

Abstract
Enzyme INtr is the first protein in the nitrogen phosphotransferase pathway. Using an array of biochemical and biophysical tools, we characterized the protein, compared its properties to that of EI of the carbohydrate PTS and, in addition, examined the effect of substitution of all nonexchangeable protons by deuterium (perdeuteration) on the properties of EINtr. Notably, we find that the catalytic function (autophosphorylation and phosphotransfer to NPr) remains unperturbed while its stability is modulated by deuteration. In particular, the deuterated form exhibits a reduction of approximately 4 °C in thermal stability, enhanced oligomerization propensity, as well as increased sensitivity to proteolysis in vitro. We investigated tertiary, secondary, and local structural changes, both in the absence and presence of PEP, using near-and far-UV circular dichroism and Trp fluorescence spectroscopy. Our data demonstrate that the aromatic residues are particularly sensitive probes for detecting effects of deuteration with an enhanced quantum yield upon PEP binding and apparent decreases in tertiary contacts for Tyr and Trp side chains. Trp mutagenesis studies showed that the region around Trp522 responds to binding of both PEP and NPr. The significance of these results in the context of structural analysis of EINtr are evaluated.
Keywords: phosphotransferase system, deuteration, conformation, protein structure
The Escherichia coli phosphoenolpyruvate:carbohydrate phosphotransferase system (PTS) is the major system for carbohydrate transport in that organism [1]. It consists of two proteins, EI and HPr, which function by sequential phosphorylation from PEP to EI, then to HPr; phospho-HPr can phosphorylate a collection of sugar-specific transporters which then go on to effect the uptake of a variety of sugar substrates. In addition to its role in sugar transport, the PTS functions in other regulatory capacities, primarily via control of the state of phosphorylation of the ~17 kDa protein IIAGlc. The structures of numerous PTS proteins have been elucidated using X-ray crystallography and NMR spectroscopy [1]. Of note, the structure of the EI component, a protein with 575 amino acid residues, has been deduced by X-ray crystallography [2] and it has been proposed that the catalytic activity of this protein involves a swiveling domain mechanism similar to that demonstrated for pyruvate phosphate dikinase [3].
The PTSNtr, a system paralogous to that of the carbohydrate PTS, is comprised of the proteins EINtr, NPr and IIANtr (homologous to EI, HPr and IIA of the carbohydrate PTS). A major role for IIANtr in regulation has been demonstrated. In this regard, it has been shown that the dephosphorylated form of IIANtr binds to and inhibits the activity of TrkA, one of the potassium transporter activities of E. coli [4]. It was demonstrated that dephospho-IIANtr also interacts with and stimulates the sensor kinase KdpD [5]. This ultimately leads to regulation of the expression of the high-affinity K+ transporter KdpFABC.
While the structures of NPr [6] and IIANtr [7] have been solved, that of EINtr is unknown. Although the primary structures of EI and EINtr are homologous, there are clear differences in their activities. In a sugar phosphorylation assay, the turnover number for EINtr is about 0.1% that for EI, which probably explains why EI is absolutely required for sugar phosphorylation in vivo [8]. Further, there is essentially absolute specificity of EINtr for NPr suggesting that specific residues of EINtr control the interaction and phosphoryl transfer between the proteins [8]. It was therefore of fundamental interest to define the three-dimensional structure of EINtr; until this time, all attempts to produce crystals of EINtr with suitable diffraction properties have been unsuccessful (Hertzberg and Peterkofsky, unpublished data). Consequently, alternative strategies (biophysical and NMR studies) to characterize EINtr were undertaken. In preparation for preliminary NMR studies, we prepared perdeuterated protein and noted changes in properties. We therefore carried out companion biophysical, enzymatic and stability studies on both the hydrogenated and perdeuterated forms of EINtr to both characterize EINtr and evaluate the suitability of the perdeuterated EINtr for structural analysis. The work described here documents similarities and differences in the two forms of the protein.
Materials and Methods
Materials
NPr, deficient in the terminal 8 amino acids (NPr 1–85), was prepared as described [6]. 32P-PEP was prepared as described [9].
Cloning and expression of EINtr
Full-length EINtr is a protein of 748 residues. This protein is homologous to the 575 residue EI of the carbon PTS but, in addition, encodes a GAF domain at its N-terminus [10]. Initial experiments indicated that the expressed full-length protein tended to aggregate. Consequently, a truncated version of EINtr that was homologous to EI was engineered in which the first 169 residues, encoding the unique GAF domain, were eliminated. This is subsequently referred to as EINtr. Details of the cloning are in Supplementary Data. The expression vector encodes residues 170–748 of EINtr preceded by MGSS(H)6SSGLVPRGSHM (see Fig. 1). This amino-terminal His-tag sequence allows for rapid purification by IMAC. Details of protein expression and purification are in Supplementary Data. The yield of protein was about 10 mg (H)/liter and 5 mg (D)/500 ml. Analysis by mass spectrometry of the active-site peptide, produced by tryptic hydrolysis, indicated that both purified proteins were approximately 80% phosphorylated. The isolated proteins were demonstrated to be active in phosphoryl transfer from PEP/Mg to purified NPr.
Fig. 1. Sequence of EINtr(H356A).

The sequence shown corresponds to the truncated form of EINtr preceded by a His-tag sequence and the aminoterminal Met deleted as described in Materials and Methods. Highlighted residues: His-tag sequence=orange; arginine residues=magenta; tryptophan residues=bright green; active site His356 changed to Ala=baby blue.
Preparation of dephosphorylated H-EINtr and D-EINtr was accomplished by incubation with pyruvate (the back reaction). To one ml samples of each form of the protein, MgCl2 and sodium pyruvate were added to a final concentration of 10 mM and the solutions were incubated for 1 h at 37 °C. The samples were then processed on a Superose 6 HR 10/30 (GE Healthcare) column equilibrated with the Tris, glycerol, NaCl, EDTA, β-mercaptoethanol buffer described above. Fractions (1 ml) were screened by SDS-PAGE and fractions containing the protein were pooled, then concentrated using Amicon Ultra (Ultrocel-3K) centrifugal filters (Millipore). Analysis by mass spectroscopy verified that the protein was completely dephosphorylated (calculated = 66711.8; observed = 66710.7).
A clone encoding EINtr with the active-site His changed to Ala (H356A) was constructed (see Fig. 1). The details are described in Supplementary Data. The protein was expressed in both H2O and D2O media and purified by the same procedure as described above for the enzyme without the mutation. The yield of purified protein was 25 mg (H) from a 1 liter culture and 7.5 mg (D) from a 500 ml culture. The expressed protein from the H2O culture was analyzed by mass spectroscopy and shown to contain a fraction with a mass of 66,646.7 Da, corresponding to the mutated protein devoid of the N-terminal Met (calculated = 66,645.7; see Fig. 1).
The purified proteins were analyzed to determine the degree of deuteration (Table I). Since the carbon source (glucose) was not deuterated, the expected level of deuteration was in the vicinity of 90% rather than 100% [11]. For the wild-type form of EINtr, the predominant form isolated was both phosphorylated at the active site and gluconoylated at the site of the His tag [12] (calculated mass of the H form = 66,969.8)(Table I). The measured mass of that form corresponded accurately to the calculation. The measured mass of the deuterated form was 70,223.8 which corresponds to 89% deuteration. This matches very well to the expected level of deuteration under the culture conditions used.
Table I.
