

Abstract
Cancer therapies that target key molecules have not fulfilled expected promises for most common malignancies. Major challenges include the incomplete understanding and validation of these targets in patients, the multiplicity and complexity of genetic and epigenetic changes in the majority of cancers, and the redundancies and cross-talk found in key signaling pathways. Collectively, the uses of single-pathway targeted approaches are not effective therapies for human malignances. To overcome these barriers, it is important to understand the molecular cross-talk among key signaling pathways and how they may be altered by targeted agents. This requires innovative approaches such as understanding the global physiological environment of target proteins and the effects of modifying them without losing key molecular details. Such strategies will aid the design of novel therapeutics and their combinations against multifaceted diseases where efficacious combination therapies will focus on altering multiple pathways rather than single proteins. Integrated network modeling and systems biology has emerged as a powerful tool benefiting our understanding of drug mechanism of action in real time. This mini-review highlights the significance of the network and systems biology-based strategy and presents a “proof-of-concept” recently validated in our laboratory using the example of a combination treatment of oxaliplatin and the MDM2 inhibitor MI-219 in genetically complex and incurable pancreatic adenocarcinoma.
Keywords: Systems Biology, Network Modeling, drug interactions, MDM2, oxaliplatin, pancreas cancer
Current Challenges of Targeted Therapy
Although partially successful in some cancers, new adjuvant targeted therapies (p53, NF-κB, EGFR, VEGF, Src etc.) for complex malignancies have met with more failure than success (1). The major reason for the low response may be related to an incomplete understanding and lack of validation of the specific molecular targets at the gene level (2). This is coupled with complexities of genetic and epigenetic changes in cancer (3), and the redundancies of cross-talk in signaling pathways that taken together may explain the observed failure of single-pathway targeted therapies. Moreover, targeted therapy faces complexities of off-targeted effects of many so-called targeted agents (4). Of the 25,000 genes representing the human genome, approximately 1,800 are involved in the etiology of numerous diseases including cancer (5). Currently available FDA approved drugs (~1200 in the market) were designed to target approximately 400 gene products (Drugome). Pharmaceutical companies handpicked the protein products of single genes in the drugome to rationally design drugs. However, contrary to the original notion, targeting individual genes in this drugome is not a straightforward task as the functional product of each gene or (Proteome) is under multiple controls, including splice variants and post translational modifications, giving rise to more than 40,000 functionally distinct proteins. Previous approaches lacked information on biologically meaningful interconnecting pathways arising from perturbations of single or a set of genes by targeted drugs. This obstacle in our understanding of drug mechanism of action highlights an urgent need for the utilization of alternate technologies that would aid in a better understanding of drug mechanism of action.
The identification of nearly a complete list of genes and gene products in the human body through the human genome project (6) has enabled researchers to draft connectivity maps between proteins (7) and gene expression profiles (8). This helped in the identification of molecules specifically associated to certain pathological processes, and have benefited the field of drug discovery. Based on these advances in molecular sciences, the initial drug design approaches placed emphasis on target selectivity and enhancement of binding affinities of novel agents to target molecules (9, 10). However, the patho-biology of diseases like cancer is often the result of an incredibly complex combination of molecular events, and despite several success stories, the reductionism approach in drug design and development has led to an unacceptable outcome (11), suggesting that newer approaches must be implemented. Numerous high affinity and specific drugs fail at the last and most costly phase in the clinic, especially in malignancies such as pancreatic adenocarcinoma, arising through the accumulation of multiple genetic alterations/mutations in crucial pathways such as p53 (12), DPC4 (12) and k-ras (13, 14). Lack of robust clinical success of such targeted drugs could be attributed to many factors including inappropriate choice of in vitro cellular models, most importantly due to inadequate knowledge of the crucial interacting pathways. This implies that removing targets from their physiological context and developing drugs solely on the basis of their increasing binding affinity and target selectivity will yield little success. Drug developers are increasingly acknowledging that the next generation pharmacology strategies to fight complex multi-faceted disease require targeting multiple pathways rather than inhibiting single proteins.
The significance of using newer methodologies in delineating therapeutic interventions and the networks involved in malignancies, along with the identification of the mechanisms of action, off-target effects of novel agents and targeted drugs are being increasingly recognized. However, lack of proper tools have hindered the in-depth understanding of the accumulated knowledge of biological processes to benefit drug discovery and clinical applications (15). In the last few years, novel and high-throughput data acquisition technologies coupled with integrated network modeling and systems biology have emerged as key components of targeted therapy research (16). These technologies have helped in understanding a drug target protein/pathway in its physiological context with the greatest molecular detail, assisting in the identification of target genes along with clinically relevant drug combinations in a cancer specific manner. Such technologies are crucial for identifying and understanding the mechanisms of potential target candidates in complex diseases such as pancreatic adenocarcinoma (17). This review presents a strong example, and provides confidence on the use of systems-level knowledge of pharmacology stemmed from extensive genomic information, which would likely increase our understanding in evaluating the efficacy of novel targeted drugs, either alone or in combination treatment. The ultimate goal of such knowledge is directed towards the development of tailored and personalized medicine that is being demanded by the experts in the field, and are being predicted to be the mainstay in the near future in the field of cancer treatment.
