

Abstract
The unfolded protein response (UPR) is a coordinated program that promotes cell survival under conditions of ER stress and is required in tumor progression as well. To date, no specific small molecule inhibitor targeting this pathway has been identified. PERK, one of the UPR transducers, is an eIF2α kinase. Compromising PERK function inhibits tumor growth in mice, suggesting that PERK may be a cancer drug target, but identifying a specific inhibitor of any kinase is challenging. The goal of this study was to identify some pair-wise receptor-ligand atomic contacts that confer selective PERK inhibition. Compounds selectively inhibiting PERK-mediated phosphorylation in vitro were identified using an initial virtual library screen, followed by structure-activity hypothesis testing. The most potent PERK selective inhibitors utilize three specific kinase active site contacts that, when absent from chemically similar compounds, abrogates the inhibition: (a) a strong van der Waals contact with PERK residue Met7, (b) interactions with the N-terminal portion of the activation loop and (c) groups providing electrostatic complementarity to Asp144. Interestingly, the activation loop contact is required for PERK selectivity to emerge. Understanding these structure-activity relationships may accelerate rational PERK inhibitor design.
Proteins fold into their native conformation and undergo a series of post-translational modifications in the endoplasmic reticulum (ER) as part of the normal process of cellular homeostasis. Disruption of cellular protein folding results in ER stress. Cells respond to ER stress by activation of the unfolded protein response (UPR) pathway in order to survive the stress. Multiple studies support the central role for UPR activation in tumor progression (1–6), presumably because the UPR allows aggressive tumor cells to survive the stresses imposed by hypoxic environments and chemotherapies they encounter in the course of progression in a patient. This observation thus suggests that manipulation of the UPR in tumors would be a novel anti-cancer approach to target one of the critical processes that hinder existing anti-tumor treatments.
The PKR-like ER protein kinase (PERK), one of the three identified UPR transducers, is a kinase that phosphorylates a single known substrate eIF2α, leading to lower levels of translation initiation, which in turn globally reduces the load of newly synthesized proteins in the ER (1, 2, 7, 8). Reduction of the overall protein folding load is an effective response to reduce ER stress. In addition, PERK-mediated eIF2α phosphorylation also induces the transcriptional activation to improve protein folding capacity, thereby further promoting cell survival in stressed cells (9–11). Among the group of three prominent UPR transducers that includes XBP1 and ATF6, PERK may have a broader range of cellular effects than other transducers, perhaps due to its unique role in regulating the general translation rate through the phosphorylation of eIF2α (6). Indeed, eIF2α phosphorylation appears to account for the entire range of the protective effects of PERK under ER stress (12). Hypoxia, a common feature in solid tumors, results in PERK activation, which protects tumor cells from hypoxic stress (2, 13). The critical role of PERK in tumor survival and growth has been established by the observation that tumors that lack PERK activity were small and exhibited a diminished capability to translate mRNAs involved in angiogenesis, tumor survival and growth (1, 14). This evidence clearly demonstrates that compromising PERK function inhibits tumor growth via lower phosphorylation of eIF2α. Inhibiting the kinase activity of PERK towards eIF2α may thus be an important and novel target for therapeutic intervention in cancer. To date, however, no specific small molecule inhibitor of PERK has been identified.
PERK is a classical serine-threonine kinase. The majority of small molecule kinase inhibitors that have been developed so far target the ATP binding site. This poses a challenge for kinase drug discovery since all these sites are designed to bind the same ATP molecule, making selectivity determinants theoretically scarce. Prior work has divided the ATP binding site into subregions: the adenine region, the ribose region, the phosphate-binding region and the hydrophobic regions I and II (15). This generic kinase pharmacophore model has been successfully used for the design and synthesis of numerous kinase inhibitors of structurally diverse classes, which have proven in some cases to be highly potent and selective (16). However, not all kinases offer selectivity determinants in these regions. In recent years the wealth of structural information available on kinases has promoted the development of pharmacophore models targeting the allosteric sites of the ATP pocket (17, 18), resulting in additional opportunities to innovate and control selectivity. The kinase activation loop strongly influences ligand binding at its adjacent ATP binding site. Unfortunately, although the activation loop differs in sequence between even closely related kinases and therefore represents a promising selectivity determinant, no pharmacophore strategy has yet been published that exploits this selectivity determinant.
Some prior drug discovery successes have used high-throughput screening (HTS) and virtual library screening (VLS), the latter of which is now widely recognized as a viable alternative and complement to HTS (19). More recently, a diversity of customized computational approaches has become accessible and proven to be efficient for drug discovery efforts with newer VLS and modeling approaches, including the use of homology models of VLS targets (20–23). No crystal structure of PERK is available, but structures of related kinases have been published. The goal of this study was to adopt a new and efficient approach to identifying the important receptor-ligand atomic contacts responsible for selective PERK inhibition using the available structural and chemistry resources. Mouse PERK was selected as the target prototype due to the ease of experimental execution and for preclinical planning (the best non-xenograft animal model for testing the inhibitors is a mouse model (24)). The extension of this prototype to human PERK or other kinases in the future is not expected to be rate limiting as all the steps in the approach are general. Thus, we first constructed two homology models of PERK catalytic domain. Subsequently, we employed an initial VLS followed by chemoinformatics hypothesis testing, in conjunction with in vitro kinase inhibition assays, to identify structural determinants of PERK inhibitor activity, selectivity and potency.
Materials and Methods
The computational work on homology modeling, VLS and chemoinformatics was done using ICM software produced by Molsoft, LLC (La Jolla, CA) (25).