Mass Spectrometric Analysis of Deuterated (D) and Hydrogenated (H) EINtr
| H | D | % Deuteration1 | |
|---|---|---|---|
| EINtr (phospho-form) | |||
| Calculated mass | 66,969.82 | 70,619.83 | |
| Measured mass | 66,969.5 | 70,223.8 | 89 |
| EINtr, Dephosphorylated 4 | |||
| Calculated mass | 66,889.85 | 70,609.83 | |
| Measured mass | 66,890.7 | 70,213.6 | 89 |
| EINtr (H356A) | |||
| Calculated mass | 66,823.75 | 70,473.73 | |
| Measured mass | 66,823.8 | 70,039.2 | 88 |
| Active Site Peptide, P350–362 6 | |||
| Calculated mass | 1,312.5 | 1,377.5 | |
| Measured mass | 1,312.5 | 1,370.0 | 88 |
Mass analysis of the active site peptide and the intact proteins was carried out as described in Methods.
The % deuteration is expressed as (Measured mass of D-calculated mass of H/calculated mass of D-calculated mass of H).
calculated mass corresponds to the protein (mass=66,711.6) which is both phosphorylated (adds 80 mass units) at the active site and gluconoylated [12] (adds 178 mass units) at the site of the His tag sequence.
The calculated mass of D is expressed as the mass of the protein in which all nonexchangeable hydrogens are replaced by deuterium (perdeuterated protein).
Dephosphorylation was accomplished by treatment with pyruvate and MgCl2 (see methods).
Calculated mass corresponds to the protein (mass=66,711.6) which is gluconoylated (adds 178 mass units).
Calculated mass corresponds to the protein (mass=66,645.7) which is gluconoylated (adds 178 mass units).
The active site peptide sequence is DGAANSHAAIMVR. The histidine residue is the site of phosphorylation.
The experiments for testing the effect of deuteration on autophosphorylation activity required the use of dephosphorylated protein. Dephosphorylation was accomplished by pyruvate treatment. Mass analysis showed that a byproduct of this treatment is that the protein can form an adduct (Schiff base) with pyruvate that increases the mass by 70 units (data not shown). The species analyzed was that of the gluconoylated form (calculated mass=66,889.8). The level of deuteration, as expected, remains at 89% (Table I).
For the form of EINtr mutated at the active site (EINtr(H356A)), the major form of the protein observed was gluconoylated (mass=66,823.7) and the level of deuteration was 88% (Table I).
We explored the possibility that some component of the measured deuteration might be due to exchangeable deuterium and that the level of nonexchangeable deuteration was less than approximately 90%. In order to evaluate this possibility, the level of deuteration of a tryptic peptide (the active site peptide) was measured (Table I); since such a peptide would be completely unfolded, all exchangeable deuterium would be replaced by hydrogen. The analysis of this peptide showed that the level of deuteration matched that seen in the protein preparations. Thus, all of the observed deuteration is in stable nonexchangeable positions.
Mass spectroscopic analysis also detected a truncated form(s) in both the H- and D- preparations of the wild-type and mutant forms of EINtr. There is a pair of arginine residues occupying the 11th and 12th positions from the C-terminal end of the protein (see Fig. 1). The calculated masses of protein products truncated at the C-terminal side of either of these arginine residues is close to the observed masses of the truncated forms of the proteins. Thus, it appears that EINtr becomes partially truncated by a trypsin-like activity in vivo. The extent of truncation is slightly greater for the deuterated species compared to the hydrogenated forms, a clue to the observed greater susceptibility of the deuterated protein to proteolysis in vitro.
Tryptophan mutants of EINtr
For mapping studies of fluorescence effects in EINtr, the 4 Trp residues of EINtr(H356A) were mutated to Phe by changing the Trp codons of W186, W270, W319 and W522 (Figure 1) to UUU, corresponding to Phe. The pET28a-EINtr(H356A) DNA was used as the template for QuikChange (Stratagene) mutagenesis. For unexplained reasons, we were unable to isolate a clone harboring the W270F mutation. The other 3 mutants were verified by mass analysis of the purified EINtr proteins, which were purified by the same methods as described for the native protein.
Arginase
Purified human arginase I was a gift from Dr. David Christianson [13]. Paste from cells expressing perdeuterated human arginase I was kindly provided by Kate Thorn from the Christianson laboratory; the perdeuterated protein was prepared according to the published procedure [13].
Synuclein
E. coli harboring a pET41a hybrid plasmid expressing full-length α-synuclein was a kind gift from Dr. Nelson Cole [14]. Expression and purification of this protein are described in Supplementary Data. In the course of examination of the fractionation properties of α-synuclein on a reverse phase column, the observation was made that the perdeuterated form of the protein eluted earlier than did the hydrogenated form (Fig. 1 of Supplementary Data). This is evidence that deuterated proteins are less hydrophobic than hydrogenated forms of the same protein.
Methods
Mass spectrometry
HPLC-mass spectrometric analysis of tryptic peptides: The assay for phosphotransfer from EINtr to NPr depended on HPLC separation of EINtr tryptic peptides followed by mass spectrometric analysis. An Agilent model 1100 HPLC with a Zorbax 300SB-C18, 1.0 × 50 mm 3.5 micron (Agilent Technologies) column flowing at 20 μl/min was used. The initial solvent was 0.05% trifluoroacetic acid with gradient elution by acetonitrile/0.05% trifluoroacetic acid (TFA) increasing at 2%/min from 0 to 70%. The effluent from the spectrophotometric detector was mixed in a tee (Upchurch M540) with 20 μl/min acetic acid pumped by another model 1100 pump, and the mixture was introduced into a model G1969A mass spectrometer (Agilent)[15] with time of flight detector. The mass range from 145 to 2000 amu was scanned at 10,000 transients/scan. Voltages were: capillary, 5000 V; fragmentor, 235 V; skimmer, 60 V; and octopole RF, 250 V. The ion source gas temperature was 350 °C, drying gas flow was 10 l/min, and nebulizer gas pressure was 10 psig. Reference mass correction utilized an infused standard of 922 amu and metal-acetic acid clusters from 173 to 537 amu generated in the electrospray [16]. For direct infusion analysis of intact proteins without a column, the only change in the HPLC-MS method was that the HPLC pumped 0.05% TFA without a gradient.
Differential scanning calorimetry (DSC)
DSC measurements were carried out using a VP-DSC calorimeter (MicroCal) as previously described [17]. The scan rate was 30 °C/h. The data was corrected for instrument baselines. Data analysis was performed using Origin software (MicroCal). Excess heat capacity (Cp) was expressed in kcal K−1 (mol monomer)−1, where 1000 cal = 4.184 J.
Isothermal Titration Calorimetry
Binding studies were performed at 20 °C in 20 mM sodium phosphate, pH 8, 100 mM NaCl, 5 mM MgCl2 in a VP-ITC titration calorimeter (MicroCal, LLC, Northampton, MA, U.S.A.) with a reaction cell volume of 1.4 ml. Typically, about 15 μM EINtr in the reaction cell was titrated with a stock solution of about 400 μM NPr contained in a 250 μl syringe. Consecutive injections corresponded to ~8–9% of the total [EINtr] in the ITC cell at 6- to 8-minute intervals, while stirring the protein solution at 260 rpm constant speed. The heat of dilution of NPr was determined by averaging the final 5–10 injections. Titration curves were analyzed with the Origin program provided by MicroCal, LLC, using one or two classes of binding sites to fit the curves.
