acknowledgments
This research was supported in part by NIH grants to D.A.B. [AG-05119] and to D.E.B. [NS-45093, NS-33376, and AG-12282], as well as support from the Keck Foundation (to D.E.B.), the Joseph Drown Foundation (to D.E.B), the Stephen D. Bechtel Foundation (to V.G.), the Alzheimer's Association (to D.E.B. and V.G.), and the UNCF/Merck Science Initiative Postdoctoral Fellowship (to R.A.S.R.). We thank Prof. Lennart Mucke for the J20 line of PDAPP mice and for the PDGF β-chain promoter-driven APPSw,In minigene.
Footnotes
This manuscript is dedicated to the life and career of William R. Markesbery, MD, a pioneer and extraordinary presence in the field of AD research, and a deep personal friend. Dr. Markesbery died January 30, 2010, leaving an exemplary record of achievement and a life well-lived.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abdul HM, Sama MA, Furman JL, Mathis DM, Beckett TL, Weidner AM, Patel ES, Baig I, Murphy MP, LeVine H, 3rd, Kraner SD, Norris CM. Cognitive decline in Alzheimer's disease is associated with selective changes in calcineurin/NFAT signaling. J Neurosci. 2009;29:12957–12969. doi: 10.1523/JNEUROSCI.1064-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abdul HM, Sultana R, St Clair DK, Markesbery WR, Butterfield DA. Oxidative damage in brain from human mutant APP/PS-1 double knock-in mice as a function of age. Free Radic Biol Med. 2008;45:1420–1425. doi: 10.1016/j.freeradbiomed.2008.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agostinho P, Lopes JP, Velez Z, Oliveira CR. Overactivation of calcineurin induced by amyloid-β and prion proteins. Neurochem Int. 2008;52:1226–1233. doi: 10.1016/j.neuint.2008.01.005. [DOI] [PubMed] [Google Scholar]
- Aitken A, Klee CB, Cohen P. The structure of the B subunit of calcineurin. Eur J Biochem. 1984;139:663–671. doi: 10.1111/j.1432-1033.1984.tb08055.x. [DOI] [PubMed] [Google Scholar]
- Ansari MA, Joshi G, Huang Q, Opii WO, Abdul HM, Sultana R, Butterfield DA. In vivo administration of D609 leads to protection of subsequently isolated gerbil brain mitochondria subjected to in vitro oxidative stress induced by amyloid β-peptide and other oxidative stressors: Relevance to Alzheimer's disease and other oxidative stress-related neurodegenerative disorders. Free Radic Biol Med. 2006;41:1694–1703. doi: 10.1016/j.freeradbiomed.2006.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blacker D, Wilcox MA, Laird NM, Rodes L, Horvath SM, Go RC, Perry R, Watson B, Jr, Bassett SS, McInnis MG, Albert MS, Hyman BT, Tanzi RE. α2-macroglobulin is genetically associated with Alzheimer disease. Nat Genet. 1998;19:357–360. doi: 10.1038/1243. [DOI] [PubMed] [Google Scholar]
- Blass JP, Gibson GE, Hoyer S. The role of the metabolic lesion in Alzheimer's disease. J Alzheimers Dis. 2002;4:225–232. doi: 10.3233/jad-2002-4312. [DOI] [PubMed] [Google Scholar]
- Boyd-Kimball D, Castegna A, Sultana R, Poon HF, Petroze R, Lynn BC, Klein JB, Butterfield DA. Proteomic identification of proteins oxidized by Aβ(1-42) in synaptosomes: Implications for Alzheimer's disease. Brain Res. 2005a;1044:206–215. doi: 10.1016/j.brainres.2005.02.086. [DOI] [PubMed] [Google Scholar]
- Boyd-Kimball D, Mohmmad Abdul H, Reed T, Sultana R, Butterfield DA. Role of phenylalanine 20 in Alzheimer's amyloid β-peptide (1-42)-induced oxidative stress and neurotoxicity. Chem Res Toxicol. 2004;17:1743–1749. doi: 10.1021/tx049796w. [DOI] [PubMed] [Google Scholar]