Homology Modeling
A search of the sequence of the mouse PERK catalytic domain (GenBank ID AAD03337; Uniprot ID Q9Z2B5) against the Protein Data Bank was performed to identify homologous structures to use as templates for homology modeling. Two crystal structures of eIF2α kinase GCN2 (PDB code: 1zy4 & 1zy5) (26) qualified on the basis of sequence similarity and structural resolution as the templates for modeling the PERK catalytic domain. The sequences were aligned using the Needleman and Wunsch alignment modified with zero end-gap penalties (27), followed by manual adjustment to avoid the insertions/deletions in the middle of the secondary structure elements (Figure 1). Two homology models, generated from two templates and thus different in side chains and loops conformation, were then built from this alignment using the ICM method (28), followed by conformational sampling of the activation loop. Loop sampling was performed using local deformation loop movements (29) and Biased Probability Monte Carlo (30). This ensemble of multiple receptor conformations (Figure 2) — each exhibiting a different activation loop conformation — was used for subsequent computational molecular docking/VLS. In the docking using ensemble of loop conformations as the target, various receptor potentials to each grid point were mapped and ligand conformations against each set of receptor potentials were subsequently scored. The residues in the models were numbered starting from the N-terminus of the catalytic domain of PERK as residue number 1, which is equivalent to Phe589 in GenBank ID AAD03337.
Figure 1.

Figure 2.
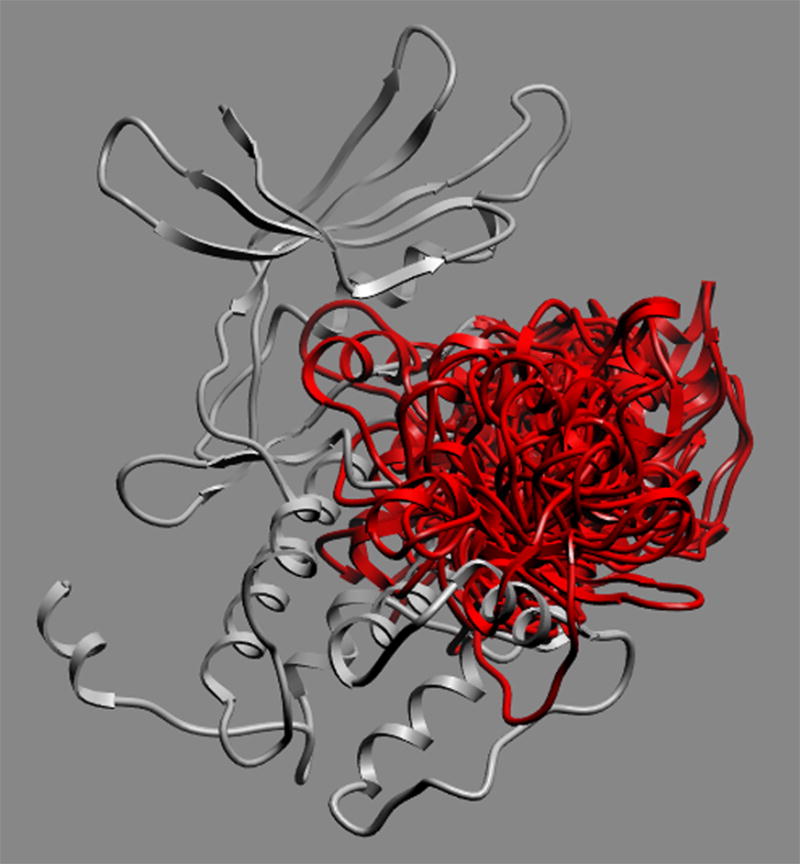
The stereochemical quality of the global structure of the models was validated in terms of Z-scores by the WHAT-IF program (31). The local energetic structural reliability of the models was assessed by calculation of the energy strain in ICM, which is represented by normalized residue energy (NRE) (32). Energy strain can detect steric clashes, exposed hydrophobic areas and other stereochemically valid, physicochemical anomalies that are invisible in graphical views of the structure (33). For conformational and physiological validation, the known ligand ATP analogue AMPPNP was docked to the PERK model and compared to its template (PDB code: 1zy5), followed by the calculation of the RMSD value for heavy atoms between the docked AMPPNP in PERK and the co-crystallized AMPPNP in GCN2. The contacts between the docked AMPPNP and PERK were further analyzed to assess the local conformational quality of the receptor.
Structure-Based Database Search
ICM–VLS rapidly docks flexible chemical ligands to a grid representation of the receptor followed by an evaluation of the docked conformation with a scoring function. The ICM scoring function takes into account conformational entropy loss and solvation electrostatic energy change (34), which are critical for accuracy of the scoring function. The algorithms have been previously validated by the successful identification of novel antagonists of the proteins in several distinct families, such as thyroid hormone receptor (35), EGFR (36), peptide deformylase (37), retinoic acid receptor (38), adenosine A2A receptor (39) and histone acetyltransferase (40). These cases are not significantly different in structural quality or scope from the PERK study done here, except for the additional challenge of utilizing a homology model instead of a crystallographic structure as the receptor. To identify PERK inhibitors, the ATP binding site in PERK homology models was initially screened against a collection of 315,102 compounds of the ChemBridge Express Library (San Diego, CA) using default ICM docking parameters (41, 42) on 3 3.0-GHz Intel Xeon processors. The VLS hits from two PERK models were merged. The resulting 8731 compounds achieving a docking score of less than or equal to −32 were further filtered by selecting those with extensive hydrogen bonds and van der Waals contacts, followed by hierarchical clustering for a diversity of scaffolds. The 72 diverse compounds chosen from different clusters were then docked to PERK models again using a full atom representation of the receptor, flexible receptor side chains and flexible ligand (ICM fully flexible docking procedure) (42). After visual inspection, 26 selected compounds were subsequently evaluated for their ability to inhibit PERK-mediated eIF2α phosphorylation in vitro. These compounds are not necessarily the ones with the best docking scores, but they were chosen because they exhibited high scores and also presented good molecular characteristics, such as van der Waals fit or hydrogen bonding within the ATP binding site. The VLS workflow is shown in Figure 8.