Fluorescence Measurements
Fluorescence measurements (excitation at 295 nm and emission at 340 nm) were made using a QM-6SE (PTI, London, Ontario) spectrofluorometer. Reaction mixtures (1 ml volume) were maintained at 37 °C under constant stirring. Sample temperature was controlled by a Peltier sample holder. All spectral measurements were performed with Glan polarizers at magic angle conditions and excitation wavelength 295 nm for the intrinsic tryptophan residue fluorescence measurements. Aliquots of concentrated stock solutions of either PEP or NPr were added as indicated in the respective figures and the fluorescence change was recorded. The changes in fluorescence were corrected for dilution resulting from addition of NPr or PEP.
Light scattering measurements
A DAWN EOS (Wyatt Technology, Santa Barbara, CA) equipped with a flow cell and a 30 mW linearly polarized GaAs laser of wavelength 690 nm was used with measurements in the in-line flow mode. An HPLC pump was used to deliver filtered solvent through a Phenomenex BioSef-SEC-S2000 gel filtration column. Protein samples were injected in a volume of 100 μl. Both the light scattering unit and the refractometer (Waters 2419 refractive index detector) were calibrated as per the manufacturer's instructions. A value of 0.185 ml/g was assumed for the dn/dc of the protein. Light scattering data were from 11 detectors ranging from 50.0° to 134° (detectors 6 through 16). The detector responses were normalized by measuring the signal from monomeric bovine serum albumin. The flow rate was maintained at 0.5 ml/min. The traces from the DAWN EOS and refractometer were analyzed with ASTRA-V software to obtain the weight-average molecular weight (Red lines).
Circular dichroism
Circular dichroism measurements were carried out as described [18].
Autophosphorylation of EINtr
For the kinetic experiments, the stock solutions of both EINtr preparations contained 21.4 pmoles of autophosphorylatable protein (dephosphorylated EINtr) in each 15 μl stop-quench shot. The EINtr solutions contained 10 mM Tris·Cl, pH 8, 10% glycerol, 100 mM NaCl, 1 mM EDTA, and 2 mM β-mercaptoethanol. The 32P-PEP solution (see Materials) contained 75 mM Tris·Cl, pH 8, 15 mM MgCl2, 15 mM β-mercaptoethanol and 300 μM PEP (specific activity ~400 cpm/pmole). The quench solution was 4× SDS Laemmli sample buffer, 30 mM EDTA. Quench-flow experiments were carried out in a KinTek Corp. Model RQF-3 Quench-Flow instrument at 37 °C where one syringe was loaded with EINtr solution, another with 32P-PEP solution and the third with quench solution. Each shot contained 15 μl of the EINtr and PEP solutions, delivered by a KdS Pico Plus syringe pump. The volume of each quenched sample was determined; 60 μl aliquots were deposited onto 10-well 4–20% Tris-glycine gradient gels (Invitrogen). Electrophoresis was carried out for 1.5 hours at 130 volts. The gels were stained for 40 min with Coomassie Blue R250, then destained for 30 min. The stained bands were excised from the gels and counted with 5 ml of scintillation fluid. From the cpm observed for the 60 μl aliquot, the volume of the original quenched sample, the specific activity of the 32P-PEP and the total pmoles EINtr (21.4), the % of phosphorylation was calculated.
Phosphotransfer from P-EINtr to NPr
Cocktails were prepared in a 200 μl volume containing: Tris·Cl, pH 7.5, 60 mM; MgCl2, 1.2 mM; KCl, 12 mM; EINtr (H or D), 11.5 μg. Aliquots (25 μl) were dispensed into tubes and reactions were initiated by the addition of NPr 1–85 (5 μl containing a 10-fold molar excess of NPr, 2.1 μg). Incubations were at room temperature. At the indicated times, reactions were terminated by the addition of 30 μl of quench solution (5 M urea, 3 M KOH). After adding 300 μl of H2O and 300 μl of 1 M Tris·Cl, pH 8, the samples were added to 100 μl of washed Talon beads and incubated with shaking for 1 h at 4 °C. The beads were then washed 4 times with 0.5 ml aliquots of 25 mM Tris·Cl, pH 8, 200 mM NaCl. The washed beads were suspended in 40 μl of 25 mM Tris·Cl, pH 8, 200 mM NaCl, 100 mM imidazole and 1 μg trypsin was added. After incubation at room temperature for 1 h with periodic shaking, the Talon beads were centrifuged and a 25 μl aliquot of the supernatant solution was transferred to a vial for analysis by mass spectrometry. The fraction of the active site peptide in the phosphorylated form is plotted against the incubation time.
Results and Discussion
1.1 Proteolysis of EINtr
Analysis of the progress of tryptic hydrolysis of EINtr(H356A) (see Fig. 1) by mass spectroscopy enabled a description of the pathway of degradation of the protein (see Fig. 2 of Supplementary Data). The initial attack on the His-tagged protein resulted in elimination of the major fraction of the His-tag (peptide with mass of 1767.84). Subsequently, the residual protein (mass of 64,894.9) was split into an N-terminal fragment (mass=39,794.4) and a C-terminal fragment (mass=25,118.5). Next, the C-terminal fragment was degraded to two fragments (masses of 12,688.9 and 12447.6). The N-terminal fragment was not further degraded during the time course of the experiment (90 min). The unambiguous identification of the location of the fragments within the EINtr sequence (Fig. 2 of Supplementary Data) was possible because of the mass accuracy of the analyses.
A previous description of proteolysis by trypsin of EI [19, 20] demonstrated the stable accumulation of the N-terminal portion of the protein (peptide 1–255, mass=27833). A local alignment of EI and EINtr in the region corresponding to loop 3 [2] of EI (Fig. 3 of Supplementary Data) shows that the region around R523 of EINtr is almost identical in the EI sequence, but the region around K255 of the EI sequence does not have a corresponding trypsin site in the EINtr sequence. Consequently, it seems reasonable to propose that, for EI, the initial cleavage site is at R358 followed by cleavage at K255 resulting in the accumulation of EIN. In the case of EINtr, the initial cleavage is at R523; since there is no trypsin site corresponding to K255 of EI, there is no further proteolysis in the N-terminal region of EINtr. Therefore, EI and EINtr have the similar property of having a stable N-terminal region; however, the two proteins differ in the precise region of the linker between the N-terminal and C-terminal domains that are susceptible to trypsin. This may point to one of the factors that determine the difference in specificity [8] of the two proteins.
Based on the mass analysis of the purified preparations of EINtr (see Materials and Methods), we suspected that EINtr in the deuterated form was more susceptible to in vivo proteolysis. Consequently, a kinetic comparison was made of H-EINtr and D-EINtr with respect to the sensitivity to limited proteolysis by trypsin (Fig. 2). In order to avoid the complication of analysis of a mixture of phospho- and dephosphopeptides, the inactive H356A mutant of EINtr (see Fig. 1) was used for the study; this active site mutant is incapable of becoming phosphorylated. The kinetics of proteolysis were followed over a period of 90 min. SDS-PAGE analysis clearly shows that the rates of disappearance of the Coomassie Blue stained band corresponding to the intact protein (uppermost arrow in Panel A) are different for the two proteins (Panel A); D-EINtr is degraded faster than is H-EINtr. Inclusion of an excess of NPr (Panel B) in the proteolysis reaction does not protect EINtr from proteolysis although the kinetics of scission, producing intermediate products (panels E through H) is somewhat influenced by the presence of NPr. This study provides further evidence that D-EINtr has a less stable structure than H-EINtr.
Fig. 2. Limited proteolysis of EINtr.