- Boyd-Kimball D, Sultana R, Mohmmad-Abdul H, Butterfield DA. Neurotoxicity and oxidative stress in D1M-substituted Alzheimer's Aβ(1-42): Relevance to N-terminal methionine chemistry in small model peptides. Peptides. 2005b;26:665–673. doi: 10.1016/j.peptides.2004.11.001. [DOI] [PubMed] [Google Scholar]
- Boyd-Kimball D, Sultana R, Poon HF, Lynn BC, Casamenti F, Pepeu G, Klein JB, Butterfield DA. Proteomic identification of proteins specifically oxidized by intracerebral injection of amyloid β-peptide (1-42) into rat brain: Implications for Alzheimer's disease. Neuroscience. 2005c;132:313–324. doi: 10.1016/j.neuroscience.2004.12.022. [DOI] [PubMed] [Google Scholar]
- Brown CR, Doxsey SJ, Hong-Brown LQ, Martin RL, Welch WJ. Molecular chaperones and the centrosome. A role for TCP-1 in microtubule nucleation. J Biol Chem. 1996;271:824–832. doi: 10.1074/jbc.271.2.824. [DOI] [PubMed] [Google Scholar]
- Bubber P, Haroutunian V, Fisch G, Blass JP, Gibson GE. Mitochondrial abnormalities in Alzheimer brain: Mechanistic implications. Ann Neurol. 2005;57:695–703. doi: 10.1002/ana.20474. [DOI] [PubMed] [Google Scholar]
- Butterfield DA, Abdul HM, Opii W, Newman SF, Joshi G, Ansari MA, Sultana R. Pin-1 in Alzheimer's disease. J Neurochem. 2006a;98:1697–1706. doi: 10.1111/j.1471-4159.2006.03995.x. [DOI] [PubMed] [Google Scholar]
- Butterfield DA, Boyd-Kimball D. Amyloid β-peptide(1-42) contributes to the oxidative stress and neurodegeneration found in Alzheimer disease brain. Brain Pathol. 2004;14:426–432. doi: 10.1111/j.1750-3639.2004.tb00087.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butterfield DA, Galvan V, Lange MB, Tang H, Sowell RA, Spilman P, Fombonne J, Gorostiza O, Zhang J, Sultana R, Bredesen DE. In vivo oxidative stress in brain of Alzheimer disease transgenic mice: Requirement for methionine 35 in amyloid β-peptide of APP. Free Radic Biol Med. 2009 doi: 10.1016/j.freeradbiomed.2009.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butterfield DA, Lange ML. Multifunctional roles of enolase in Alzheimer's disease brain: Beyond altered glucose metabolism. J Neurochem. 2009;111:915–933. doi: 10.1111/j.1471-4159.2009.06397.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butterfield DA, Lauderback CM. Lipid peroxidation and protein oxidation in Alzheimer's disease brain: Potential causes and consequences involving amyloid β-peptide-associated free radical oxidative stress. Free Radic Biol Med. 2002;32:1050–1060. doi: 10.1016/s0891-5849(02)00794-3. [DOI] [PubMed] [Google Scholar]
- Butterfield DA, Perluigi M, Sultana R. Oxidative stress in Alzheimer's disease brain: New insights from redox proteomics. Eur J Pharmacol. 2006b;545:39–50. doi: 10.1016/j.ejphar.2006.06.026. [DOI] [PubMed] [Google Scholar]
- Butterfield DA, Stadtman ER. Protein oxidation processes in aging brain. Adv Cell Aging Gerontol. 1997;2:161–191. [Google Scholar]
- Butterfield DA, Sultana R. Identification of 3-nitrotyrosine-modified brain proteins by redox proteomics. Methods Enzymol. 2008;440:295–308. doi: 10.1016/S0076-6879(07)00819-1. [DOI] [PubMed] [Google Scholar]