Figure 8.

Identification of Structural Determinants of PERK inhibition from compounds tested in vitro
Ligands were manually inspected in their docked pose for receptor residues making contact with different chemical groups in the ligand for each of the active and inactive compounds. The Wilcoxon rank sum test was performed to identify the difference in contact area for certain residues between the two groups.
Confirmation of Structural Determinants of PERK Inhibition
Compounds with similar chemical scaffolds but little or no Met7 contact were identified by substructure search through the ChemBridge library, ICM MolCart and the PubChem database using the part of the compound corresponding to its scaffold as a query. The scaffolds were defined to include all of the chemical architecture not making Met7 contact in each active compound. The compounds retrieved from the substructure search were then screened against the PERK homology models, followed by the calculation of Met7 contact area. All the small molecules with a Met7 contact area lower than 10Å2 were subsequently clustered with the six active compounds identified from the initial VLS. The small molecules with the least Tanimoto distance to their corresponding active compounds that were also commercially available were selected for experimental testing. A similar procedure was performed to identify a chemically similar, contact-negative comparison group for Asp144.
Identification of Derivatives conferring PERK Inhibitor Selectivity
A substructure search in ChemBridge library using the A4 chemical group making contact with the PERK activation loop in A4’s docked pose was performed to get all compounds potentially using this contact in the databases. Given that the conformational rearrangement of the activation loop upon ligand binding is crucial for kinase activity, this sublibrary of compounds were subsequently docked to the ensemble of multiple loop conformations of PERK. The chemicals with a Met7 contact area more than 20 Å2 were further screened by hydrogen binding and van der Waals contacts with the activation loop. Nine compounds making extensive contacts with both the activation loop and Met7 in their docked poses were selected for experimental testing.
in vitro Kinase Inhibition Assay
All the compounds tested in the PERK inhibition assay were purchased from the following commercial vendors: ChemBridge Corporation (San Diego, CA), ChemDiv (San Diego, CA), Ryan Scientific, Inc. (Mt. Pleasant, MC) and Nanosyn (San Diego, CA). The purity of all the compounds is ≥ 99%. 5 nM PERK protein was pre-incubated with varying concentrations of compound at room temperature for 10 minutes. The kinase reaction was initiated by the addition of 10 μM eIF2α, 2 μM ATP, 0.2 μM γ-[32P]ATP and allowed to proceed at room temperature for 15 minutes (43). The phosphorylated eIF2α was then separated on 14% SDS-PAGE and visualized by autoradiography. Only the compounds showing consistent results in three independent experiments were considered true actives or true inactives. To determine the selectivity of the compounds, their ability to inhibit the protein kinase PKA was also evaluated. Briefly, five nM PKA was pre-incubated with the compound at 40 μM, the highest concentration used in the PERK assay, for 10 minutes. Then 10 μM GSTag, 10 μM ATP and 0.2 μM γ-[32P]ATP were added, followed by the incubation at room temperature for 15 minutes.
Results
Homology Modeling
A crystallographic structure of the PERK catalytic domain has not been published, so homology models of PERK were required in order to perform this study. Two structures of the catalytic domain of eIF2α kinase GCN2 (PDB code: 1zy4 & 1zy5) were selected as the templates as they have a highly statistically significant structural homology (P = 10e-21) (27). In addition, these two crystal structures have high resolution and low temperature factors in the ATP binding site, suggesting the good structural quality of these two templates at that location. The activation loops in these models were conformationally sampled, and an ensemble of multiple activation loop conformations was generated and used for docking. The structural quality of the models, analyzed using the WHAT-IF program, energy strain analysis and cross docking with a known ligand, indicated that the structural quality in the ATP binding site in the PERK models was reliable for VLS (data not shown).
Initial Virtual Library Screening
Our objective was to uncover specific structure-activity relationships in the active site of PERK, including the identification of the structural determinants of ligand activity, selectivity and potency. An initial VLS of 315,102 compounds in the ChemBridge library against the two PERK homology models was carried out. All compounds with a docking score less than or equal to −32 (38) were further filtered by hydrogen bonds and van der Waals contacts, followed by hierarchical clustering, ICM fully flexible docking and visual inspection. Twenty-six (26) compounds surviving these criteria were then tested for their ability to inhibit PERK-mediated eIF2α phosphorylation in vitro. Six compounds inhibited PERK in vitro, while the other six compounds were found to be inactive (Table 1). The mean of the six van der Waals contact areas, recorded from the docked poses of the active compounds, with Met7 in PERK was significantly different from the mean of the six van der Waals contact areas, recorded from the docked poses of the inactive compounds, with Met7 in PERK (p value = 0.0022) (Table 1, Figure 3A & 3C), suggesting that van der Waals contact of a compound with Met7 in PERK might be critical for inhibition activity.
Table 1.
Met7 Contact Area in the active and inactive compounds from initial screening
| Compound | Structure | Activity | Met7 Contact Area (Å2) |
|---|---|---|---|
| A4 | 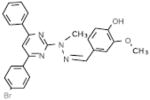 |
active | 21.6 |
| B2 | 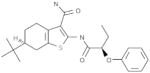 |
active | 22.9 |
| B6 | 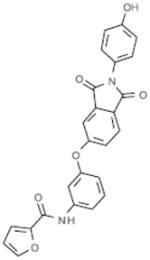 |
active | 22.1 |
| C1 | 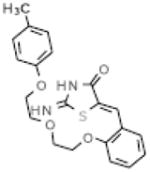 |
active | 22.8 |
| D1 | 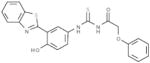 |
active | 32.6 |
| E1 |  |
active | 29.7 |
| A1 | 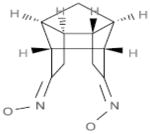 |
inactive | 11.1 |
| A2 | inactive | 5.3 | |
| A3 | 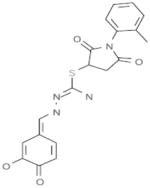 |
inactive | 2.2 |
| C3 | 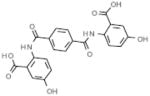 |
inactive | 11.5 |
| C6 | 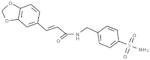 |
inactive | 14.7 |
| D4 | 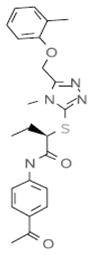 |
inactive | 0 |
Figure 3.

