

Abstract
Smad3 is a key signal transducer of transforming growth factor-β (TGF-β) and activin, and is known to be a DNA-binding transcriptional regulator. Here we report a novel property of Smad3 in regulating the proteasomal degradation of the human enhancer of filamentation 1 (HEF1), which is a member of the Cas family of cytoplasmic docking proteins. Our studies revealed that Smad3 interacts with HEF1 and triggers the proteasomal degradation of HEF1 in overexpression systems. In addition, TGF-β stimulation induces rapid proteasomal degradation of endogenous HEF1 in different TGF-β-responsive cell lines. Interestingly, the degradation of HEF1 protein in epithelial cells is followed closely by an increase in HEF1 mRNA, resulting in a time-dependent increase in HEF1 protein level in TGF-β-treated cells. Furthermore, we observed that an elevated HEF1 protein level inhibits TGF-β-induced Smad3-mediated gene responses. These data provide the first evidence for a novel cytoplasmic activity of Smad3 in regulating proteasomal degradation of HEF1 and also suggest a role for HEF1 in a negative feedback mechanism of the TGF-β signaling pathway.
Keywords: HEF1/proteasome/signal transduction/Smad3/TGF-β
Introduction
Transforming growth factor-β (TGF-β) is the prototype of the TGF-β superfamily and is well known for its biological activities such as inhibition of cell proliferation, induction of extracellular matrix synthesis and immunosuppressive function (Letterio and Roberts, 1998; Massague, 1998). TGF-β signals through its distinct type I and type II receptor serine/threonine kinases (Massague, 1998). The Smad family proteins have been shown to mediate signaling events downstream of the various type I receptors of the TGF-β family ligands (Derynck et al., 1998; Massague, 1998; Wrana, 2000). Upon phosphorylation on their C-terminal SSXS motif by the activated TGF-β and activin type I receptors, Smad3 and Smad2 subsequently form a complex with the co-Smad, Smad4, and then translocate from the cytoplasm into the nucleus (Derynck et al., 1998; Massague, 1998; Wrana, 2000). In the nucleus, Smad2, Smad3 and Smad4 mediate gene regulation via interaction with different DNA-binding transcriptional activators, repressors and the transcriptional co-activator CBP/p300 and co-repressor TGIF (Derynck et al., 1998; Wrana, 2000). Smad3 and Smad4 also exhibit sequence-specific DNA-binding activities (Derynck et al., 1998; Massague, 1998; Wrana, 2000). Based upon these and similar observations of the bone morphogenetic protein (BMP)-regulated Smad1, the Smad family proteins are currently considered to be DNA-binding transcription factors.
Here we report the identification of a novel cytoplasmic activity of Smad3. We found that Smad3 interacts with a cytoplasmic multidomain docking protein HEF1 (the human enhancer of filamentation 1) (Law et al., 1996; Minegishi et al., 1996). HEF1, also known as Cas-L, belongs to a family of multidomain docking proteins including p130Cas and another structurally related protein Efs (or Sin), all of which play important roles in coordinating cell adhesion with cell response to multiple extracellular stimuli (O’Neill et al., 2000). Like p130Cas, HEF1 consists of multiple protein–protein interaction domains: an N-terminal SH3 domain followed by a stretch of YXXP tyrosine phosphorylation sites, a central serine-rich region and a highly conserved C-terminal novel domain (Law et al., 1996; Minegishi et al., 1996). Pre dominantly expressed in lymphocytes and epithelial cells, HEF1 has been implicated to function as an adaptor protein in the signaling pathways of integrin, T-cell antigen receptor (TCR), B-cell antigen receptor (BCR) and the G-protein-coupled calcitonin receptor (Minegishi et al., 1996; Manie et al., 1997; Sattler et al., 1997; Ohashi et al., 1998; Zhang et al., 1999). However, the exact nature of the signaling events associated with HEF1 has yet to be defined.
Our studies of the physical interaction between Smad3 and HEF1 led us to uncover several novel aspects of Smad3-mediated signaling. (i) We found that Smad3 interaction with HEF1 triggers the proteasomal degradation of HEF1. (ii) The ability of Smad3 to induce HEF1 degradation resides within the MH1 domain but not the MH2 domain. (iii) Smad3 couples TGF-β type I receptor activation to enhanced HEF1 degradation. (iv) Rapid HEF1 degradation upon TGF-β stimulation in epithelial cell lines is followed by a rapid increase in HEF1 mRNA, leading to a highly elevated HEF1 protein level. (v) An increased HEF1 protein level is inhibitory to TGF-β-induced Smad3-mediated gene activation. These data together indicate a novel cytoplasmic activity of Smad3 in regulating the proteasomal degradation of HEF1 and also suggest a role for HEF1 in a negative feedback mechanism for Smad3-mediated signaling events.
Results
HEF1 interacts with Smad3 in the yeast two-hybrid system and mammalian cells
To understand the regulation or signaling mechanisms of Smad3, we applied the yeast two-hybrid system to isolate Smad3-interacting proteins. By screening 7.5 × 105 clones of a human fetal brain cDNA library, we obtained 16 different cDNA species from 133 positives. HEF1 was one of the 16 Smad3 interactors isolated, and exhibited specific and strong binding to Smad3 in the yeast two-hybrid system (Figure 1A).

Fig. 1. Smad3 interacts with HEF1 and regulates HEF1 stability via proteasomal degradation. (A) HEF1 is a specific interactor of Smad3 in the yeast two-hybrid system. B42-HEF1(506–834) fusion protein (isolated from the yeast two-hybrid screen) was tested for interaction with different LexA fusion proteins as indicated. R4C contains the cytoplasmic domain of the TGF-β type I receptor. Max is a dimerization partner for the oncoprotein c-Myc. B42 activation domain alone served as a negative control. Yeast transformants were grown in Ura–His–Trp– glucose medium for 12 h before they were spotted onto Ura–His–Trp– X-Gal plates containing either glucose or galactose/raffinose. The expression of B42 and B42 fusion proteins is under the control of the GAL1 promoter and both fusion proteins are expressed only when yeast transformants are grown on galactose plates, as shown. All transformants were white on glucose plates (not shown). (B) Smad3 interacts with HEF1 in mammalian cells. HEF1 was transfected alone or co-transfected with Flag-tagged Smad1, Smad2, Smad3 or Smad4 into 293 cells. Cell lysates were immunoprecipitated with α-Flag (α-F) antibody, followed by western blotting with α-HEF1 antibody (top panel, lanes 2–5). Expression levels of transfected Smads and HEF1 were determined by western blotting using α-F and α-HEF1 antibodies, respectively (middle and bottom panels). The solid arrowhead indicates a non-specific band detected by the α-HEF1 antibody. (C) Increasing Smad3 protein level induces proteasomal degradation of HEF1, and co-expression of the TGF-β and activin type I receptors further enhances HEF1 degradation. HEF1 was transfected alone or co-transfected with Flag-tagged Smad3 in the absence or presence of the TGF-β and activin type I receptors into 293 cells (lanes 2–7). R4, wild-type TGF-β type I receptor; R4TD, constitutively active mutant TGF-β type I receptor R4T204D; R2, wild-type activin type I receptor; R2TD, constitutively active activin type I receptor R2T206D. An identical set of transfections was carried out and cells were treated with 50 µM LLnL for 9 h before harvest (lanes 8–13). Lysates were prepared and blotted with α-HEF1 antibody. The solid arrowhead indicates a non-specific band detected by the α-HEF1 antibody. Additional bands just below p105HEF1, as marked by an asterisk, may represent other proteolytic intermediates of HEF1 stabilized by LLnL. (D) Smad3-mediated HEF1 degradation in the absence or presence of receptor activation can be inhibited by the proteasome-specific inhibitor lactacystin but not by the caspase inhibitor ZVAD. Combinations of HEF1, Smad3 and the constitutively active TGF-β type I receptor R4TD were transfected into 293 cells as indicated. Cells were either untreated, or treated with 10 µM lactacystin or 100 µM ZVAD for 9 h before harvest. Cell lysates were prepared and blotted with α-HEF1 antibody. (E) p115HEF1 and p105HEF1 exhibit differential phosphorylation on tyrosine and threonine residues. HEF1 was transfected into 293 cells and immunoprecipitated with α-p130Cas monoclonal antibody that cross-reacts with HEF1. Immunoprecipitated HEF1 was then blotted with antibodies that specifically recognize α-phosphotyrosine, α-phosphoserine and α-phosphothreonine, respectively. The expression level of HEF1 protein was assayed by α-HEF1 western blotting (top panel).