Approximately 65 μg of either H-EINtr(H356A) or D-EINtr(H356A) was incubated with 0.1 μg trypsin in 10 mM Tris·Cl (pH 8), 100 mM NaCl, 2 mM β-mercaptoethanol, 1 mM EDTA and 10 % glycerol in a total volume of 75 μl at room temperature. A companion incubation mixture was supplemented with 56 μg of NPr 1–85. At the indicated times (0 to 90 mins, shown above the lanes), 7.5 μl aliquots were removed and mixed with 7.5 μl of 2× Laemmli sample buffer. The samples were run on 4–20% SDS-PAGE gradient gels (Invitrogen), then stained with Coomassie Blue R250 (panels A & B). Lanes labeled H correspond to H-EINtr and those labeled D correspond to D-EINtr. The stained gels were scanned in an Odyssey near infrared fluorescence scanner (Li-Cor Biosciences) and then the fluorescence of Coomassie Blue [41] was quantitated by the Odyssey software. The intensity of the bands (panels C through H) were measured after subtraction of the background of the gels. Panels C & D, patterns of the degradation of the intact protein, designated by the topmost arrowhead on the left side of panel A; panels E & F, relative amounts of the ~40 kDa peptide (identified as including residues 170–523, mass of 39,794.4, see fig. 2), designated by the center arrowhead; panels G & H, relative amounts of the ~25 kDa peptide (G524-L748, see fig. 2), designated by the lowest arrowhead. Solid lines with squares = H-EINtr; dashed lines with circles = D-EINtr.Panels A, C, E, and G correspond to proteolysis in the absence of NPr; panels B, D, F and H correspond to proteolysis in the presence of NPr.
There is precedent for some perdeuterated proteins exhibiting less stability in proteolysis experiments. In 1965, Hattori et al [21] reported that phycocyanin with deuterated side-chains exhibited a destabilization resulting in an increased sensitivity to in vitro proteolysis. They argued that deuteration of nonexchangeable hydrogen positions decreases nonpolar side-chain interactions with an effect on the conformational integrity of proteins. This was the result of less crowding, leading to reduced hydrophobic interactions and, possibly, increased dynamics. They proposed that the initial step in tryptic hydrolysis required substrate denaturation necessary to overcome the steric hindrance to hydrolysis.
In another study, H- and D-forms of the 26 kDa homodimeric glutathione S-transferase were compared for susceptibility to proteolysis by chymotrypsin; the perdeuterated protein was hydrolyzed considerably faster than the hydrogenated form [11]. It was suggested that barriers to local unfolding, presumably a necessary requirement for proteolysis, are reduced in the deuterated protein. In summary, there are several precedents for the observation that perdeuteration of some proteins may be coupled to increased susceptibility to proteolysis.
1.2 Proteolysis of Human Arginase
We posed the question of whether a protein that has been shown to have an unchanged three dimensional structure upon perdeuteration would also show an increased susceptibility to proteolysis. Human arginase I, in complex with the substrate analog 2(S)-amino-6-boronohexanoic acid (ABH) has been crystallized in both the unlabeled and perdeuterated forms [13]. The findings were that the structure of the arginase I-ABH complex was not changed by perdeuteration. We studied the sensitivity to tryptic hydrolysis of the two forms of arginase I. Initial studies of the proteolysis by trypsin indicated that both forms of the protein (a gift from Dr. David Christianson) were essentially completely resistant to hydrolysis. The protein has been shown to contain a binuclear manganese cluster [22] which plays an important role in stabilizing the structure [23]. Thus, the form of the protein which was crystallized was stabilized by both manganese as well as the substrate analog. However, when we tested the hydrogenated and perdeuterated forms of arginase for proteolysis by trypsin in the presence of 1 mM EDTA, which released manganese from the arginase, hydrolysis was observed (Fig. 4 of Supplementary Data). Similar to the case with EINtr, the perdeuterated form of the protein exhibited a greater sensitivity to trypsin than did the normal form of arginase. As pointed out by Brockwell et al [11], the stability of stable proteins appears unaffected by deuteration. However, when arginase was somewhat destabilized, it was possible to detect a stability difference associated with deuteration. This is in keeping with the notion that hydrophobic interactions are diminished by deuteration.
1.3 Proteolysis of α-synuclein
The observation that perdeuteration of both EINtr and arginase resulted in a destabilization as reflected in sensitivity to tryptic hydrolysis introduced the possibility that deuterated polypeptides were intrinsically better substrates for trypsin. To evaluate this possibility, we studied the properties of α-synuclein, which has been shown to be an intrinsically disordered protein in solution [24]. Full-length α-synuclein, expressed in E. coli, was prepared in both hydrogenated and perdeuterated forms (see Materials and Methods and Supplementary Data). The kinetic study of tryptic hydrolysis (Fig. 5 of Supplementary Data) showed that the rates are identical for the two forms of the protein. Thus, the possibility that a deuterated polypeptide is a better substrate for trypsin than a hydrogenated peptide was eliminated. Taken together, the experiments reported herein support the idea that perdeuteration of EINtr, as well as arginase, results in structural destabilization as evidenced by a change in sensitivity to trypsin digestion. How the structural destabilization affects the three-dimensional structure of a protein is uncertain and may vary from protein to protein.
2.1 Thermal Unfolding of EINtr
Previous studies by differential scanning calorimetry of EI unfolding described an unfolding pattern with a midpoint at about 52 °C [17]. A comparable study was performed with EINtr and a similar unfolding profile was observed, suggesting that the stability of EI and EINtr is similar (Fig. 3). Then, a comparison was made of the unfolding properties of perdeuterated EINtr (D) and hydrogenated EINtr (H). Thermal unfolding was studied through the range of 20 to 70 °C. The Tm values for unfolding were calculated by determination of the mid-points of the unfolding curves. For the fluorescence measurements (Panel A), D-EINtr = 48.88 °C; H-EINtr = 52.58 °C; deuterium dependent destabilization = 3.7 °C. For the calorimetry experiment (Panel B), the deuterium dependent destabilization was 3.4 °C. The effect of interaction of EINtr with the ligand PEP on the stability of the two forms was also evaluated (panel A). D−P = 52.84 °C; stabilization due to PEP binding = 3.96 °C; H−P = 56.62 °C; stabilization due to PEP binding = 4.04 °C. Unfolding led to aggregation and precipitation of the proteins. Effects of perdeuteration of proteins on thermal stability have previously been noted. P450cam displays a reduction of 4–5 °C [25], glutathione-S-transferase [11] a reduction of 3.9 °C and phycocyanin [21] a reduction of 7 °C. Thus, similar to the case with sensitivity to proteolysis, the effect of perdeuteration on unfolding of some proteins is demonstrable with an effect on thermal stability.
Figure 3. Thermal unfolding of EINtr.

Panel A: Thermally induced changes in fluorescence. EINtr(H356A) in the H and D forms (25 μg/ml) in 20 mM Tris·Cl (pH 8), 2 mM MgCl2, 5 mM NaCl, 0.1 mM β-mercaptoethanol, 0.05 mM EDTA and 0.5% glycerol in a total volume of 1 ml were scanned through the indicated temperature range at a scan rate of 30 °C/h. Where indicated (H–P and D–P), the samples also contained 150 μM PEP. Excitation was at 295 nm and emission at 340 nm. Panel B: DSC scans (see Materials and Methods) of EINtr(H356A) H and D forms. The proteins were dialyzed against 10 mM NaPO4, pH 8 and adjusted to a concentration of 300 μg/ml.