A Strong Met7 van der Waals Contact is a Structural Determinant of PERK inhibition
To further validate the hypothesis that Met7 contact is a structural determinant of PERK inhibition in vitro, a set of corresponding chemically similar compounds to the active compounds, which demonstrated little or no Met7 contact upon docking, were identified from chemical databases. These compounds were inactive in vitro (Figure 3B &3D), suggesting the indispensable role of strong van der Waals contact (> 20 Å2) with Met7 in PERK inhibitor design. Notably, none of the six, Met7 dependent, active compounds in the initial VLS was selective for PERK as compared to PKA (Figure 3E), Thus, Met7 is a structural determinant to distinguish between the active and inactive compounds for PERK, but is not itself a determinant of selectivity for PERK.
The N-terminal portion of the PERK Activation Loop Area is a Structural Determinant of Selectivity for PERK Inhibitors
Among the six active compounds in the initial VLS, A4 and D1 showed relatively stronger inhibition. However, compound A4 displayed the inhibition within a narrow range of concentration, while D1 exhibited an activity varying over a range of 1–25 μM (data not shown). Interestingly, the analysis of the predicted structural interactions of these two compounds with PERK showed that A4 makes hydrogen bonds with Leu147 and Val148 at the origin of the activation loop, while D1 interacts with the backbone of the hinge region via the hydrogen bonding with Gln78 and Cys80 (Figure 4). We hypothesized that A4 might exhibit a slight cooperative effect in PERK inhibition due to its interaction with the stem of the activation loop. The activation loop, adjacent to the ATP binding pocket, is critical for the transition between active and inactive conformation of kinases. Activation loops in kinases have been known to influence activity at active sites in dimerized or oligomerized domains nearby by cross-domain contact (44), suggesting a structural basis for cooperativity in an inhibition that contacts the activation loop. Moreover, this area, flanking the ATP binding site, is less conserved (Ala150 to Glu157, Val160 to His170, Gln173) compared to other regions of ATP pocket.
Figure 4.

A screen for new small molecules in the chemical databases that make contacts in the area in the PERK model near the activation loop was designed and carried out in order to test this hypothesis. Nine compounds making extensive contacts with the N-terminal portion of the activation loop, including hydrogen bonding and van der Waals contacts, were subsequently selected for in vitro test and were found to be active. Notably, none of these compounds showed significant inhibition of PKA, while the inhibition of PERK was maintained (Figure 5, Table 2 row 1 – row 9).
Figure 5.

Table 2.
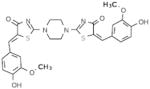
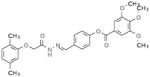
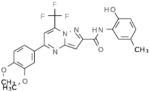
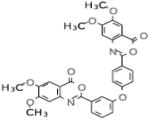
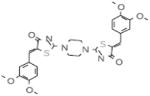
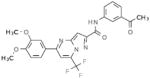
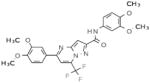
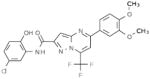
Selective PERK Inhibitors Identified
| Structure | IC50 (μM) | No. of Hbonds with the Activation Loop | Total vdw Contact Areas with the Activation Loop (Å2) |
|---|---|---|---|
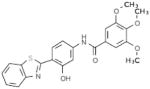 |
5 | 0 | 53 |
 |
1 | 2 | 35 |
 |
2.5 | 0 | 57 |
 |
1 | 0 | 48 |
 |
30 | 0 | 53 |
| 8 | 0 | 48 | |
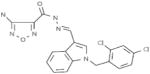 |
5 | 2 | 24 |
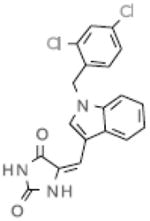 |
1.5 | 1 | 58 |
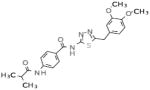 |
NA* | 3 | 20 |
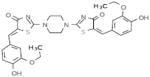 |
4 | 1 | 30 |
 |
2.5 | 2 | 61 |
 |
6 | 0 | 45 |
 |
1.5 | 2 | 83 |
 |
1.5 | 0 | 47 |
NA — not tested
Electrostatic Complementarity to Asp144 Side Chain is a Structural Determinant of PERK Inhibitor Potency
The IC50 of the PERK-selective compounds were determined. Interestingly, seven showed an IC50 less than 10 μM, while one compound displayed an IC50 of 30 μM (Table 2 row 1–row 9). To identify the structural determinant accounting for this potency difference, the atomic interactions with the surrounding residues within 6 Å of each ligand were further analyzed. All the compounds with an IC50 less than 10 μM have a nitrogen-centered partial positive charge in the midsection of the compound scaffold, providing electrostatic complementarity to the negative charge of the side-chain carboxyl group displayed in the PERK active site by Asp144 (Figure 6A). On the contrary, the one compound with an IC50 of 30 μM exhibited a partial negative charge at this location in its scaffold midsection (Figure 6B). Asp144 is part of the DFG motif—a crucial triad of residues for kinase activation. Thus, we speculated that electrostatic complementarity to Asp144 side chain might be a structural determinant of PERK inhibitor potency. To validate this hypothesis, seven corresponding compounds with similar chemical groups but partial negative charges in their scaffold midsections were subsequently generated. Five were inactive in vitro (Figure 6C), while two were weakly active with an IC50 more than 25 μM (data not shown). This finding indicates that electrostatic complementarity to Asp144 side chain is important for PERK inhibitory potency as it is for many kinases, which is consistent with a recent study on the identification of a novel inhibitor of the JAK2 tyrosine kinase (45).