To verify the ability of HEF1 to interact with Smad3 in mammalian cells, HEF1 was transiently co-expressed with Smad1, Smad2, Smad3 or Smad4 in 293 cells. Western blot analyses of the transfected 293 cells detected two different forms of HEF1, p115HEF1 and p105HEF1, both of which have been described previously (Law et al., 1996, 1998; Minegishi et al., 1996). The difference in the mobility of p115HEF1 and p105HEF1 is a result of differential phosphorylation of HEF1, since p115HEF1 can be converted to p105HEF1 by λ phosphatase treatment (Law et al., 1998). No functional difference of these two forms has been reported.
Co-immunoprecipitation assays detected p115HEF1 to be the predominant form that associated with Smad3 (Figure 1B, top panel, lane 4) and, to a lesser extent, with Smad1 and Smad2 (Figure 1B, top panel, lanes 2 and 3), although p115HEF1 and p105HEF1 were expressed at a similar level when HEF1 was transfected alone (Figure 1B, top panel, lane 1). Neither p115HEF1 nor p105HEF1 was detected in the immunoprecipitates of Smad4 (Figure 1B, top panel, lane 5). Therefore, p115HEF1 interacts strongly with Smad3, weakly with Smad1 and Smad2, but does not interact with Smad4 in mammalian cells.
Smad3 induces the proteasomal degradation of p115HEF1 while TGF-β and activin type I receptors enhance this effect of Smad3
In the above overexpression system, we observed that the steady-state levels of both forms of HEF1, especially that of p115HEF1, were reduced in cells co-expressing Smad3 (Figure 1B, bottom panel, lane 4). This phenomenon was then examined more closely. While the steady-state level of p105HEF1 was only slightly decreased, that of p115HEF1 was dramatically reduced upon co-expression of Smad3 alone (Figure 1C, lane 3). The reduction of both p105HEF1 and p115HEF1 was more pronounced when Smad3 and the type I receptors of TGF-β and activin were co-transfected with HEF1 (Figure 1C, lanes 4–7).
Since p115HEF1 and p105HEF1 are derived from differential phosphorylation of the same protein (Law et al., 1998), the differential reduction of these two forms of HEF1 upon Smad3 co-expression suggests a post-translational mechanism contributing to the reduction. Studies of Smad-interacting proteins in the signaling pathways of BMPs have recently revealed a novel physical and functional connection between Smad1 and the 26S proteasome (Kim et al., 2000; Y.Lin, J.Martin, C.Gruendler, X.Meng, B.-Y.Li, R.Lechleider, C.Huff, R.H.Kim, W.Grasser, V.Paralkar and T.Wang, submitted). Thus, we tested whether the Smad3-induced HEF1 reduction is mediated by proteasomal degradation. Indeed, a peptidyl aldehyde proteasome inhibitor LLnL (N-acetyl-l-leucinyl-l-leucinal-l-norleucinal) efficiently blocked the Smad3-induced decrease in HEF1 protein level (Figure 1C, lane 9). Furthermore, LLnL was able to rescue the level of HEF1 in the presence of the TGF-β and activin type I receptors (Figure 1C, lanes 10–13). In addition to LLnL, a highly specific proteasome inhibitor, lactacystin, also blocked the reduction of HEF1 upon Smad3 co-expression, in the absence or presence of the activated TGF-β type I receptor (Figure 1D, lanes 5 and 6). Since caspase-dependent cleavage of HEF1 has been reported (Law et al., 1998), we also tested the effect of the caspase inhibitor ZVAD. In contrast to the protective effect of lactacystin and LLnL, ZVAD failed to inhibit the decrease in HEF1 in the presence of Smad3 (Figure 1D, lanes 7 and 8). These data suggest that proteasome-mediated degradation is involved specifically in the reduction of the steady-state levels of HEF1, especially that of p115HEF1, upon co-expression of Smad3 and the TGF-β and activin type I receptors.
Since p115HEF1 is more sensitive to Smad3-induced proteasomal degradation than p105HEF1, it is important to determine the phosphorylation differences between p115HEF1 and p105HEF1. We therefore analyzed immunoprecipitated HEF1 with antibodies specific for phosphotyrosine, phosphoserine and phosphothreonine (Figure 1E). While the expression levels of p115HEF1 and p105HEF1 were comparable (Figure 1E, top panel), there was more phosphotyrosine signal detected in p105HEF1 (Figure 1E, second panel), but more phosphothreonine signal detected in p115HEF1 (Figure 1E, bottom panel). No major difference was observed in the serine phosphorylation of the two HEF1 forms (Figure 1E, third panel). Thus, p115HEF1 differs from p105HEF1 by containing more phosphothreonine but less phosphotyrosine.
The MH1 domain of Smad3 is responsible for inducing HEF1 degradation while the MH2 domain has a role in receptor-enhanced HEF1 degradation
Smad3 can be divided into three subdomains: an N-terminal MH1 domain, a central linker region and a C-terminal MH2 domain. We investigated the role of MH1 and MH2 domains in Smad3-induced HEF1 degradation. When HEF1 was co-expressed with MH1, MH2 or the full-length Smad3, the MH1 domain mimicked the full-length Smad3, albeit more weakly, in reducing the level of p115HEF1 (Figure 2A, lanes 3 and 4). LLnL also blocked the Smad3 N-terminus-mediated reduction in p115HEF1 (Figure 3A, top panel, lane 6). In contrast, the MH2 domain failed to alter the protein level of p115HEF1 (Figure 2A, lane 5). Therefore, the activity of Smad3 in inducing the proteasomal degradation of HEF1 resides primarily within its MH1 domain.