- Castegna A, Aksenov M, Aksenova M, Thongboonkerd V, Klein JB, Pierce WM, Booze R, Markesbery WR, Butterfield DA. Proteomic identification of oxidatively modified proteins in Alzheimer's disease brain. Part I: Creatine kinase BB, glutamine synthase, and ubiquitin carboxy-terminal hydrolase L-1. Free Radic Biol Med. 2002a;33:562–571. doi: 10.1016/s0891-5849(02)00914-0. [DOI] [PubMed] [Google Scholar]
- Castegna A, Aksenov M, Thongboonkerd V, Klein JB, Pierce WM, Booze R, Markesbery WR, Butterfield DA. Proteomic identification of oxidatively modified proteins in Alzheimer's disease brain. Part II: Dihydropyrimidinase-related protein 2, α-enolase, and heat shock cognate 71. J Neurochem. 2002b;82:1524–1532. doi: 10.1046/j.1471-4159.2002.01103.x. [DOI] [PubMed] [Google Scholar]
- Castegna A, Thongboonkerd V, Klein JB, Lynn B, Markesbery WR, Butterfield DA. Proteomic identification of nitrated proteins in Alzheimer's disease brain. J Neurochem. 2003;85:1394–1401. doi: 10.1046/j.1471-4159.2003.01786.x. [DOI] [PubMed] [Google Scholar]
- Celsi F, Svedberg M, Unger C, Cotman CW, Carri MT, Ottersen OP, Nordberg A, Torp R. β-Amyloid causes downregulation of calcineurin in neurons through induction of oxidative stress. Neurobiol Dis. 2007;26:342–352. doi: 10.1016/j.nbd.2006.12.022. [DOI] [PubMed] [Google Scholar]
- Chen Q, Wang S, Thompson SN, Hall ED, Guttmann RP. Identification and characterization of PEBP as a calpain substrate. J Neurochem. 2006;99:1133–1141. doi: 10.1111/j.1471-4159.2006.04160.x. [DOI] [PubMed] [Google Scholar]
- Chen X, Sullivan DS, Huffaker TC. Two yeast genes with similarity to TCP-1 are required for microtubule and actin function in vivo. Proc Natl Acad Sci U S A. 1994;91:9111–9115. doi: 10.1073/pnas.91.19.9111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruts M, van Duijn CM, Backhovens H, Van den Broeck M, Wehnert A, Serneels S, Sherrington R, Hutton M, Hardy J, St George-Hyslop PH, Hofman A, Van Broeckhoven C. Estimation of the genetic contribution of presenilin-1 and -2 mutations in a population-based study of presenile Alzheimer disease. Hum Mol Genet. 1998;7:43–51. doi: 10.1093/hmg/7.1.43. [DOI] [PubMed] [Google Scholar]
- Dahiyat M, Cumming A, Harrington C, Wischik C, Xuereb J, Corrigan F, Breen G, Shaw D, St Clair D. Association between Alzheimer's disease and the NOS3 gene. Ann Neurol. 1999;46:664–667. doi: 10.1002/1531-8249(199910)46:4<664::aid-ana18>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- Demuro A, Mina E, Kayed R, Milton SC, Parker I, Glabe CG. Calcium dysregulation and membrane disruption as a ubiquitous neurotoxic mechanism of soluble amyloid oligomers. J Biol Chem. 2005;280:17294–17300. doi: 10.1074/jbc.M500997200. [DOI] [PubMed] [Google Scholar]
- Drake J, Link CD, Butterfield DA. Oxidative stress precedes fibrillar deposition of Alzheimer's disease amyloid β-peptide (1-42) in a transgenic Caenorhabditis elegans model. Neurobiol Aging. 2003;24:415–420. doi: 10.1016/s0197-4580(02)00225-7. [DOI] [PubMed] [Google Scholar]
- Foster TC, Sharrow KM, Masse JR, Norris CM, Kumar A. Calcineurin links Ca2+ dysregulation with brain aging. J Neurosci. 2001;21:4066–4073. doi: 10.1523/JNEUROSCI.21-11-04066.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galvan V, Gorostiza OF, Banwait S, Ataie M, Logvinova AV, Sitaraman S, Carlson E, Sagi SA, Chevallier N, Jin K, Greenberg DA, Bredesen DE. Reversal of Alzheimer's-like pathology and behavior in human APP transgenic mice by mutation of Asp664. Proc Natl Acad Sci U S A. 2006;103:7130–7135. doi: 10.1073/pnas.0509695103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goate A, Chartier-Harlin MC, Mullan M, Brown J, Crawford F, Fidani L, Giuffra L, Haynes A, Irving N, James L, et al. Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer's disease. Nature. 1991;349:704–706. doi: 10.1038/349704a0. [DOI] [PubMed] [Google Scholar]
