

Abstract
The matrix protein VP40 from Ebola virus is targeted to the plasma membrane, where it is thought to induce assembly and budding of virions through its association with the lipid bilayer. Ebola virus VP40 is expressed as a monomeric molecule in solution, consisting of two loosely associated domains. Here we show that a C-terminal truncation of seven residues destabilizes the monomeric closed conformation and induces spontaneous hexamerization in solution, as indicated by chemical cross-linking and electron microscopy. Three-dimensional reconstruction of electron microscopy images shows ring-like structures consisting of the N-terminal domain along with evidence for flexibly attached C-terminal domains. In vitro destabilization of the monomer by urea treatment results in similar hexameric molecules in solution. In addition, we demonstrate that membrane association of wild-type VP40 also induces the conformational switch from monomeric to hexameric molecules that may form the building blocks for initiation of virus assembly and budding. Such a conformational change induced by bilayer targeting may be a common feature of many viral matrix proteins and its potential inhibition may result in new anti-viral therapies.
Keywords: Ebola virus/matrix protein/membrane association/oligomerization/VP40
Introduction
Filoviridae are negative-stranded non-segmented RNA viruses (Mononegavirales) comprising the two species Marburg and Ebola virus. Filoviruses infect a number of cell types, such as monocytes/macrophages, fibroblasts and hepatocytes, as well as endothelial cells (Becker et al., 1995; Feldmann et al., 1996; Takada et al., 1997; Zaki and Peters, 1997), through interactions of the viral glycoprotein with an as yet unknown cellular receptor, most likely followed by endocytosis (Geisbert and Jahrling, 1995; Wool-Lewis and Bates, 1998). In particular, endothelial cell infection and subsequent destruction have been linked to the severe hemorrhages that are observed at a late stage of a mostly fatal disease (Schnittler et al., 1993; Feldmann and Klenk, 1996; Yang et al., 1998). Infection is established by fusion of viral and cellular membranes mediated by the viral fusion protein Gp2 (Weissenhorn et al., 1998; Volchkov, 1999) and filoviruses replicate in the cytoplasm (Feldmann and Kiley, 1999), leading to the assembly and budding of new particles at the plasma membrane (Feldmann et al., 1996).
Most enveloped viruses express a matrix protein that is instrumental for the proper assembly of viral particles. The matrix protein VP40 is the most abundant protein in filovirus particles, and like matrix proteins from other enveloped viruses (Garoff et al., 1998), it forms a layer beneath the viral envelope that contributes to the structural integrity of viral particles (Geisbert and Jahrling, 1995). The matrix proteins associate with cellular membranes and interact at the site of assembly with the cytoplasmic tails of viral glycoproteins (Metsikkö and Simons, 1986; Sanderson et al., 1994; Jin et al., 1997; Schnell et al., 1998; Mebatsion et al., 1999; Schmitt et al., 1999), as well as with the ribonucleoprotein particle (RNP) (Garoff et al., 1998). The concerted interactions of these complexes trigger assembly and budding of infectious viral particles (Garoff et al., 1998), which in some cases seem to be further regulated by cellular proteins (Garnier et al., 1996; Yasuda and Hunter, 1998; Harty et al., 1999).
Some matrix proteins, including Ebola virus VP40, have been suggested to bind preferentially to membranes containing high amounts of negatively charged phospholipids (Zakowski et al., 1981; Hill et al., 1996; Provitera et al., 2000; Ruigrok et al., 2000a,b). The virus envelope contains a defined lipid composition, which may play an important role during assembly (Lenard and Compans, 1974). Incorporation of viral glycoproteins, matrix proteins, or both, into lipid rafts may be implicated in the acquisition of specific lipids in the viral envelope (Scheiffele et al., 1999; Manié et al., 2000; Zhang et al., 2000).
The crystal structure of monomeric VP40 shows an elongated, two-subunit assembly, with each domain folded into a β-sandwich. The domains are connected by a flexible linker and are only loosely packed against each other (Dessen et al., 2000a). Trypsin cleaves VP40 at position 212 in a loop connecting β-strands 7 and 8 in the C-terminal domain (Ruigrok et al., 2000b). This leads to the spontaneous dissociation of the remaining part of the C-terminal domain as well as to the hexamerization of N-terminal domains, which form ring-like structures with 3-fold symmetry (Ruigrok et al., 2000b). In contrast, the C-terminal domain contains large hydrophobic patches and is absolutely required for membrane association, since C-terminally truncated ring-shaped molecules (containing residues 31–212) do not interact with liposomes in vitro (Ruigrok et al., 2000b).
Here we show that the matrix protein VP40 from Ebola virus can adopt a hexameric ring-like structure by either destabilizing the inter-domain interactions with a seven-residue truncation at the C-terminus or by urea treatment of wild-type VP40. The core of the hexamers forms ring-like structures similar to those observed for the N-terminal domain of VP40 alone, as seen by electron microscopy (EM). Association of full-length VP40 with liposomes induces a similar pattern of hexamerization, as detected by chemical cross-linking and EM, which indicates that the ring-like structure observed in vitro may be an intermediate structure in the virus assembly process. This is the first observation of a conformational change in a viral matrix protein that induces oligomerization and membrane binding in vitro. Our data indicate that similar changes in matrix protein conformations may play a role during the life cycle of other members of the Mononegavirales or even more distantly related enveloped virus families. Our data also suggest that the molecular transition from monomer to oligomer may be prevented by the use of small molecules that bind in a prominent pocket between the N- and C-terminal domains of VP40, thus potentially blocking virus particle formation.
Results
Oligomerization of a C-terminally truncated mutant of VP40
The crystal structure of VP40 showed that the C-terminal end is positioned towards the inter-domain interface, which is held together by a flexible linker and is loosely connected by a few hydrogen bonds and hydrophobic interactions (Dessen et al., 2000a). In order to analyze the role of the C-terminal end in bilayer targeting, we expressed in Escherichia coli and purified a C-terminally truncated construct missing the last seven residues, VP40(31–319). In the crystal structure, only two of these seven residues were ordered, while no clear electron density could be observed for residues 322–326 (Figure 1A and B; Dessen et al., 2000a). VP40(31–319) elutes from a gel filtration column as both a monomeric protein whose size is comparable to monomeric full-length VP40 (data not shown) and an oligomeric form, which elutes at a similar position to 440 kDa ferritin (Figure 2A). This observation indicates that the removal of the last seven residues destabilizes monomeric VP40. This can be further enhanced by incubating VP40(31–319) in 1 M urea, which results only in an oligomeric form of VP40(31–319), as shown by gel filtration chromatography (Figure 2B). To establish the oligomerization state of VP40(31–319), we performed chemical cross-linking on the peak, which eluted as an oligomer from the gel filtration column (see Figure 2A and B). Chemical cross-linking was performed in 1% n-octyl-β-d-glucopyranoside to avoid extensive aggregation of VP40(31–319). Incubation of VP40 (31–319) with increasing amounts of cross-linking reagent leads to the appearance of bands which may correspond to dimeric (∼70 kDa), trimeric (∼110 kDa), tetrameric (∼150 kDa) and hexameric forms of VP40(31–319) (Figure 2C). The major cross-linked species migrates close to the 250 kDa marker protein, which is approximately in accordance with a hexameric form of the 37 kDa monomer. A few faint higher molecular weight bands indicate the tendency of the protein to form higher molecular weight aggregates. Negatively stained electron micrographs of VP40(31–319) reveal ring-like structures (Figure 2D) similar to those observed with C-terminally truncated VP40(31–212) (Ruigrok et al., 2000b).