Figure 6.

Active Compounds Act as ATP Competitive Inhibitors
Our small molecular inhibitors were designed to block PERK activity via binding to the ATP binding site. To confirm that they act as ATP competitive inhibitors, we evaluated the inhibitory potency of A4B2 and A4B4, two compounds with an IC50 of 1 μM in vitro, on PERK activity by introduction of different concentrations of ATP. Lineweaver Burke plots for PERK inhibition by A4B2 and A4B4 with respect to different ATP concentrations showed that increasing the concentration of inhibitors results in a family of lines with a common y-intercept but with different slopes, which is the indicative of competitive inhibition with ATP (Figure 7).
Figure 7.

Discussion
Structure-Activity Hypothesis Testing of Specific Structural Determinants of PERK Inhibition Led to the Discovery of Selective PERK Inhibitors
In this study, we identified several diverse near-nanomolar selective PERK inhibitors using a method distinct from VLS or HTS. We initially identified six active compounds using homology modeling and structure-based VLS. Given the correlations of the structural analyses with the experimental results in this initial screening, two hypotheses on structural determinants of PERK inhibitor activity and selectivity were proposed. These two hypotheses were further validated using substructure search, docking and loop sampling. Nine selective compounds were then found by exploiting these two structural determinants. The third hypothesis on the structural determinant of PERK inhibitor potency was identified by the analysis of the selective compounds and the subsequent validation. In total, fourteen selective PERK inhibitors were identified from 42 compounds tested in total. The two most potent PERK selective inhibitors have IC50s of 1 μM. Thus, identification of some of the structural determinants of compound activity, selectivity and potency led to the discovery of PERK inhibitors with some selectivity. The workflow of this study is shown in Figure 8. Surprisingly, none of these 14 PERK inhibitors interact with the hinge region, indicating that contact with the hinge region may not be critical for all PERK inhibitors, a surprising finding given that most prior binders at the ATP site (including known kinase inhibitors) contact the hinge region.
A Pharmacophore Model for Selective PERK Inhibitors
This is the first report on a particular pharmacophore for PERK inhibitors with some selectivity. We identified three structural determinants of PERK inhibition. A chemical scaffold that fits reasonably well into the PERK active site and a strong van der Waals contact of the aromatic/hydrophobic group(s) in the docked chemical with PERK Met7 appears to be a minimal set of requirements for PERK inhibition using the pharmacophore space we identified. Additional contact(s) via hydrogen bonding or van der Waals with the N-terminal portion of the activation loop is required for such a chemical to achieve selective inhibition of PERK compared to other kinase. Finally, an additional chemically complementary contact with PERK Asp144 residue can improve the potency of the selective PERK inhibitor. These three findings draw out a novel, minimal pharmacophore model for PERK inhibitors (Figure 9).
Figure 9.
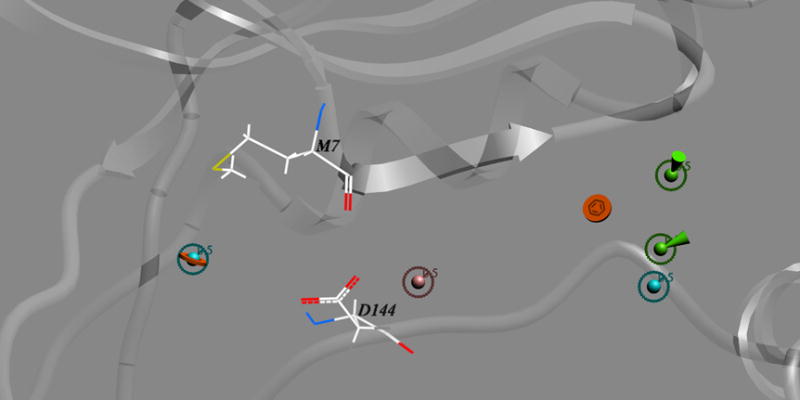
This newly identified pharmacophore departs in several original ways from current kinase pharmacophore models. Structural studies on the relatively potent and selective kinase inhibitors that are showing promise in clinical trials have revealed several routes to selectivity, including targeting less conserved adjacent pockets (46). A number of inhibitors have achieved selectivity by exploiting the two less conserved pockets hydrophobic region I and II, which are not occupied by any parts of the ATP molecule, through the hydrogen bonding and van der Waals contacts (47–51). In the case of PERK, however, the entry to the hydrophobic region I is blocked in our homology models by the bulky gatekeeper residue Met77, preventing the exploitation of this pocket in specific inhibitor design. Instead we found a way to exploit the less sequence-conserved segment at the activation loop area for PERK inhibitor design. Although the unstructured nature of the kinase activation loops hampers rational drug design approaches, we have shown that a systematic approach, with loop conformational sampling, can succeed in targeting this area. Regions partly formed by the activation loop may thus represent important new frontiers for specific kinase inhibitor design beyond the traditional hydrophobic regions I and II. Because of the variability of the activation loop, the pharmacophore space presented here is only one of the several achievable by this or other strategies. It remains to be seen which of these alternatives will turn out to be the most productive at the lead optimization and preclinical phases of translating these and other leads into a clinically useful PERK inhibitor.
The experimental validation of our proposed 3D pharmacophore model consists of 1) the significant differences in eIF2α phosphorylation observed between matched sets of chemically similar compounds that differ only in the groups that are predicted by docking to interact with the key contact points; and 2) the Lineweaver-Burke ATP competition results (Figure 7) showing that the compounds act competitively at the kinase ATP binding site. The correlation of the docking results with the activity to such a high significance for three different contact points is unlikely to occur without the binding geometry predicted by docking being at least moderately accurate, thereby validating our study as a novel method to identify specific PERK leads. Nevertheless, future work to crystallize the compounds with PERK is the gold standard validation for an optimized PERK inhibitor, and the results may reveal additional important features of the ultimately successful pharmacophore model.