3.1 Oligomerization State of EINtr
Previous studies on EI of the carbohydrate PTS, a paralog of EINtr [10], have demonstrated that the protein exhibits a monomer-dimer interconversion [17, 26, 27] and that dimerization is promoted by PEP. Early in purification attempts, differences in behavior of D-EINtr were observed. A change of buffer from pH 8 to pH 7 frequently led to precipitation of D-EINtr; long term storage of D-EINtr (at 4 °C in 25 mM phosphate pH 8, 100 mM NaCl, 5 mM MgCl2) also resulted in protein precipitation. In contrast, H-EINtr was stable under these conditions. Consequently, we examined the oligomerization and aggregation properties of the two forms of EINtr (Fig. 4). SDS-PAGE analysis (data not shown) indicated that both forms were essentially homogeneous under denaturing conditions. Gel filtration chromatography coupled to light scattering analysis of H-EINtr (Fig. 4) showed that, at the protein concentration studied (about 20 times higher than the fluorescence study of Fig. 3A), H-EINtr was mainly monomeric. When the gel filtration column was pre-equilibrated with 150 μM PEP, H-EINtr became mainly dimeric (see red lines). It therefore appears that EINtr, similar to EI, exhibits a monomer-dimer equilibrium which is shifted in the direction of dimer by PEP. Gel filtration analysis of D-EINtr (Fig. 4 inset) demonstrated another significantly different property of that protein. It was obviously polydisperse with most of the protein eluting in the excluded volume. The conclusion from this study is that deuteration of EINtr converts the protein from a relatively simple mixture of the monomeric and dimeric forms to a collection of many other species of oligomers. At this concentration and pH, addition of PEP to perdeuterated protein results in further aggregation and tendency to precipitate (data not shown).
Figure 4. Oligomerization state of EINtr.

Light scattering analysis of H-EINtr was carried out as described in Materials and Methods. The gel filtration column was equilibrated with 20 mM Tris pH 8, 2 mM MgCl2, 150 mM NaCl. 100 μl of protein at 0.5 mg/ml was injected. When the protein was run in the presence of PEP, the column was pre-equilibrated with the buffer described above supplemented with 150 μM PEP. An aliquot (100 μl of protein at 0.5 mg/ml in the PEP supplemented buffer) was injected onto the column. The masses of the different regions (denoted by the red line) of the eluate were determined. Inset: Light scattering analysis of D-EINtr. A 100 μl sample of protein (0.5 mg/ml) was injected onto the column equilibrated with the Tris, MgCl2, NaCl buffer described above. The horizontal scale is time and the vertical scale is light scattering intensity.
Previous results dealing with the effect of perdeuteration on the associative properties of proteins showed a nonuniform picture. In the case of phycocyanin [21], deuteration resulted in a decreased tendency to associate into trimer and hexamer forms; in contrast, for glutathione-S-transferase [11], deuteration promoted dimer association. Consequently, it appears that the effect of perdeuteration on protein associative properties is unpredictable.
4.1 Secondary and Tertiary Structure of EINtr
Possible changes in secondary and tertiary structure associated with deuteration of EINtr were explored by circular dichroism (CD) spectroscopy. The typical far UV spectroscopy study showed little difference in the two forms of the protein indicating no major secondary structure differences (Fig. 6 of Supplemental Data). In contrast, the near UV region (Fig. 5A) showed significant spectral differences suggesting changes to the tyrosine and tryptophan tertiary contacts, but not for the phenylalanine residues. The addition of PEP/Mg resulted in similar spectra (Fig. 5B) of the two forms of the protein. These data are consistent with the interpretation that the deuteration-dependent conformational change is due to changes in tertiary structure in the vicinity of one or more Trp and Tyr residues.
Figure 5. Near UV circular dichroism of EINtr.

The H and D forms of EINtr at 0.6 mg/ml were scanned (16 accumulations) in a Jasco J-715 spectrometer. In panel B, PEP (150 μM) and MgCl2 (2 mM) were added.
The previous work on P450cam [25] showed that deuteration led to a decrease in α-helix content and an increase in the unordered content; the authors nevertheless concluded that there was little effect on the structure and dynamics of the protein associated with deuteration.
5.1 Enzymatic Activities of EINtr
5.1.1 Autophosphorylation
In light of the preceding observations concerning the decrease in stability of EINtr associated with deuteration, it was of interest to explore the effect of deuteration on the catalytic activity of EINtr. In contrast to the previous experiments which utilized the active site mutant of EINtr, this experiment required the wild-type protein. The H and D forms of the wild-type protein were expressed and purified in the same manner as were the mutated forms. Since the proteins as isolated were mainly in the phospho-form (Table 1), they were subjected to a dephosphorylation procedure (see Materials and Methods). A kinetic study of the autophosphorylation by 32P-PEP of the two forms was carried out using a stop-quench instrument (Fig. 6). Half-maximal autophosphorylation was achieved in approximately 120 msec. The results indicate no discernable difference in the kinetics of autophosphorylation of either form of the protein. The experiment was carried out at 37 °C, a temperature well below that at which unfolding takes place. Thus, although D-EINtr is considerably less stable than H-EINtr, catalytic activity for autophosphorylation appears to be unaffected. This is not surprising, since numerous studies [28, 29] have indicated variable or no effects on catalytic activity associated with structural variation.
Figure 6. Autophosphorylation of EINtr.

Preliminary autophosphorylation experiments (with 5 and 10 min incubation periods) were carried out to determine the maximum autophosphorylation of EINtr stocks of the dephosphorylated (see Materials and Methods) H and D forms. Kinetic studies were carried out as described in Methods. Filled circles, H-EINtr; filled squares, D-EINtr. The inset to the figure displays the same data with the time scale plotted logarithmically to clarify the kinetics.
5.1.2 Phosphotransfer to NPr
In addition to the ability of EINtr to be autophosphorylated by PEP, the phosphorylated protein is able to perform phosphotransfer to an active site histidine of NPr. Studies were carried out to evaluate the effect of deuteration on the capability of EINtr to interact with and make a phosphotransfer to the acceptor, NPr.
As isolated, the purified preparations of both H- and D-EINtr were at least 80% phosphorylated. These preparations were therefore suitable for in vitro experiments designed to measure the phosphotransfer to NPr (Fig. 7). The experiments used a 10-fold molar excess of NPr to drive the phosphotransfer reaction. Equilibrium was reached in approximately one min. It was noteworthy that the phosphotransfer reaction was very much slower than was the autophosphorylation reaction. Fifty percent completion of phosphotransfer was accomplished in approximately 20 sec. There was no detectable difference in the kinetics of the phosphotransfer reaction for the two forms of EINtr.
Figure 7. Phosphotransfer from P-EINtr to NPr.
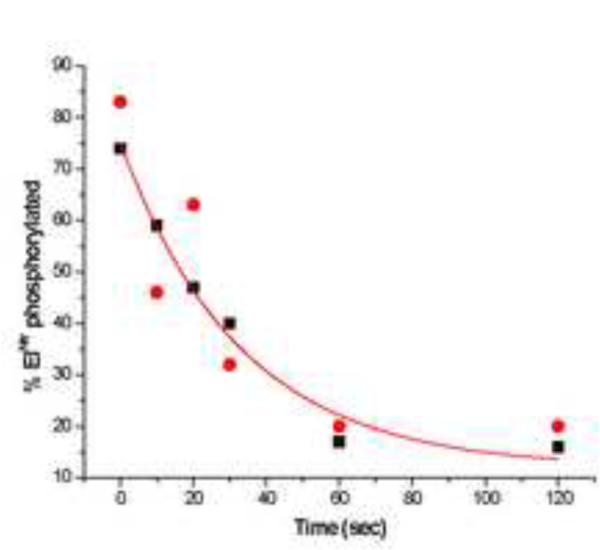
Kinetics of the phosphotransfer were carried out as described in Methods. Filled circles, D- EINtr; filled squares, H-EINtr.