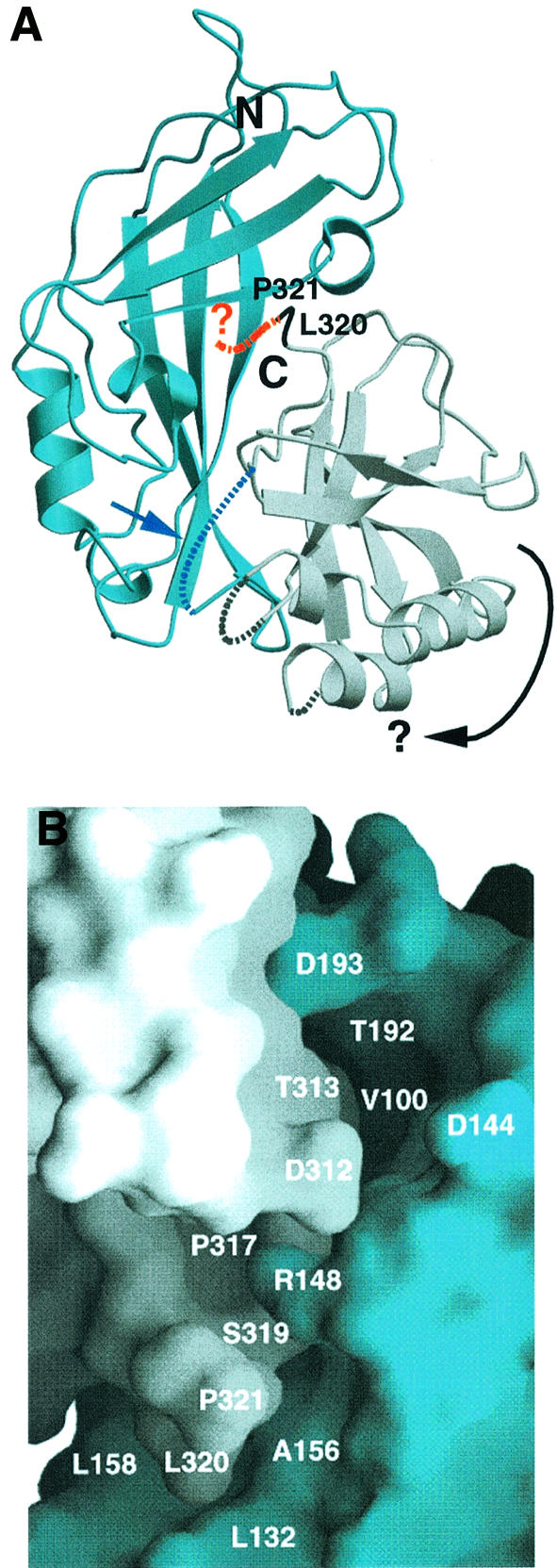
Fig. 1. Ribbon diagram [generated with the program Molscript (Kraulis, 1991)] and surface representation of VP40 [generated with the program Grasp (Nicholls et al., 1991)]. The N-terminal domain is shown in blue and the C-terminal domain in gray. (A) C-terminal residues L320 and P321 are shown in black, and the disordered residues 322–326 are indicated as a red dashed line. Three other disordered loop regions, including the connection between the N- and C-terminal domains, are also shown as dashed lines. The connection between the N- and C-terminal domains may constitute a hinge region (blue arrow and dashed line) and may allow the rotation of the C-terminal domain (indicated by a black arrow), thus opening up potential hexamerization interfaces. (B) Close-up of the interface of the N- and C-terminal domains around the position of P321. This view shows VP40 rotated anti-clockwise by 180° compared with the one shown in (A). Residues lining potential pockets for the binding of small molecule(s) at the interface are indicated (L132 and P317 are part of an ∼14 × 7 × 6.5 Å deep pocket, and V100 and T192 are at the bottom of an ∼8 × 8 × 10 Å deep pocket). Removal of residues 320–326 leads to spontaneous hexamerization of VP40(31–319) and to membrane binding in vitro.
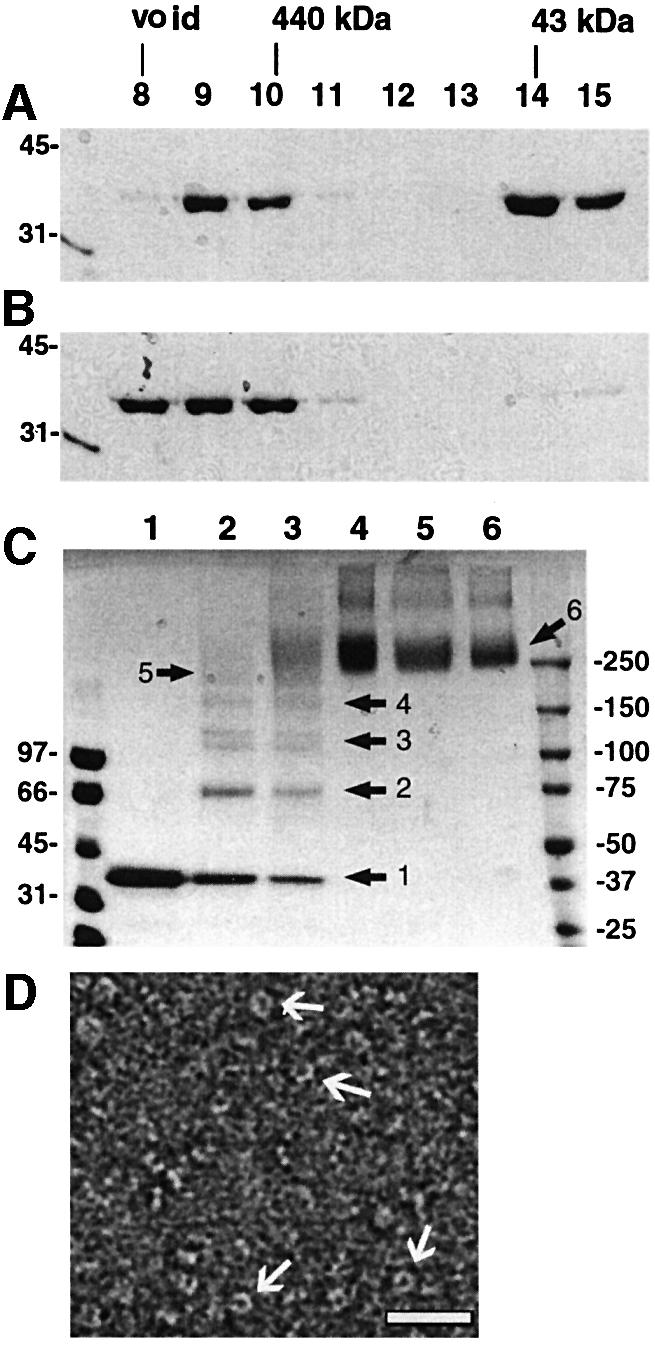
Fig. 2. (A) Gel filtration profile of VP40(31–319). Samples of fractions corresponding to elution volumes of 8–15 ml were separated by 12% SDS–PAGE and stained with Coomassie Blue. (B) Gel filtration profile of VP40(31–319) after incubation in 1 M urea. Molecular weight markers and the position of the elution of molecular weight markers from the gel filtration column are indicated. (C) Chemical cross-linking of VP40(31–319) (fraction 10, A). Lane 1, no cross-linking reagent; lane 2, 0.05 mM EGS; lane 3, 0.1 mM EGS; lane 4, 0.5 mM EGS; lane 5, 1 mM EGS; lane 6, 5 mM EGS. Molecular weight standards and the positions of intermediate cross-linking products are indicated. The samples were separated by 5–12% gradient SDS–PAGE and bands were visualized by Coomassie Blue staining. (D) Electron micrographs of VP40(31–319); the protein was stained with 1% SST and photo graphed under low-dose conditions. Several ring structures with an outer diameter of ∼90 Å are indicated by arrows. These micrographs were used for the 3D reconstruction. The white bar corresponds to 300 Å.
Three-dimensional reconstruction of VP40(31–319)
The VP40(31–319) ring structures were examined by single-particle image analysis and their three-dimensional (3D) reconstruction was calculated as described (Materials and methods). Figure 3A shows a model of VP40(31–319) displayed at the same cut-off density as used for the VP40(31–212) model calculation (Figure 3B; Ruigrok et al., 2000b), which results in very similar models for both constructs. Comparison of images of class averages (Figure 3C, middle panel) and reprojections of the VP40(31–319) model (Figure 3C, upper panel) with the images of the class averages for VP40(31–212) (Figure 3C, lower panel; Ruigrok et al., 2000b) shows the intense density corresponding to the core structure of VP40(31–319) together with additional, lower density around the central ring (Figure 3C, upper and middle panel). A corresponding model, which is displayed with a three times lower cut-off value, is shown in Figure 3D. The display results in slightly bigger central ring structures with the visualization of extra domains placed above and below the ring structures, corresponding to the averaged and lower threshold density (Figure 3D). Since the N-terminal domains are capable of forming ring structures on their own (Ruigrok et al., 2000b), the extra density most probably corresponds to the C-terminal domains of VP40(31–319) that are flexibly linked to the N-terminal domains. This also indicates that the VP40(31–319) monomers are associated in an anti-parallel fashion.