We achieved proof-of-principle of this drug discovery approach by our novel workflow. The method identifies very specific contact points within the inhibitor pocket and correlates them with inhibitor potency and selectivity. The identification of the key locations in the pocket (pharmacophore features) may be more challenging than identifying a compound specifically complementary with the amino acid at specific pharmacophore locations. Thus, having identified the Met 7 location as the important location, the human PERK kinase with its Leucine at position 7, should be readily targeted using another iteration of our method. Notably, many known kinase inhibitors make specific contact with the amino acid located at the position of Met7 in their respective kinases (52) demonstrating that, despite the chemical difference between Met and Leu (and other amino acids) at this position, identification of complementary chemistry for the different amino acid chemistry is not rate limiting. This study suggests the more important finding that this location is indeed important for PERK. We chose to prove the principle using the mouse model system because future preclinical prototyping of PERK inhibition in mouse model systems is expected to be more rate-limiting in proving that PERK inhibitors can be clinically useful drugs than adapting the approach after the mouse prototype is proven at the cell and animal level to accommodate the few amino acid differences in human PERK.
Conclusions
We have developed the first pharmacophore model for PERK inhibitors with some selectivity. Specifically, a strong van der Waals contact with Met7, interactions with the N-terminal portion of the activation loop and electrostatic complementarity to Asp144 are three important structural determinants of PERK inhibition by a small molecule compound. Notably, these results were achieved utilizing a homology model of PERK along with conformational loop sampling of the PERK activation loop. Further exploiting these methods and the structural determinants we have identified may inform PERK inhibitor lead optimization.
Acknowledgments
The authors would like to thank Dr. Ruben Abagyan, Dr. Kent Kirshenbaum and Dr. Emeline Maillet for their helpful comments. This work was supported by the NIH grants (DPOD004631 to TC and DK075311 and DK047119 to DR). In addition we acknowledge Yuhong Zhang, Dr. Tony Chin and Dr. Ester Zito in DR’s lab for their experimental help.
References
- 1.Bi M, Naczki C, Koritzinsky M, Fels D, Blais J, Hu N, et al. ER stress-regulated translation increases tolerance to extreme hypoxia and promotes tumor growth. EMBO J. 2005;24:3470–81. doi: 10.1038/sj.emboj.7600777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Koumenis C, Naczki C, Koritzinsky M, Rastani S, Diehl A, Sonenberg N, et al. Regulation of protein synthesis by hypoxia via activation of the endoplasmic reticulum kinase PERK and phosphorylation of the translation initiation factor eIF2alpha. Mol Cell Biol. 2002;22:7405–16. doi: 10.1128/MCB.22.21.7405-7416.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ameri K, Lewis CE, Raida M, Sowter H, Hai T, Harris AL. Anoxic induction of ATF-4 through HIF-1-independent pathways of protein stabilization in human cancer cells. Blood. 2004;103:1876–82. doi: 10.1182/blood-2003-06-1859. [DOI] [PubMed] [Google Scholar]
- 4.Romero-Ramirez L, Cao H, Nelson D, Hammond E, Lee AH, Yoshida H, et al. XBP1 is essential for survival under hypoxic conditions and is required for tumor growth. Cancer Res. 2004;64:5943–7. doi: 10.1158/0008-5472.CAN-04-1606. [DOI] [PubMed] [Google Scholar]
- 5.Shuda M, Kondoh N, Imazeki N, Tanaka K, Okada T, Mori K, et al. Activation of the ATF6, XBP1 and grp78 genes in human hepatocellular carcinoma: a possible involvement of the ER stress pathway in hepatocarcinogenesis. J Hepatol. 2003;38:605–14. doi: 10.1016/s0168-8278(03)00029-1. [DOI] [PubMed] [Google Scholar]
- 6.Blais J, Bell JC. Novel therapeutic target: the PERKs of inhibiting the integrated stress response. Cell Cycle. 2006;5:2874–7. doi: 10.4161/cc.5.24.3597. [DOI] [PubMed] [Google Scholar]
- 7.Shi Y, Vattem KM, Sood R, An J, Liang J, Stramm L, et al. Identification and characterization of pancreatic eukaryotic initiation factor 2 alpha-subunit kinase, PEK, involved in translational control. Mol Cell Biol. 1998;18:7499–509. doi: 10.1128/mcb.18.12.7499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Harding HP, Novoa I, Zhang Y, Zeng H, Wek R, Schapira M, et al. Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol Cell. 2000;6:1099–108. doi: 10.1016/s1097-2765(00)00108-8. [DOI] [PubMed] [Google Scholar]
- 9.Lu PD, Harding HP, Ron D. Translation reinitiation at alternative open reading frames regulates gene expression in an integrated stress response. J Cell Biol. 2004;167:27–33. doi: 10.1083/jcb.200408003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wu J, Kaufman RJ. From acute ER stress to physiological roles of the Unfolded Protein Response. Cell Death Differ. 2006;13:374–84. doi: 10.1038/sj.cdd.4401840. [DOI] [PubMed] [Google Scholar]