Studies on the effect of deuteration on the catalytic properties of other proteins have shown a variety of differences, if any. Glutathione-S-transferase showed an increased rate of catalysis associated with deuteration [11]. In the case of ribulose bisphosphate carboxylase [30], the activity of the deuterated form was only 28% that of the hydrogenated form and this functional defect was recovered by the addition of a chaperone. It was concluded that the deuterated enzyme has a defective folding relative to the hydrogenated form. In contrast, deuteration had no significant effect on the reactivity of cytochrome c peroxidase [31] or human arginase I [13].
6.1 Ligand Binding Studies
6.1.1 Interaction with PEP
Although the capability of EINtr to be phosphorylated by PEP was unaffected by deuteration (Fig. 6), additional biophysical methods were used to study the interaction of EINtr with PEP. In order to avoid the complication of PEP binding followed by phosphorylation, the active site mutant, H356A, which is capable of binding PEP but not phosphorylation, was again used.
EINtr(H356A) contains 4 Trp residues, located at positions 186, 270, 319, and 522 (numbering as for the intact full-length protein; see Figure 1). Based on the likely similarity to the structure of EI of the carbohydrate PTS [2], Trp 270 was assumed to be in the NPr binding domain, Trp 186 and 319 in the His domain and Trp 522 in the PEP binding domain. Measurements of intrinsic Trp fluorescence have been widely used to monitor protein environment and conformational changes [32–34]. The emission properties are particularly sensitive to solvent polarity, microenvironment viscosity and molecular mobility.
At 0.5 μM, the two forms of the H356A protein exhibit similar fluorescence properties: λmax~340 nm and comparable quantum yields (see legend to Fig. 8). The effect of PEP concentration on Trp fluorescence of H- and D-EINtr is shown in Fig. 8A. Upon sequential additions of PEP at 37 °C, Trp fluorescence intensities increase and reach saturation indicating complete binding. Interestingly, the titration data show a significant difference in the relative fluorescence increase in the two proteins upon PEP binding. The fluorescence change of D-EINtr was approximately twice as great as that of H-EINtr. Further, the dissociation constants (Kd) suggest that the binding affinities could be modulated by deuteration (Kd(H-EINtr)=21 μM vs.Kd (D-EINtr)=33 μM). While the exact nature of the differences in quantum yield changes are generally difficult to determine directly, the greater fluorescence increase associated with PEP binding to D-EINtr could reflect restriction of local mobility of one or more Trp residues in D-EINtr. Alternatively, changes in local conformation can lead to differences in nonradiative relaxation (i.e., solvent exposure). Notably, the PEP effect on Trp fluorescence is highly dependent on the protein concentration (Fig. 8C); higher protein concentrations result in smaller PEP-dependent increases in fluorescence. It is likely that the Trp residues are sensitive to the monomer-dimer equilibrium and the apparent differences in PEP interaction may be attributable to changes in the oligomerization state.
Figure 8. Effect of PEP and NPr concentration on EINtr fluorescence.



(A) PEP titration: Reaction mixtures (1 ml volume) contained: Tris·Cl, pH 8, 20 mM; MgCl2, 2 mM, EINtr(H356A), 0.5 μM. The baseline fluorescence intensities were: H, 4960; D, 5150. Fluorescence at 340 nm was measured at 37 °C after the indicated additions of PEP. The increase in fluorescence is plotted against the PEP concentration. An incubation mixture without added EINtr showed little fluorescence and no change in fluorescence after addition of PEP (data not shown). H form, squares; D form, circles. The measured fluorescence intensities were expressed as the % increase in fluorescence over the baseline. (B) NPr titration: Reaction mixtures (1 ml volume) contained: Tris·Cl, pH 8, 20 mM; MgCl2, 2 mM, EINtr(H356A), 0.5 μM. The baseline fluorescence intensities were: H, 5985; D, 4986. Fluorescence at 340 nm was measured at 37 °C after the indicated additions of NPr. The increase in fluorescence is plotted against the NPr concentration. An incubation mixture without added EINtr showed some fluorescence change after addition of NPr (data not shown). NPr has no Trp or Tyr residues but does have 3 Phe residues. The sequence is: MTVKQTVEITNKLGMHARPAMKLFELMQGFDAEVLLRNDEGTEAEANSVIALLMLDSA KGRQIEVEATGPQEEEALAAVIALFNS. The data were corrected for this change due to the NPr fluorescence. H-EINtr, filled squares; D-EINtr, open circles. The measured fluorescence intensities were expressed as the % increase in fluorescence over the baseline. (C) Effect of H-EINtr concentration on the PEP-dependent fluorescence change. Experimental conditions were as described above, except that PEP, where added, was at 2 mM. The EINtr(H356A) concentration was varied, as indicated.
Fluorescence quenching [35, 36] has been widely used to determine the solvent exposure of fluorophores. It is assumed that quenching cannot occur when a fluorophore like Trp is located in the interior of a protein. Further insight into the relative conformational changes produced by interaction of PEP with either H- or D-EINtr was gained by a study of quenching by iodide under various conditions (Fig. 9). The effect of iodide at concentrations up to 1 M on Trp fluorescence in the absence or presence of 2 mM PEP was measured. In both cases (absence or presence of PEP), the sensitivity of D-EINtr to iodide quench was greater than for H-EINtr. This indicates that at least one of the 4 Trp residues of EINtr is more solvent exposed in D-EINtr than in H-EINtr, further evidence for a conformational difference associated with deuteration.
Figure 9. Iodide quench of the fluorescence of EINtr.

Incubation mixtures (150 μl total volume) contained Tris·Cl, pH 8, 20 mM; KI and KCl, the sum of which was always 1 M; and EINtr(H356A) in the H form (0.98 μM) or D form (1.36 μM). Fluorescence was measured with excitation at 295 nm and emission at 340 nm. Fluorescence quenching was analyzed according to the Stern-Vollmer relationship (F0/F1) [35] where F0 is the fluorescence in the absence of quencher and F1 is the fluorescence in the presence of the indicated quencher concentration. Filled circles, D-EINtr; filled squares, H-EINtr. Upper panel, absence of PEP; lower panel, presence of 2 mM PEP and 2 mM MgCl2.
To gather some insight into the region of the protein involved with PEP binding, further fluorescence measurements were made using Trp to Phe mutants of EINtr (Fig. 10A). As predicted above, the region in the vicinity of Trp522 appears to be important in the interaction with PEP. Mutations of Trp residues 186 or 319 have little effect on the fluorescence increase associated with PEP binding, while mutation of Trp522 essentially abolishes the PEP effect on fluorescence.
Figure 10. Fluorescence effects on PEP and NPr binding to Trp mutants of EINtr.


(A) PEP titration: Incubation conditions were as described for Fig. 13, except that the final concentrations of all the EINtr proteins used was 82 μg/ml. The baseline fluorescence intensities were: wild-type (filled squares), 42622; W186F (inverted triangles), 41324; W319F (triangles), 37487; W522F (circles), 28078. (B) NPr titration: incubation conditions were as in (A). The baseline fluorescence intensities were: wild-type, 42622; W186F, 72761; W319F, 87741; W522F, 51338.
