

Abstract
Diploid strains of the budding yeast Saccharomyces cerevisiae change the pattern of cell division from bipolar to unipolar when switching growth from the unicellular yeast form (YF) to filamentous, pseudohyphal (PH) cells in response to nitrogen starvation. The functions of two transmembrane proteins, Bud8p and Bud9p, in regulating YF and PH cell polarity were investigated. Bud8p is highly concentrated at the distal pole of both YF and PH cells, where it directs initiation of cell division. Asymmetric localization of Bud8p is independent of the Rsr1p/Bud1p GTPase. rsr1/bud1 mutations are epistatic to bud8 mutations, placing Rsr1p/Bud1p downstream of Bud8p. In YF cells, Bud9p is also localized at the distal pole, yet deletion of BUD9 favours distal bud initiation. In PH cells, nutritional starvation for nitrogen efficiently prevents distal localization of Bud9p. Because Bud8p and Bud9p proteins associate in vivo, we propose Bud8p as a landmark for bud initiation at the distal cell pole, where Bud9p acts as inhibitor. In response to nitrogen starvation, asymmetric localization of Bud9p is averted, favouring Bud8p-mediated cell division at the distal pole.
Keywords: BUD8/BUD9/cell polarity/pseudohyphal development/yeast
Introduction
Control of cell polarity is fundamental for the development of many organisms. The yeast Saccharomyces cerevisiae is a simple model for studying the molecular basis underlying establishment of cell polarity and oriented cell division. Yeast cells divide by budding and choose cell division sites in different spatial patterns that are under genetic control of their cell type (Freifelder, 1960; Hicks et al., 1977; Chant and Pringle, 1995). Haploid a or α cells bud in an axial pattern, where mother and daughter cells bud adjacent to the cell pole that defined the previous mother–daughter junction. This region of the yeast cell surface is also referred to as the proximal pole or the birth end of the cell. Diploid a/α yeast cells bud in a bipolar pattern, where buds form either at the proximal pole or at the site opposite to it, called the distal pole.
Yeast cell polarity and corresponding budding patterns are affected by extracellular stimuli, such as pheromones or nutrients. For instance, haploid cells that have been exposed to a concentration gradient of pheromone of the opposite mating partner redirect their axis of polarity and start to form mating projections in the direction of the presumed mating partner (Segall, 1993). In addition, budding patterns of haploid cells can be altered by nutritional starvation (Madden and Snyder, 1992; Chant and Pringle, 1995). Diploid cells starved for nitrogen switch their budding pattern from bipolar to unipolar distal, where most of the buds emerge at the distal cell pole (Gimeno et al., 1992; Kron et al., 1994). The unipolar distal budding programme is essential for the formation of multicellular filaments called pseudohyphae (PH), whose development is induced when diploid cells are starved for nitrogen, subsequently change cell morphology and show substrate-invasive growth behaviour. Unipolar distal budding is a prerequisite for the establishment of filamentous structures and, therefore, can be viewed as a process regulated by nutritional signals and guiding the direction of the growing PH filaments.
In yeast, selection of cell division sites is regulated by at least three different classes of genes and corresponding proteins (for recent reviews see Madden and Snyder, 1998; Chant, 1999). One class of genes is required for axial and bipolar budding and includes RSR1/BUD1, BUD2 and BUD5 (Bender and Pringle, 1989; Chant and Herskowitz, 1991; Chant et al., 1991). Mutations in these genes cause random budding patterns in haploid and diploid yeast form (YF) cells. Rsr1p/Bud1p, Bud2p and Bud5p constitute a GTPase signalling module that is thought to help direct bud formation components to the selected cell division site (Park et al., 1993, 1997). A second class of genes is required specifically for axial budding of haploids without affecting the bipolar pattern of diploids. Genes of this class include AXL1, BUD10/AXL2, BUD3 and BUD4 (Chant and Herskowitz, 1991; Fujita et al., 1994; Halme et al., 1996; Roemer et al., 1996). A third class of genes is required for the bipolar budding pattern of diploid yeast cells but not for haploid axial budding. Many genes of this class have been identified by genetic screening and include AIP3/BUD6, BUD7, BUD8, BUD9, BNI1, PEA2 and SPA2 (Snyder, 1989; Valtz and Herskowitz, 1996; Zahner et al., 1996). Mutations in most of these genes cause a random budding pattern only in diploids without affecting axial budding in haploids. Only two genes of this class, BUD8 and BUD9, have been described that shift the bipolar pattern to a unipolar pattern and therefore appear to have the most specific effects on bipolar budding. Mutations in BUD8 cause a unipolar proximal budding pattern in diploids, whereas bud9 mutants bud with high frequency from the distal cell pole (Zahner et al., 1996). Therefore, Bud8p and Bud9p have been proposed to act as bipolar landmarks that might recruit components of the common budding factors, e.g. Bud2p, Bud5p or Rsr1p/Bud1p, to either of the two cell poles (Chant, 1999).
Most studies that have addressed the function of genes controlling bud site selection were performed under nutrient-rich conditions, where S.cerevisiae will grow and divide in the unicellular YF. Little is known about the molecular mechanisms that control changes in cell polarity in response to nutritional starvation. Because nitrogen starvation causes a switch in the budding pattern from bipolar to unipolar distal in diploid cells, pseudohyphal development is an ideal model to study factors that control oriented cell division in response to external signals. To date, no class of genes has been identified that is specifically required for the unipolar distal pattern of PH cells without affecting bipolar budding of YF cells. An initial study has identified Rsr1p/Bud1p to be required for pseudohyphal development, because expression of a dominant-negative form of RSR1/BUD1, RSR1Asn16, suppresses filament formation in response to nitrogen starvation (Gimeno et al., 1992). A genetic screen directed at the identification of genes specifically required for pseudohyphal development has uncovered several of the bipolar specific bud site selection genes, including BUD8, BNI1, PEA2/DFG9 and SPA2 (Mösch and Fink, 1997).
In this study, we investigated the requirement and subcellular localization of Bud8p and Bud9p proteins during both YF growth in nutrient-rich media and PH filamentous growth under nitrogen starvation conditions. Our study suggests that Bud8p acts as a landmark for bud initiation at the distal cell pole, whereas Bud9p appears to be an inhibitor of distal budding that might interfere with Bud8p functions in YF cells. In PH cells, Bud9p is prevented from being localized at the distal cell pole, causing a switch in cell polarity from bipolar to unipolar budding.
Results
Bud8p and Bud9p are asymmetrically localized at the distal pole of YF cells
Previous genetic studies have suggested that Bud8p and Bud9p might act as landmarks for the selection of cell division sites. Therefore, we first determined the subcellular localization of Bud8p and Bud9p in diploid cells. GFP–BUD8 and GFP–BUD9 fusion genes expressing the GFP–Bud8p and GFP–Bud9p fusion proteins (where GFP is green fluorescent protein) from the endogenous BUD8 and BUD9 promoters were constructed and expressed in YF cells from low-copy-number plasmids. Low levels of GFP–Bud8p or GFP–Bud9p did not produce fluorescent signals that were detectable by GFP fluorescence microscopy, although the highly fluorescent GFPuv (cycle 3) variant was used (Crameri et al., 1996). Importantly, low-copy-number expression of GFP fusion genes complemented the budding defects of diploid bud8 or bud9 mutant strains, demonstrating that GFP fusion proteins were produced at levels sufficient for function but not for visual detection. GFP fusion proteins were detectable when GFP–Bud8p and GFP–Bud9p were expressed from high-copy-number plasmids. Localization was first analysed in exponentially growing cultures in nutrient-rich media, when strains grow predominantly in the YF form and develop the bipolar budding pattern (Figure 1A). GFP–Bud8p was found to be localized at both the tip of the growing daughter cell and the mother side of the mother–daughter neck. The concentration of GFP–Bud8p was more pronounced at the mother-bud neck than at the bud tip of small-budded YF cells. However, this difference was no longer detectable in large-budded YF cells. Surprisingly, GFP–Bud9p was also found to be highly concentrated at the tip of the growing bud throughout cell division. In contrast with GFP–Bud8p, no GFP–Bud9p was detectable at the mother-bud neck of small-budded cells, and only a weak fluorescent signal was detectable in this region in large-budded cells. Moreover, GFP–Bud9p was already found to be highly concentrated at the distal pole of unbudded cells, indicating that Bud9p concentrates at the site of the incipient bud in G1.