- 11.Blais JD, Filipenko V, Bi M, Harding HP, Ron D, Koumenis C, et al. Activating transcription factor 4 is translationally regulated by hypoxic stress. Mol Cell Biol. 2004;24:7469–82. doi: 10.1128/MCB.24.17.7469-7482.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lu PD, Jousse C, Marciniak SJ, Zhang Y, Novoa I, Scheuner D, et al. Cytoprotection by pre-emptive conditional phosphorylation of translation initiation factor 2. EMBO J. 2004;23:169–79. doi: 10.1038/sj.emboj.7600030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Koritzinsky M, Magagnin MG, van den Beucken T, Seigneuric R, Savelkouls K, Dostie J, et al. Gene expression during acute and prolonged hypoxia is regulated by distinct mechanisms of translational control. EMBO J. 2006;25:1114–25. doi: 10.1038/sj.emboj.7600998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Blais JD, Addison CL, Edge R, Falls T, Zhao H, Wary K, et al. Perk-dependent translational regulation promotes tumor cell adaptation and angiogenesis in response to hypoxic stress. Mol Cell Biol. 2006;26:9517–32. doi: 10.1128/MCB.01145-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Traxler P, Furet P. Strategies toward the design of novel and selective protein tyrosine kinase inhibitors. Pharmacol Ther. 1999;82:195–206. doi: 10.1016/s0163-7258(98)00044-8. [DOI] [PubMed] [Google Scholar]
- 16.Fabbro D, Ruetz S, Buchdunger E, Cowan-Jacob SW, Fendrich G, Liebetanz J, et al. Protein kinases as targets for anticancer agents: from inhibitors to useful drugs. Pharmacol Ther. 2002;93:79–98. doi: 10.1016/s0163-7258(02)00179-1. [DOI] [PubMed] [Google Scholar]
- 17.Liu Y, Gray NS. Rational design of inhibitors that bind to inactive kinase conformations. Nat Chem Biol. 2006;2:358–64. doi: 10.1038/nchembio799. [DOI] [PubMed] [Google Scholar]
- 18.Zuccotto F, Ardini E, Casale E, Angiolini M. Through the “gatekeeper door”: exploiting the active kinase conformation. J Med Chem. 53:2681–94. doi: 10.1021/jm901443h. [DOI] [PubMed] [Google Scholar]
- 19.Marrero-Ponce Y, Castillo-Garit JA, Olazabal E, Serrano HS, Morales A, Castanedo N, et al. TOMOCOMD-CARDD, a novel approach for computer-aided ‘rational’ drug design: I. Theoretical and experimental assessment of a promising method for computational screening and in silico design of new anthelmintic compounds. J Comput Aided Mol Des. 2004;18:615–34. doi: 10.1007/s10822-004-5171-y. [DOI] [PubMed] [Google Scholar]
- 20.Chao WR, Yean D, Amin K, Green C, Jong L. Computer-aided rational drug design: a novel agent (SR13668) designed to mimic the unique anticancer mechanisms of dietary indole-3-carbinol to block Akt signaling. J Med Chem. 2007;50:3412–5. doi: 10.1021/jm070040e. [DOI] [PubMed] [Google Scholar]
- 21.Zotchev SB, Stepanchikova AV, Sergeyko AP, Sobolev BN, Filimonov DA, Poroikov VV. Rational design of macrolides by virtual screening of combinatorial libraries generated through in silico manipulation of polyketide synthases. J Med Chem. 2006;49:2077–87. doi: 10.1021/jm051035i. [DOI] [PubMed] [Google Scholar]
- 22.Zauhar RJ, Moyna G, Tian L, Li Z, Welsh WJ. Shape signatures: a new approach to computer-aided ligand- and receptor-based drug design. J Med Chem. 2003;46:5674–90. doi: 10.1021/jm030242k. [DOI] [PubMed] [Google Scholar]
- 23.Grassy G, Calas B, Yasri A, Lahana R, Woo J, Iyer S, et al. Computer-assisted rational design of immunosuppressive compounds. Nat Biotechnol. 1998;16:748–52. doi: 10.1038/nbt0898-748. [DOI] [PubMed] [Google Scholar]
- 24.Ozcan U, Ozcan L, Yilmaz E, Duvel K, Sahin M, Manning BD, et al. Loss of the tuberous sclerosis complex tumor suppressors triggers the unfolded protein response to regulate insulin signaling and apoptosis. Mol Cell. 2008;29:541–51. doi: 10.1016/j.molcel.2007.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Abagyan R, Totrov M, Kuznetsov D. ICM – A New Method for Protein Modeling and Design: Applications to Docking and Structure Prediction from the Distorted Native Conformation. Journal of Computational Chemistry. 1994;15:488–506. [Google Scholar]
- 26.Padyana AK, Qiu H, Roll-Mecak A, Hinnebusch AG, Burley SK. Structural basis for autoinhibition and mutational activation of eukaryotic initiation factor 2alpha protein kinase GCN2. J Biol Chem. 2005;280:29289–99. doi: 10.1074/jbc.M504096200. [DOI] [PubMed] [Google Scholar]
- 27.Abagyan RA, Batalov S. Do aligned sequences share the same fold? J Mol Biol. 1997;273:355–68. doi: 10.1006/jmbi.1997.1287. [DOI] [PubMed] [Google Scholar]
- 28.Cardozo T, Totrov M, Abagyan R. Homology modeling by the ICM method. Proteins. 1995;23:403–14. doi: 10.1002/prot.340230314. [DOI] [PubMed] [Google Scholar]
- 29.Abagyan RA, Mazur AK. New methodology for computer-aided modelling of biomolecular structure and dynamics. 2. Local deformations and cycles. J Biomol Struct Dyn. 1989;6:833–45. doi: 10.1080/07391102.1989.10507740. [DOI] [PubMed] [Google Scholar]
- 30.Abagyan R, Totrov M. Biased probability Monte Carlo conformational searches and electrostatic calculations for peptides and proteins. J Mol Biol. 1994;235:983–1002. doi: 10.1006/jmbi.1994.1052. [DOI] [PubMed] [Google Scholar]