Fig. 2. Smad3 regulates HEF1 degradation via its MH1 domain and couples the type I receptor activation to enhanced HEF1 degradation via a mechanism independent of Smad3 phosphorylation at the SSVS motif. (A) The ability of Smad3 to induce HEF1 degradation resides within its MH1, but not its MH2 domain. HEF1 was transfected alone or co-transfected with full-length Smad3 (FL), Smad3 MH1 domain (N) or Smad3 MH2 domain (C) into 293 cells. HEF1 protein level was monitored by western blotting with α-HEF1 antibody. The solid arrowhead indicates the non-specific band detected by α-HEF1 antibody, as shown in Figure 1B. (B) HEF1 degradation upon activation of the TGF-β type I receptor can be inhibited by the Smad3 MH2 domain. 293 cells were transfected with an equal amount of the indicated constructs. HEF1 protein level was determined by western blotting using α-HEF1 antibody. (C) The kinase activities of the TGF-β and activin type I receptors are necessary for receptor-induced proteasomal degradation of HEF1. HEF1 was transfected in the absence or presence of the wild-type or mutant TGF-β and activin type I receptors into 293 cells. HEF1 expression was assayed by α-HEF1 western blotting. R4KR is a mutant R4 that lacks the kinase activity due to a point mutation K230R within its ATP-binding site. R2KR is a mutant R2 that lacks the kinase activity due to a similar point mutation K234R. LLnL (50 µM) was added 9 h before cells were lysed. Additional bands just below p105HEF1, as marked by an asterisk, may represent other proteolytic intermediates of HEF1. (D) Receptor-mediated phosphorylation of Smad3 on its C-terminal serine residues is not essential for coupling receptor activation to proteasomal degradation of HEF1 in an overexpression system. F-Smad3SA is a Flag-tagged Smad3 mutant in which all three C-terminal serines were substituted by alanines. Experimental methods are the same as above.

Fig. 3. Smad3 and HEF1 interact with each other through their N- and C-terminal domains. (A) Smad3 binds to HEF1 via both Smad3N (MH1) and Smad3C (MH2) domains, but only the N-terminal domain induces the degradation of HEF1. HEF1 was transfected alone or co-transfected with Flag-tagged full-length (FL), the N-terminal MH1 domain (N) or C-terminal MH2 domain (C) of Smad3 into 293 cells, untreated or treated with 50 µM LLnL for 9 h before harvest. Cell lysates were prepared and blotted with α-HEF1 and α-F antibodies (top and middle panels, respectively). Lysates were also immunoprecipitated with α-F antibody and blotted with α-HEF1 antibody (bottom panel). (B) The N-terminus of HEF1 preferentially binds to the Smad3 N-terminal domain. 293 cells were transfected with HEF1(1–505) alone or in combination with Flag-tagged full-length (FL), or the N- or C-terminal domains of Smad3. Lysates were immunoprecipitated with α-F antibody and blotted with α-HEF1 antibody (top panel, lanes 2–5). Lane 1 is the western blot for HEF1(1–505), which was expressed in two forms, designated as tHEF1(1–505) and bHEF1(1–505), respectively. Expression levels of HEF1(1–505) and Smad3 constructs are shown in the middle and bottom panels. (C) The C-terminus of HEF1 binds to the C-terminus of Smad3. T7-tagged HEF1 deletion constructs were transfected alone or co-transfected with Flag-tagged full-length (FL), or the N- or C-terminal domains of Smad3 into 293 cells. For the top panel, lysates were immunoprecipitated with α-F antibody and blotted with α-T7 antibody to detect the co-immunoprecipitation of HEF1 subdomains with Smad3 subdomains (lanes 1–16). Expression of the HEF1 subdomains was detected by western blotting in the left panel to serve as the molecular weight markers for the right panel. The protein levels of the transfected constructs in each lysate were monitored by western blotting using α-T7 and α-F antibodies, respectively (middle and bottom panels). (D) Summary of the HEF1 deletion constructs used in (C) and the result of the domain-mapping studies. (E) A cartoon that illustrates the mutual interaction between Smad3 and HEF1. Smad3 and HEF1 interact with each other through their N- and C-terminal domains.
Although co-expression of the type I receptors enhances HEF1 degradation (Figure 1C), it is not clear whether the enhancement is mediated through Smad3. To test this, we co-expressed either full-length Smad3 or the Smad3 MH2 domain with the receptors. While the full-length Smad3 further enhanced the degradation of HEF1 (Figure 2B, lanes 4 and 5), the MH2 domain exhibited a dominant-negative activity and completely blocked the receptor-induced HEF1 degradation (Figure 2B, lanes 6 and 7). These data indicate the critical role of Smad3 in coupling TGF-β receptor activation to HEF1 degradation.
The receptor-enhanced HEF1 degradation requires the receptor kinase activities but is not dependent upon the receptor-mediated phosphorylation of the SSVS motif of Smad3
The observation that overexpressed wild-type TGF-β and activin type I receptors also induce HEF1 degradation was surprising, since endogenous TGF-β type I receptor serine/threonine kinase remains inactive before the addition of TGF-β (Wrana et al., 1994). One possibility is that there was leaky activation of receptor kinase activities as a result of overexpression. We therefore compared wild-type type I receptors with kinase-inactive mutant type I receptors in terms of their abilities to destabilize HEF1. As shown in Figure 2C, the wild-type and the constitutively active TGF-β and activin type I receptors caused an LLnL-sensitive reduction in p115HEF1 (lanes 1–3, 5–8 and 10–11), while the kinase-inactive TGF-β and activin type I receptors lost their ability to destabilize p115HEF1 (lanes 4 and 9). Thus, the kinase activities of the TGF-β and activin type I receptors are required for receptor-induced proteasomal degradation of HEF1.
It is known that the TGF-β type I receptor kinase directly phosphorylates Smad3 at the C-terminal SSVS motif. We thus tested whether such phosphorylation mediates the enhancement effect of the type I receptor on HEF1 degradation by overexpressing a phosphorylation-defective mutant Smad3 (Smad3SA) in which all three C-terminal serines were substituted by alanines. Smad3SA was still able to induce HEF1 degradation upon its overexpression and failed to block type I receptor-induced HEF1 degradation (Figure 2D). Therefore, the enhancement effect of the type I receptors is via a phosphorylation event(s) different from SSVS phosphorylation.
HEF1 interacts with Smad3 via two separate domains on each protein
To understand how Smad3 interacts with HEF1, we carried out deletion analyses to determine the domains of interaction on Smad3 and HEF1. First, full-length HEF1 was tested against full-length Smad3, MH1 (Smad3N) and MH2 (Smad3C), using co-immunoprecipitation assays (Figure 3A). Smad3 and its deletions all exhibited the ability to bind HEF1, especially to p115HEF1 (Figure 3A, bottom panel). Although the strongest signal of HEF1 was detected in the Smad3C immunoprecipitates in the absence of any proteasome inhibitors, the highest amount of HEF1 was found in Smad3 immunoprecipitates when LLnL was added to stabilize HEF1 (Figure 3A, bottom panel, lanes 3 and 8). Such a difference is consistent with the observed HEF1 degradation in response to full-length Smad3 but not to Smad3C.