Network and Systems Biology- a powerful new tool in the field of medicine
Systems biology is a science that defines the physical and functional relationships between components responsible for shaping-up a biological system (18). This technology allows real-time simulation of how biological molecules function in coordination to achieve a particular outcome, consequently providing tremendous power of predicting the drug response in terms of the effect of modulating the function of a given protein or pathway. A network perspective of complex cancers has direct implications in drug discovery process since it changes the target entity from a single protein to entire molecular pathways and or cellular networks. In recent years, the applicability of these powerful tools is increasingly being recognized in the clinical setting and researchers are beginning to change the way they think of a complex disease from gene-centric to a network-centric view, (19) although with skepticism. Such an approach identifies a collection of modifiable drug targets (instead of one protein) in their entirety and provides ample/optimal points for therapeutic intervention (20, 21). This is the key to a successful therapy for disease states that are known to be inherently resistant to drug treatment due to the maintenance of back-up or alternate survival mechanism such as typically observed in pancreatic adenocarcinoma (22). Combining high-throughput bioinformatics, followed by molecular network and systems level analysis, one can predict genes associated with cancers, biomarkers of response, as well as novel druggable targets. Such an approach may be applied to case-control cohorts for disease susceptibility or to study individual responses to drugs and their most clinically beneficially combinations. Several inroads have been made in our understanding of cancer development and progression utilizing such network-centric approaches. Examples where such technology showed benefit include the identification of novel genes associated with increased breast cancer risk, through disease-network analysis of BRCA1 (23). However, the role of these mutational variations in drug response has not been determined in cancers and most importantly, no such information is available for tumors with complex genetic make-up. Therefore, it is believed that this technology can be effectively utilized for the management of most cancers where, at present, specific biomarkers or targets have not been clearly defined. In the following sections, we will discuss our recent experience in the use of a systems-biology approach in analyzing the synergistic interaction of two anti-cancer drugs.
Systems understanding of drug action
The goal of applying integrated network modeling and systems biology in medicine is to identify drugs that can be prescribed together, and discover a combination of targets and modulators to produce synergistic effects. However, prior to applying a systems approach one needs to understand the complex and multi-tiered interactions between various scales of organization starting from a very basic molecular and cellular network, to tissue organization, and finally to organ interaction in the organism. Drug effects on patho-physiology that are manifested at the organism level are measured by clinical parameters, laboratory studies, and radiological measurements that aid in designing tailored therapy in order to achieve maximal treatment outcome. The other view (also called zoom-in view) stems from laboratory research, where response to drug can be studied at the gene, protein or cellular level by utilizing high throughput technologies such as integrated genomic microarray expression profiling coupled with pathway network modeling. Generally, a systems approach that integrates knowledge from analyses across multiple zoom levels is likely to uncover new target(s) in a pathway of interest that probably would have been left undiscovered using traditional techniques.
Most targeted drugs currently used in the clinic have been designed to affect a single protein, or in some cases multiple kinases. Unfortunately, even with the most specific drugs, additional proteins or related pathways may also be involved (off-target effects). Systems pharmacology categorizes these off-targets into two types: 1) off-targets that result in unwanted drug effects and 2) secondary targets that enhance desired drug effect (24). These secondary targets exist within a complex network that determines the balance between therapeutic and adverse effects. Understanding the beneficial secondary targets of targeted drugs is likely to provide valuable information for designing personalized therapies based on the host’s molecular make-up and eventually aid in the rational design of multi-targeted therapies using multiple drugs selected on the basis of synergy, or near-synergy in their effects. Such an understanding requires cell-based studies coupled with robust, computational tools to obtain irrevocably strong proof for the integration of pathways involved in the observed synergy. One such approach involves the use of network modeling that provides mathematically and statistically robust information regarding the involvement of effector networks in the interaction between multiple drugs. These network models can also predict key secondary targets of drug interactions, thus, uncovering previously unrecognized targets that may be useful for future drug development in cancer. Based on network modeling and systems level analyses, more than 100 drug synergistic cases have been recently reported or are currently being commercialized. For example, in a recent study, the identification of expression signatures predictive of sensitivity to the Bcl-2 family member inhibitor ABT-263 in small cell lung carcinoma (SCLC) and leukemia/lymphoma cell lines has been documented (25). This study revealed that global expression data could identify key gene expression patterns for sensitivity to ABT-263 in SCLC and leukemia/lymphoma and may provide guidance in the selection of patients’ in future clinical trials. Other rationally designed studies based on extensive knowledge of the pathways have recently been reported for metastatic breast cancer. In this case, the role of epidermal growth factor receptor (EGF-R), the cyclo-oxygenase (COX-2) and the matrix metalloproteases 1 and 2 (MMP-1, MMP-2) were found as the key genes that trigger lung metastasis, and further documented a significant reduction in lung metastasis when the cells were treated with anti-EGFR antibody cetuximab, the COX-2 inhibitor celecoxib, and the broad-spectrum MMP inhibitor GM6001 (26). Supported by this and other excellent examples, we undertook a systems level approach in combination with network biology to identify and validate potential targets in the synergistic combination of a novel small molecule inhibitor of MDM2 and oxaliplatin in pancreatic adenocarcinoma.