- Good PF, Werner P, Hsu A, Olanow CW, Perl DP. Evidence of neuronal oxidative damage in Alzheimer's disease. Am J Pathol. 1996;149:21–28. [PMC free article] [PubMed] [Google Scholar]
- Goto S, Matsukado Y, Mihara Y, Inoue N, Miyamoto E. Calcineurin in human brain and its relation to extrapyramidal system. Immunohistochemical study on postmortem human brains. Acta Neuropathol. 1986;72:150–156. doi: 10.1007/BF00685977. [DOI] [PubMed] [Google Scholar]
- Gupta RS. Evolution of the chaperonin families (Hsp60, Hsp10 and Tcp-1) of proteins and the origin of eukaryotic cells. Mol Microbiol. 1995;15:1–11. doi: 10.1111/j.1365-2958.1995.tb02216.x. [DOI] [PubMed] [Google Scholar]
- Hata R, Masumura M, Akatsu H, Li F, Fujita H, Nagai Y, Yamamoto T, Okada H, Kosaka K, Sakanaka M, Sawada T. Up-regulation of calcineurin Aβ mRNA in the Alzheimer's disease brain: Assessment by cDNA microarray. Biochem Biophys Res Commun. 2001;284:310–316. doi: 10.1006/bbrc.2001.4968. [DOI] [PubMed] [Google Scholar]
- Hemenway CS, Heitman J. Calcineurin. Structure, function, and inhibition. Cell Biochem Biophys. 1999;30:115–151. doi: 10.1007/BF02737887. [DOI] [PubMed] [Google Scholar]
- Hsia AY, Masliah E, McConlogue L, Yu GQ, Tatsuno G, Hu K, Kholodenko D, Malenka RC, Nicoll RA, Mucke L. Plaque-independent disruption of neural circuits in Alzheimer's disease mouse models. Proc Natl Acad Sci U S A. 1999;96:3228–3233. doi: 10.1073/pnas.96.6.3228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joerchel S, Raap M, Bigl M, Eschrich K, Schliebs R. Oligomeric β-amyloid(1-42) induces the expression of Alzheimer disease-relevant proteins in cholinergic SN56.B5.G4 cells as revealed by proteomic analysis. Int J Dev Neurosci. 2008;26:301–308. doi: 10.1016/j.ijdevneu.2008.01.004. [DOI] [PubMed] [Google Scholar]
- Klee CB, Ren H, Wang X. Regulation of the calmodulin-stimulated protein phosphatase, calcineurin. J Biol Chem. 1998;273:13367–13370. doi: 10.1074/jbc.273.22.13367. [DOI] [PubMed] [Google Scholar]
- Lee MS, Tsai LH. Cdk5: One of the links between senile plaques and neurofibrillary tangles? J Alzheimers Dis. 2003;5:127–137. doi: 10.3233/jad-2003-5207. [DOI] [PubMed] [Google Scholar]
- Levy-Lahad E, Lahad A, Wijsman EM, Bird TD, Schellenberg GD. Apolipoprotein E genotypes and age of onset in early-onset familial Alzheimer's disease. Ann Neurol. 1995;38:678–680. doi: 10.1002/ana.410380420. [DOI] [PubMed] [Google Scholar]
- Lian Q, Ladner CJ, Magnuson D, Lee JM. Selective changes of calcineurin (protein phosphatase 2B) activity in Alzheimer's disease cerebral cortex. Exp Neurol. 2001;167:158–165. doi: 10.1006/exnr.2000.7534. [DOI] [PubMed] [Google Scholar]
- Lu KP, Liou YC, Vincent I. Proline-directed phosphorylation and isomerization in mitotic regulation and in Alzheimer's Disease. Bioessays. 2003;25:174–181. doi: 10.1002/bies.10223. [DOI] [PubMed] [Google Scholar]