Fig. 3. (A) 3D reconstruction of VP40(31–319). The threshold was set to visualize only the N-terminal domain (see Materials and methods). The 3D reconstruction is shown in the end view orientation (left) and side view orientations (middle and right). The right-most view is the middle view rotated by 60° along the 3-fold axis. The measurements of the particle are approximately as follows: outside diameter ∼90 Å; inside diameter ∼35 Å; height ∼25–30 Å; width of the dimer ∼55 Å. (B) 3D reconstruction of the VP40(31–212) ring structures (Ruigrok et al., 2000b). The reconstruction shown was calculated with a threshold corresponding to the volume of the N-terminal domain. The dimensions of the particle are similar to those shown in (A). (C) Model reprojections and class-averaged views used for the reconstruction calculations of VP40(31–319) (upper and middle panel, respect ively) and class-averaged views used to calculate the reconstruction of VP40(31–212) (lower panel; Ruigrok et al., 2000b). The reprojections and average views are shown from end views (left) to side views (right) every 10°. The arrows in the reprojections of VP40(31–319) reconstructions indicate extra density that is not visible in the VP40(31–212) images. These densities are also visible in the averaged images in the middle panel, but not in the class-averaged views of VP40(31–212) (lower panel) missing most of the C-terminal domain (Ruigrok et al., 2000b). (D) 3D reconstruction of VP40(31–319) with a threshold value that allows for visualization of the C-terminal domains represented by lower density. As a result of lowering the threshold, the N-terminal part has also increased in size.
Membrane association of VP40(31–319)
The removal of seven C-terminal residues does not affect membrane association of VP40. In fact, the efficiency of binding of VP40(31–319) to liposomes containing a high amount of negatively charged phospholipids in vitro seems to be enhanced (Figure 4A) compared with the wild-type molecule VP40(31–326) (Figure 4B), which generally shows only 50–70% membrane association in vitro. VP40(31–319) does not float up in a sucrose gradient with no liposomes added (Figure 4C).
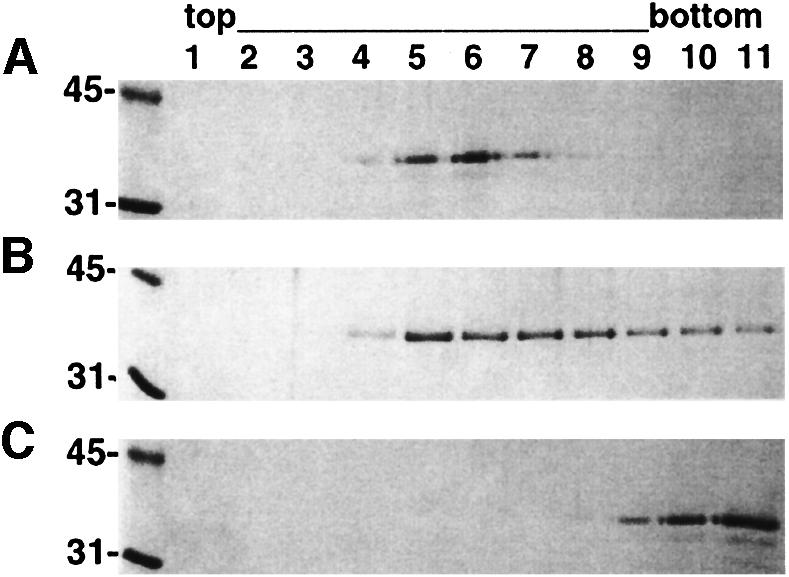
Fig. 4. Membrane association of VP40(31–319). Floatation of (A) VP40(31–319) and (B) VP40(31–326) with liposomes containing 50% l-α-phosphatidyl-l-serine. (C) Floatation of VP40(31–319) with no liposomes added. Samples from each fraction were separated by 12% SDS–PAGE and stained with Coomassie Blue. Bottom fractions 9–11 contain VP40 in solution. Molecular weight markers are shown.
VP40(31–326) inter-domain destabilization results in hexameric structures induced by a conformational change
VP40(31–326), like full-length VP40 [VP40(1–326)], is monomeric in solution (Dessen et al., 2000a). To determine whether we can destabilize the weak interactions between the two domains as seen in the crystal structure (Dessen et al., 2000a), and thus induce a conformational change to obtain intermediate hexameric structures, we incubated VP40(31–326) with increasing amounts of urea. The incubation in 1 and 2 M urea had no effect as the protein elutes from a gel filtration column in a peak corresponding to the monomer (Figure 5B and C), which is comparable to the elution profile of VP40(31–326) with no urea treatment (Figure 5A). However, urea concentrations of 3 and 4 M induced partial (Figure 5D) and complete (Figure 5E) oligomerization with an elution profile similar to that obtained for oligomeric VP40(31–319) (Figure 2A and B). In addition, gel filtration of VP40(31–326) in a buffer containing 1% n-octyl-β-d-glucopyranoside did not alter its monomeric conformation (Figure 5F). Chemical cross-linking performed on VP40(31–326) destabilized in 3 M urea shows that it forms hexameric structures. Increasing concentrations of cross-linking reagent produce several intermediate forms and a major band migrating slightly slower than the 250 kDa marker band (Figure 6A). The profile of the intermediate bands and the size of the final cross-linked product are consistent with a hexameric form comparable to that shown for the mutant VP40(31–319) (Figure 2D). A higher molecular weight band(s) is also visible, indicating the protein’s tendency to aggregate even in 1% detergent.
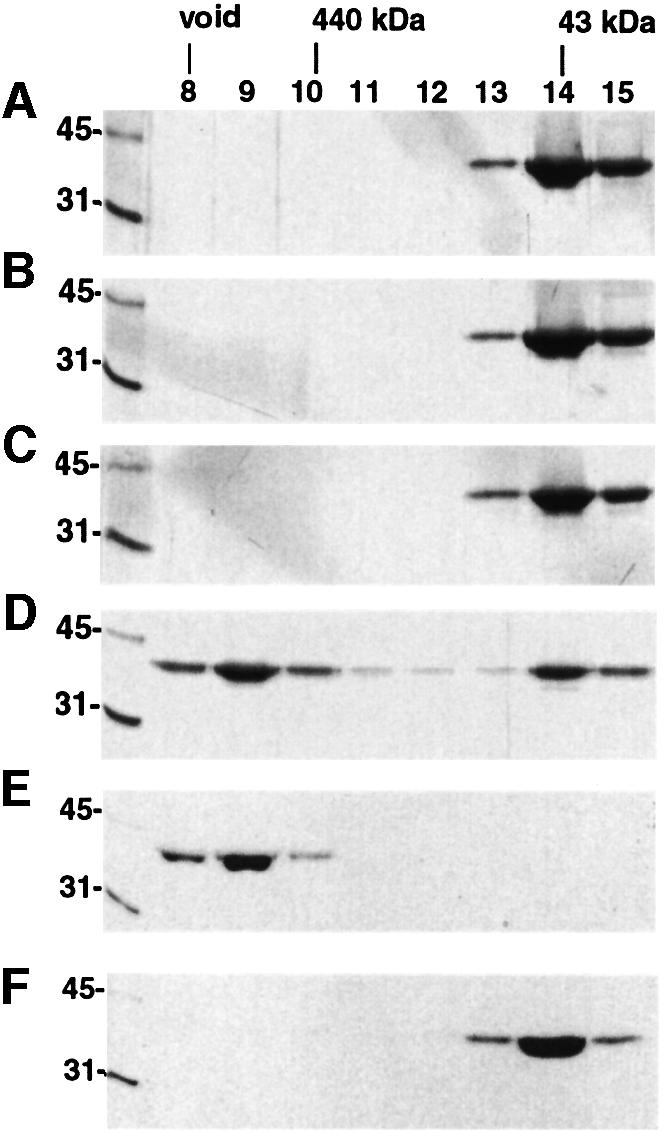
Fig. 5. Destabilization of the interaction of the N- and C-terminal domains leads to hexamerization. (A) Gel filtration profile of VP40(31–326). Samples of fractions corresponding to elution volumes of 8–15 ml were separated by 12% SDS–PAGE and stained with Coomassie Blue. Molecular weight markers are shown and the elution position of molecular weight markers from the gel filtration column are indicated. Gel filtration profiles of VP40(31–326) after incubation in (B) 1 M urea, (C) 2 M urea, (D) 3 M urea, (E) 4 M urea and (F) a buffer containing 1% n-octyl-β-d-glucopyranoside.