Proof of Concept: using a systems-biology based approach to predict potent drug combinations
Our laboratory has been focused on delineating the molecular mechanism(s) of action in pancreatic adenocarcinoma, and recently has been working on a specific small molecule inhibitor (MI-219) of murine double minute 2 protein (MDM2). The MI-219 is a specific, orally active, MDM2 inhibitor that binds to the p53 binding pocket of MDM2 and disrupts the MDM2-p53 interaction leading to apoptosis [the structure of MI-219 is given in Figure 1 and reported previously; (27–29)]. Recently we have observed that MDM2 inhibitor synergizes with chemotherapy leading to enhanced growth inhibition and apoptosis in pancreatic adenocarcinoma cells. Interestingly, 50% of wt-p53 tumor-bearing mice treated with this combination remained tumor free without recurrence for 120 days (30). Our most recent study also showed a synergistic enhancement of MI-219 activity in the presence of zinc (31). Overall our published results indicate that MI-219 can be used in conjunction with chemotherapy; however, the precise mechanism of this synergy has not been fully characterized at the molecular level. Given the complex network of interaction between MDM2 and p53 that ultimately governs apoptosis, it is reasonable to speculate that analyzing the local network of crucial members may influence or help predict the cellular response to MI-219. In the case of MI-219, elucidating important key protein/pathways will help us in identifying patients who are more likely to respond to MI-219 treatment, which will provide molecular guidance for conducting rationally designed combination trials. We have used this model to validate the applicability of a systems approach in predicting potent drug combinations in pancreatic adenocarcinoma and obtained critical information toward understanding the mechanism for this synergy, which serves as a “proof-of-concept” for launching any future clinical studies.
Figure 1.
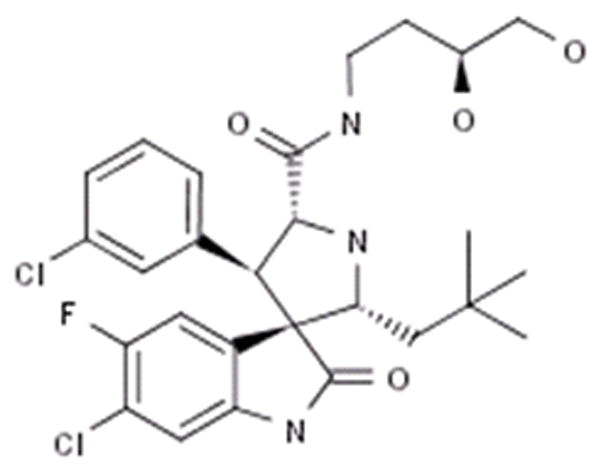
Structure of the MDM2 inhibitor MI-219.