- Maillet M, Robert SJ, Cacquevel M, Gastineau M, Vivien D, Bertoglio J, Zugaza JL, Fischmeister R, Lezoualc'h F. Crosstalk between Rap1 and Rac regulates secretion of sAPPα. Nat Cell Biol. 2003;5:633–639. doi: 10.1038/ncb1007. [DOI] [PubMed] [Google Scholar]
- Maki M, Matsukawa N, Yuasa H, Otsuka Y, Yamamoto T, Akatsu H, Okamoto T, Ueda R, Ojika K. Decreased expression of hippocampal cholinergic neurostimulating peptide precursor protein mRNA in the hippocampus in Alzheimer disease. J Neuropathol Exp Neurol. 2002;61:176–185. doi: 10.1093/jnen/61.2.176. [DOI] [PubMed] [Google Scholar]
- Markesbery WR. Oxidative stress hypothesis in Alzheimer's disease. Free Radic Biol Med. 1997;23:134–147. doi: 10.1016/s0891-5849(96)00629-6. [DOI] [PubMed] [Google Scholar]
- Mattson MP, Chan SL. Dysregulation of cellular calcium homeostasis in Alzheimer's disease: Bad genes and bad habits. J Mol Neurosci. 2001;17:205–224. doi: 10.1385/JMN:17:2:205. [DOI] [PubMed] [Google Scholar]
- Merat DL, Cheung WY. Calmodulin-dependent protein phosphatase: Isolation of subunits and reconstitution to holoenzyme. Methods Enzymol. 1987;139:79–87. doi: 10.1016/0076-6879(87)39076-7. [DOI] [PubMed] [Google Scholar]
- Mohmmad Abdul H, Sultana R, Keller JN, St Clair DK, Markesbery WR, Butterfield DA. Mutations in amyloid precursor protein and presenilin-1 genes increase the basal oxidative stress in murine neuronal cells and lead to increased sensitivity to oxidative stress mediated by amyloid β-peptide (1-42), H2O2 and kainic acid: Implications for Alzheimer's disease. J Neurochem. 2006;96:1322–1335. doi: 10.1111/j.1471-4159.2005.03647.x. [DOI] [PubMed] [Google Scholar]
- Mohmmad Abdul H, Wenk GL, Gramling M, Hauss-Wegrzyniak B, Butterfield DA. APP and PS-1 mutations induce brain oxidative stress independent of dietary cholesterol: Implications for Alzheimer's disease. Neurosci Lett. 2004;368:148–150. doi: 10.1016/j.neulet.2004.06.077. [DOI] [PubMed] [Google Scholar]
- Mucke L, Masliah E, Yu GQ, Mallory M, Rockenstein EM, Tatsuno G, Hu K, Kholodenko D, Johnson-Wood K, McConlogue L. High-level neuronal expression of Aβ 1-42 in wild-type human amyloid protein precursor transgenic mice: Synaptotoxicity without plaque formation. J Neurosci. 2000;20:4050–4058. doi: 10.1523/JNEUROSCI.20-11-04050.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norris CM, Kadish I, Blalock EM, Chen KC, Thibault V, Porter NM, Landfield PW, Kraner SD. Calcineurin triggers reactive/inflammatory processes in astrocytes and is up-regulated in aging and Alzheimer's models. J Neurosci. 2005;25:4649–4658. doi: 10.1523/JNEUROSCI.0365-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ojika K. Hippocampal cholinergic neurostimulating peptide. Seikagaku. 1998;70:1175–1180. [PubMed] [Google Scholar]
- Owen JB, Di Domenico F, Sultana R, Perluigi M, Cini C, Pierce WM, Butterfield DA. Proteomics-determined differences in the concanavalin-A-fractionated proteome of hippocampus and inferior parietal lobule in subjects with Alzheimer's disease and mild cognitive impairment: Implications for progression of AD. J Proteome Res. 2009;8:471–482. doi: 10.1021/pr800667a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pappolla MA, Chyan YJ, Poeggeler B, Bozner P, Ghiso J, LeDoux SP, Wilson GL. Alzheimer β protein mediated oxidative damage of mitochondrial DNA: Prevention by melatonin. J Pineal Res. 1999;27:226–229. doi: 10.1111/j.1600-079x.1999.tb00619.x. [DOI] [PubMed] [Google Scholar]