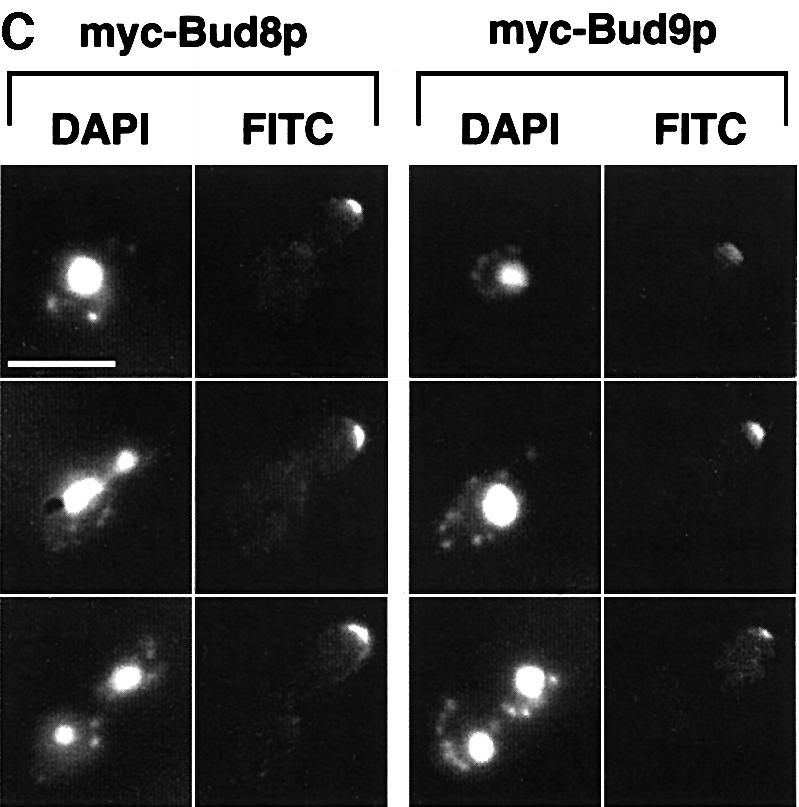
Fig. 1. Subcellular localization of Bud8p and Bud9p in YF cells. (A) Representative cells of wild-type strain RH2447 expressing either GFP–Bud8p from plasmid pME1772 or GFP–Bud9p from plasmid pME1777. Strains were grown in high ammonium media to exponential phase. Living cells at different stages of the cell cycle were chosen for photography according to their bud size and were viewed by differential interference contrast microscopy (DIC) or by fluorescence microscopy (GFP). Identical results were obtained when expressing GFP–Bud8p or GFP–Bud9p under the control of the MET25 promoter using plasmid pME1773 or pME1778, respectively. Scale bar applies to (A), (B) and (C) and represents 5 µm. (B) Immunofluorescence microscopy. Strain RH2447 expressing myc-Bud8p (pME1775) or myc-Bud9p (pME1780) was grown to exponential phase and prepared for anti-myc immunofluorescence. Shown are representative cells that were viewed for nuclear DNA with DAPI imaging (DAPI) or for anti-myc immunofluorescence (FITC). (C) Anti-myc immunofluorescence microscopy of strains expressing myc-Bud8p (RH2491) or myc-Bud9p (RH2493) at endogenous levels. Shown are representative cells viewed for nuclear DNA with DAPI imaging (DAPI) or for anti-myc immunofluorescence (FITC).
The subcellular localization of epitope-tagged versions of Bud8p and Bud9p was analysed by indirect immunofluorescence microscopy to corroborate the data found with GFP fusion proteins. A triple myc epitope tag was inserted just after the start codons of BUD8 and BUD9. The corresponding fusion genes were either expressed from high-copy-number plasmids or were integrated into the genome of wild-type as well as bud8 or bud9 mutant strains to obtain endogenous expression levels of tagged proteins. Phenotypic analysis of tagged versions of BUD8 and BUD9 in bud8 and bud9 diploid mutant strains revealed no difference when compared with non-tagged versions. The localization pattern of myc-Bud8p was similar to that observed using GFP–Bud8p throughout the cell cycle (Figure 1B and C). In contrast to GFP–Bud8p, however, myc-Bud8p was found predominantly at the tip of growing cells and only very weak staining was visible at the mother-bud neck. Similar results were found for myc-Bud9p when compared with GFP–Bud9p. The epitope-tagged version of Bud9p was highly concentrated at the site of the incipient bud in unbudded cells and at the tip of the growing daughter cells.
Expression of BUD8 and BUD9 is highly regulated during the cell cycle, with BUD8 showing peak expression in M phase and BUD9 peaking in G1 (Spellman et al., 1998). Therefore, GFP–Bud8p and GFP–Bud9p subcellular localization was analysed further when expressed from the MET25 promoter to test whether cell cycle-specific-expression is important for localization of Bud8p or Bud9p. However, no differences were found when compared with GFP–BUD8 or GFP–BUD9 under the control of the endogenous BUD8 or BUD9 promoters.
Localization of Bud8p and Bud9p was further measured in haploid strains using GFP fusions and myc-tagged versions. Interestingly, localization and expression patterns of Bud8p and Bud9p in haploids were found to be identical to those obtained in diploids, although haploid strains displayed an axial budding pattern (data not shown). This suggests that in haploid cells asymmetrically localized Bud8p and Bud9p proteins are not sufficient for induction of bipolar budding, most likely due to the presence of the haploid-specific budding proteins that might override functions of Bud8p and Bud9p.
In summary, subcellular localization studies show that both Bud8p and Bud9p are asymmetrically localized at the tip of growing cells throughout cell division, indicating an important function of both proteins at the distal cell pole.
Selection of the distal pole as site of cell division requires the presence of Bud8p and is favoured by the absence of Bud9p
Previous studies have addressed the function of BUD8 and BUD9 by use of only point mutations or partial gene disruptions (Zahner et al., 1996; Mösch and Fink, 1997). Therefore, we constructed homozygous diploid strains carrying full deletions of BUD8 or BUD9, and analysed their budding patterns in both YF and in PH cells by staining of bud scars (Figure 2). In addition, time-lapse microscopy was used, in order to distinguish between unipolar proximal (at the birth end of the cell) and unipolar distal (at the site opposite to the birth end) budding patterns (Figure 3). Full deletion of BUD8 caused a unipolar proximal budding pattern in both YF cells and PH filaments, whereas bud site selection of a control strain was bipolar in YF cells and switched to unipolar distal in PH filaments (Figures 2 and 3). In agreement with earlier observations, full deletion of BUD8 completely suppressed the formation of pseudohyphal filaments when tested on nitrogen starvation media (Figure 4). A detailed analysis of pseudohyphal sub-phenotypes revealed that the formation of long pseudohyphal cells and substrate-invasive growth were similar in wild-type and bud8 diploid mutants (Table I). Thus, changes of cell shape or switching from surface to invasive growth do not require BUD8. The budding-specific function of Bud8p is supported by the fact that overexpression of BUD8 from high-copy-number plasmids or from the MET25 promoter significantly enhanced the frequency of distal budding in YF cells without affecting cell morphology or invasive growth (data not shown). Deletion of BUD9 led to preferentially unipolar distal budding in YF cells (Figure 2). In contrast to BUD8, however, we could not detect significant alterations in bud site selection patterns by overexpression of BUD9. In PH cells, the unipolar distal pattern was not influenced by the absence of BUD9 (Figures 2 and 3). As a consequence, bud9/bud9 diploids produce regular amounts of pseudohyphae when grown on nitrogen starvation media (Figure 4). As found for BUD8, BUD9 was not required for changes in cell morphology or invasive growth behaviour during pseudohyphal development (Table I). This is in agreement with the fact that bud9/bud9 mutant strains do not produce pseudohyphal filaments on nitrogen-rich media, although the absence of BUD9 already induces unipolar distal budding in the YF.