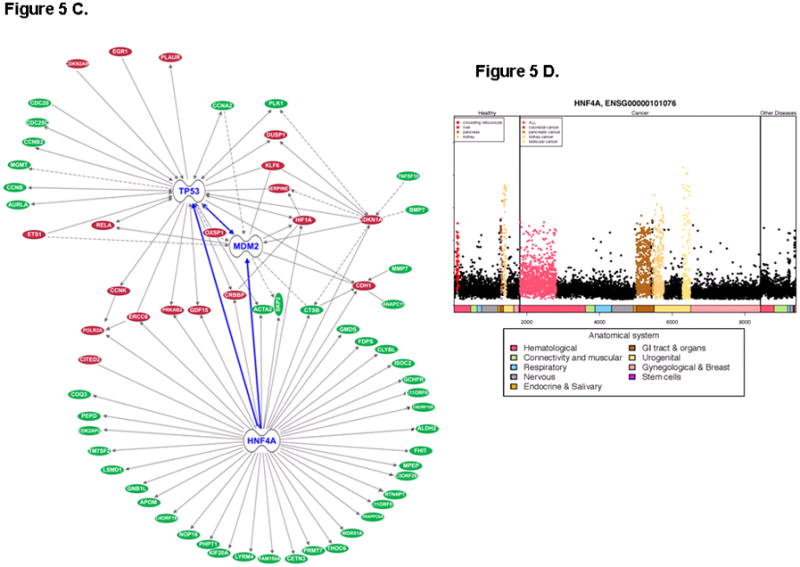
Microarray profiling of a wt-p53 containing pancreatic adenocarcinoma cell line (Capan-2) treated with either MI-219, or oxaliplatin, or their combination, revealed some very interesting results that may have clinical implications. Capan-2 cells were purchased from American Type Culture Collection (ATCC). The cell line have been tested and authenticated in our core facility, Applied Genomics Technology Center at Wayne State University, as late as March 13, 2009. The method used for testing was short tandem repeat (STR) profiling using the PowerPlexR 16 System from Promega (Madison, WI). Global analysis of genes showed that MI-219 treatment resulted in the alteration of only 48 genes (which highlights the targeted nature of MDM2 inhibitor MI-219). On the other hand, oxaliplatin is a cytotoxic agent and caused alteration of 761 genes. The combination of MI-219 with oxaliplatin resulted in 767 genes being altered. The most important aspect of this finding is the emergence of 286 synergy-specific unique genes that were not found in the MI-219 alone or in the oxaliplatin treated group (Fig 2A). This finding confirms that the synergy between MI-219 and oxaliplatin is at the gene level. Principle component analysis showed that the global gene signatures between single treatments vs. combination were non-overlapping and could be differentiated at different time points (Fig 2B). Molecular Network modeling of a total of 767 gene associated pathways revealed a total of 22 statistically enriched functional groups that were linked to biologically distinct functional pathways (Fig 3A). Interestingly, network modeling of the 286 synergy unique genes showed statistical enrichment of 14 disease (cancer) relevant pathways (Fig 3B). This suggests that these pathways are relevant to cancer, further indicating that the combination synergy between MI-219 and oxaliplatin is at the gene level comprising distinct biologically meaningful processes. Further analysis of the combination treatment network revealed the presence of several local networks, or hubs, rather than a single hub of activity interconnecting MDM2-p53 (Fig 4). Central players such as CREBBP (i.e. ubiquitously expressed gene) that is involved in the transcriptional co-activation of many different transcription factors including p53 (32), CARF that is responsible for p53 stability (33) and NF-κB and EGR1 tumor suppressor module (34), all of which are known to positively affect the p53 re-activation, which in principle would drive cells toward increased apoptosis. Most importantly, these observed gene changes could also be validated at the mRNA and protein level (data not shown and beyond the scope of this review article).
Figure 2. Gene expression microarray profiling and molecular network modeling predicts synergy between two drugs at the gene level. [A].
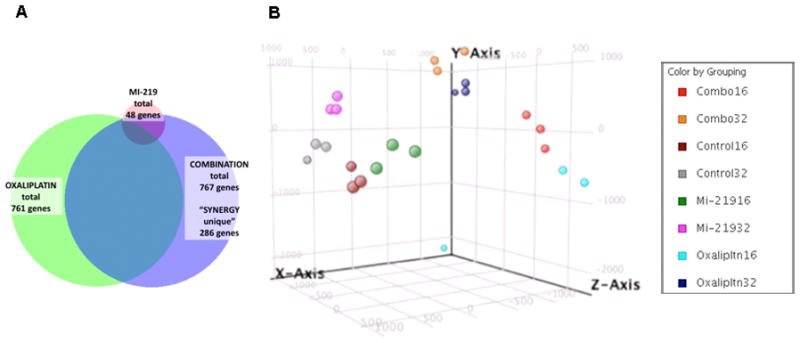
Venn Diagram showing synergy between MDM2 inhibitor MI-219 and oxaliplatin. Note: emergence of 286 synergy unique genes in the combination group. [B] Principle component analysis showing global gene patterns post drug treatments: single treatment vs combination at different time points. Analysis is representative of biological triplicates of Capan-2 pancreatic adenocarcinoma cells (i) untreated; (ii) MI-219 treated (15 μM); (iii) oxaliplatin treated (15 μM); and (iv) MI-219 (15 μM) + oxaliplatin (15μM) combination treatment for 16 and 32 hrs. RNA quality was assessed by Agilent Bioanalyzer 2100 RIN analysis. Expression levels at each time point and treatment were determined by microarray analyses using the human HT12 array. Data were processed for quality control and normalized across compared arrays by quantile normalization. Genes with 1.7 or greater expression fold-change at any time point in the series were included in Ingenuity Pathway Analyses. Cluster analysis of expression profiles was performed with Bayesian analysis using CAGED software. Canonical pathways analysis identified the pathways from the Ingenuity Pathways Analysis library of canonical pathways that were most significant to the data set. Molecules from the data set that met the 1.7 fold-change cut-off and were associated with a canonical pathway in Ingenuity’s Knowledge Base were considered for the analysis. The significance of the association between the data set and the canonical pathway was measured in 2 ways: 1) a ratio of the number of molecules from the dataset that map to the pathway divided by the total number of molecules that map to the canonical pathway; 2) Fisher’s exact test was used to calculate a p-value determining the probability that the association between the genes in the dataset and the canonical pathway is explained by chance alone.