- Pastorino L, Sun A, Lu PJ, Zhou XZ, Balastik M, Finn G, Wulf G, Lim J, Li SH, Li X, Xia W, Nicholson LK, Lu KP. The prolyl isomerase Pin-1 regulates amyloid precursor protein processing and amyloid-β production. Nature. 2006;440:528–534. doi: 10.1038/nature04543. [DOI] [PubMed] [Google Scholar]
- Price DL, Tanzi RE, Borchelt DR, Sisodia SS. Alzheimer's disease: Genetic studies and transgenic models. Annu Rev Genet. 1998;32:461–493. doi: 10.1146/annurev.genet.32.1.461. [DOI] [PubMed] [Google Scholar]
- Resende R, Moreira PI, Proenca T, Deshpande A, Busciglio J, Pereira C, Oliveira CR. Brain oxidative stress in a triple-transgenic mouse model of Alzheimer disease. Free Radic Biol Med. 2008;44:2051–2057. doi: 10.1016/j.freeradbiomed.2008.03.012. [DOI] [PubMed] [Google Scholar]
- Schagger H, Ohm TG. Human diseases with defects in oxidative phosphorylation. 2. F1F0 ATP-synthase defects in Alzheimer disease revealed by blue native polyacrylamide gel electrophoresis. Eur J Biochem. 1995;227:916–921. doi: 10.1111/j.1432-1033.1995.tb20219.x. [DOI] [PubMed] [Google Scholar]
- Schuller E, Gulesserian T, Seidl R, Cairns N, Lube G. Brain T-complex polypeptide 1 (TCP- 1) related to its natural substrate β1 tubulin is decreased in Alzheimer's disease. Life Sci. 2001;69:263–270. doi: 10.1016/s0024-3205(01)01126-2. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ. Amyloid β-protein and the genetics of Alzheimer's disease. J Biol Chem. 1996;271:18295–18298. doi: 10.1074/jbc.271.31.18295. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ. Alzheimer's disease: Genes, proteins, and therapy. Physiol Rev. 2001;81:741–766. doi: 10.1152/physrev.2001.81.2.741. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ. Soluble oligomers of the amyloid β-protein impair synaptic plasticity and behavior. Behav Brain Res. 2008;192:106–113. doi: 10.1016/j.bbr.2008.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shankar GM, Leissring MA, Adame A, Sun X, Spooner E, Masliah E, Selkoe DJ, Lemere CA, Walsh DM. Biochemical and immunohistochemical analysis of an Alzheimer's disease mouse model reveals the presence of multiple cerebral Abeta assembly forms throughout life. Neurobiol Dis. 2009;36:293–302. doi: 10.1016/j.nbd.2009.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slooter AJ, Cruts M, Kalmijn S, Hofman A, Breteler MM, Van Broeckhoven C, van Duijn CM. Risk estimates of dementia by apolipoprotein E genotypes from a population-based incidence study: The Rotterdam Study. Arch Neurol. 1998;55:964–968. doi: 10.1001/archneur.55.7.964. [DOI] [PubMed] [Google Scholar]
- Smith MA, Richey Harris PL, Sayre LM, Beckman JS, Perry G. Widespread peroxynitrite-mediated damage in Alzheimer's disease. J Neurosci. 1997;17:2653–2657. doi: 10.1523/JNEUROSCI.17-08-02653.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith MA, Richey PL, Taneda S, Kutty RK, Sayre LM, Monnier VM, Perry G. Advanced Maillard reaction end products, free radicals, and protein oxidation in Alzheimer's disease. Ann N Y Acad Sci. 1994;738:447–454. doi: 10.1111/j.1749-6632.1994.tb21836.x. [DOI] [PubMed] [Google Scholar]
- Sultana R, Boyd-Kimball D, Cai J, Pierce WM, Klein JB, Merchant M, Butterfield DA. Proteomics analysis of the Alzheimer's disease hippocampal proteome. J Alzheimers Dis. 2007;11:153–164. doi: 10.3233/jad-2007-11203. [DOI] [PubMed] [Google Scholar]