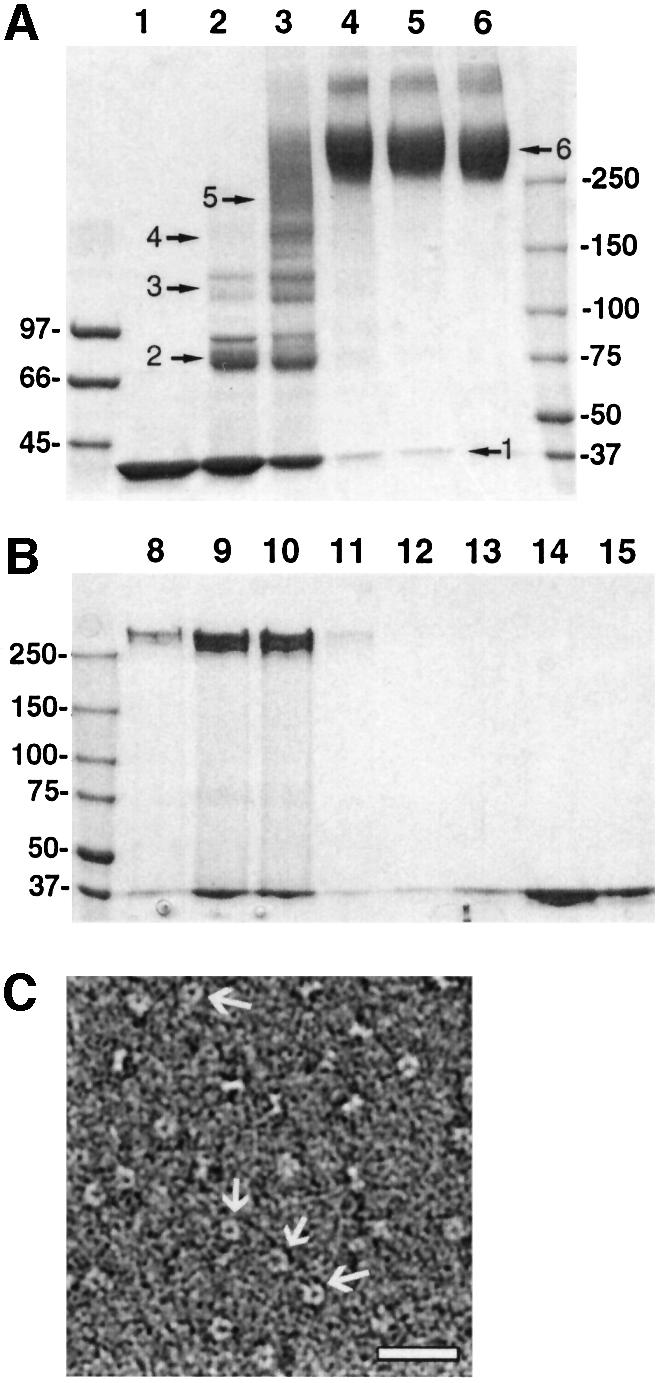
Fig. 6. Urea destabilization of VP40(31–326) induces a conformational change. (A) Chemical cross-linking of VP40(31–326) destabilized by incubation in 3 M urea and separated on a gel filtration column (Figure 5D, fraction 10). Lane 1, no cross-linking reagent; lane 2, 0.05 mM EGS; lane 3, 0.1 mM EGS; lane 4, 0.5 mM EGS; lane 5, 1 mM EGS; lane 6, 5 mM EGS. Samples were separated by 5–12% gradient SDS–PAGE and bands were visualized by Coomassie Blue. (B) SDS resistance of hexameric VP40(31–326). Gel filtration profiles of VP40(31–326) after incubation in 3 M urea; samples corresponding to fractions of the elution volume from 8 to 15 ml were separated by 5–12% gradient SDS–PAGE under reducing conditions and not boiled. Bands were visualized by Coomassie Blue staining. Molecular weight standards are indicated. (C) Electron micrograph of VP40(31–326) destabilized in 3 M urea and separated by gel filtration chromatography (Figure 5D, fraction 10); the protein was stained with 1% SST and photographed under low-dose conditions. Ring structures with an outer diameter of ∼90 Å are indicated by arrows and the white bar corresponds to 300 Å.
The oligomerization state of VP40 can also be analyzed by its resistance to SDS. SDS–PAGE of non-boiled VP40(31–326) destabilized by urea shows bands migrating at ∼260 kDa, similar to the hexameric bands obtained by chemical cross-linking (Figures 6A and 7A). Only fractions corresponding to high molecular weight VP40(31–326) show SDS-resistant hexamers (Figure 6B, lanes 8–10), while VP40(31–326) eluting as a monomer from the gel filtration column remains monomeric when separated on SDS–PAGE under non-boiling conditions (Figure 6B, lanes 13–15). This indicates that 100% of oligomeric VP40(31–326) forms hexamers in solution, which have a tendency to aggregate, as indicated by the gel filtration elution profile. In addition, in negative staining EM, oligomeric particles of VP40(31–326), generated by urea treatment (Figure 6C), were indistinguishable from the ring-like structures formed by the mutant VP40(31–319) (Figure 2D).

Fig. 7. Oligomerization of VP40(31–326) induced by lipid bilayer association. (A) VP40(31–326) was incubated with liposomes containing 25% l-α-phosphatidylcholine, 50% l-α-phosphatidyl-l-serine and 25% cholesterol. Liposomes containing VP40(31–326) were solubilized in detergent and VP40(31–326) was purified by gel filtration chromatography prior to chemical cross-linking. Lane 1, no cross-linking reagent; lane 2, 0.05 mM EGS; lane 3, 0.1 mM EGS; lane 4, 0.5 mM EGS; lane 5, 1 mM EGS; lane 6, 5 mM EGS. Samples were separated by 5–12% gradient SDS–PAGE and bands were visualized by western blotting using a VP40-specific antiserum. Molecular weight standards are shown. (B and C) Electron micro graphs of VP40(31–326) bound to lipid vesicles. The protein was stained with 1% SST and photographed under low-dose conditions. The bar corresponds to 300 Å and the electron micrograph of a ring structure of soluble, hexameric VP40(31–326) (Figure 6B) is shown in a white box for comparison. (C) Liposomes were incubated with a lower amount of protein that results in a sparse binding pattern. Few ring structures can be seen on the top surface of the small liposome and on the side/edge of the liposome.
VP40(31–326) membrane association induces hexamerization
In order to determine whether the hexamerization of VP40 in solution observed in vitro may be induced by lipid bilayer interaction of monomeric VP40, we separated VP40(31–326) bound to liposomes containing 50% negatively charged phospholipids (l-α-phosphatidyl-l-serine) and unbound VP40(31–326) by sucrose gradient floatation (Ruigrok et al., 2000b). The fractions containing liposomes and VP40(31–326) were pooled, and the liposomes were solubilized in detergent. Chemical cross-linking of treated VP40(31–326) shows the same hexamerization profile (Figure 7A) as observed for the mutant protein VP40(31–319) and VP40(31–326) destabilized with 3 M urea (Figures 2C and 6A). Increasing concentrations of cross-linking reagent induce the formation of higher molecular weight bands, with a major species again migrating close to the 250 kDa marker protein (Figure 7A). Urea-destabilized and cross-linked VP40(31–326) (Figure 6A, lane 6) migrates at exactly the same position as the hexameric form shown in Figure 7A (lane 6). The cross-linking experiment also shows that 100% of VP40(31–326) bound to liposomes is hexameric as no monomeric or intermediate bands can be detected at a concentration of 5 mM ethylene glycol-bis(succinimidylsuccinate) (EGS) reagent (Figure 7A). Chemical cross-linking of VP40(31–326) from the bottom fractions of the sucrose gradient experiment (Figure 4B, lanes 9–11) revealed that free VP40 stays monomeric under these conditions (data not shown). In addition, EM of negatively stained VP40(31–326) bound to liposomes reveals the same ring structures as observed for hexameric VP40 in solution (Figure 7B and C) without, however, the formation of a defined protein lattice. VP40’s tendency to form higher molecular weight aggregates is underlined by the necessity of detergent to prevent higher molecular weight oligomerization. This can be observed in cross-linking experiments with no detergent present (data not shown) as well as by gel filtration chromatography (Figures 2A and 5; lane 8 corresponding to the void volume). Therefore, our results clearly demonstrate that VP40 can adopt an intermediate hexameric structure in solution either artificially induced by mutagenesis, denaturant or by lipid bilayer interaction. These results strongly indicate that the interaction of VP40(31–326) with bilayers may trigger a conformational change upon which hexamerization and efficient membrane association can take place.