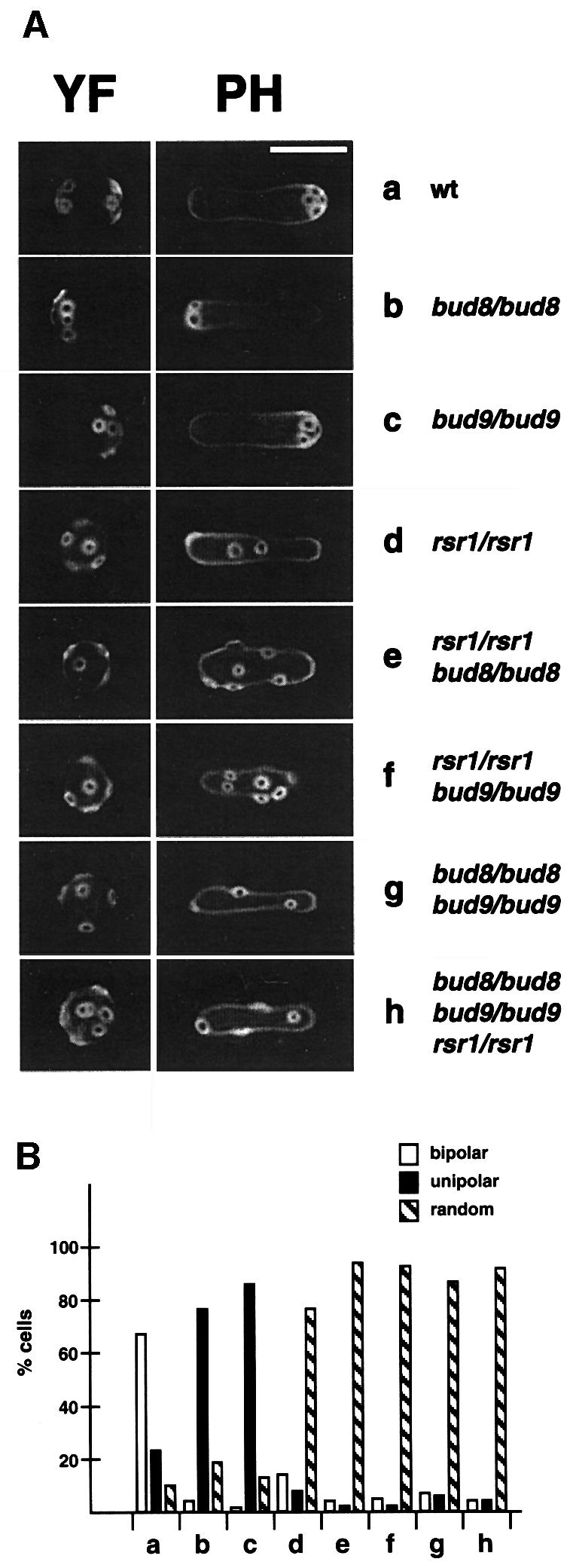
Fig. 2. Regulation of bud site selection by BUD8, BUD9 and RSR1/BUD1 in diploid YF and PH cells. (A) Fluorescence imaging of bud scar distribution of YF and PH cells after staining bud scars with calcofluor. Representative cells of strains RH2447 (wt) (a), RH2449 (bud8/bud8) (b), RH2450 (bud9/bud9) (c), RH2448 (rsr1/rsr1) (d), RH2451 (rsr1/rsr1 bud8/bud8) (e), RH2452 (rsr1/rsr1 bud9/bud9) (f), RH2453 (bud8/bud8 bud9/bud9) (g), RH2454 (rsr1/rsr1 bud8/bud8 bud9/bud9) (h). For YF cells, strains were transformed with plasmid pRS316 and grown to exponential phase in high ammonium media before staining with calcofluor. For PH cells, strains were transformed with plasmid pCG38 overexpressing PHD1 and grown in SLAD/LA media for 15 h. Scale bar represents 5 µm. (B) Quantitative analysis of bud scar distribution. At least 200 YF cells of strains described in (A) were analysed for bud scar distribution (see Materials and methods). Bars represent the percentage of cells exhibiting a bipolar (white bars), unipolar (black bars) or random (hatched bars) budding pattern.
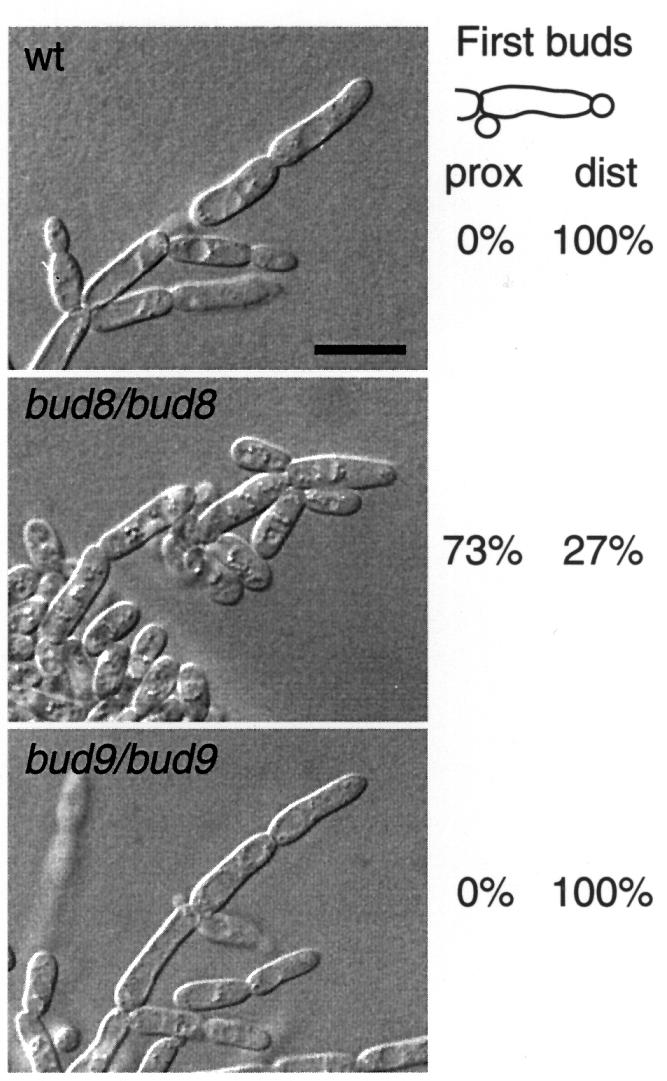
Fig. 3. Time-lapse observation of PH development. Saccharomyces cerevisiae strains RH2447 (wt), RH2449 (bud8/bud8) and RH2450 (bud9/bud9), all carrying plasmid pCG38, were analysed for selection patterns of first buds of virgin pseudohyphal cells. For each strain, at least 70 cell divisions were observed using a chamber for high magnification imaging of yeast growth on solid SLAD media (Kron et al., 1994). Numbers given indicate the percentage of virgin PH cells producing their first bud at either their birth end (proximal site) or opposite to their birth end (distal pole). After 3 days of growth, pseudohyphal development of cells at the edges of the colonies was visualized under the microscope using Nomarski optics. Scale bar represents 5 µm.

Fig. 4. Regulation of pseudohyphal development by RSR1/BUD1, BUD8 and BUD9. Diploid strains homozygous for the indicated genotype and transformed with pRS316 were grown on nitrogen starvation media: wt (RH2447), bud8 (RH2449), bud9 (RH2450), rsr1 (RH2448), rsr1 bud8 (RH2451), rsr1 bud9 (RH2452), bud8 bud9 (RH2453), rsr1 bud8 bud9 (RH2454). After 4 days of growth, pseudohyphal development of strains was visualized under a microscope and photographed. Scale bar represents 100 µm.
Table I. Regulation of pseudohyphal development by RSR1/BUD1, BUD8 and BUD9.
| Strain | Relevant genotype |
Invasion | Cell shape (%) |
||||
|---|---|---|---|---|---|---|---|
| RSR1/BUD1 | BUD8 | BUD9 | Long PH | Oval YF | Round YF | ||
| RH2447 | + | + | + | +++ | 25 | 61 | 14 |
| RH2448 | – | + | + | +++ | 26 | 67 | 7 |
| RH2449 | + | – | + | + | 22 | 65 | 13 |
| RH2450 | + | + | – | +++ | 29 | 64 | 7 |
| RH2451 | – | – | + | ++ | 25 | 63 | 12 |
| RH2452 | – | + | – | +++ | 25 | 66 | 9 |
| RH2453 | + | – | – | ++ | 21 | 60 | 19 |
| RH2454 | – | – | – | ++ | 17 | 71 | 12 |
In summary, selection of the distal pole as the site of cell division requires the presence of Bud8p and is favoured by the absence of Bud9p. This suggests that Bud8p acts as a landmark for bud initiation at the distal pole, whereas Bud9p appears to inhibit distal budding.
Bud8p and Bud9p proteins associate in vivo
Because Bud8p and Bud9p proteins are co-localized at the distal bud and both affect distal bud site selection, we tested whether Bud8p and Bud9p proteins physically interact in vivo. For this purpose, in-frame fusions between glutathione S-transferase (GST) and BUD8 or GST and BUD9 were constructed and expressed from the GAL1 promoter together with myc epitope-tagged versions of either BUD8 or BUD9. Fusion proteins were induced, and purified with glutathione beads to isolate each fusion and any associated proteins. Proteins purified by glutathione–agarose were analysed by western blot analysis using polyclonal anti-GST antibodies or monoclonal anti-myc epitope antibodies. We found that myc-Bud8p co-purifies with GST–Bud9p, but not with GST alone (Figure 5). Vice versa, myc-Bud9p is associated with GST–Bud8p, but not with the GST control. Thus, Bud8p and Bud9p proteins are associated in vivo, suggesting that Bud8p and Bud9p might influence each other’s function.
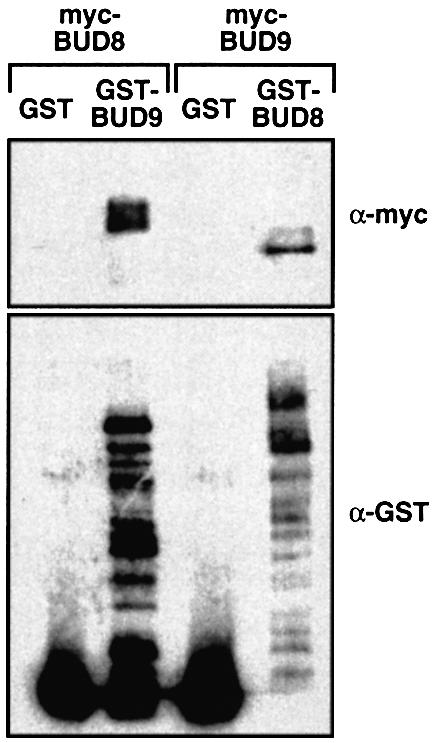
Fig. 5. Co-purification of GST–Bud9p with myc-Bud8p and of GST–Bud8p with myc-Bud9p. Total protein extracts were prepared from strain RH2495 carrying either of the plasmid pairs pME1937 (myc-BUD8) and pYGEX-2T (GST), pME1937 (myc-BUD8) and pME1941 (GST-BUD9), pME1939 (myc-BUD9) and pYGEX-2T (GST), or pME1939 (myc-BUD9) and pME1940 (GST-BUD8).GST and GST fusion proteins were purified as described. Equivalent amounts of each sample were subjected to SDS–PAGE, transferred to nitrocellulose and probed with a monoclonal anti-myc antibody(α-myc) or a polyclonal anti-GST antibody (α-GST).