- Sultana R, Boyd-Kimball D, Poon HF, Cai J, Pierce WM, Klein JB, Markesbery WR, Zhou XZ, Lu KP, Butterfield DA. Oxidative modification and down-regulation of Pin-1 in Alzheimer's disease hippocampus: A redox proteomics analysis. Neurobiol Aging. 2006a;27:918–925. doi: 10.1016/j.neurobiolaging.2005.05.005. [DOI] [PubMed] [Google Scholar]
- Sultana R, Boyd-Kimball D, Poon HF, Cai J, Pierce WM, Klein JB, Merchant M, Markesbery WR, Butterfield DA. Redox proteomics identification of oxidized proteins in Alzheimer's disease hippocampus and cerebellum: An approach to understand pathological and biochemical alterations in AD. Neurobiol Aging. 2006b;27:1564–1576. doi: 10.1016/j.neurobiolaging.2005.09.021. [DOI] [PubMed] [Google Scholar]
- Sultana R, Poon HF, Cai J, Pierce WM, Merchant M, Klein JB, Markesbery WR, Butterfield DA. Identification of nitrated proteins in Alzheimer's disease brain using a redox proteomics approach. Neurobiol Dis. 2006c;22:76–87. doi: 10.1016/j.nbd.2005.10.004. [DOI] [PubMed] [Google Scholar]
- Sultana R, Ravagna A, Mohmmad-Abdul H, Calabrese V, Butterfield DA. Ferulic acid ethyl ester protects neurons against amyloid β-peptide(1-42)-induced oxidative stress and neurotoxicity: Relationship to antioxidant activity. J Neurochem. 2005;92:749–758. doi: 10.1111/j.1471-4159.2004.02899.x. [DOI] [PubMed] [Google Scholar]
- Thongboonkerd V, Luengpailin J, Cao J, Pierce WM, Cai J, Klein JB, Doyle RJ. Fluoride exposure attenuates expression of Streptococcus pyogenes virulence factors. J Biol Chem. 2002;277:16599–16605. doi: 10.1074/jbc.M200746200. [DOI] [PubMed] [Google Scholar]
- Ursic D, Sedbrook JC, Himmel KL, Culbertson MR. The essential yeast Tcp1 protein affects actin and microtubules. Mol Biol Cell. 1994;5:1065–1080. doi: 10.1091/mbc.5.10.1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viola KL, Velasco PT, Klein WL. Why Alzheimer's is a disease of memory: The attack on synapses by Aβ oligomers (ADDLs) J Nutr Health Aging. 2008;12:51S–57S. doi: 10.1007/BF02982587. [DOI] [PubMed] [Google Scholar]
- Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS, Rowan MJ, Selkoe DJ. Naturally secreted oligomers of amyloid β protein potently inhibit hippocampal long-term potentiation in vivo. Nature. 2002;416:535–539. doi: 10.1038/416535a. [DOI] [PubMed] [Google Scholar]
- Watson GS, Craft S. Modulation of memory by insulin and glucose: Neuropsychological observations in Alzheimer's disease. Eur J Pharmacol. 2004;490:97–113. doi: 10.1016/j.ejphar.2004.02.048. [DOI] [PubMed] [Google Scholar]
- Wu SK, Zeng K, Wilson IA, Balch WE. Structural insights into the function of the Rab GDI superfamily. Trends Biochem Sci. 1996;21:472–476. doi: 10.1016/s0968-0004(96)10062-1. [DOI] [PubMed] [Google Scholar]
- Yoo BC, Kim SH, Cairns N, Fountoulakis M, Lubec G. Deranged expression of molecular chaperones in brains of patients with Alzheimer's disease. Biochem Biophys Res Commun. 2001;280:249–258. doi: 10.1006/bbrc.2000.4109. [DOI] [PubMed] [Google Scholar]
- Zhu M, Gu F, Shi J, Hu J, Hu Y, Zhao Z. Increased oxidative stress and astrogliosis responses in conditional double-knockout mice of Alzheimer-like presenilin-1 and presenilin-2. Free Radic Biol Med. 2008;45:1493–1499. doi: 10.1016/j.freeradbiomed.2008.08.027. [DOI] [PubMed] [Google Scholar]