Discussion
The matrix protein from Ebola virus consists of two structurally related β-sandwich domains, which are connected by a flexible linker (Dessen et al., 2000a). Here we show that the removal of the last seven residues by mutagenesis destabilizes the interaction between the N- and the C-terminal domains, which leads to oligomerization of VP40. Chemical cross-linking results, SDS resistance analysis as well as the 3D reconstruction based on negatively stained images of VP40(31–319) are in agreement with the previously reported hexameric ring-like structures formed by VP40(31–212) missing most of the C-terminal domain (Ruigrok et al., 2000b). However, the averaged EM images of VP40(31–319) and their 3D reconstruction show that the C-terminal domains are poorly visible, indicating their flexible linkage to the N-terminal units that constitute the hexamerization module (Ruigrok et al., 2000b). The flexibility of the C-terminal domain may also contribute to the observation that hexameric VP40 elutes from a gel filtration column with a higher molecular weight (∼440 kDa) than its calculated one (∼230 kDa). In addition, it has a high tendency to form aggregates, as seen in gel filtration elution profiles. However, the C-terminal domain contains VP40’s membrane-targeting activity (Ruigrok et al., 2000b) and it is therefore conceivable that this domain may adopt a fixed conformation with respect to the N-terminal domain upon binding to its target. The removal of the last seven residues does not abolish VP40’s binding to membranes in vitro, indicating that this sequence is not directly involved in lipid bilayer binding.
In order to test the stability of the inter-domain interactions of wild-type VP40, we incubated VP40(31–326) with increasing amounts of urea, which indeed induced the same hexamerization pattern as observed for the mutant VP40(31–319). The larger amount of urea (4 M) necessary to induce complete oligomerization of VP40(31–326) compared with the mutant VP40(31–319) underlines the role of residues 320–326 in stabilizing the association of the N- and C-terminal domains. We also demonstrate that the conformational switch that leads to VP40 hexamerization is induced by membrane association, as detected by gel filtration chromatography and chemical cross-linking of membrane-bound VP40 solubilized in detergent, as well as in the direct visualization of VP40 ring structures on liposomes by EM. We show further that hexameric VP40 is resistant to SDS when separated by SDS–PAGE under non-boiling conditions, and we present evidence that only hexameric VP40 is associated with membranes while monomeric VP40 remains in the bottom of sucrose gradients. However, the fact that VP40(31–326) usually gives rise to an efficiency of ∼50–80% liposome association, while artificially induced hexameric VP40(31–319) usually shows 100% binding, also indicates that our liposome composition may not be ideal to induce the switch from monomer to hexamer with 100% efficiency, which would also result in complete bilayer interaction. Incubation with liposomes containing low amounts of l-α-phosphatidyl-l-serine showed only inefficient association (Ruigrok et al., 2000b; Figure 5D) and VP40(31–326), which did not float with liposomes, remained monomeric (data not shown), indicating that the conformational switch and membrane association are probably interdependent processes. These results strongly imply that the switch from a monomeric to a hexameric form can be induced by destabilization of the few inter-domain interactions observed in the VP40 crystal structure (Dessen et al., 2000a) either artificially, by urea, or in a physiologically relevant manner by membranes.
VP40(31–212), an N-terminal fragment in which all but the first 12 residues of the C-terminal domain are absent (Dessen et al., 2000a), hexamerizes in solution, indicating that the N-terminal domain of VP40 is responsible for oligomerization (Ruigrok et al., 2000b). The loose association of the domains favors a model in which the C-terminal domain sterically prevents the transition from a monomer to a hexamer. Therefore, a simple movement of the C-terminal domain (see Figure 1A) may allow the N-terminal unit to hexamerize by opening up the protein interfaces necessary both for oligomerization and membrane binding. The conformational change may be induced by specific lipid–protein interactions, which may explain the preferred lipid composition reported to be necessary for some matrix protein–membrane interactions (Zakowski et al., 1981; Provitera et al., 2000; Ruigrok et al., 2000a,b). Furthermore, lipid rafts implicated in virus assembly (Scheiffele et al., 1999; Manié et al., 2000; Zhang et al., 2000) may also play a role in the conformational switch by providing the right lipids.
The more efficient in vitro membrane association of the mutant protein VP40(31–319) is consistent with the idea that hexameric VP40(31–319) is already in an active membrane-binding conformation. Our data suggest that hexameric VP40 may be a structural module of the matrix protein in the assembly process of filoviruses. Although chemical cross-linking of VP40 bound to membranes shows high molecular weight aggregates (data not shown), which indirectly indicates that ring structures contact each other, we do not observe the formation of an ordered lattice upon bilayer interaction. VP40 ring structures are generally loosely ordered on liposomes despite their dense packing. However, the formation of a protein lattice may play a crucial role in Ebola virus assembly. Therefore, matrix protein interactions with the short cytoplasmic tails of the glycoproteins and/or with a second membrane-associated protein VP24 as well as the RNP complexes may contribute to a defined lattice formation, initiating assembly and budding of new virus particles at the plasma membrane (Feldmann et al., 1996).
Three-dimensional reconstruction of VP40(31–319) indicates that the N-terminal domains form an anti-parallel dimer with their C-termini pointing in opposite directions. This arrangement places the C-terminal domains above and below the N-terminal ring-like structure. The maximum density corresponding to the C-terminal domains shown is only one-third of that of the N-terminal domains, which underlines the fact that the C-terminal domains are flexibly linked to the N-terminal part. This is also apparent from the raw images where the C-terminal domains are not in fixed positions. We have shown previously that the C-terminal domains are important for membrane binding (Ruigrok et al., 2000b). However, the EM images indicate that the C-terminal domains might have an additional function, as both layers of C-terminal domains cannot bind to membranes simultaneously, despite their flexible linkage to the N-terminal part. Other viral proteins, like a second membrane-associated filovirus protein VP24 (Feldmann and Kiley, 1999) or the nucleocapsid proteins, may interact with the second layer of C-terminal domains. However, we also cannot completely exclude the possibility that virus assembly and budding may induce a different oligomerization pattern of VP40 through interactions with other viral proteins.
It is generally difficult to work with viral matrix proteins in vitro since they all show a high tendency to aggregate, which may reflect their function in virus assembly. Therefore, this is a first report of a solubilized intermediate of a matrix protein in a defined oligomeric state that is generated by a conformational change. Indirect evidence for hexameric forms of a virus matrix protein arose from crystallization of lentiviral matrix proteins from HIV and SIV, whose crystalline lattices suggested a defined oligomerization pattern (Rao et al., 1995; Hill et al., 1996). Further evidence for a hexameric intermediate structure of the HIV capsid protein came from EM studies of ordered arrays of protein (Barklis et al., 1998). Membrane binding of HIV Gag involves a cluster of basic residues in the matrix protein that preferentially binds to negatively charged phospholipids. In addition, an N-terminal myristoylate signal seems to play an important role in membrane binding as well as stabilization by a switch mechanism (Göttlinger et al., 1989; Bryant and Ratner, 1990; Zhou and Resh, 1996; Paillart and Göttlinger, 1999). It may be interesting to compare these results with our observation of hexameric Ebola virus matrix protein in vitro. However, it is necessary to consider that the retroviral assembly process includes the proteolytic cleavage of a precursor molecule (Gag) into matrix, capsid, nucleocapsid and p6 proteins during a final maturation step (Henderson et al., 1990), which is controlled by a conformational switch (Gross et al., 2000). Furthermore, immature capsids of HIV and Moloney murine leukemia virus show Gag molecules arranged with local order in spherical assemblies, but with no apparent icosahedral/hexagonal symmetry (Fuller et al., 1997; Yeager et al., 1998). However, this may not completely exclude a role for the hexameric arrays of retroviral Gag observed in vitro (Nermut et al., 1993; Barklis et al., 1997, 1998).