To understand how MH1 and MH2 can both bind to HEF1, we tested the abilities of full-length Smad3, Smad3N and Smad3C to interact with different deletion mutants of HEF1. First, we tested their interaction with HEF1(1–505), which contains the SH3 domain, all of the SH2-binding sites and the serine-rich region. Like full-length HEF1, HEF1(1–505) was also expressed in two different forms, designated as tHEF1(1–505) and bHEF1(1–505), respectively (Figure 3B, top panel, lane 1). In the immunoprecipitates of full-length Smad3 and Smad3N, strong signals of tHEF1(1–505) but not bHEF1(1–505) were detected (Figure 3B, top panel, lanes 3 and 4). A much weaker signal of tHEF1(1–505) was also detected in the immunoprecipitates of Smad3C (Figure 3B, top panel, lane 5). Thus, the N-terminal domain of HEF1 preferentially associates with Smad3 and Smad3N compared with Smad3C. Next we tested the abilities of Smad3, Smad3N and Smad3C to bind to the C-terminal subdomains of HEF1. A set of four deletion constructs, named M2, M2/N, M2/M and M2/C as illustrated in Figure 3D, were used. All four tested HEF1 deletions strongly interacted with Smad3C, but not with Smad3N or full-length Smad3 (Figure 2C, top panel). In summary, these domain-mapping studies revealed a unique feature in the mutual interaction between Smad3 and HEF1. The two proteins appear to interact with each other through their modular domains, with the MH1 domain (Smad3N) contacting the HEF1 N-terminal domain and the MH2 domain (Smad3C) binding to the HEF1 C-terminal domain, as summarized in Figure 3D and illustrated by a cartoon in Figure 3E.
HEF1 degradation is dependent upon Smad3 interaction with HEF1(1–505)
Since HEF1(1–505) is sufficient to bind to the full-length Smad3, HEF1(1–505) may act as a dominant-negative mutant to compete with full-length HEF1 for Smad3 interaction and block HEF1 degradation induced by Smad3. Indeed, overexpression of HEF1(1–505) efficiently blocked the Smad3-induced proteasomal degradation of HEF1 in a dose-dependent manner (Figure 4A, lanes 3 and 4). Interestingly, higher molecular weight ladders of HEF1 were detected when HEF1 was fully stabilized by excess HEF1(1–505) (Figure 4A, lane 4). Overexpression of HEF1(1–505) also blocked the degradation of HEF1 induced by Smad3N (Figure 4B, lane 5), by co-expression of Smad3 with the TGF-β type I receptor (Figure 4B, lanes 10 and 11) and, to a lesser extent, by co-expression of Smad3 with the activin type I receptor (Figure 4B, lanes 12 and 13). Again, higher molecular weight ladders of HEF1 were detected (Figure 4B, lanes 3, 5 and 10–12). We suspected that the higher molecular weight ladders represented the ubiquitylated forms of HEF1, thus the ubiquitylation of HEF1 was tested in the absence or presence of Smad3. HEF1 was constitutively ubiquitylated (Figure 4C, top panel, lane 3) and co-expression of Smad3 led to the reduction in p115HEF1 level (Figure 4C, bottom panel, lane 4) as well as the disappearance of the ubiquitylated ladder of HEF1 (Figure 4C, top panel, lane 4). LLnL treatment partially restored the protein level of p115HEF1 as well as the ubiquitylated forms of HEF1 (Figure 4C, lane 6). Thus, Smad3 appears to destabilize the ubiquitylated forms of HEF1, which become stabilized upon overexpression of HEF1(1–505), possibly due to inhibition of the interaction between the N-terminal domains of Smad3 and HEF1.

Fig. 4. Overexpression of the Smad3-binding N-terminal HEF1(1–505) blocks proteasomal degradation of HEF1. (A) Smad3-induced HEF1 degradation is blocked by the overexpression of HEF1(1–505) in a dose-dependent manner. 293 cells were transfected with HEF1 alone or together with Flag-tagged Smad3 in the absence or presence of increasing amounts of HEF1(1–505). Expression of HEF1 and HEF1(1–505) was determined by western blotting using α-HEF1 antibody. Higher molecular weight ladders of HEF1, possibly the ubiquitylated forms of HEF1, are indicated. The right panel is a longer exposure of the left panel for better resolution of the higher molecular weight ladders of HEF1. The arrowhead indicates a non-specific band recognized by the α-HEF1 antibody. (B) Overexpression of HEF1(1–505) also blocks p115HEF1 degradation induced by Smad3N or Smad3 co-expression with the TGF-β and activin type I receptors. 293 cells were transfected with the indicated expression vectors, and expression of HEF1 and HEF1(1–505) was determined by western blotting using α-HEF1 antibody. The right panel is a longer exposure of the left panel for better resolution of the higher molecular weight ladders of HEF1. (C) HEF1 is constitutively ubiquitylated, and overexpressed Smad3 induces proteasomal degradation of the ubiquitylated HEF1. 293 cells were transfected with the indicated expression vectors. Ubiquitin was tagged with T7 epitope. The top panel is the result of immunoprecipitation of HEF1 followed by western blotting with α-T7 antibody to detect ubiquitylated HEF1. The bottom panel is the result of western blotting using α-HEF1 antibody to detect the expression level of transfected HEF1 in each lysate. In lanes 5 and 6, cells were treated with LLnL for 8 h before harvest. The arrowhead indicates a non-specific band recognized by the α-HEF1 antibody.
TGF-β stimulation induces a time-dependent reduction in endogenous HEF1 protein levels in T-lymphoblastoid H9 cells
Since HEF1 is expressed predominantly in lymphocytes and epithelial cells, we tested whether stimulation with TGF-β ligand could cause endogenous HEF1 degradation in these cells. We first tested the change in endogenous HEF1 protein level in the human T-lymphoblastoid H9 cells, since HEF1 has been implicated to function as an adaptor protein in the signaling events induced by VLA-4 integrin ligation and the activation of TCR in this cell line (Minegishi et al., 1996; Sattler et al., 1997; Ohashi et al., 1998). H9 cells were either untreated or treated with TGF-β for different time periods. Cells were then harvested at the same time and HEF1 protein level was determined by western blotting using α-HEF1 antibody (Figure 5A). The predominant form of HEF1 in H9 cells is p105HEF1. The steady-state levels of both p115 and p105 HEF1 started to decrease after 30 min exposure to TGF-β (Figure 5A, lane 4), which was manifested at 1 h (lane 5). After 3 h of TGF-β treatment, p115HEF1 was almost undetectable whereas p105HEF1 was greatly reduced (Figure 5A, lane 6). The levels of p115HEF1 and p105HEF1 were quantified and are shown in Figure 5B. These results demonstrated that TGF-β stimulation induces a time-dependent reduction in endogenous p115HEF1 and p105HEF1 protein levels in H9 cells.

Fig. 5. TGF-β rapidly induces the degradation of endogenous HEF1 in the T-lymphoblastoid H9 cell line in a time-dependent fashion. (A) Endogenous p115HEF1 and p105HEF1 are reduced upon TGF-β stimulation in H9 cells. At various time points after exposure to 400 pM TGF-β, lysates were prepared from H9 cells and immunoprecipitated with α-HEF1 antibody. Precipitated HEF1 was revealed subsequently by western blotting with the same antibody. (B) Quantification of the endogenous p115HEF1 and p105HEF1 protein levels in (A). The levels of both p115HEF1 and p105HEF1 in (A) were quantified using Imagemaster 1D (Pharmacia) and plotted as integrated optical density (IOD) using Microsoft Excel.
TGF-β-induced degradation of endogenous HEF1 occurs more rapidly in epithelial cells and is followed by rapid restoration above the basal level
We also tested the degradation of endogenous HEF1 in TGF-β-responsive epithelial cells. A549 lung carcinoma cells, which predominantly express p115HEF1, were exposed to TGF-β for different time periods and then harvested at the same time. Endogenous HEF1 protein levels were detected by anti-HEF1 western blotting (Figure 6A) and then quantified (Figure 6B). In contrast to H9 cells, A549 cells exhibited a much more rapid reduction in endogenous p115HEF1, as early as 15 min after TGF-β stimulation (Figure 6A and B, lane 3). The change of p105HEF1 in response to TGF-β was also observed, but was not as apparent as that of p115HEF1 due to the very low level of p105HEF1 in this cell line. Surprisingly, the protein level of p115HEF1, and possibly also of p105HEF1, started to increase after the 30 min time point and was completely recovered after TGF-β treatment for 3 h (Figure 6A and B, lanes 4–6). Similar results were also obtained in several other epithelial cell lines, such as HepG2 and HaCaT (data not shown).