Diploid bud8Δ bud9Δ null mutants produce a random budding pattern comparable with rsr1Δ/bud1Δ strains
BUD8 and BUD9 are as yet the only known genes encoding proteins that function as diploid-specific landmarks at cell poles. It has been observed that diploid bud8 bud9 double mutants exhibit the unipolar proximal budding pattern of bud8 single mutants, suggesting the existence of further factors acting as bipolar landmarks (Zahner et al., 1996). However, the above study was performed with strains carrying point mutations in BUD8 and BUD9. Therefore, we constructed homozygous diploid bud8 bud9 double mutant strains carrying full deletions of the BUD8 and BUD9 open reading frames to re-examine these results. We found that diploid bud8 bud9 null mutants behave differentially to single mutants and are similar to rsr1/bud1 strains, because they produce a random budding pattern in YF and PH cells (Figure 2). As controls, we also constructed homozygous diploid rsr1/bud1 single mutant and rsr1/bud1 bud8 bud9 triple mutant strains. We found that bud8 bud9 double mutants are indistinguishable from the rsr1/bud1 single or rsr1/bud1 bud8 bud9 triple mutants with respect to bud site selection patterns or pseudohyphal development (Figures 2 and 4; Table I). These results suggest that in diploid cells BUD8 and BUD9 encode the only gene products that act as bipolar landmarks at the cell poles of YF or PH cells.
Mutations in RSR1/BUD1 are epistatic to mutations in BUD8 and BUD9, and Rsr1p/Bud1p is not required for unipolar localization of Bud8p or Bud9p
Both Bud8p and Bud9p have been suggested to function as landmarks at the cell poles of diploid yeast cells that might recruit or locally activate the Rsr1p/Bud1p GTP-binding protein (Chant, 1999). To test this hypothesis, we constructed homozygous diploid bud8 and bud9 mutant strains in combination with mutations in RSR1/BUD1. We predicted that if RSR1/BUD1 acts downstream of BUD8 and BUD9 in the bud site selection pathway, mutations in RSR1/BUD1 should be epistatic over mutations in either BUD8 or BUD9. As described above, rsr1/bud1 bud8 and rsr1/bud1 bud9 double mutant strains were assayed for bud site selection patterns in YF and PH cells (Figure 2). In addition, pseudohyphal filament formation along with a detailed analysis of PH cell morphogenesis and substrate-invasive growth was investigated (Figure 4; Table I). We found that both the rsr1/bud1 bud8 and rsr1/bud1 bud9 double mutants were indistinguishable from the rsr1/bud1 single mutant with respect to all phenotypes measured. This result argues for RSR1/BUD1 acting downstream of BUD8 or BUD9 in the control of bud site selection of both YF and PH cells.
Our genetic analysis indicated that RSR1/BUD1 acts downstream of BUD8 and BUD9. This assumption could be verified if Rsr1p/Bud1p was not required for asymmetric localization of Bud8p and Bud9p. Therefore, subcellular localization of GFP–Bud8p and GFP–Bud9p was analysed in a homozygous diploid rsr1/bud1 mutant strain (Figure 6A). Indeed, no obvious difference in the subcellular localization of GFP–Bud8p or GFP–Bud9p was observed in an rsr1/bud1 mutant when compared with a control strain, further confirming that Rsr1p/Bud1p acts downstream of Bud8p and Bud9p. Similarly, we tested the localization of GFP–Bud8p in diploid bud9 mutants and GFP–Bud9p in diploid bud8 strains (Figure 6B). Again, no differences could be detected when compared with the localization of the GFP fusion proteins in a control strain. These findings are in agreement with the genetic studies predicting that in the absence of Bud9p, Bud8p should be normally localized at the distal bud site thereby allowing the unipolar distal pattern found in the bud9 single mutant. Vice versa, the absence of Bud8p was not expected to affect the localization of Bud9p, because the absence of both Bud8p and Bud9p (in a bud8 bud9 double mutant strain) was found to cause random and not unipolar proximal budding, as exhibited by the bud8 single mutant.

Fig. 6. Unipolar localization of GFP–Bud8p and GFP–Bud9p is independent of RSR1/BUD1, BUD8 or BUD9. (A) Living cells of strain RH2448 (rsr1Δ/rsr1Δ) expressing GFP–Bud8p (pME1772) or GFP–Bud9p (pME1777) grown in high ammonium media to exponential phase were viewed by DIC or by fluorescence microscopy (GFP). Identical results were obtained when expressing GFP–Bud8p or GFP–Bud9p under the control of the MET25 promoter using plasmid pME1773 or pME1778, respectively. (B) Subcellular localization of GFP–Bud8p and GFP–Bud9p in strains RH2450 (bud9Δ/bud9Δ) and RH2449 (bud8Δ/bud8Δ). Scale bar applies to (A) and (B) and represents 5 µm.
Nitrogen starvation initiates unipolar distal cell divisions in pseudohyphal filaments by preventing localization of Bud9p, but not Bud8p, at the distal cell pole
During switching from the YF to the PH filamentous form, the budding pattern of diploid strains switches from bipolar to unipolar distal. Therefore, we wanted to know whether Bud8p or Bud9p is directly involved in this process. Our genetic data and localization studies suggest that Bud8p is a landmark at the distal cell pole that is required for distal bud site selection. In contrast, Bud9p appears to act as an inhibitor of distal bud site selection, because absence of Bud9p (in a bud9 mutant) favours unipolar distal budding in YF cells. For this reason, PH cells can be viewed as YF cells lacking Bud9p with respect to their budding pattern. We reasoned that PH cells might differ from YF cells by their expression patterns of the BUD9 gene or by the subcellular localization of the Bud9 protein. Therefore, subcellular localization of GFP–Bud8p and GFP–Bud9p was measured under nitrogen starvation conditions that favour pseudohyphal development. In addition, expression levels of BUD8 and BUD9 genes as well as intracellular levels of GFP–Bud8p and GFP–Bud9p proteins were determined under these conditions. Nitrogen starvation did not significantly alter the subcellular localization of GFP–Bud8p (Figure 7) when compared with non-starved cells (Figure 1). GFP–Bud8p was still found to be concentrated at the tip of the growing bud as well as to the mother-bud neck region. Expression of GFP–Bud8p in nitrogen-starved cultures hardly changed, but a 2-fold induction of steady-state BUD8 mRNA was measured under these conditions (Figure 8). However, localization of GFP–Bud9p to the distal cell pole was completely suppressed when cultures were starved for nitrogen (Figure 7). In contrast, intracellular protein levels of GFP–Bud9p or BUD9 mRNA levels in these cultures did not decrease more than by a factor of roughly two (Figure 8). Thus, although significant levels of GFP–Bud9p were still present in nitrogen-starved cells, none of this protein was found to be concentrated at the distal bud tip during cell division. Similar results were obtained with the epitope-tagged myc-Bud8p and myc-Bud9p proteins (data not shown). These findings suggest that the starvation-induced switch of cell polarity from bipolar budding of YF cells to unipolar distal budding of PH cells is achieved by a mechanism that prevents Bud9p from being localized at the distal cell pole.

Fig. 7. Subcellular localization of GFP–Bud8p and GFP–Bud9p in living PH cells. Representative cells of wild-type strain RH2447 expressing either GFP–Bud8p from plasmid pME1772 or GFP–Bud9p from plasmid pME1777. Saccharomyces cerevisiae strains were grown in low ammonium media (SLAD/LA) for 15 h. Living cells were viewed by DIC or by fluorescence microscopy (GFP). Scale bar represents 5 µm.