Figure 3. Synergy between MI-219 and oxaliplatin is a consequence of multiple closely knit pathway interactions.
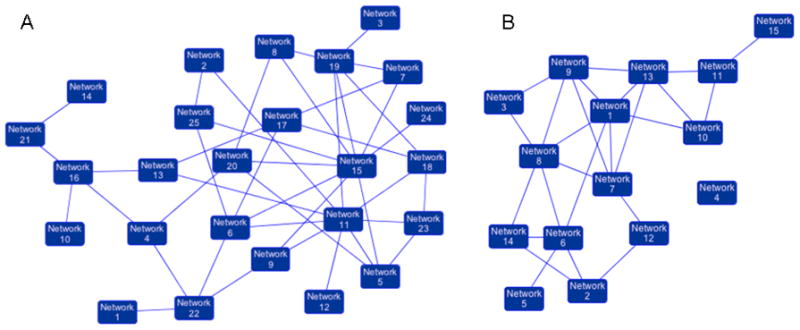
[A] Network analysis of a total of 767 genes showing 22 statistically enriched and biologically meaningful pathways. [B] Network analysis of 286 synergy unique genes showing statistical enrichment of 14 biologically meaningful pathways.
Figure 4. A p53 hub is activated by MI-219-oxaliplatin in Capan-2 cells.
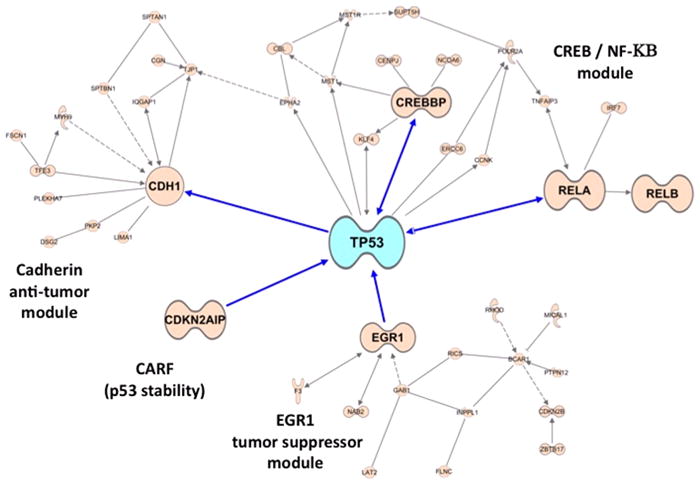
Pathway network modeling of statistically enriched local networks involved in the re-activation of p53 in Capan-2 cells post 32 hr treatment as described in Fig-1. Note: interaction with cadherin anti-tumor module, NF-κB, p53 stabilizing protein EGR1 and CREBBP, and the MDM2 negative regulator CARF (known to further drives p53 reactivation and apoptosis).
Taken together, these results show a rich pattern of interactions between MI-219, oxaliplatin and their targets, and further confirmed that the observed synergy is indeed at the gene level. Such a vast amount of information regarding the mechanism involved could be useful in predicting response to MI-219-oxaliplatin synergy, and certainly validates the applicability of this technology in understanding drug target gene signatures. It is believed that such information will aid in the design of clinically successful drug combinations for complex diseases such as pancreatic adenocarcinoma, and will ultimately improve the overall survival of patients.
Implications for the treatment of pancreatic adenocarcinoma
Our intended goal in using network modeling and systems analysis was to demonstrate as a “proof of concept” the applicability of such an approach in understanding the synergy between MI-219 and oxaliplatin at the gene level, and to further identify crucial driver pathways that augment p53 re-activation mediated events. Consistent with our intended goals, we observed that in addition to the genes mentioned above, our analysis revealed a prominent role for hepatocyte nuclear factor 4 alpha (HNF4α) (35) that modulated a totally distinct yet p53-linked set of proteins driving apoptosis (Fig 5A & B; MI-219 single treatment 16 and 32 hrs). In the combination network analysis, significant down-regulation of HNF4α target genes was observed (Fig 5C shown in green). This was concomitant with up-regulation of CARF, EGR1, HIF-1α, ETS transcription factor and E-cadherin. The identification of HNF4α as a key player was interesting because it has not been well defined in pancreatic adenocarcinoma cells used in this study (Capan-2 cells with wt-p53 gene). Nevertheless, published data have shown that HNF4α is highly expressed in pancreatic tumors compared to their normal counterpart (Fig 5D) (36). The HNF4α is known to interact with the p53 positive regulator CREBBP, (37) which underscores its role in augmenting apoptotic effects in this synergic combination. In summary, these findings support newer applications of this systems approach in determining the drug target gene signatures and providing information on potential newer targets that could be further studied either as biomarkers or molecules worthy of therapeutic targeting.