Fig. 6. TGF-β-induced degradation of endogenous HEF1 occurs more rapidly in epithelial cells and is followed by rapid restoration above the basal level. (A and B) HEF1 protein levels rapidly decrease in A549 cells upon TGF-β stimulation, which are subsequently restored upon longer ligand exposure. (A) An equal amount of total proteins from each time point upon TGF-β (375 pM) stimulation was analyzed by SDS–PAGE and western blotting with α-HEF1 antibody. (B) Quantifi cation of the endogenous p115HEF1 protein level in (A). (C and D) The TGF-β-induced degradation of endogenous HEF1 in A549 cells is sensitive to proteasome inhibitor LLnL while the subsequent increase in HEF1 protein level requires new protein synthesis. (C) A time-course study of HEF1 protein level in A549 cells after TGF-β treatment was performed as in (A) except that two groups of cells were pre-treated with LLnL (50 µM) or cycloheximide (10 µg/ml) for 15 min before being exposed to TGF-β for 15 or 30 min, as indicated. (D) Quantification of the endogenous p115HEF1 protein level in (C). The integrated optical density (IOD) of p115HEF1 in lane 8 is placed next to that in lane 3 for direct comparison of the effect of LLnL.
To determine the nature of the reduction in HEF1 level after 15 min of TGF-β treatment, a similar time course was performed in A549 cells except that a duplicate dish was pre-treated with LLnL before being exposed to TGF-β for 15 min (Figure 6C and D, lane 8). The reduction in HEF1 level at the 15 min time point was completely blocked by LLnL treatment (Figure 6C, lane 8, and D, open bar), suggesting that the rapid reduction in endogenous HEF1 in A549 cells was mediated by the proteasome. The increase in HEF1 protein level after 30 min of TGF-β exposure was again observed (Figure 6C and D, lane 4) and continued upon prolonged TGF-β treatment (Figure 6C and D, lanes 4–7). The steady-state levels of both forms of HEF1 after 6 h of TGF-β stimulation (Figure 6C and D, lane 7) were much higher than those from untreated cells (Figure 6C and D, lane 1). To determine whether such an increase in HEF1 level was due to new protein synthesis or increased protein stability, A549 cells were exposed to TGF-β for 30 min in the presence (Figure 6C, lane 9) or absence (Figure 6C, lane 4) of the protein synthesis inhibitor cycloheximide. Cells treated with both TGF-β and cycloheximide lost all HEF1 signals (Figure 6C, lane 9), suggesting that the increase in HEF1 protein level was due to an increased translation of HEF1.
The post-degradation increase in endogenous HEF1 protein level results from an induction of mRNA synthesis and is inhibitory to Smad3-mediated gene responses of TGF-β
We observed that actinomycin D could inhibit the post-degradation increase in HEF1 protein level (data not shown); thus, the level of HEF1 mRNA was monitored in TGF-β-treated cells. A time-course northern blot was carried out on HEF1 mRNA isolated from A549 cells treated with TGF-β for different time periods. The level of HEF1 mRNA exhibited a dramatic increase as early as 1 h after TGF-β stimulation and continued until 24 h (Figure 7A). Thus, TGF-β not only downregulates HEF1 protein level via proteasomal degradation but also up regulates HEF1 mRNA, possibly through transcriptional activation of the HEF1 gene, which leads to the increase in HEF1 protein level upon longer TGF-β exposure.
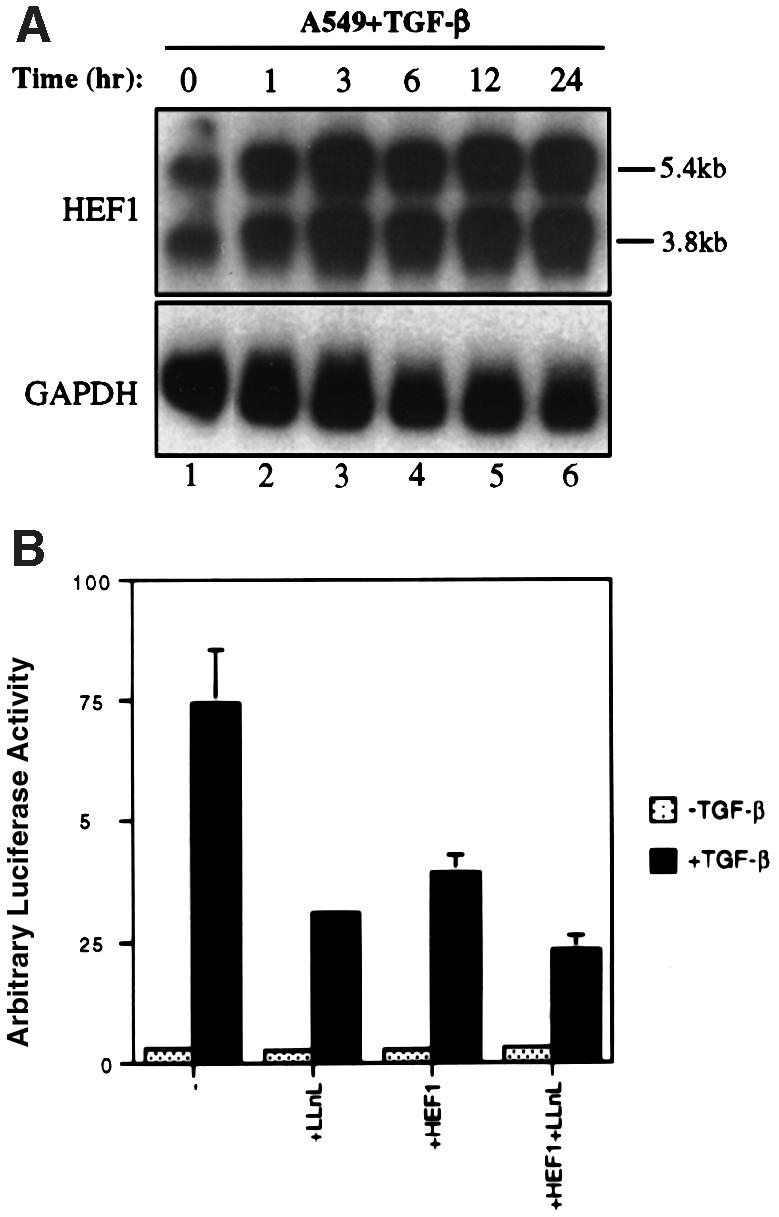
Fig. 7. The post-degradation increase in the endogenous HEF1 protein level is due to an increase in HEF1 mRNA and elevated HEF1 protein level is inhibitory to Smad3-mediated gene responses of TGF-β. (A) TGF-β induces a rapid increase in HEF1 mRNA level in A549 cells. Cells were exposed to 400 pM TGF-β and mRNA was isolated and subjected to northern blotting using 32P-labeled HEF1 as a probe (top panel). The 5.4 and 3.8 kb HEF1 transcripts are indicated. The GAPDH level was used to monitor the total amount of mRNA in each lane (bottom panel). (B) Increased HEF1 protein level is inhibitory to TGF-β-induced activation of the Smad3-responsive reporter CAGA-luc. A549 cells were transiently transfected in duplicate with CAGA-luc (0.15 µg) and the internal control CMV-RL Renilla (0.05 µg) in the absence or presence of HEF1 expression vector (0.3 µg), as indicated. The following day, cells were either untreated or treated with 100 pM TGF-β in the absence or presence of 50 µM LLnL for 9 h before cells were harvested for luciferase assays.