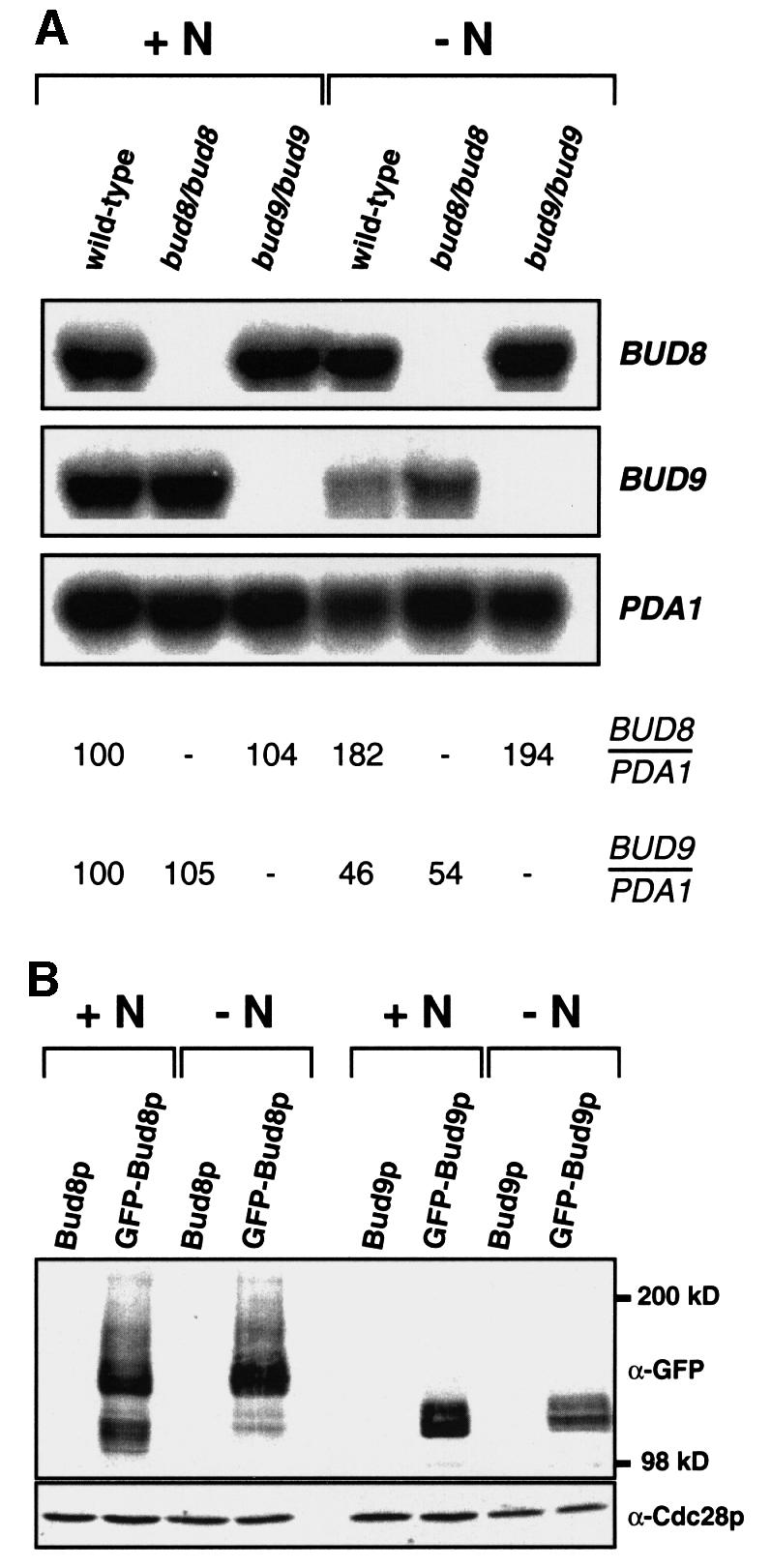
Fig. 8. Regulation of BUD8 and BUD9 expression by nitrogen availability. (A) Autoradiogram showing steady-state levels of BUD8 and BUD9 mRNA of strains RH2447 (wild type), RH2449 (bud8/bud8) and RH2450 (bud9/bud9), all carrying plasmid pRS316 for Ura+ prototrophy and either grown to exponential phase in high ammonium media (+N) or grown in low ammonium media (–N) for 15 h. Expression of the PDA1 gene served as an internal control. Relative expression levels of BUD8 (BUD8/PDA1) and BUD9 (BUD9/PDA1) are shown below and were obtained using a Phosphor-Imaging scanner. Numbers represent mean values of three independent measurements and were obtained by normalizing BUD gene transcript levels with respect to PDA1 and to BUD gene expression in wild-type strain RH2447 grown on high ammonium (+N). The standard deviation was below 20%. (B) Regulation of GFP–Bud8p and GFP–Bud9p fusion protein levels by nitrogen. Total protein extracts were prepared from strain RH2447 carrying plasmid pME1783 (Bud8p), pME1772 (GFP–Bud8p), pME1784 (Bud9p) or pME1777 (GFP–Bud9p) grown in high ammonium (+N) or low ammonium (–N) media. Extracts were analysed for expression of GFP fusion proteins by western blot analysis using a polyclonal anti-GFP antibody (α-GFP). As an internal control, protein levels of Cdc28p were measured in the same extracts using a polyclonal anti-Cdc28p antibody (α-Cdc28p).
Discussion
During nitrogen starvation, diploid yeast cells switch polarity from the bipolar to the unipolar distal pattern that is a prerequisite for the formation of linear filaments during pseudohyphal growth (Gimeno et al., 1992; Kron et al., 1994). Our study provides novel evidence for a molecular model that explains how Bud8p and Bud9p regulate the polarity of diploid yeast cells in response to nutrients (Figure 9). We propose that Bud8p is a cortical tag at the distal pole of both YF and PH cells, where it directs bud initiation. When nutrients are available, Bud9p is also localized at the distal pole, where it significantly reduces the potential of distal bud site selection. As a consequence, YF cells develop the bipolar budding pattern. In response to nitrogen starvation, Bud9p (but not Bud8p) is mislocalized and therefore absent at the distal cell pole. This leads to unipolar distal cell divisions in PH cells, a budding pattern that can be mimicked in YF cells by deletion of BUD9. Because Bud8p and Bud9p associate in vivo, Bud9p might be an inhibitor of Bud8p-mediated distal bud site selection.

Fig. 9. Model for regulation of bud site selection at the distal cell pole of S.cerevisiae. In YF cells, Bud9p is localized at the distal cell pole and interferes with Bud8p-mediated bud site selection via the Rsr1p/Bud1p–Bud5p–Bud2p GTPase module. In PH cells, nutritional starvation for nitrogen prevents distal localization of Bud9p, allowing efficient Bud8p-mediated distal budding.
Both Bud8p and Bud9p are predicted to be transmembrane proteins. As previously discussed (Chant, 1999), Bud8p and Bud9p consist of N-terminal extracellular domains (515 and 460 amino acids, respectively), membrane-spanning domains, short cytoplasmic loops (42 and 38 amino acids), second membrane-spanning domains and short (3 and 2 amino acids) extracellular domains at the C-terminus (Figure 9). Membrane association of Bud8p and Bud9p is in agreement with the fact that membrane-dissolving detergents are required for full extraction of both proteins (see Materials and methods). In addition, deletion of the predicted transmembrane domains of Bud8p inhibits both its function and proper intracellular localization (our unpublished results). Surprisingly, neither Bud8p nor Bud9p has a predicted signal sequence, although their N-terminal domains contain a number of predicted N-glycosylation sites. Both Bud8p and Bud9p might be glycosylated, because their apparent molecular weights are much higher than calculated when analysed by SDS–PAGE (Figure 8). However, whether Bud8p and Bud9p are glycoproteins that are delivered to the cell surface via the secretory pathway, similar to Axl2p/Bud10p (Halme et al., 1996; Roemer et al., 1996; Sanders et al., 1999), remains to be elucidated.
Our study defines the distal pole as the main site of action for Bud8p and Bud9p in YF and PH cells. However, highly concentrated amounts of GFP–Bud8p and weak amounts of GFP–Bud9p were also detectable at the bud neck in living cells. Because the bud neck is positioned between the distal cell pole of the mother and the proximal pole of the daughter, careful analysis of bud neck staining of a given protein is necessary for interpretation of its function. Our localization studies clearly show that GFP–Bud8p and GFP–Bud9p proteins residing at the bud neck are asymmetrically concentrated at the mother side of the bud neck (Figure 1). Moreover, dividing cells were never observed with GFP–Bud8p concentrated at the distal pole of the growing bud and at the proximal pole of the mother. Thus, we prefer the view that this portion of Bud8p and Bud9p is localized at the distal cell pole of the mother, and not at the proximal pole of the daughter. This argues for Bud8p and Bud9p functioning not only at the distal pole of the new daughter, but also at the distal pole of the mother.
How does Bud8p act as a cortical tag? Our study provides genetic evidence that the general budding factor Rsr1p/Bud1p might be recruited to, or locally be activated at the distal pole through Bud8p, because rsr1/bud1 mutants display a random budding pattern independent of Bud8p. Vice versa, Bud8p is localized to the distal bud site irrespective of the presence or absence of Rsr1p/Bud1p. However, a direct interaction between Bud8p and Rsr1p/Bud1p seems unlikely, because Rsr1p/Bud1p is distributed uniformly around the plasma membrane (Michelitch and Chant, 1996). Thus, Bud8p might control distal bud site selection via the regulatory proteins of Rsr1p/Bud1p, e.g. Bud2p or Bud5p (Park et al., 1999).