6.1.2 Interaction with NPr
A fluorescence experiment similar to that shown in Fig. 8A was carried out; in this case, the interaction of the two forms of EINtr with NPr was studied. The results with NPr interaction (Fig. 8B) were different than those for interaction with PEP (Fig. 8A). The fluorescence increases for binding of NPr to either form of the protein were similar. It is noteworthy that the extent of fluorescence change due to addition of NPr was similar to that seen for the addition of PEP to H-EINtr (Fig. 8A); the major difference in fluorescence changes was associated with the effect of PEP on D-EINtr. The calculated Kd values for binding NPr to EINtr forms did differ, however; for H-EINtr, the Kd was 23 μM and for D-EINtr, it was 43 μM. Thus, overall, it seemed that deuteration of EINtr has a more profound effect on the interaction with PEP than for interaction with NPr.
Previous studies on EI of the carbohydrate PTS indicated that the isolated amino-terminal half of the protein (termed EIN) had the capability to bind the phosphoacceptor protein HPr [2, 37]. We established that the comparable EIN construct of EINtr was capable of binding NPr (unpublished studies of AP and E. Fodor) A study was therefore carried out (Fig. 10B) to determine the effect of NPr concentration on the fluorescence of the collection of Trp-Phe mutants that were used for the analogous experiments for PEP binding (Fig. 10A). Contrary to expectation, the Trp mutants in the amino-terminal half of the EINtr demonstrated binding curves similar to the wild-type protein; only the W522F protein had lost the capability to show a Trp fluorescence response to NPr binding. These data suggest that the binding of NPr to the aminoterminal region of EINtr promotes a conformational change in the loop 3 region encompassing W522.(see Figure 3 of Supporting Data). It is noteworthy that binding of HPr to EIN is not associated with a significant structural change in either protein [37]. Therefore, the absence of fluorescence effects on W186 or W319 attendant on NPr binding is not surprising. A further exploration of the characteristics of interaction of NPr with both H- (Fig. 11A) and D- (Fig. 11B) EINtr was carried out by isothermal titration calorimetry. In both cases, the binding data fit reasonably well to a model for one binding site (N=~0.7) with a Kd of ~7 μM. Thus, little difference was detected between the two forms of EINtr in either phosphotransfer activity or in calorimetric measurements of the binding of NPr.
Figure 11. Titration Calorimetry of EINtr(H356A) with NPr.

Isothermal titration calorimetry was carried out as described in Materials and Methods. Panel A: The concentration of H-EINtr was 14 μM and that of NPr in the injection syringe was 380 μM. Panel B: The concentration of D-EINtr was 16.4 μM and that of NPr in the injection syringe was 380 μM.
7.1 Structural Considerations
Results in the literature concerning the effect of perdeuteration on structure do not present a uniform picture [38]. In some cases (human arginase I [13] and carbonic anhydrase [39]), it was concluded that deuteration has no discernible effect on structure. There are, however, reports detailing deuteration effects on structure. P450cam [25] showed a change in α-helix content associated with deuteration. Haloalkane dehalogenase [40] assumed similar conformations in both protein forms, but there were crucial differences in the active site. As yet, there have been no three-dimensional structures reported for EINtr in either H- or D-forms. Since the protein is large and multi-domained, it is likely that a careful analysis of the structure of the two forms would reveal significant differences.
The collection of data presented here suggests that the locus of sensitivity to proteolysis (Fig. 2) and differential fluorescence effects upon ligand binding to perdeuterated EINtr (Fig. 8) is in the flexible loop 3 region (Fig. 3 of Supporting Data) of the protein that links the two domains of the protein. This suggests the possibility that mutagenic manipulation of the amino acid sequence in this loop might minimize the difference in biophysical properties of the two protein forms. In such a case, structural data derived from NMR studies of EINtr could be interpreted with more confidence.
Concluding Remarks
The biophysical and stability properties of EINtr have been characterized. A collection of studies (proteolysis, unfolding, oligomerization, near UV CD, ligand binding measured by Trp fluorescence) comparing the biophysical properties of the hydrogenated and perdeuterated forms of EINtr provide evidence for structural and stability differences associated with perdeuteration. These differences may be explained by differences in molecular dynamics of the two forms of the protein. The data suggest potential complications in analyzing structural data using the perdeuterated form of this protein. The data further suggest that the loop 3 region of EINtr is a favorable target for exploring structural changes that might stabilize the protein; such a form might provide less ambiguous structural information. It is worthy of note that the truncated EINtr protein used in these studies lacks the normally occurring GAF domain and may differ in properties from the full-length protein; however, its structure should be quite analogous to its paralog, EI, and therefore serves as a good experimental model. The function of the GAF domain of EINtr remains to be elucidated.
Supplementary Material
Acknowledgements
The authors acknowledge helpful discussions with Drs. Edward Korn (NIH), Osnat Herzberg and John Moult (Center for Advanced Research in Biotechnology), expert guidance in the use of the stop-quench instrument by Dr. James Sellers (NIH) as well as the excellent technical assistance of Nancy Wehr (NIH). We thank Dr. David Christianson (University of Pennsylvania) for gifts of hydrogenated and perdeuterated human arginase and Dr. Nelson Cole (NIH) for a gift of α-synuclein. YJS was supported by the National Research Foundation Grant (NRF 2010-0017384) funded by MEST, Korea.
This work was supported by the Intramural Research Program of the National Heart, Lung and Blood Institute.
Abbreviations
- PTS
phosphotransferase system
- EI
Enzyme I of the carbohydrate PTS
- PTSNtr
nitrogen PTS
- EINtr
Enzyme I of the nitrogen PTS
- H-EINtr
the hydrogenated form of EINtr
- D-EINtr
the deuterated form of EINtr
- perdeuterated protein
protein in which nonexchangeable, carbon-bound protons are replaced by deuterium
- PEP
phosphoenolpyruvate
- GAF domains
small-molecule-binding domains that recognize cGMP-binding cyclic nucleotide phosphodiesterases, adenylyl cyclases, and the transcription factor FhlA
- TROSY
transverse relaxation optimized spectroscopy
- amu
atomic mass unit
- IMAC
immobilized metal affinity chromatography
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
This work is dedicated to the memory of Ann Ginsburg who died on February 25, 2008. She was an important long-term collaborator on joint studies of proteins of the carbohydrate PTS and served as an Associate Editor of Archives of Biochemistry and Biophysics.