Although HEF1 has been implicated to function as an adaptor protein in multiple signaling pathways, the nature of the signaling pathways downstream of HEF1 is not known. Thus, currently, it is not feasible for us to test directly the effect of TGF-β on a specific HEF1-regulated signaling event. However, the observation that the degradation of HEF1 upon TGF-β treatment is followed by a dramatic increase in HEF1 protein level prompted us to determine the effect of an increased HEF1 protein level on the TGF-β signaling pathway. Therefore, we overexpressed HEF1 in A549 cells together with the CAGA-luc reporter, which has been shown to be activated by TGF-β through direct binding of Smad3 to the CAGA sequences upstream of the luciferase reporter (Dennler et al., 1998). HEF1 overexpression inhibited the TGF-β-induced gene response (Figure 7B). The proteasome inhibitor LLnL, which stabilizes HEF1, also efficiently blocked the gene response and further enhanced the inhibitory effect of HEF1 (Figure 7B).
Discussion
The Smad family signal transducers of the TGF-β family are currently considered to be DNA-binding transcriptional regulators. Our studies of the interaction between Smad3 and an adaptor protein HEF1 have revealed a novel cytoplasmic activity of Smad3. We found that Smad3 regulates the stability of HEF1, especially that of p115HEF1, via the proteasomal degradation pathway. Interestingly, such an activity of Smad3 resides within its MH1, but not its MH2 domain. The ability of Smad3 to regulate HEF1 degradation is enhanced upon activation of the TGF-β and activin type I receptors. The enhancement is dependent upon intact kinase activities of the type I receptors, suggesting that a phosphorylation event mediated by the receptor serine/threonine kinases is involved. We tested whether the well-known phosphorylation of Smad3 at its C-terminal SSVS motif by the type I receptors accounts for the enhancement effect of the receptors. Since the Smad3SA mutant, which lacks all three C-terminal serine residues for type I receptor phosphorylation, was still capable of inducing the degradation of HEF1 upon its overexpression and failed to block the receptor-induced enhancement of HEF1 degradation, it is likely that serine phosphorylation within the SSVS motif of Smad3 does not mediate the enhancement of Smad3-induced HEF1 degradation by the receptors. It thus remains to be determined whether there is a different phosphorylation event that underlies the effect of the type I receptors on HEF1 degradation. HEF1 or HEF1-associated protein(s) could be a direct substrate(s) for the type I receptors. Alternatively, the type I receptors may trigger phosphorylation of Smad3 at sites other than the SSVS motif. It is clear, however, that Smad3 couples the activation of the type I receptors to the degradation of HEF1, since the receptor activation-induced degradation of HEF1 is inhibited by overexpression of the Smad3 MH2 domain.
How does Smad3 destabilize p115HEF1? So far, we have established the importance of the physical interaction between Smad3 and HEF1 in HEF1 degradation. The ability of Smad3 to regulate HEF1 stability directly correlates with its ability to interact with HEF1. Full-length HEF1 has two different forms due to differential phosphorylation (Law et al., 1998). We found that p115HEF1, which has more phosphothreonine but less phosphotyrosine than p105HEF1, is more sensitive to Smad3-induced proteasomal degradation, consistent with the fact that Smad3 preferentially associates with p115HEF1. Domain-mapping studies revealed that HEF1 interlocks with Smad3, with Smad3N binding to the HEF1 N-terminal domain and Smad3C binding to the HEF1 C-terminal domain. The interaction between Smad3N and the N-terminal HEF1(1–505) is crucial to the degradation of p115HEF1, since uncoupling this interaction by overexpressing HEF1(1–505) rendered p115HEF1 immune to proteasomal degradation induced by Smad3, Smad3N and the receptors. It is still unclear how Smad3 destabilizes HEF1 via proteasomal degradation upon binding to HEF1. We have observed that Smad3 has the ability to bind to ubiquitin, several E3 ubiquitin ligases and to a proteasome β-subunit (S.Guedes, X.Liu and T.Wang, unpublished). Future studies of the biochemical details of Smad3 interaction with HEF1 or with other Smad3 interactors implicated in the proteasomal degradation pathway will provide new insights into the novel role of Smad3 in regulating HEF1 stability.
Since the ability of Smad3 to regulate HEF1 degradation and the ability of the activated TGF-β and activin type I receptors to enhance HEF1 degradation were observed initially in the 293 overexpression system, we felt it necessary to extend these observations and to test directly whether HEF1 degradation can be regulated by TGF-β in non-overexpression systems. So far, we have detected TGF-β-induced HEF1 degradation in four different cell lines: the human T-lymphoblastoid H9 cell line and the epithelial cell lines A549, HepG2 and HaCaT. HEF1 has been implicated to function as an adaptor protein in multiple signaling pathways, such as those mediated by integrin, TCR, BCR and the calcitonin receptor (Nojima et al., 1992; Minegishi et al., 1996; Manie et al., 1997; Sattler et al., 1997; Ohashi et al., 1998; Zhang et al., 1999). Thus, the degradation of HEF1 stimulated by TGF-β and activin will most probably have an impact on these signaling pathways. Unfortunately, the exact nature of the signaling events mediated by HEF1 in these systems is unclear at present, thus precluding the possibility of any direct test of the effects of TGF-β/activin on specific signaling events of HEF1. Our observations, however, have suggested a new cross-talk mechanism between TGF-β/activin pathways and multiple HEF1-involved pathways through Smad3-regulated HEF1 degradation.