An important finding of our study is that neither protein levels nor subcellular localization of Bud8p undergo significant changes when cells are starved of nitrogen and switch to the PH form. Yet, BUD8 is absolutely required for pseudohyphal development. Thus, Bud8p does not appear to be a prime regulator that controls the switch from bipolar to unipolar cell division in response to nitrogen starvation. Rather, the efficiency of Bud8p as a cortical tag might be altered in PH cells by, for example, post-translational modification or interaction with an inhibitor. We favour Bud9p being a negative regulator of distal budding, whose subcellular localization is under the control of nitrogen starvation. This view of Bud9p is supported by several observations. (i) Bud9p is highly concentrated at the distal pole of YF cells, whereas the absence of Bud9p (in bud9 deletion strains) favours distal bud initiation. This finding per se defines a negative function for Bud9p at the distal pole, given the assumption that the main localization of Bud9p reflects its major site of action. (ii) Bud8p and Bud9p co-purify, indicating physical interaction in vivo. (iii) PH cells display a unipolar distal pattern much like YF bud9 mutant cells, thus naturally reflecting the artificial situation created by deletion of BUD9. (iv) In PH cells, Bud9p is prevented from being localized to its presumed site of negative action, the distal cell pole. How does Bud9p fulfil such a negative function? As discussed above, the Bud9p sequence predicts an extracellular domain of 460 amino acids at the N-terminus and a short loop of 38 amino acids at the inside of the cell. Thus, Bud9p might interact directly with Bud8p and prevent Bud8p from recruiting downstream factors to the distal pole. Alternatively, Bud9p might compete with Bud8p for these downstream proteins, but act negatively on their function. It remains to be determined whether such a mechanism involves post-translational modifications of Bud9p or alterations of the machinery that recognizes and asymmetrically localizes Bud9p to the distal pole.
Our study provides novel evidence of how distal pole selection is regulated in diploid yeast cells. However, it is not clear how the proximal pole is tagged. Surprisingly, we found that full deletion of both BUD8 and BUD9 causes random budding and not, as might be expected, unipolar proximal bud site selection. Although we have no detailed model explaining this finding, one might imagine that Bud8p and Bud9p could have overlapping functions in the general establishment of cell polarity.
In summary, our findings support the view that Bud8p is a positive determinant at the distal pole, where it recruits the machinery required for bud initiation in YF and PH cells. In contrast, Bud9p is a negative determinant at the distal pole and interferes with distal budding by inhibition of Bud8p or by negatively regulating the budding machinery. In addition, Bud9p function is under nutritional control, because nitrogen starvation suppresses asymmetric localization of Bud9p to the distal pole. This regulatory mechanism controls the cell polarity switch from bipolar to unipolar distal budding in diploid yeast cells.
Materials and methods
Yeast strains and growth conditions
All yeast strains used in this study are congenic to the Σ1278b genetic background (Table II). bud8Δ::HIS3, bud9Δ::HIS3 and rsr1Δ::kanR deletion mutations were introduced using plasmids pME1767, pME1768 and pME1766 (Table III). Strains RH2491, RH2492, RH2493 and RH2494, all expressing myc-epitope-tagged versions of either BUD8 or BUD9 at endogenous levels, were obtained by integration of linearized plasmids pME1936 or pME1938 into the ura3-52 locus. Standard methods for genetic crosses and transformation were used and standard yeast culture YPD, YNB and SC media were prepared essentially as described (Guthrie and Fink, 1991). Low ammonium medium (SLAD) was prepared as described (Gimeno et al., 1992). Solid SLAD 2% agar medium was used for qualitative and quantitative pseudohyphal growth assays. Strains were grown in liquid SLAD layered over SLAD 2% agar in Petri plates (SLAD/LA) essentially as described (Kron et al., 1994) for bud scar staining and GFP fluorescence microscopy of PH cells, as well as for isolation of RNA and protein extracts from PH cells. The PHD1 PH inducer was overexpressed from plasmid pCG38 in strains used for bud scar staining and time-lapse microscopy to obtain a high proportion of PH cells required for these measurements.
Table II. Strains used in this study.
| Strain | Genotype | Source |
|---|---|---|
| RH2447 | MATa/MATα, ura3-52/ura3-52, leu2::hisG/LEU2, trp1::hisG/TRP1 | this study |
| RH2448 | MATa/MATα, rsr1Δ::kanR/rsr1Δ::kanR, ura3-52/ura3-52, leu2::hisG/LEU2, trp1::hisG/TRP1 | this study |
| RH2449 | MATa/MATα, bud8Δ::HIS3/bud8Δ::HIS3, ura3-52/ura3-52, his3::hisG/his3::hisG, leu2::hisG/LEU2, trp1::hisG/TRP1 | this study |
| RH2450 | MATa/MATα, bud9Δ::HIS3/bud9Δ::HIS3, ura3-52/ura3-52, his3::hisG/his3::hisG, leu2::hisG/LEU2, trp1::hisG/TRP1 | this study |
| RH2451 | MATa/MATα, rsr1Δ::kanR/rsr1Δ::kanR, bud8Δ::HIS3/bud8Δ::HIS3, ura3-52/ura3-52, his3::hisG/his3::hisG,leu2::hisG/LEU2, trp1::hisG/TRP1 | this study |
| RH2452 | MATa/MATα, rsr1Δ::kanR/rsr1Δ::kanR, bud9Δ::HIS3/bud9Δ::HIS3, ura3-52/ura3-52, his3::hisG/his3::hisG,leu2::hisG/LEU2, trp1::hisG/TRP1 | this study |
| RH2453 | MATa/MATα, bud8Δ::HIS3/bud8Δ::HIS3, bud9Δ::HIS3/bud9Δ::HIS3, ura3-52/ura3-52, his3::hisG/his3::hisG,leu2::hisG/LEU2, trp1::hisG/TRP1 | this study |
| RH2454 | MATa/MATα, rsr1Δ::kanR/rsr1Δ::kanR, bud8Δ::HIS3/bud8Δ::HIS3, bud9Δ::HIS3/bud9Δ::HIS3ura3-52/ura3-52,his3::hisG/his3::hisG, leu2::hisG/LEU2, trp1::hisG/TRP1 | this study |
| RH2491 | MATa/MATα, myc-BUD8-URA3/myc-BUD8-URA3, ura3-52/ura3-52, leu2::hisG/LEU2, trp1::hisG/TRP1 | this study |
| RH2492 | MATa/MATα, myc-BUD8-URA3/myc-BUD8-URA3, bud8Δ::HIS3/bud8Δ::HIS3, ura3-52/ura3-52,his3::hisG/his3::hisG, leu2::hisG/LEU2, trp1::hisG/TRP1 | this study |
| RH2493 | MATa/MATα, myc-BUD9-URA3/myc-BUD9-URA3, ura3-52/ura3-52, leu2::hisG/LEU2, trp1::hisG/TRP1 | this study |
| RH2494 | MATa/MATα, myc-BUD9-URA3/myc-BUD9-URA3, bud9Δ::HIS3/bud9Δ::HIS3, ura3-52/ura3-52,his3::hisG/his3::hisG,leu2::hisG/LEU2, trp1::hisG/TRP1 | this study |
| RH2495 | MATa/MATα, ura3-52/ura3-52, leu2::hisG/leu2::hisG, his3::hisG/HIS3, trp1::hisG/TRP1 | this study |
Table III. Plasmids used in this study.
| Plasmid | Description | Reference |
|---|---|---|
| pME1766 | rsr1Δ::kanR cassette for full deletion of RSR1 | this study |
| pME1767 | bud8Δ::HIS3 cassette for full deletion of BUD8 | this study |
| pME1768 | bud9Δ::HIS3 cassette for full deletion of BUD9 | this study |
| pME1769 | 4.0 kb fragment containing BUD8 in pRS316 | this study |
| pME1770 | 5.6 kb fragment containing BUD9 in pRS316 | this study |
| YCp(RSR1) | 1.6 kb fragment containing RSR1 in YCp50 | Ruggieri et al. (1992) |
| pME1771 | BUD8prom–GFP–BUD8 fusion in pRS316 | this study |
| pME1772 | BUD8prom–GFP–BUD8 fusion in pRS426 | this study |
| pME1773 | MET25prom–GFP–BUD8 fusion in pRS426MET25 | this study |
| pME1775 | BUD8prom–myc3–BUD8 fusion in pRS426 | this study |
| pME1776 | BUD9prom–GFP–BUD9 fusion in pRS316 | this study |
| pME1777 | BUD9prom–GFP–BUD9 fusion in pRS426 | this study |
| pME1778 | MET25prom–GFP–BUD9 fusion in pRS426MET25 | this study |
| pME1780 | BUD9prom–myc3–BUD9 fusion in pRS426 | this study |
| pME1783 | 3.1 kb fragment containing BUD8 in pRS426 | this study |
| pME1784 | 5.6 kb fragment containing BUD9 in pRS426 | this study |
| pME1936 | BUD8prom–myc3–BUD8 fusion in pRS306 | this study |
| pME1937 | BUD8prom–myc3–BUD8 fusion in pRS425 | this study |
| pME1938 | BUD9prom–myc3–BUD9 fusion in pRS306 | this study |
| pME1939 | BUD9prom–myc3–BUD9 fusion in pRS425 | this study |
| pME1940 | GAL1prom–GST–BUD8 fusion in pYGEX-2T | this study |
| pME1941 | GAL1prom–GST–BUD9 fusion in pYGEX-2T | this study |
| pRS316 | URA3-marked centromere vector | Sikorski and Hieter (1989) |
| pRS426 | URA3-marked 2 µm vector | Christianson et al. (1992) |
| p426MET25 | pRS426 containing MET25 promoter and CYC1 terminator | Mumberg et al. (1994) |
| pCG38 | 2.6 kb fragment containing PHD1 in pRS202 | Gimeno and Fink (1994) |
| pYGEX-2T | URA3-marked 2 µm GAL1prom–GST fusion vector | Schlenstedt et al. (1995) |
Plasmids
Plasmids pME1766, pME1767 and pME1768 carrying the rsr1Δ::kanR, bud8Δ::HIS3 and bud9Δ::HIS3 deletion cassettes were created by replacement of the coding sequences of RSR1/BUD1, BUD8 and BUD9 for either the HIS3 selectable marker or the kanR kanamycin resistance gene using a PCR-based three-step cloning strategy. To obtain genomic fragments carrying BUD8 and BUD9, plasmids pRS202-BUD8 and pRS202-BUD9 were isolated from a yeast genomic library in pRS202 (from P.Hieter, University of British Columbia, Vancouver, Canada) using colony hybridizations and 32P-radiolabelled probes for BUD8 and BUD9.