- 31.Vriend G. WHAT IF: a molecular modeling and drug design program. J Mol Graph. 1990;8:52–6. 29. doi: 10.1016/0263-7855(90)80070-v. [DOI] [PubMed] [Google Scholar]
- 32.Maiorov V, Abagyan R. Energy strain in three-dimensional protein structures. Fold Des. 1998;3:259–69. doi: 10.1016/S1359-0278(98)00037-6. [DOI] [PubMed] [Google Scholar]
- 33.Cardozo T, Abagyan R. Druggability of SCF ubiquitin ligase-protein interfaces. Methods Enzymol. 2005;399:634–53. doi: 10.1016/S0076-6879(05)99042-3. [DOI] [PubMed] [Google Scholar]
- 34.Cavasotto CN, Abagyan RA. Protein flexibility in ligand docking and virtual screening to protein kinases. J Mol Biol. 2004;337:209–25. doi: 10.1016/j.jmb.2004.01.003. [DOI] [PubMed] [Google Scholar]
- 35.Schapira M, Raaka BM, Das S, Fan L, Totrov M, Zhou Z, et al. Discovery of diverse thyroid hormone receptor antagonists by high-throughput docking. Proc Natl Acad Sci U S A. 2003;100:7354–9. doi: 10.1073/pnas.1131854100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cavasotto CN, Ortiz MA, Abagyan RA, Piedrafita FJ. In silico identification of novel EGFR inhibitors with antiproliferative activity against cancer cells. Bioorg Med Chem Lett. 2006;16:1969–74. doi: 10.1016/j.bmcl.2005.12.067. [DOI] [PubMed] [Google Scholar]
- 37.Howard MH, Cenizal T, Gutteridge S, Hanna WS, Tao Y, Totrov M, et al. A novel class of inhibitors of peptide deformylase discovered through high-throughput screening and virtual ligand screening. J Med Chem. 2004;47:6669–72. doi: 10.1021/jm049222o. [DOI] [PubMed] [Google Scholar]
- 38.Schapira M, Raaka BM, Samuels HH, Abagyan R. Rational discovery of novel nuclear hormone receptor antagonists. Proc Natl Acad Sci U S A. 2000;97:1008–13. doi: 10.1073/pnas.97.3.1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Katritch V, Jaakola VP, Lane JR, Lin J, Ijzerman AP, Yeager M, et al. Structure-based discovery of novel chemotypes for adenosine A(2A) receptor antagonists. J Med Chem. 53:1799–809. doi: 10.1021/jm901647p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bowers EM, Yan G, Mukherjee C, Orry A, Wang L, Holbert MA, et al. Virtual ligand screening of the p300/CBP histone acetyltransferase: identification of a selective small molecule inhibitor. Chem Biol. 17:471–82. doi: 10.1016/j.chembiol.2010.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Abagyan RTM, Kuznetsov D. ICM-A new method for protein modeling and design: Applications to docking and structure prediction from the distorted native conformation. J Comput Chem. 1994;15:488–506. [Google Scholar]
- 42.MolSoft. ICM 2.7 Program Manual. San Diego: MolSoft; 1998. [Google Scholar]
- 43.Marciniak SJ, Garcia-Bonilla L, Hu J, Harding HP, Ron D. Activation-dependent substrate recruitment by the eukaryotic translation initiation factor 2 kinase PERK. J Cell Biol. 2006;172:201–9. doi: 10.1083/jcb.200508099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Taylor SS, Haste NM, Ghosh G. PKR and eIF2alpha: integration of kinase dimerization, activation, and substrate docking. Cell. 2005;122:823–5. doi: 10.1016/j.cell.2005.09.007. [DOI] [PubMed] [Google Scholar]
- 45.Kiss R, Polgar T, Kirabo A, Sayyah J, Figueroa NC, List AF, et al. Identification of a novel inhibitor of JAK2 tyrosine kinase by structure-based virtual screening. Bioorg Med Chem Lett. 2009;19:3598–601. doi: 10.1016/j.bmcl.2009.04.138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Noble ME, Endicott JA, Johnson LN. Protein kinase inhibitors: insights into drug design from structure. Science. 2004;303:1800–5. doi: 10.1126/science.1095920. [DOI] [PubMed] [Google Scholar]
- 47.Davies TG, Bentley J, Arris CE, Boyle FT, Curtin NJ, Endicott JA, et al. Structure-based design of a potent purine-based cyclin-dependent kinase inhibitor. Nat Struct Biol. 2002;9:745–9. doi: 10.1038/nsb842. [DOI] [PubMed] [Google Scholar]
- 48.De Azevedo WF, Jr, Mueller-Dieckmann HJ, Schulze-Gahmen U, Worland PJ, Sausville E, Kim SH. Structural basis for specificity and potency of a flavonoid inhibitor of human CDK2, a cell cycle kinase. Proc Natl Acad Sci U S A. 1996;93:2735–40. doi: 10.1073/pnas.93.7.2735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tong L, Pav S, White DM, Rogers S, Crane KM, Cywin CL, et al. A highly specific inhibitor of human p38 MAP kinase binds in the ATP pocket. Nat Struct Biol. 1997;4:311–6. doi: 10.1038/nsb0497-311. [DOI] [PubMed] [Google Scholar]
- 50.Nagar B, Hantschel O, Young MA, Scheffzek K, Veach D, Bornmann W, et al. Structural basis for the autoinhibition of c-Abl tyrosine kinase. Cell. 2003;112:859–71. doi: 10.1016/s0092-8674(03)00194-6. [DOI] [PubMed] [Google Scholar]
- 51.Mohammadi M, McMahon G, Sun L, Tang C, Hirth P, Yeh BK, et al. Structures of the tyrosine kinase domain of fibroblast growth factor receptor in complex with inhibitors. Science. 1997;276:955–60. doi: 10.1126/science.276.5314.955. [DOI] [PubMed] [Google Scholar]
- 52.Wang H, Cardozo TJ. Thesis. New York: New York University; 2010. Rational Design of Modulators of the Unfolded Protein Response; pp. 72–8. [Google Scholar]