Despite the lack of well-established functional readout for HEF1, our recent studies led to the unexpected finding that HEF1 is not simply a target for Smad3 to allow TGF-β/activin to regulate HEF1-involved pathways, but itself has an important function in the known signaling events of TGF-β in epithelial cells. First, we observed that the time course of the degradation of endogenous HEF1 differs between epithelial cells and T cells. Unlike the time course of HEF1 degradation in H9 T cells, endogenous HEF1 degradation occurs more quickly in epithelial cells (as early as 15 min after TGF-β treatment). Furthermore, the protein level of HEF1 is restored rapidly. The recovery of the HEF1 level involves a rapid increase in HEF1 mRNA in these cells. Interestingly, longer TGF-β exposure leads to a continued increase in HEF1 protein level that far exceeds the basal level of HEF1. We then tested the effect of increased HEF1 level on the TGF-β-induced Smad3-mediated gene response, which was monitored by a reporter gene (CAGA-luc) that is regulated directly by Smad3. Interestingly, HEF1 exhibits a potent inhibitory effect on the TGF-β-induced activation of the CAGA-luc reporter. The ability of HEF1 to inhibit the activation of this reporter suggests that HEF1 is a cytoplasmic inhibitor of Smad3. Thus, the degradation of HEF1 may be an integral step in the activation of Smad3 after TGF-β stimulation. The further elevated HEF1 protein level after HEF1 degradation could therefore serve to restore the inhibitory effect of HEF1 in the cytoplasm and finally to turn off Smad3 signaling in the nucleus. This phenomenon is very interesting, since a very similar observation was reported recently for a different Smad3 interactor, the oncoprotein SnoN (Stroschein et al., 1999; Sun et al., 1999). SnoN is a nuclear transcriptional repressor that binds to both Smad3 and Smad4 (Stroschein et al., 1999). It has been proposed that SnoN functions as a nuclear repressor of TGF-β-responsive genes in the absence of TGF-β stimulation, but its repression activity is abolished by Smad3-induced proteasomal degradation of SnoN upon TGF-β stimulation (Stroschein et al., 1999). Strikingly, the mRNA of SnoN also increases rapidly (after 1 h), which leads to a further elevated SnoN protein level (after 2 h), with a very similar time course to that which we observed for HEF1. Our data thus extended this observation to suggest further that Smad3 can regulate proteasomal degradation of both cytoplasmic and nuclear repressors of the TGF-β pathway, and that these degradation events are followed by negative feedbacks in the cytoplasm and the nucleus to shut off the nuclear signaling activities of Smad3 efficiently. Interestingly, both feedback mechanisms are linked to proteasomal degradation of Smad3 interactors.
The ability of Smad3 to regulate proteasomal degradation may not be unique to Smad3, since we also discovered the ability of two other Smads, Smad1 and Smad4, to regulate proteasomal degradation of SNIP1, a novel CBP/p300 repressor (Kim et al., 2000; Y.Lin, J.Martin, C.Gruendler, X.Meng, B.-Y.Li, R.Lechleider, C.Huff, R.H.Kim, W.Grasser, V.Paralkar and T.Wang, submitted). Thus, Smad-regulated proteasomal degradation of cytoplasmic and nuclear interactors may be an important signaling mechanism of TGF-β family ligands.
Materials and methods
Antibodies and reagents
α-HEF1 (SB) polyclonal antibody has been described previously (Law et al., 1998). Anti-p130Cas and α-phosphotyrosine (α-pTyr, clone PY20) monoclonal antibodies were purchased from Transduction Laboratories. Anti-phosphoserine (α-pSer) and α-phosphothreonine (α-pThr) polyclonal antibodies were obtained from Zymed Laboratories. Lactacystin was obtained from Dr E.J.Corey (Harvard University). ZVAD was kindly provided by Dr Honglin Li (Harvard Medical School).
Plasmids
The pCMV-HEF1 expression vector has been described previously (Law et al., 1998). All the constructs for HEF1 and Smad3 were constructed by PCR using Pfu polymerase (Stratagene) and standard subcloning procedures (Ausubel et al., 1994). Smad3N and Smad3C encode amino acids 1–144 and 229–424, respectively. For HEF1 C-terminal deletions, M2, M2/N, M2/M and M2/C encode amino acids 651–834, 651–759, 695–759 and 695–832, respectively. The type I receptors of TGF-β (R4), activin (R2) and the mutant receptors R4TD (R4T204D), R4KR (R4K230R), R2TD (R2T206D) and R2KR (R2K234R) were described previously (He et al., 1993; Bassing et al., 1994; Antisano et al., 1996). T7-Ub was constructed by PCR amplification of ubiquitin and subcloning into EcoRI–SalI sites of a modified pCMV6 vector containing a T7 epitope upstream of the multiple cloning sites.
Yeast two-hybrid screening
Full-length human Smad3 fused to the LexA DNA-binding domain was used as bait to screen a human fetal brain library for interacting proteins, using the ‘Protein Trap’ yeast two-hybrid system (Zervos et al., 1993). A total of 7.5 × 105 yeast transformants were screened. cDNAs from candidate clones were rescued, grouped by PCR restriction mapping and tested for specificity of interaction by transformation into yeast strains expressing different bait proteins. All baits were fused in-frame with the LexA DNA-binding domain by subcloning into the EcoRI–XhoI sites of the pEG202 vector. Preys were fused in-frame with the B42 activation domain by subcloning into the EcoRI–XhoI sites of the pJG4-5 vector.
Immunoprecipitation and western blotting
293 cells were transfected by the standard CaPO4 procedure (Ausubel et al., 1994) and cells were harvested 24 h after transfection. Immuno precipitation and western blotting were performed as described, with slight modifications (Law et al., 1998). All the steps were carried out at 4°C. Briefly, cells were washed twice with ice-cold phosphate-buffered saline (PBS) and harvested into buffer A [50 mM HEPES pH 7.5, 50 mM NaCl, 5 mM EDTA, 1% Triton X-100, 50 mM NaF, 10 mM Na4P2O7, 1 mM Na3VO4, 1 mM phenylmethylsulfonyl fluoride (PMSF), 10 µg/ml aprotinin, 10 µg/ml leupeptin, 1 µg/ml pepstatin A and 10 mM β-glycerol phosphate]. Lysates were rotated at 4°C for 30 min. Cell debris was pelleted by spinning in a microfuge at 12 000 g for 10 min, and supernatant was saved for immunoprecipitation and western blot analysis. A 750 µg aliquot of cell lysates was immunoprecipitated overnight with 2.5 µl of α-F antibody or 7 µl of α-p130Cas antibody in the presence of 38 µl of 50% protein G–Sepharose (Pharmacia) at 4°C. Alternatively, lysates were immunoprecipitated with 10 µl of α-HEF1 antibody or 3 µl of α-T7 antibody and 38 µl of 50% protein A–agarose (Pharmacia). Beads were washed once with buffer A and three times with modified buffer A containing 0.1% Triton X-100. Precipitated proteins were eluted with 2× SDS sample buffer (100 mM Tris–HCl pH 6.8, 4% SDS, 0.2% bromophenol blue and 20% glycerol) and boiled for 2 min. Supernatant was supplemented with 100 mM dithiothreitol (DTT) and analyzed by western blotting. Antibodies were diluted as follows for western blot analysis: α-HEF1 (1:500), α-F (1:1000), α-T7 (1:2000), α-pTyr (1:1000), α-pSer (1:500) and α-pThr (1:500). To test the stability of the endogenous HEF1 protein in response to TGF-β, cells were first switched to medium containing 0.2% fetal bovine serum (FBS) before TGF-β (375 pM for A549 cells; 400 pM for H9 cells) was added directly into the medium at different time points. Proteasome inhibitor LLnL (50 µM) or protein synthesis inhibitor cycloheximide (10 µg/ml) was added 15 min before the addition of TGF-β as necessary. Cells were then harvested at the same time. Cell lysates were prepared as described above and an equal amount of total proteins was loaded onto SDS–PAGE gel. After transferring the proteins onto PVDF membrane (Millipore), western blotting was carried out using α-HEF1 antibody (1:500).
Northern blot analysis
A549 cells were serum starved for 2 h in medium containing 0.2% FBS, after which 400 pM TGF-β was added at different time points. Cells were harvested and mRNA was isolated using the QuickPrep Micro mRNA Purification Kit (Pharmacia). Northern blotting was performed by standard procedures (Ausubel et al., 1994) using a 32P-labeled HEF1 probe. The blot was stripped and reprobed with a GAPDH probe to show equal sample loading.