Plasmid pME1769 was obtained by subcloning of a 4.0 kb BamHI–XhoI genomic fragment from pRS202-BUD8 into pRS316 (Sikorski and Hieter, 1989), and plasmid pME1783 by subcloning of a 3.1 kb BamHI–ScaI fragment from pRS202-BUD8 into pRS426 (Christianson et al., 1992). Plasmids pME1771 and pME1772, both expressing GFP–Bud8p from the BUD8 promoter, were constructed by introducing a BglII site in front of the second codon of BUD8 and insertion of a 750 bp BglII fragment encoding the GFPuv variant of GFP that was amplified from plasmid pBAD-GFPuv (Clontech, Heidelberg, Germany). Plasmid pME1773 expressing GFP–Bud8p from the MET25 promoter was obtained by subcloning of a 3.3 kb EcoRV–ScaI fragment carrying GFP–BUD8 from pME1771 into p426MET25 (Mumberg et al., 1994). Plasmids pME1775, pME1936 and pME1937, all expressing a triple myc epitope-tagged version of Bud8p under the control of the BUD8 promoter, were obtained by insertion of a 120 bp BamHI fragment carrying the triple myc epitope (myc3) after the start codon of BUD8. Plasmids pME1770 and pME1784 were obtained by subcloning of a 5.6 kb EcoRI genomic fragment from pRS202-BUD9 into either pRS316 or pRS426. Plasmids pME1776 and pME1777, both expressing GFP–Bud9p from the BUD9 promoter, were constructed by introducing a BamHI site in front of the second codon of BUD9 and insertion of the GFPuv cassette described above. pME1778 expressing GFP–Bud9p from the MET25 promoter was obtained by subcloning of a 3 kb BamHI–EcoRI BUD9 fragment into p426MET25 and insertion of the GFPuv BglII cassette. Plasmids pME1780, pME1938 and pME1939, expressing a triple myc epitope-tagged version of Bud9p under the control of the BUD9 promoter, were obtained by insertion of the triple myc epitope after the start codon of BUD9. Plasmids pME1940 and pME1941 were obtained by N-terminal fusion of BUD8 and BUD9 open reading frames to GST in vector pYGEX-2T (Schlenstedt et al., 1995).
Qualitative and quantitative assays of pseudohyphal growth
Qualitative assays for pseudohyphal development were performed as described previously (Mösch et al., 1996). After 3 days of growth on solid SLAD medium, pseudohyphal colonies were viewed with a Zeiss Axiovert microscope and photographed using a Xillix Microimager digital camera and the Improvision Openlab software (Improvision, Coventry, UK). Quantitative assays for PH growth, including determination of substrate invasion and cell shape, were performed following the protocols described earlier (Mösch and Fink, 1997).
Bud scar staining and determination of budding patterns
Bud scar staining was performed on YF and PH cells grown to exponential phase. YF cells in exponential phase were prepared by growing strains in liquid YNB medium at 30°C to an OD600 of 0.6. PH cells in exponential phase were obtained by growth on SLAD/LA medium. Routinely, 5 × 105 cells were inoculated into 10 ml of SLAD liquid medium layered over 10 ml of SLAD 2% agar in Petri dishes and incubated at 30°C. After 15 h, cells were suspended and collected by centrifugation in conical polystyrene tubes. YF and PH cell suspensions were fixed at room temperature for 2 h in 3.7% formaldehyde. Samples were rinsed twice in water and resuspended in 200 µl of a fresh stock of 1 mg/ml calcofluor white (Fluorescent Brightener F-6259; Sigma). Bud scars were visualized by fluorescence microscopy using a Zeiss Axiovert microscope and photographed using a Xillix Microimager digital camera and the Improvision Openlab software (Improvision, Coventry, UK). Cells with between 2 and 10 obvious bud scars were divided into three classes: bipolar, cells with two or more bud scars with at least one scar at each end of the cell (the birth end and the free end); unipolar, cells with all bud scars at one end of the cell immediately adjacent to one another; random, cells with bud scar distributions other than bipolar or unipolar. Numbers in the tables represent the percentage of cells in each class for a sample of at least 200 cells.
Time-lapse microscopy
Bud site selection of growing PH filaments was determined by using a chamber for high magnification imaging of yeast growth as described previously (Kron et al., 1994). Positions of bud site emergence were determined by direct microscopic observation. For each strain measured, at least 70 cell divisions were observed.
GFP fluorescence and indirect immunofluorescence microscopy
Yeast strains harbouring plasmids encoding GFP–Bud8p or GFP–Bud9p were grown to exponential phase in high or low ammonium media as described for bud scar staining. Cells from 1 ml of the cultures were harvested by centrifugation and immediately viewed in vivo on a Zeiss Axiovert microscope by either differential interference contrast microscopy (DIC) or fluorescence microscopy using a GFP filter set (AHF Analysentechnik AG, Tübingen, Germany). Cells were photographed using a Xillix Microimager digital camera and the Improvision Openlab software (Improvision, Coventry, UK). For immunofluorescence microscopy, cells were cultured as for GFP microscopy, fixed in 3.7% formaldehyde and spheroblasts were prepared as described (Pringle et al., 1991). 4′,6-diamidino-2-phenylindole (DAPI) staining and monoclonal mouse anti-myc antibodies (9E10) together with an Alexa 488- conjugated goat anti-mouse antibody (Molecular Probes, Eugene, OR) were used for visualization of nuclei and myc epitope-tagged proteins, respectively. Cells were viewed and photographed as described above using standard DAPI and fluorescein isothiocyanate (FITC) filter sets.
Northern blot analysis
Total RNA was prepared from cultures grown in high or low ammonium media exactly as described for bud scar staining and according to the method described previously (Cross and Tinkelenberg, 1991). Total RNA was separated on a 1.4% agarose gel containing 3% formaldehyde and transferred onto nylon membranes as described earlier (Mösch et al., 1992). BUD8, BUD9 and PDA1 transcripts were detected using gene specific 32P-radiolabelled DNA probes. Hybridizing signals were quantified using a BAS-1500 Phosphor-Imaging scanner (Fuji).
Protein analysis
Whole-cell extracts. Extracts were prepared from cultures grown to exponential phase in high or low ammonium medium as described above. Briefly, cultures were washed in ice-cold buffer R (50 mM Tris–HCl pH 7.5, 1 mM EDTA, 50 mM dithiothreitol), lysed with glass beads in 200 µl of buffer R + PIM (1 mM each phenylmethylsulfonyl fluoride, tosyl-L-lysine-chloromethylketone, tosyl-l-phenylalanine-chloromethylketone, p-aminobenzamidine–HCl and o-phenanthroline) + 3% Triton X-100 + 0.8% SDS at 4°C, and spun at 3500 r.p.m. for 5 min to remove glass beads and large cell debris. Extracts (10 µl) were removed to determine total protein concentration using a protein assay kit from (Bio-Rad, München, Germany). SDS loading dye was added to the remaining total extracts and proteins were denatured by heating at 37°C for 5 min. Equal amounts of proteins were then subjected to SDS–PAGE and transferred to nitrocellulose membranes. GFP fusion proteins and Cdc28p were detected using ECL technology (Amersham, UK) after incubation of membranes with either a rabbit polyclonal anti-GFP antibody (Clontech, Heidelberg, Germany) or rabbit polyclonal anti-Cdc28p antibodies (a kind gift of S.Irniger, Georg August University Göttingen, Germany) and a peroxidase-coupled goat anti-rabbit IgG secondary antibody (Dianova, Hamburg, Germany).
Purification of GST fusions. Extracts of strains expressing GST fusion proteins together with myc-tagged versions of Bud8p or Bud9p were prepared after growth on galactose medium for 6 h exactly as previously described (Roberts et al., 1997). Extracts were incubated with glutathione–agarose overnight at 4°C, and beads were repeatedly washed and collected to purify GST fusions and any associated proteins. Samples were denatured by heating at 60°C for 5 min in SDS loading dye, and equal amounts of each sample were analysed by western blot analysis as described above using either polyclonal anti-GST antibodies (Santa Cruz Biotechnologies, Santa Cruz, CA) or the monoclonal mouse anti-myc antibody (9E10).