Figure 5. Hepatocyte nuclear factor 4 alpha (HNF4α) is a novel target in MI-219 response network.
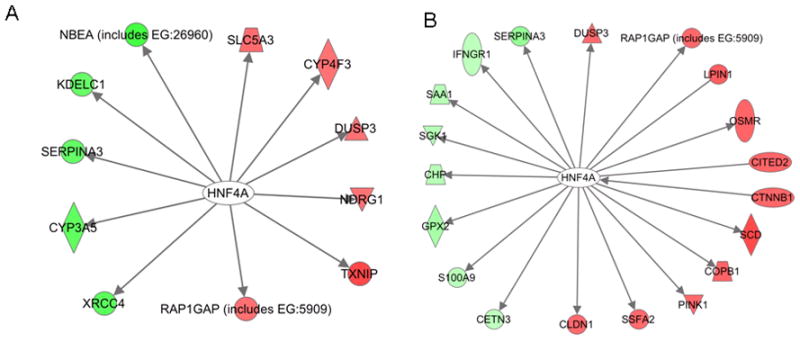
[A & B] Pathway network modeling of MI-219 treated at 16 and 32 hrs showing down-regulation of HNF4α and related network genes; [C] Pathway network modeling of combination treatment showing down-regulation of HNF4α target genes. Red (Genes that are up-regulated); Green (Genes that are down-regulated); [D] Gene expression profile of 48 healthy tissues and 68 cancer types reveals HNF4α high expression specific to pancreatic tumors.
Conclusion
Biological interaction networks have been available to the scientific community for nearly a decade, but only in the last five years has the concept of network biology found its application in the area of drug discovery. Despite being imperfect and error-prone, the initial version of human interactome networks (38, 39) are of sufficient quality to provide clinically useful information. Such integrated analyses may lead to the identification of pathways, and targeting of these may lead to synergy between drugs, or add to the effect of a given drug. Thus far, network analysis has facilitated the prediction of possible molecules affected by specified perturbations of up and downstream targets by different drugs. Such predictions can be applied for developing clinically relevant drug combinations, and was recently validated by a systems based approach using MI-219 (in this review) and ABT-263 (25). Network modeling and systems biology are still in their infancy, but have the potential to provide a major contribution to the advancement of personalized medicine. It is expected that the completion of the disease related interaction maps and the phenotypic effects of targeting multiple proteins in model organisms with chemical probes will soon permit refinement of systems-biology models to the point where they can be routinely applied to some of the most important areas of drug development. In conclusion, we advocate the use of this systems biology approach to further understand multiple drug combinations currently being tested in different cancers. Such cutting-edge knowledge is anticipated to improve rational design of combination therapy, and will hopefully improve the overall outcome of treatment, especially for pancreatic cancer-a disease for which better treatment is so urgently needed.
Acknowledgments
Grant support and Acknowledgements: National Cancer Institute, NIH grant R01CA109389 (R.M. Mohammad) and NIH grant 5R01CA101870 (F.H. Sarkar) are acknowledged. We thank Dr. Craig Giroux and the Systems biology core for their services in the network modeling and systems analysis. We deeply acknowledge Dajun Yang, Shaomeng Wang and Ascenta Therapeutics for providing MI-219. We sincerely thank the Guido foundation for their support. Dr. Tony Shields at KCI is gratefully acknowledged for his financial assistance and support in completing the systems biology studies. Shadan Ali is gratefully acknowledged for her help in the improvement of graphics. The authors also deeply acknowledge Asma S. Azmi for carefully editing and proof reading this manuscript.
Grant support and Acknowledgements: National Cancer Institute, NIH grant R01CA109389 (R.M. Mohammad) and NIH grant 5R01CA101870 (F.H. Sarkar) are acknowledged.
Abbreviations
- MDM2
Murine Double Minute Two
- HNF4α
Hepatocyte Nuclear Factor Four Alpha
- CREBBP
CREB Binding Proteins
- CARF
Collaborates/Cooperates with ARF
- EGR1
Early Growth Response Protein
Footnotes
Potential Conflict of Interest: There are no potential conflicts of interest for any of the authors.