Luciferase assay
A549 cells in 6-well plates were transfected in duplicate by Fugene (Boehringer Mannheim) with 0.15 µg of CAGA-luc reporter, 0.05 µg of pCMV-RL Renilla luciferase construct and 0.3 µg of pCMV-HEF1 or pCMV6 empty vector. The following day, cells were untreated or treated with 100 pM TGF-β for 9 h in the absence or presence of 50 µM LLnL. Cells were lysed and luciferase activities were determined using a luminometer.
Acknowledgments
Acknowledgements
We are grateful to Drs A.Roberts for TGF-β and Smad1 cDNA; R.Derynck for human Smad3; J.Wrana and L.Antisano for R2T206D, R2K234R, Smad2 and Smad4; X.-F.Wang for R4T230R; A.Schneyer for activin; J.-M.Gauthier for CAGA-luc reporter; and N.E.Fusenig for the HaCaT cell line. We also thank C.Gruendler and W.Weber for data quantification. This work was supported by the Claflin Distinguished Scholar Award from Massachusetts General Hospital (T.W.), a grant from the National Institutes of Health GM53710 (T.W.) and by the Virginia Mason Research Center.
References
- Antisano L., Wrana,J.L., Montalvo,E. and Massague,J. (1996) Activation of signaling by the activin receptor complex. Mol. Cell. Biol., 16, 1066–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ausubel F.M., Brent,R., Kingston,R.E., Moore,D.D., Seidman,J.G., Smith,J.A. and Struhl,K. (1994) In Janssen,K. (ed.), Current Protocols in Molecular Biology. John Wiley & Sons, New York, NY. [Google Scholar]
- Bassing C.H., Yingling,J.M., Howe,D.J., Wang,T., He,W.W., Gustafson,M.L., Shah,P., Donahoe,P.K. and Wang,X.-F. (1994) A transforming growth factor β type I receptor that signals to activated gene expression. Science, 263, 87–89. [DOI] [PubMed] [Google Scholar]
- Dennler S., Itoh,S., Vivien,D., ten Dijke,P., Huet,S. and Gauthier,J.-M. (1998) Direct binding of Smad3 and Smad4 to critical TGFβ-inducible elements in the promoter of human plasminogen activator inhibitor-type 1 gene. EMBO J., 17, 3091–3100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derynck R., Zhang,Y. and Feng,X.H. (1998) Smads: transcriptional activators of TGF-β responses. Cell, 95, 737–740. [DOI] [PubMed] [Google Scholar]
- He W.W., Gustafson,M.L., Hirobe,S. and Donahoe,P. (1993) Develop mental expression of four novel serine/threonine kinase receptors homologous to the activin/transforming growth factor-β type II receptor family. Dev. Dyn., 196, 133–142. [DOI] [PubMed] [Google Scholar]
- Kim R.H. et al. (2000) A novel smad nuclear interacting protein, SNIP1, suppresses p300-dependent TGF-β signal transduction. Genes Dev., 14, 1605–1616. [PMC free article] [PubMed] [Google Scholar]
- Law S.F., Estojak,J., Wang,B., Mysliwiec,T., Kruh,G. and Golemis,E.A. (1996) Human enhancer of filamentation 1, a novel p130cas-like docking protein, associates with focal adhesion kinase and induces pseudohyphal growth in Saccharomyces cerevisiae. Mol. Cell. Biol., 16, 3327–3337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Law S.F., Zhang,Y.Z., Klein-Szanto,A.J.P. and Golemis,E.A. (1998) Cell cycle-regulated processing of HEF1 to multiple protein forms differentially targeted to multiple subcellular compartments. Mol. Cell. Biol., 18, 3540–3551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letterio J.J. and Roberts,A.B. (1998) Regulation of immune responses by TGF-β. Annu. Rev. Immunol., 16, 137–161. [DOI] [PubMed] [Google Scholar]
- Manie S.N. et al. (1997) Involvement of p130(Cas) and p105(HEF1), a novel Cas-like docking protein, in a cytoskeleton-dependent signaling pathway initiated by ligation of integrin or antigen receptor on human B cells. J. Biol. Chem., 272, 4230–4236. [DOI] [PubMed] [Google Scholar]
- Massague J. (1998) TGF-β signal transduction. Annu. Rev. Biochem., 67, 753–791. [DOI] [PubMed] [Google Scholar]
- Minegishi M., Tachibana,K., Sato,T., Iwata,S., Nojima,Y. and Morimoto,C. (1996) Structure and function of Cas-L, a 105-kD Crk-associated substrate-related protein that is involved in β1 integrin-mediated signaling in lymphocytes. J. Exp. Med., 184, 1365–1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nojima Y., Rothstein,D.M., Sugita,K., Schlossman,S.F. and Morimoto,C. (1992) Ligation of VLA-4 on T cells stimulates tyrosine phosphorylation of a 105-kD protein. J. Exp. Med., 175, 1045–1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Neill G.M., Fashena,S.J. and Golemis,E.A. (2000) Integrin signalling: a new Cas(t) of characters enters the stage. Trends Cell Biol., 10, 111–119. [DOI] [PubMed] [Google Scholar]
- Ohashi Y., Tachibana,K., Kamiguchi,K., Fujita,H. and Morimoto,C. (1998) T cell receptor-mediated tyrosine phosphorylation of Cas-L, a 105-kDa Crk-associated substrate-related protein and its association of Crk and C3G. J. Biol. Chem., 273, 6446–6451. [DOI] [PubMed] [Google Scholar]
- Sattler M., Salgia,R., Shrikhande,G., Verma,S., Uemura,N., Law,S.F., Golemis,E.A. and Griffin,J.D. (1997) Differential signaling after β1 integrin ligation is mediated through binding of CRKL to p120(CBL) and p110(HEF1). J. Biol. Chem., 272, 14320–14326. [DOI] [PubMed] [Google Scholar]
- Stroschein S.L., Wang,W., Zhou,S., Zhou,Q. and Luo,K. (1999) Negative feedback regulation of TGF-β signaling by the SnoN oncoprotein. Science, 286, 771–774. [DOI] [PubMed] [Google Scholar]
- Sun Y., Liu,X., Ng-Eaton,E., Lodish,H.F. and Weinberg,R.A. (1999) SnoN and Ski proto-oncoproteins are rapidly degraded in response to transforming growth factor β signaling. Proc. Natl Acad. Sci. USA, 96, 12442–12447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wrana J.L. (2000) Regulation of Smad activity. Cell, 100, 189–192. [DOI] [PubMed] [Google Scholar]
- Wrana J.L., Attisano,L., Weiser,R., Ventura,F. and Massague,J. (1994) Mechanism of activation of the TGF-β receptor. Nature, 370, 341–347. [DOI] [PubMed] [Google Scholar]
- Zervos A.S., Gyuris,J. and Brent,R. (1993) Mxi1, a protein that specifically interacts with Max to bind Myc–Max recognition sites. [Published erratum appears in Cell (1994), 79] Cell, 72, 223–232. [DOI] [PubMed] [Google Scholar]
- Zhang Z., Hernandez-Lagunas,L., Horne,W.C. and Baron,R. (1999) Cytoskeleton-dependent tyrosine phosphorylation of the p130(Cas) family member HEF1 downstream of the G protein-coupled calcitonin receptor. Calcitonin induces the association of HEF1, paxillin and focal adhesion kinase. J. Biol. Chem., 274, 25093–25098. [DOI] [PubMed] [Google Scholar]