Viral matrix proteins from members of the Mononegavirales have been shown to polymerize in vitro (Heggeness et al., 1982; McCreedy et al., 1990; Gaudin et al., 1997) and in vivo with the observation of crystalline lattices on the plasma membrane (Büechi and Bächi, 1982). The self-assembly capability also becomes evident from the release of virus-like particles or vesicles when matrix proteins are expressed by themselves (Li et al., 1993; Justice et al., 1995; Coronel et al., 1999; Sakaguchi et al., 1999). Interestingly, this feature is shared by retroviral Gag precursor molecules (Gonzalez et al., 1993), which also assemble into hexagonal arrays on membranes in vitro (Barklis et al., 1998). These data indicate that some matrix proteins constitute a minimal assembly and budding machinery. However, there is likewise evidence that the concerted interaction of other viral proteins, including the cytoplasmic tails of the glycoproteins (Metsikkö and Simons, 1986; Sanderson et al., 1994; Jin et al., 1997; Schnell et al., 1998; Mebatsion et al., 1999; Schmitt et al., 1999), the RNP particle (Garoff et al., 1998) and/or cellular factors, influences the efficiency of particle formation (Garnier et al., 1996; Harty et al., 1999; Yasuda and Hunter, 1998).
Although the mode of assembly may differ substantially between virus families, our data suggest that viral matrix proteins may have to undergo a molecular switch from a soluble form to a less soluble membrane-interacting conformation, a process that will thus help to explain their versatile functions (Garoff et al., 1998). Matrix proteins are often found in an insoluble form associated with membranes or in a soluble form in infected and transfected cells (reviewed by Lenard, 1996). In addition, the observation of soluble and insoluble membrane-bound VSV matrix proteins in transfected cells (Chong and Rose, 1993), as well as the detection of two isoforms of the Sendai virus matrix protein (De Melo et al., 1992), are both in agreement with the two conformations described for the matrix protein of Ebola virus. The proposed requirement for a conformational switch may be also exploited for the development of new antiviral therapies. The structure of VP40 shows prominent pocket(s) lined by hydrophobic residues and shared by the N- and C-terminal domains, with the C-terminal end protruding out in the middle (Figure 1B). It is conceivable that small molecules that may be targeted to this pocket(s) may prevent domain dissociation, and thus membrane association and virus particle production.
Materials and methods
Expression and purification of VP40
VP40(31–326) was expressed and purified as described (Dessen et al., 2000b). The DNA fragment encoding VP40 residues 31–319 was generated by standard PCR techniques and DNA fragments were cloned into expression vector pRset (Invitrogen) containing an N-terminal His tag by standard procedures [VP40(31–319)]. The sequence was verified by DNA sequencing. Expression was induced with 1 mM isopropyl-β-d-thiogalactopyranoside for 3 h. Cells were harvested by centrifugation and lysed by sonication in lysis buffer (50 mM Tris pH 8.8, 100 mM NaCl, 10 mM β-mercaptoethanol). The supernatant was cleared by centrifugation and applied to a Ni2+–Sepharose column, pre-equilibrated in lysis buffer. VP40 constructs were eluted with a linear imidazole gradient in lysis buffer, concentrated, and further purified by size exclusion chromatography (Superdex 200 column; Pharmacia) in a buffer containing 20 mM bicine pH 9.3, 100 mM NaCl, 10 mM dithiothreitol (DTT). The protein concentration was determined by Bio-Rad assay.
In order to destabilize the inter-domain interactions between the N- and the C-terminal domains, proteins (at 1 mg/ml) were equilibrated with the indicated urea concentration in a buffer containing 20 mM bicine pH 9.3, 100 mM NaCl, 10 mM DTT for 20 min at room temperature. The protein was then separated by gel filtration chromatography (Superdex 200; Pharmacia) in 20 mM bicine pH 9.3, 100 mM NaCl, 10 mM DTT buffer. Fractions of 0.5 ml were collected, samples were separated by 12% SDS–PAGE, and bands were stained with Coomassie Brilliant Blue.
Chemical cross-linking and SDS resistance
For all chemical cross-linking experiments, the protein sample buffer was exchanged by gel filtration chromatography (Superdex 200; Pharmacia) to 50 mM HEPES pH 9.3, 100 mM NaCl, 1% n-octyl-β-d-glucopyranoside. Fractions of 0.5 ml were collected and the VP40 protein in the fraction corresponding to an elution volume of 10 ml was immediately used for cross-linking with EGS (Pierce). EGS was dissolved to 50 mM in DMSO and different dilutions were added to the protein solution as indicated. Reactions were incubated at room temperature for 20 min and subsequently quenched with 50 mM Tris pH 8.8 for 5 min. Samples were then separated on an SDS–PAGE gradient (12–5%) under reducing conditions. VP40-specific bands were detected by Coomassie Blue staining or by western blotting with a VP40-specific antiserum. To test VP40’s SDS resistance, samples were mixed with standard reducing SDS loading buffer, not boiled, and separated on an SDS–PAGE gradient (12–5%). Gels were stained with Coomassie Blue.
Liposome preparation and membrane floatation experiments
Liposomes containing 25% l-α-phosphatidylcholine (Sigma), 50% l-α-phosphatidyl-l-serine (Sigma) and 25% cholesterol (Sigma) were produced as described (Ruigrok et al., 2000b). One hundred micrograms of VP40(31–319) or VP40(31–326) were incubated with 100 µl of liposomes (2 mg/ml lipid) for 1 h at 37°C. Sucrose gradients were generated in 25 mM HEPES pH 8.9, 100 mM NaCl as described previously (Ruigrok et al., 2000b). Fractions from the sucrose gradients were separated by 12% SDS–PAGE and bands were detected with Coomassie Blue staining.
Fractions containing VP40(31–326) bound to liposomes were pooled. Liposomes were dissolved in a buffer containing 20 mM bicine pH 9.3, 100 mM NaCl, 10 mM DTT, 1% n-octyl-β-d-glucopyranoside, 0.02% Triton X-100 and subsequently separated by gel filtration chromatography (Superdex 200; Pharmacia) in 50 mM HEPES pH 9.2, 100 mM NaCl, 1% n-octyl-β-d-glucopyranoside. The fraction corresponding to the elution volume of 10 ml was then used for chemical cross-linking as described above.
Electron microscopy
Negative staining. Protein at a concentration of ∼0.1 mg/ml was adsorbed to the clean face of carbon on mica (the carbon–mica interface), which was floated onto a solution of 1% sodium silicotungstate (SST). A grid was placed on top of the carbon film, which was subsequently picked up from the top with a small piece of paper and air-dried. The samples were photographed in a JEOL 1200 EXII electron microscope at 100 kV under low-dose conditions (<10 e–/Å2) at a nominal magnification of 40 000. The images were recorded on Kodak SO163 films, which were developed in 1/3 strength D19 developer for 9 min.
Image processing and 3D reconstruction. The quality of the EM negatives was checked on an optical bench and selected negatives were digitized on an Optronics microdensitometer, coupled to a PC, with a step size of 12.5 µm, which corresponds to a pixel size of 3.1 Å on the specimen. A total of 4800 views (half of them side views) were selected using Ximdisp (Crowther et al., 1996). They were then cut out into squares of 128 × 128 pixels, band pass filtered between 150 and 20 Å without CTF correction (the first zero was beyond 20 Å), and subsequently normalized to the same mean and standard deviation with the program Spider (Frank et al., 1996).
Two different starting models were used for solving the structure of VP40(31–319) employing the projection-matching procedure implemented in Spider (Frank et al., 1996). The structure of VP40(31–212) (Ruigrok et al., 2000b) and one centered side view were used. The side view was back-projected using the 3-fold symmetry that was determined for the VP40(31–212) structure (Ruigrok et al., 2000b; Schoehn et al., 2000). After 20 cycles of the projection-matching technique, both starting models led to the same solution in terms of orientation angles, center co-ordinates and optimal matching projections (less than 1° and 1 pixel difference). The final reconstruction was calculated by back-projecting 66 classes equally distributed in the asymmetric unit and by imposing a 3-fold symmetry. The Fourier Shell Correlation (FSC) (0.5 level) gave a resolution of 32 Å for the map.