References
- 1.Wilson TR, Johnston PG, Longley DB. Anti-apoptotic mechanisms of drug resistance in cancer. Curr Cancer Drug Targets. 2009;9:307–19. doi: 10.2174/156800909788166547. [DOI] [PubMed] [Google Scholar]
- 2.Newell DR. How to develop a successful cancer drug--molecules to medicines or targets to treatments? Eur J Cancer. 2005;41:676–82. doi: 10.1016/j.ejca.2004.12.024. [DOI] [PubMed] [Google Scholar]
- 3.Heng HH, Bremer SW, Stevens JB, Ye KJ, Liu G, Ye CJ. Genetic and epigenetic heterogeneity in cancer: a genome-centric perspective. J Cell Physiol. 2009;220:538–47. doi: 10.1002/jcp.21799. [DOI] [PubMed] [Google Scholar]
- 4.Le TC, Stathis A, Vidal L, Moore MJ, Siu LL. Choice of starting dose for molecularly targeted agents evaluated in first-in-human phase I cancer clinical trials. J Clin Oncol. 2010;28:1401–7. doi: 10.1200/JCO.2009.25.9606. [DOI] [PubMed] [Google Scholar]
- 5.Wist AD, Berger SI, Iyengar R. Systems pharmacology and genome medicine: a future perspective. Genome Med. 2009;1:11. doi: 10.1186/gm11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lander ES. Genomics: launching a revolution in medicine. J Law Med Ethics. 2000;28:3–14. [PubMed] [Google Scholar]
- 7.Stelzl U, Worm U, Lalowski M, et al. A human protein-protein interaction network: a resource for annotating the proteome. Cell. 2005;122:957–68. doi: 10.1016/j.cell.2005.08.029. [DOI] [PubMed] [Google Scholar]
- 8.Barrett T, Troup DB, Wilhite SE, et al. NCBI GEO: archive for high-throughput functional genomic data. Nucleic Acids Res. 2009;37:D885–D890. doi: 10.1093/nar/gkn764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Drews J. Pharmaceuticals: Classes, Therapeutic Agents, Areas of Application Volume 4. Drug Discov Today. 2001;6:1100. doi: 10.1016/s1359-6446(01)02027-x. [DOI] [PubMed] [Google Scholar]
- 10.Drews J. Drug discovery: a historical perspective. Science. 2000;287:1960–4. doi: 10.1126/science.287.5460.1960. [DOI] [PubMed] [Google Scholar]
- 11.Glaser B. Genetic analysis of complex disease--a roadmap to understanding or a colossal waste of money. Pediatr Endocrinol Rev. 2010;7:258–65. [PubMed] [Google Scholar]
- 12.Moore PS, Sipos B, Orlandini S, et al. Genetic profile of 22 pancreatic carcinoma cell lines. Analysis of K-ras, p53, p16 and DPC4/Smad4. Virchows Arch. 2001;439:798–802. doi: 10.1007/s004280100474. [DOI] [PubMed] [Google Scholar]
- 13.Rozenblum E, Schutte M, Goggins M, et al. Tumor-suppressive pathways in pancreatic carcinoma. Cancer Res. 1997;57:1731–4. [PubMed] [Google Scholar]
- 14.Radulovich N, Qian JY, Tsao MS. Human pancreatic duct epithelial cell model for KRAS transformation. Methods Enzymol. 2008;439:1–13. doi: 10.1016/S0076-6879(07)00401-6. [DOI] [PubMed] [Google Scholar]
- 15.Kramer R, Cohen D. Functional genomics to new drug targets. Nat Rev Drug Discov. 2004;3:965–72. doi: 10.1038/nrd1552. [DOI] [PubMed] [Google Scholar]
- 16.Russell RB, Aloy P. Targeting and tinkering with interaction networks. Nat Chem Biol. 2008;4:666–73. doi: 10.1038/nchembio.119. [DOI] [PubMed] [Google Scholar]
- 17.Jones S, Zhang X, Parsons DW, et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science. 2008;321:1801–6. doi: 10.1126/science.1164368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pujol A, Mosca R, Farres J, Aloy P. Unveiling the role of network and systems biology in drug discovery. Trends Pharmacol Sci. 2010;31:115–23. doi: 10.1016/j.tips.2009.11.006. [DOI] [PubMed] [Google Scholar]
- 19.Faratian D, Clyde RG, Crawford JW, Harrison DJ. Systems pathology--taking molecular pathology into a new dimension. Nat Rev Clin Oncol. 2009;6:455–64. doi: 10.1038/nrclinonc.2009.102. [DOI] [PubMed] [Google Scholar]
- 20.Stein A, Pache RA, Bernado P, Pons M, Aloy P. Dynamic interactions of proteins in complex networks: a more structured view. FEBS J. 2009;276:5390–405. doi: 10.1111/j.1742-4658.2009.07251.x. [DOI] [PubMed] [Google Scholar]
- 21.Pache RA, Zanzoni A, Naval J, Mas JM, Aloy P. Towards a molecular characterisation of pathological pathways. FEBS Lett. 2008;582:1259–65. doi: 10.1016/j.febslet.2008.02.014. [DOI] [PubMed] [Google Scholar]