Reference List
- [1].Deutscher J, Francke C, Postma PW. Microbiology and Molecular Biology Revs. 2006;70:939–1031. doi: 10.1128/MMBR.00024-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Teplyakov A, Lim K, Zhu P-P, Kapadia G, Chen CC, Schwartz J, Howard A, Reddy P, Peterkofsky A, Herzberg O. Proc. Natl. Acad. Sci. USA. 2006;103:16218–16223. doi: 10.1073/pnas.0607587103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Herzberg O, Chen CC, Kapadia G, McGuire M, Carroll LJ, Noh SJ, Dunaway-Mariano D. Proc. Natl. Acad. Sci. , USA. 1996;93:2652–2657. doi: 10.1073/pnas.93.7.2652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Lee C-R, Cho S-H, Yoon M-J, Peterkofsky A, Seok Y-J. Proc. Natl. Acad. Sci. USA. 2007;104:4124–4129. doi: 10.1073/pnas.0609897104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Lüttmann D, Heermann R, Zimmer B, Hillmann A, Rampp IS, Jung K, Görke B. Mol. Microbiol. 2009;72:978–994. doi: 10.1111/j.1365-2958.2009.06704.x. [DOI] [PubMed] [Google Scholar]
- [6].Li X, Peterkofsky A, Wang G. Amino. Acids. 2008;35:531–539. doi: 10.1007/s00726-008-0079-9. [DOI] [PubMed] [Google Scholar]
- [7].Wang G, Peterkofsky A, Keifer PA, Li X. Protein Sci. 2005;14:1082–1090. doi: 10.1110/ps.041232805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Rabus R, Reizer J, Paulsen I, Saier MH., Jr. J. Biol. Chem. 1999;274:26185–26191. doi: 10.1074/jbc.274.37.26185. [DOI] [PubMed] [Google Scholar]
- [9].Roossien FF, Brink J, Robillard GT. Biochim. Biophys. Acta. 1983;760:185–187. doi: 10.1016/0304-4165(83)90141-1. [DOI] [PubMed] [Google Scholar]
- [10].Peterkofsky A, Wang G, Seok Y-J. Arch. Biochem. Biophys. 2006;453:101–107. doi: 10.1016/j.abb.2006.01.004. [DOI] [PubMed] [Google Scholar]
- [11].Brockwell D, Yu L, Cooper S, McCleland S, Cooper A, Attwood D, Gaskell SJ. J. Barber, Protein Sci. 2001:572–580. doi: 10.1110/ps.46001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Geoghegan KF, Dixon HB, Rosner PJ, Hoth LR, Lanzetti AJ, Borzilleri KA, Marr ES, Pezzullo LH, Martin LB, LeMotte PK, McColl AS, Kamath AV, Stroh JG. Anal. Biochem. 1999;267:169–184. doi: 10.1006/abio.1998.2990. [DOI] [PubMed] [Google Scholar]
- [13].DiConstanzo L, Moulin M, Haertlein M, Meilleur F, Christianson DW. Arch. Biochem. Biophys. 2007;465:82–89. doi: 10.1016/j.abb.2007.04.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Cole NB, Murphy DB, Grider T, Reuter S, Brasaemle D, Nussbaum RL. J Biol Chem. 2002;277:6344–6352. doi: 10.1074/jbc.M108414200. [DOI] [PubMed] [Google Scholar]
- [15].Levine RL. Rapid Commun. Mass Spectrom. 2006;20:1828–1830. doi: 10.1002/rcm.2519. [DOI] [PubMed] [Google Scholar]
- [16].Apffel A, Fischer S, Goldberg G, Goodley PC, Kuhlmann FE. J. Chromatogr. A. 1995;712:177–190. doi: 10.1016/0021-9673(95)00175-m. [DOI] [PubMed] [Google Scholar]
- [17].Dimitrova MN, Peterkofsky A, Ginsburg A. Protein Sci. 2003;12:2047–2056. doi: 10.1110/ps.0352103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Ginsburg A, Szczepanowski RH, Ruvinov SB, Nosworthy NJ, Sondej M, Umland TC, Peterkofsky A. Protein Sci. 2000;9:1085–1094. doi: 10.1110/ps.9.6.1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].LiCalsi C, Crocenzi TS, Freire E, Roseman S. J. Biol. Chem. 1991;266:19519–19527. [PubMed] [Google Scholar]
- [20].Lee BR, Lecchi P, Pannell L, Jaffe H, Peterkofsky A. Arch. Biochem. Biophys. 1994;312:121–124. doi: 10.1006/abbi.1994.1289. [DOI] [PubMed] [Google Scholar]
- [21].Hattori H, Crespi HL, Katz JJ. Biochemistry. 1965;4:1213–1225. doi: 10.1021/bi00883a002. [DOI] [PubMed] [Google Scholar]
- [22].Kanyo ZF, Scolnick LR, Ash DE, Christianson DW. Nature. 1996;383:554–557. doi: 10.1038/383554a0. [DOI] [PubMed] [Google Scholar]
- [23].Diez AM, Campo ML, Soler G. Int. J. Biochem. 1992;24:1925–1932. doi: 10.1016/0020-711x(92)90288-c. [DOI] [PubMed] [Google Scholar]
- [24].Uversky VN, Eliezer D. Curr. Protein Pept. Sci. 2009;10:483–499. doi: 10.2174/138920309789351921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Meilleur F, Contzen J, Myles DA, Jung C. Biochemistry. 2004;43:8744–8753. doi: 10.1021/bi049418q. [DOI] [PubMed] [Google Scholar]
- [26].Ginsburg A, Peterkofsky A. Arch. Biochem. Biophys. 2002;397:273–278. doi: 10.1006/abbi.2001.2603. [DOI] [PubMed] [Google Scholar]
- [27].Dimitrova MN, Szczepanowski RH, Ruvinov SB, Peterkofsky A, Ginsburg A. Biochemistry. 2002;41:906–913. doi: 10.1021/bi011801x. [DOI] [PubMed] [Google Scholar]
- [28].Beernink PT, Yang R, Graf R, King DS, Shah SS, Schachman HK. Protein Sci. 2001;10:528–537. doi: 10.1110/ps.39001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Gong S, Blundell TL. PLOS one. 2010;5:1–12. doi: 10.1371/journal.pone.0009186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Yokogaki S, Unno K, Oku N, Okada S. Plant Cell Physiol. 2009;36:419–423. [Google Scholar]
- [31].Savenkova MI, Satterlee JD, Erman JE, Siems WF, Helms GL. Biochemistry. 2001;40:12123–12131. doi: 10.1021/bi0111000. [DOI] [PubMed] [Google Scholar]
- [32].Beechem JM, Brand L. Annu. Rev Biochem. 1985;54:43–71. doi: 10.1146/annurev.bi.54.070185.000355. [DOI] [PubMed] [Google Scholar]
- [33].Eftink MR. Methods Biochem Anal. 1991;35:127–205. doi: 10.1002/9780470110560.ch3. [DOI] [PubMed] [Google Scholar]
- [34].Callis PR. Methods Enzymol. 1997;278:113–150. doi: 10.1016/s0076-6879(97)78009-1. [DOI] [PubMed] [Google Scholar]
- [35].Eftink MR, Ghiron CA. Biochemistry. 1976;15:672–680. doi: 10.1021/bi00648a035. [DOI] [PubMed] [Google Scholar]
- [36].Lakowicz J. Principles of Fluorescence Spectroscopy. Kluver Academic Publisher; Dordrecht, The Netherlands: 1999. [Google Scholar]
- [37].Garrett DS, Seok Y-J, Peterkofsky A, Gronenborn AM, Clore GM. Nature Struct. Biol. 1999;6:166–173. doi: 10.1038/5854. [DOI] [PubMed] [Google Scholar]
- [38].Fisher SJ, Helliwell JR. Acta Crystallographica Section A. 2008;A64:359–367. doi: 10.1107/S0108767308004807. [DOI] [PubMed] [Google Scholar]
- [39].Budayova-Spano M, Fisher SZ, Dauvergne MT, gbandje-McKenna M, Silverman DN, Myles DA, McKenna R. Acta Crystallogr Sect F Struct Biol Cryst Commun. 2006:9. doi: 10.1107/S1744309105038248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Liu X, Hanson BL, Langan P, Viola RE. Acta Crystallogr D Biol Crystallogr. 2007:1000–1008. doi: 10.1107/S0907444907037705. [DOI] [PubMed] [Google Scholar]
- [41].Luo S, Wehr NB, Levine RL. Anal. Biochem. 2006;350:233–238. doi: 10.1016/j.ab.2005.10.048. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.