Comparison of the class average images with those of the previous analysis of VP40(31–212) suggested that the VP40(31–319) structure had a very similar core with regions of lower density around this core (see the arrows in Figure 3C). Therefore, the visualization of the map with WEB (Frank et al., 1996) was performed in several ways. (i) A density threshold was chosen that resulted in the same sized middle hole of VP40(31–319) (∼35 Å) as calculated for VP40(31–212) (Ruigrok et al., 2000b). This procedure allowed us to display the correct density for the N-terminal part of the protein (Figure 3A and B). (ii) Displaying VP40(31–319) particles with a volume corresponding to the mass of the N-terminal domain alone [i.e. the mass of the VP40(31–212) particle] gave the same result as in (i) (data not shown). (iii) In order to visualize the smeared out density, probably corresponding to the C-terminal domains, a lower threshold was applied (Figure 3D). The maximum density for the C-terminal parts appeared to be three times lower than that for the N-terminal domains. Although the application of a lower density threshold results in significantly larger N-terminal domains than the corresponding protein volume, it allows visualization of averaged positions of the expected C-terminal domains.
Acknowledgments
Acknowledgements
We thank Dr V.Volchkow and Dr O.Dolnik for the VP40 antiserum, and Dr N.Ranson and Dr H.Saibil (Birkbeck College, London) for providing the latest versions of the single-particle analysis procedure running on Spider. The EM negatives were digitized at the Institut de Biologie Structurale, Grenoble. This work was supported in part by Deutsche Forschungsgemeinschaft SFB 286.
References
- Barklis E. et al. (1997) Structural analysis of membrane-bound retrovirus capsid proteins. EMBO J., 16, 1199–1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barklis E., McDermott,J., Wilkens,S., Fuller,S. and Thompson,D. (1998) Organization of HIV-1 capsid proteins on a lipid monolayer. J. Biol. Chem., 273, 7177–7180. [DOI] [PubMed] [Google Scholar]
- Becker S., Spiess,M. and Klenk,H.D. (1995) The asialoglycoprotein receptor is a potential liver-specific receptor for Marburg virus. J. Gen. Virol., 76, 393–399. [DOI] [PubMed] [Google Scholar]
- Bryant M. and Ratner,L. (1990) Myristoylation-dependent replication and assembly of human immunodeficiency virus 1. Proc. Natl Acad. Sci. USA, 87, 523–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Büechi M. and Bächi,Th. (1982) Microscopy of internal structures of Sendai virus associated with the cytoplasmic surface of host membranes. Virology, 120, 349–359. [DOI] [PubMed] [Google Scholar]
- Chong L.D. and Rose,J.K. (1993) Membrane association of functional vesicular stomatitis virus matrix protein in vivo. J. Virol., 67, 407–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coronel E.C., Murti,K.G., Takimoto,T. and Portner,A. (1999) Human parainfluenza virus type 1 matrix and nucleoprotein genes transiently expressed in mammalian cells induce the release of virus-like particles containing nucleocapsid-like structures. J. Virol., 73, 7035–7038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crowther R.A., Henderson,R. and Smith,J.M. (1996) MRC image processing programs. J. Struct. Biol., 116, 9–16. [DOI] [PubMed] [Google Scholar]
- De Melo M., Mottet,G., Orvell,C. and Roux,L. (1992) Sendai virus M protein is found in two distinct isoforms defined by monoclonal antibodies. Virus Res., 24, 47–64. [DOI] [PubMed] [Google Scholar]
- Dessen A., Volchkov,V., Dolnik,O., Klenk,H.-D. and Weissenhorn,W. (2000a) Crystal structure of the matrix protein VP40 from Ebola virus. EMBO J., 19, 4228–4236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dessen A., Forest,E., Volchkov,V., Dolnik,O., Klenk,H.-D. and Weissenhorn,W. (2000b) Crystallization and preliminary X-ray analysis of the matrix protein from Ebola virus. Acta Crystallogr. D, 56, 758–760. [DOI] [PubMed] [Google Scholar]
- Feldmann H. and Kiley,M.P. (1999) Classification, structure and replication of filoviruses. Curr. Top. Microbiol. Immunol., 235, 1–21. [DOI] [PubMed] [Google Scholar]
- Feldmann H. and Klenk,H.-D. (1996) Marburg and Ebola viruses. Adv. Virus Res., 47, 1–52. [DOI] [PubMed] [Google Scholar]
- Feldmann H., Bugany,H., Mahner,F., Klenk,H.-D., Drenckhahn,D. and Schnittler,H.J. (1996) Filovirus induced endothelial leakage triggered by infected monocytes/macrophages. J. Virol., 70, 2208–2214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank J., Radermacher,M., Penczek,P., Zhu,J., Li,Y., Ladjadj,M. and Leith,A. (1996) SPIDER and WEB: processing and visualisation of images in 3D electron microscopy and related fields. J. Struct. Biol., 116, 190–199. [DOI] [PubMed] [Google Scholar]
- Fuller S.D., Wilk,T., Gowen,B.E., Krausslich,H.-G. and Vogt,V.M. (1997) Cryo-electron microscopy reveals ordered domains in the immature HIV-1 particle. Curr. Biol., 7, 729–738. [DOI] [PubMed] [Google Scholar]
- Garnier L., Wills,J.W., Verderame,M.F. and Sudol,M. (1996) WW domains and retrovirus budding. Nature, 381, 744–745. [DOI] [PubMed] [Google Scholar]
- Garoff H., Hewson,R. and Opstelten,D.-J.E. (1998) Virus maturation by budding. Microbiol. Mol. Biol. Rev., 62, 1171–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaudin Y., Sturgis,L., Doumith,M., Barge,A., Robert,B. and Ruigrok,R.W.H. (1997) Conformational flexibility and polymerization of vesicular stomatitis virus matrix protein. J. Mol. Biol., 274, 816–825. [DOI] [PubMed] [Google Scholar]
- Geisbert T.W. and Jahrling,P.B. (1995) Differentiation of filoviruses by electron microscopy. Virus Res., 39, 129–150. [DOI] [PubMed] [Google Scholar]
- Gonzalez S.A., Affranchino,J.L., Gelderblom,H.R. and Burny,A. (1993) Assembly of the matrix protein of simian immunodeficiency virus into virus like particles. Virology, 194, 548–556. [DOI] [PubMed] [Google Scholar]
- Göttlinger H.G., Sodroski,J.G. and Haseltine,W.A. (1989) Role of capsid precursor processing and myristoylation in morphogenesis and infectivity of human immunodeficiency virus type 1. Proc. Natl Acad. Sci. USA, 86, 5781–5785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross I., Hohenberg,H., Wilk,T., Wiegers,K., Grättinger,M., Müller,B., Fuller,S. and Kräusslich,H.-G. (2000) A conformational switch controlling HIV-1 morphogenesis. EMBO J., 19, 103–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harty R.N., Paragas,J., Sudol,M. and Palese,P. (1999) A proline-rich motif within the matrix protein of vesicular stomatitis virus and rabies virus interacts with WW domains of cellular proteins: implications for viral budding. J. Virol., 73, 2921–2929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heggeness M.H., Smith,P.R. and Choppin,P.W. (1982) In vitro assembly of the nonglycosylated membrane protein (M) of Sendai virus. Proc. Natl Acad. Sci. USA, 79, 6232–6236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson L.E., Sowder,R.C., Copeland,T.D., Oroszlan,S. and Benveniste,R.E. (1990) Gag precursors of HIV and SIV are cleaved into six proteins found in mature virions. J. Med. Primatol., 19, 411–419. [PubMed] [Google Scholar]
- Hill C.P., Worthylake,D., Bancroft,D.P., Christensen,A.M. and Sundquist,W.I. (1996) Crystal structures of the trimeric human immunodeficiency virus type 1 matrix protein: implications for membrane association and assembly. Proc. Natl Acad. Sci. USA, 93, 3099–3104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin H., Leser,G.P., Zhang,J. and Lamb,R.A. (1997) Influenza virus hemagglutinin and neuraminidase cytoplasmic tails control particle shape. EMBO J., 16, 1236–1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Justice P.A., Sun,W., Li,Y., Ye,Z., Grigera,P.R. and Wagner,R.R. (1995) Membrane vesiculation function and exocytosis of wild-type and mutant matrix proteins of vesicular stomatitis virus. J. Virol., 69, 3156–3160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraulis P. (1991) MOLSCRIPT: a program to produce both detailed and schematic plots of protein structures. J. Appl. Crystallogr., 24, 924–950. [Google Scholar]