- 22.Kitano H. Cancer robustness: tumour tactics. Nature. 2003;426:125. doi: 10.1038/426125a. [DOI] [PubMed] [Google Scholar]
- 23.Pujana MA, Han JD, Starita LM, et al. Network modeling links breast cancer susceptibility and centrosome dysfunction. Nat Genet. 2007;39:1338–49. doi: 10.1038/ng.2007.2. [DOI] [PubMed] [Google Scholar]
- 24.Berger SI, Iyengar R. Network analyses in systems pharmacology. Bioinformatics. 2009;25:2466–72. doi: 10.1093/bioinformatics/btp465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jia J, Zhu F, Ma X, Cao Z, Li Y, Chen YZ. Mechanisms of drug combinations: interaction and network perspectives. Nat Rev Drug Discov. 2009;8:111–28. doi: 10.1038/nrd2683. [DOI] [PubMed] [Google Scholar]
- 26.Gupta GP, Nguyen DX, Chiang AC, et al. Mediators of vascular remodelling co-opted for sequential steps in lung metastasis. Nature. 2007;446:765–70. doi: 10.1038/nature05760. [DOI] [PubMed] [Google Scholar]
- 27.Shangary S, Wang S. Small-molecule inhibitors of the MDM2-p53 protein-protein interaction to reactivate p53 function: a novel approach for cancer therapy. Annu Rev Pharmacol Toxicol. 2009;49:223–41. doi: 10.1146/annurev.pharmtox.48.113006.094723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shangary S, Qin D, McEachern D, et al. Temporal activation of p53 by a specific MDM2 inhibitor is selectively toxic to tumors and leads to complete tumor growth inhibition. Proc Natl Acad Sci U S A. 2008;105:3933–8. doi: 10.1073/pnas.0708917105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Brown CJ, Lain S, Verma CS, Fersht AR, Lane DP. Awakening guardian angels: drugging the p53 pathway. Nat Rev Cancer. 2009;9:862–73. doi: 10.1038/nrc2763. [DOI] [PubMed] [Google Scholar]
- 30.Azmi AS, Aboukameel A, Banerjee S, et al. MDM2 inhibitor MI-319 in combination with cisplatin is an effective treatment for pancreatic cancer independent of p53 function. Eur J Cancer. 2010 doi: 10.1016/j.ejca.2010.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Azmi AS, Philip PA, Beck FW, et al. MI-219-zinc combination: a new paradigm in MDM2 inhibitor-based therapy. Oncogene. 2010 doi: 10.1038/onc.2010.403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Livengood JA, Scoggin KE, Van OK, et al. p53 Transcriptional activity is mediated through the SRC1-interacting domain of CBP/p300. J Biol Chem. 2002;277:9054–61. doi: 10.1074/jbc.M108870200. [DOI] [PubMed] [Google Scholar]
- 33.Abida WM, Gu W. p53-Dependent and p53-independent activation of autophagy by ARF. Cancer Res. 2008;68:352–7. doi: 10.1158/0008-5472.CAN-07-2069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu J, Grogan L, Nau MM, Allegra CJ, Chu E, Wright JJ. Physical interaction between p53 and primary response gene Egr-1. Int J Oncol. 2001;18:863–70. doi: 10.3892/ijo.18.4.863. [DOI] [PubMed] [Google Scholar]
- 35.Chartier FL, Bossu JP, Laudet V, Fruchart JC, Laine B. Cloning and sequencing of cDNAs encoding the human hepatocyte nuclear factor 4 indicate the presence of two isoforms in human liver. Gene. 1994;147:269–72. doi: 10.1016/0378-1119(94)90079-5. [DOI] [PubMed] [Google Scholar]
- 36.Perilhou A, Tourrel-Cuzin C, Zhang P, et al. The MODY1 gene for hepatocyte nuclear factor 4alpha and a feedback loop control COUP-TFII expression in pancreatic beta cells. Mol Cell Biol. 2008;28:4588–97. doi: 10.1128/MCB.01191-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yoshida E, Aratani S, Itou H, et al. Functional association between CBP and HNF4 in trans-activation. Biochem Biophys Res Commun. 1997;241:664–9. doi: 10.1006/bbrc.1997.7871. [DOI] [PubMed] [Google Scholar]
- 38.Rual JF, Venkatesan K, Hao T, et al. Towards a proteome-scale map of the human protein-protein interaction network. Nature. 2005;437:1173–8. doi: 10.1038/nature04209. [DOI] [PubMed] [Google Scholar]
- 39.Stelzl U, Wanker EE. The value of high quality protein-protein interaction networks for systems biology. Curr Opin Chem Biol. 2006;10:551–8. doi: 10.1016/j.cbpa.2006.10.005. [DOI] [PubMed] [Google Scholar]