Acknowledgments
Acknowledgements
We thank Maria Meyer for excellent technical assistance during the course of this work. We are grateful to Alan Bender, Martin Funk, Philip Hieter and Stefan Irniger for generously providing plasmids and reagents, and Olav Grundmann for critical reading of the manuscript. This work was supported by grants from the Deutsche Forschungsgemeinschaft, the Volkswagenstiftung and Fonds der Chemischen Industrie.
References
- Bender A. and Pringle,J.R. (1989) Multicopy suppression of the cdc24 budding defect in yeast by CDC42 and three newly identified genes including the ras-related gene RSR1. Proc. Natl Acad. Sci. USA, 86, 9976–9980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chant J. (1999) Cell polarity in yeast. Annu. Rev. Cell Dev. Biol., 15, 365–391. [DOI] [PubMed] [Google Scholar]
- Chant J. and Herskowitz,I. (1991) Genetic control of bud site selection in yeast by a set of gene products that constitute a morphogenetic pathway. Cell, 65, 1203–1212. [DOI] [PubMed] [Google Scholar]
- Chant J. and Pringle,J.R. (1995) Patterns of bud-site selection in the yeast Saccharomyces cerevisiae. J. Cell Biol., 129, 751–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chant J., Corrado,K., Pringle,J.R. and Herskowitz,I. (1991) Yeast BUD5, encoding a putative GDP–GTP exchange factor, is necessary for bud site selection and interacts with bud formation gene BEM1. Cell, 65, 1213–1224. [DOI] [PubMed] [Google Scholar]
- Christianson T.W., Sikorski,R.S., Dante,M., Shero,J.H. and Hieter,P. (1992) Multifunctional yeast high-copy-number shuttle vectors. Gene, 110, 119–122. [DOI] [PubMed] [Google Scholar]
- Crameri A., Whitehorn,E.A., Tate,E. and Stemmer,W.P. (1996) Improved green fluorescent protein by molecular evolution using DNA shuffling. Nature Biotechnol., 14, 315–319. [DOI] [PubMed] [Google Scholar]
- Cross F.R. and Tinkelenberg,A.H. (1991) A potential positive feedback loop controlling CLN1 and CLN2 gene expression at the start of the yeast cell cycle. Cell, 65, 875–883. [DOI] [PubMed] [Google Scholar]
- Freifelder D. (1960) Bud position in Saccharomyces cerevisiae. J. Bacteriol., 80, 567–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita A., Oka,C., Arikawa,Y., Katagai,T., Tonouchi,A., Kuhara,S. and Misumi,Y. (1994) A yeast gene necessary for bud-site selection encodes a protein similar to insulin-degrading enzymes. Nature, 372, 567–570. [DOI] [PubMed] [Google Scholar]
- Gimeno C.J. and Fink,G.R. (1994) Induction of pseudohyphal growth by overexpression of PHD1, a Saccharomyces cerevisiae gene related to transcriptional regulators of fungal development. Mol. Cell. Biol., 14, 2100–2112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gimeno C.J., Ljungdahl,P.O., Styles,C.A. and Fink,G.R. (1992) Unipolar cell divisions in the yeast S. cerevisiae lead to filamentous growth: regulation by starvation and RAS. Cell, 68, 1077–1090. [DOI] [PubMed] [Google Scholar]
- Guthrie C. and Fink,G.R. (1991) Guide to yeast genetics and molecular biology. Methods Enzymol., 194. [PubMed] [Google Scholar]
- Halme A., Michelitch,M., Mitchell,E.L. and Chant,J. (1996) Bud10p directs axial cell polarization in budding yeast and resembles a transmembrane receptor. Curr. Biol., 6, 570–579. [DOI] [PubMed] [Google Scholar]
- Hicks J.B., Strathern,J.N. and Herskowitz,I. (1977) Interconversion of yeast mating types III. Action of the homothallism (HO) gene in cells homozygous for the mating type locus. Genetics, 85, 395–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kron S.J., Styles,C.A. and Fink,G.R. (1994) Symmetric cell division in pseudohyphae of the yeast Saccharomyces cerevisiae. Mol. Biol. Cell, 5, 1003–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madden K. and Snyder,M. (1992) Specification of sites for polarized growth in Saccharomyces cerevisiae and the influence of external factors on site selection. Mol. Biol. Cell, 3, 1025–1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madden K. and Snyder,M. (1998) Cell polarity and morphogenesis in budding yeast. Annu. Rev. Microbiol., 52, 687–744. [DOI] [PubMed] [Google Scholar]
- Michelitch M. and Chant,J. (1996) A mechanism of Bud1p GTPase action suggested by mutational analysis and immunolocalization. Curr. Biol., 6, 446–454. [DOI] [PubMed] [Google Scholar]
- Mösch H.-U. and Fink,G.R. (1997) Dissection of filamentous growth by transposon mutagenesis in Saccharomyces cerevisiae. Genetics, 145, 671–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mösch H.-U., Graf,R. and Braus,G.H. (1992) Sequence-specific initiator elements focus initiation of transcription to distinct sites in the yeast TRP4 promoter. EMBO J., 11, 4583–4590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mösch H.-U., Roberts,R.L. and Fink,G.R. (1996) Ras2 signals via the Cdc42/Ste20/mitogen-activated protein kinase module to induce filamentous growth in Saccharomyces cerevisiae. Proc. Natl Acad. Sci. USA, 93, 5352–5356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mumberg D., Müller,R. and Funk,M. (1994) Regulatable promoters of Saccharomyces cerevisiae: comparison of transcriptional activity and their use for heterologous expression. Nucleic Acids Res., 22, 5767–5768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park H.O., Chant,J. and Herskowitz,I. (1993) BUD2 encodes a GTPase-activating protein for Bud1/Rsr1 necessary for proper bud-site selection in yeast. Nature, 365, 269–274. [DOI] [PubMed] [Google Scholar]
- Park H.O., Bi,E., Pringle,J.R. and Herskowitz,I. (1997) Two active states of the Ras-related Bud1/Rsr1 protein bind to different effectors to determine yeast cell polarity. Proc. Natl Acad. Sci. USA, 94, 4463–4468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park H.O., Sanson,A. and Herskowitz,I. (1999) Localization of Bud2p, a GTPase-activating protein necessary for programming cell polarity in yeast to the presumptive bud site. Genes Dev., 13, 1912–1917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pringle J.R., Adams,A.E., Drubin,D.G. and Haarer,B.K. (1991) Immunofluorescence methods for yeast. Methods Enzymol., 194, 565–602. [DOI] [PubMed] [Google Scholar]
- Roberts R.L., Mösch,H.-U. and Fink,G.R. (1997) 14-3-3 proteins are essential for RAS/MAPK cascade signaling during pseudohyphal development in S. cerevisiae. Cell, 89, 1055–1065. [DOI] [PubMed] [Google Scholar]
- Roemer T., Madden,K., Chang,J. and Snyder,M. (1996) Selection of axial growth sites in yeast requires Axl2p, a novel plasma membrane glycoprotein. Genes Dev., 10, 777–793. [DOI] [PubMed] [Google Scholar]
- Ruggieri R., Bender,A., Matsui,Y., Powers,S., Takai,Y., Pringle,J.R. and Matsumoto,K. (1992) RSR1, a ras-like gene homologous to Krev-1 (smg21A/rap1A): role in the development of cell polarity and interactions with the Ras pathway in Saccharomyces cerevisiae. Mol. Cell. Biol., 12, 758–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders S.L., Gentzsch,M., Tanner,W. and Herskowitz,I. (1999) O-glycosylation of Axl2/Bud10p by Pmt4p is required for its stability, localization and function in daughter cells. J. Cell Biol., 145, 1177–1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlenstedt G., Saavedra,C., Loeb,J.D., Cole,C.N. and Silver,P.A. (1995) The GTP-bound form of the yeast Ran/TC4 homologue blocks nuclear protein import and appearance of poly(A)+ RNA in the cytoplasm. Proc. Natl Acad. Sci. USA, 92, 225–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segall J.E. (1993) Polarization of yeast cells in spatial gradients of α mating factor. Proc. Natl Acad. Sci. USA, 90, 8332–8336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sikorski R.S. and Hieter,P. (1989) A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics, 122, 19–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snyder M. (1989) The SPA2 protein of yeast localizes to sites of cell growth. J. Cell Biol., 108, 1419–1429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spellman P.T., Sherlock,G., Zhang,M.Q., Iyer,V.R., Anders,K., Eisen,M.B., Brown,P.O., Botstein,D. and Futcher,B. (1998) Comprehensive identification of cell cycle-regulated genes of the yeast Saccharomyces cerevisiae by microarray hybridization. Mol. Biol. Cell, 9, 3273–3297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valtz N. and Herskowitz,I. (1996) Pea2 protein of yeast is localized to sites of polarized growth and is required for efficient mating and bipolar budding. J. Cell Biol., 135, 725–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zahner J.E., Harkins,H.A. and Pringle,J.R. (1996) Genetic analysis of the bipolar pattern of bud site selection in the yeast Saccharomyces cerevisiae. Mol. Cell. Biol., 16, 1857–1870. [DOI] [PMC free article] [PubMed] [Google Scholar]