- Lenard J. (1996) Negative-strand virus M and retrovirus MA proteins: all in a family? Virology, 216, 289–298. [DOI] [PubMed] [Google Scholar]
- Lenard J. and Compans,R.W. (1974) The membrane structure of lipid-containing viruses. Biochim. Biophys. Acta, 344, 51–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y., Luo.L., Schubert,M., Wagner,R.R. and Kang,C.Y. (1993) Viral liposomes released from insect cells infected with recombinant baculovirus expressing the matrix protein of vesicular stomatitis virus. J. Virol., 67, 4415–4420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manié S.N., Debreyne,S., Vincent,S. and Gerlier,D. (2000) Measles virus structural components are enriched into lipid raft microdomains: a potential cellular location for virus assembly. J. Virol., 74, 305–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCreedy B.J., McKinnon,K.P. and Lyles,D.S. (1990) Solubility of vesicular stomatitis virus M protein in the cytosol of infected cells or isolated from virions. J. Virol., 64, 902–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mebatsion T., Weiland,F. and Conzelmann,K.K. (1999) Matrix protein of rabies virus is responsible for the assembly and budding of bullet-shaped particles and interacts with the transmembrane spike glycoprotein G. J. Virol., 73, 242–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metsikkö K. and Simons,K. (1986) The budding mechanism of spikeless vesicular stomatitis virus particles. EMBO J., 5, 1913–1920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nermut M.V., Grief,C., Hashmi,S. and Hockley,D.J. (1993) Further evidence of icosahedral symmetry in human and simian immunodeficiency virus. AIDS Res. Hum. Retroviruses, 9, 929–938. [DOI] [PubMed] [Google Scholar]
- Nicholls A., Sharp,K.A. and Honig,B. (1991) Protein folding and association: insights from the interfacial and thermodynamic properties of hydrocarbons. Proteins, 11, 281–296. [DOI] [PubMed] [Google Scholar]
- Paillart J.C. and Göttlinger,H.G. (1999) Opposing effects of human immunodeficiency virus type 1 matrix mutations support a myristyl switch model of gag membrane targeting. J. Virol., 73, 2604–2612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Provitera P., Bouamr,F., Murray,D., Carter,C. and Scarlata,S. (2000) Binding of equine infectious anemia virus matrix protein to membrane bilayers involves multiple interactions. J. Mol. Biol., 296, 887–898. [DOI] [PubMed] [Google Scholar]
- Rao Z., Belyaev,A.S., Fry,E., Roy,P., Jones,I.A. and Stuart,D.I. (1995) Crystal structure of SIV matrix antigen and implications for viral assembly. Nature, 378, 743–747. [DOI] [PubMed] [Google Scholar]
- Ruigrok R.W.H., Barge,A., Durrer,P., Brunner,J., Ma,K. and Whittaker,G.R. (2000a) Membrane interaction of influenza virus M1 protein. Virology, 267, 289–298. [DOI] [PubMed] [Google Scholar]
- Ruigrok R.W.H., Schoehn,G., Dessen,A., Forest,E., Volchkov,V., Dolnik,O., Klenk,H.-D. and Weissenhorn,W. (2000b) Structural characterization and membrane binding properties of the matrix protein VP40 of Ebola. J. Mol. Biol., 300, 103–112. [DOI] [PubMed] [Google Scholar]
- Sakaguchi T., Uchiyama,T., Fujii,Y., Kiyotani,K., Kato,A., Nagai,Y., Kawai,A. and Yoshida,T. (1999) Double-layered membrane vesicles released from mammalian cells infected with Sendai virus expressing the matrix protein of vesicular stomatitis virus. Virology, 263, 230–243. [DOI] [PubMed] [Google Scholar]
- Sanderson C.M., Wu,H.H. and Nayak,D.P. (1994) Sendai virus M protein binds independently to either the F or the HN glycoprotein in vivo. J. Virol., 68, 69–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheiffele P., Rietveld,A., Wilk,T. and Simons,K. (1999) Influenza virus select ordered lipid domains during budding from the plasma membrane. J. Biol. Chem., 274, 2038–2044. [DOI] [PubMed] [Google Scholar]
- Schmitt A.P., He,B. and Lamb,R.A. (1999) Involvement of the cytoplasmic domain of the hemagglutinin-neuraminidase protein in assembly of the paramyxovirus simian virus 5. J. Virol., 73, 8703–8712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnell M.J., Buonocore,L., Boritz,E., Ghosh,H.P., Chernish,R. and Rose,J.K. (1998) Requirement for a non-specific glycoprotein cytoplasmic domain sequence to drive efficient budding of vesicular stomatitis virus. EMBO J., 17, 1289–1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnittler H.J., Mahner,F., Drenckhahn,D., Klenk,H.-D. and Feldmann,H. (1993) Replication of Marburg virus in human endothelial cells. A possible mechanism for the development of viral hemorrhagic disease. J. Clin. Invest., 91, 1301–1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoehn G., Quaite-Randall,E., Jiménez,J.L., Joachimiak,A. and Saibil,H.R. (2000) Three conformations of an archaeal chaperonin, TF55 from Sulfolobus shibatae. J. Mol. Biol., 296, 813–819. [DOI] [PubMed] [Google Scholar]
- Takada A., Robison,C., Goto,H., Sanchez,A., Murti,K.G., Whitt,M.A. and Kawaoka,Y. (1997) A system for functional analysis of Ebola virus glycoprotein. Proc. Natl Acad. Sci. USA, 94, 14764–14769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volchkov V. (1999) Processing of the Ebola virus glycoprotein. Curr. Top. Microbiol. Immunol., 235, 35–47. [DOI] [PubMed] [Google Scholar]
- Weissenhorn W., Carfi,A., Lee,K.-H., Skehel,J.J. and Wiley,D.C. (1998) Crystal structure of the Ebola virus membrane fusion subunit, GP2, from the envelope glycoprotein subunit ectodomain. Mol. Cell, 2, 605–616. [DOI] [PubMed] [Google Scholar]
- Wool-Lewis R.J. and Bates,P. (1998) Characterization of Ebola virus entry by using pseudotyped viruses: identification of receptor-deficient cell lines. J. Virol., 72, 3155–3160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z., Delgado,R., Xu,L., Todd,R.F., Nabel,E.G., Sanchez,A. and Nabel,G.J. (1998) Distinct cellular interactions of secreted and transmembrane Ebola virus glycoproteins. Science, 279, 1034–1037. [DOI] [PubMed] [Google Scholar]
- Yasuda J. and Hunter,E. (1998) A proline-rich motif (PPPY) in the Gag polyprotein of Mason–Pfizer monkey virus plays a maturation-independent role in virion release. J. Virol., 72, 4095–4103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeager M., Wilson-Kubalek,E.M., Weiner,S.G., Brown,P.O. and Rein,A. (1998) Supramolecular organization of immature and mature murine leukemia virus revealed by electron cryo-microscopy: implications for retroviral assembly mechanisms. Proc. Natl Acad. Sci. USA, 95, 7299–7304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaki S.R. and Peters,C.J. (1997) Viral hemorraghic fevers. In Connor,D.H., Schwartz,D.A., Manz,H.J. and Lack,E.E. (eds), Diagnostic Pathology of Infectious Diseases. Appleton and Lange, Stamford, CT, pp. 347–364. [Google Scholar]
- Zakowski J.J., Petri,W.A. and Wagner,R.R. (1981) Role of matrix protein in assembling the membrane of vesicular stomatitis virus: reconstitution of matrix protein with negatively charged phospholipid vesicles. Biochemistry, 20, 3902–3907. [DOI] [PubMed] [Google Scholar]
- Zhang J., Pekosz,A. and Lamb,R.A. (2000) Influenza virus assembly and lipid raft microdomains: a role for the cytoplasmic tails of the spike glycoproteins. J. Virol., 74, 4634–4644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou W. and Resh,M.D. (1996) Differential membrane binding of the human immunodeficiency virus type 1 matrix protein. J. Virol., 70, 8540–8548. [DOI] [PMC free article] [PubMed] [Google Scholar]