

Abstract
The β subunits of voltage-gated Ca2+ channels are best known for their roles in regulating surface expression and gating of voltage-gated Ca2+ channel α1 subunits. Recent evidence, however, indicates that these proteins have a variety of Ca2+ channel-independent functions. For example, on the molecular level, they regulate gene expression, and on the whole animal level, they regulate early cell movements in zebrafish development. In the present study, an alternatively spliced, truncated β4 subunit (β4c) is identified in the human brain and shown to be highly expressed in nuclei of vestibular neurons. Pull-down assays, nuclear magnetic resonance, and isothermal titration calorimetry demonstrate that the protein interacts with the chromo shadow domain (CSD) of heterochromatin protein 1γ. Site-directed mutagenesis reveals that the primary CSD interaction occurs through a β4c C-terminal PXVXL consensus motif, adding the β4c subunit to a growing PXVXL protein family with epigenetic responsibilities. These proteins have multiple nuclear functions, including transcription regulation (TIF1α) and nucleosome assembly (CAF1). An NMR-based two-site docking model of β4c in complex with dimerized CSD is presented. Possible roles for the interaction are discussed.
Keywords: Calcium Channels, Calorimetry, Chromatin, Neurobiology, Protein Structure, Alternative Splicing, Nuclear Magnetic Resonance
Introduction
Voltage-gated Ca2+ channels play critical roles in a number of cellular events including excitation-contraction coupling, neurotransmitter release, and Ca2+-dependent gene transcription (1). Voltage-gated Ca2+ channels are multisubunit proteins composed of a pore-forming α1 subunit, a two-part α2δ/γ integral membrane subunit, and a soluble cytosolic β subunit (2). The auxiliary β subunit functions both as a chaperone in trafficking the channel complex to the plasma membrane and as a major regulator of channel gating (opening and closing). These functions are thought to occur mainly through a specific interaction between the β subunit and the α1 interaction domain of the intracellular loop between α1 repeat motifs I and II (3). More recently, reports of Ca2+ channel-independent functions for β subunits have begun to emerge that appear to be centered in the nucleus. We showed recently, for example, that zebrafish embryos lacking β4 subunits failed to initiate epiboly and that the underlying mechanism for this phenomenon may be related to the high levels of β4 expression in yolk syncytial nuclei (4). Other studies have shown that β subunit functions in the nucleus may be cell type- and splice variant-specific (5).
Voltage-gated Ca2+ channel β4 subunit structures are composed of multiple domains. Sequence comparisons led to the initial discovery that Ca2+ channel β subunits are membrane-associated guanylate kinase proteins consisting of core Src homology 3 and guanylate kinase (GK)2 domains connected by a large variable loop (HOOK) (6). The core is flanked by highly variable N- and C-terminal domains. High resolution x-ray crystallographic studies further confirmed the structure of the β subunit core domains and determined that the interaction between the β and α1 subunits occurs via GK domain binding with high affinity to the α1 interaction domain of α1 (7–10).
The β subunits are encoded by four non-allelic genes, and multiple isoforms resulting from alternative splicing of each gene have been identified (11–13). Alternative splicing of different full-length β subunit isoforms not only generates variable coding sequences for N-terminal, HOOK, and C-terminal domains but also generates shorter variants with truncated Src homology 3 and/or GK domains. These variants result from splicing-induced frameshifts and subsequent early stop codons (12, 14). Interestingly, a previous yeast two-hybrid study (14) showed that a truncated β4 splice variant, β4c, isolated from chicken cochlear sensory epithelia (but not full-length β4a), interacted with nuclear protein heterochromatin protein 1 (HP1). The interaction was shown to attenuate the gene silencing activity by HP1 in vitro. This preliminary study did not identify the specific binding mechanism and concluded, based on RT-PCR, that the protein was not expressed in the rat brain. Consequently, there have been no subsequent studies of the β4c-HP1γ interaction. Our results show that alternative splicing of exon 9 of β4 RNA does indeed occur in the human brain, that the resultant human β4c protein is nearly identical to chicken β4c, and that β4c is almost exclusively found in the nuclei of vestibular and deep cerebellar neurons. Using multiple in vitro biochemical and biophysical techniques, we show that β4c interacts with the chromo shadow domains (CSDs) of HP1 dimers and that the interaction occurs via two β4c sites, the primary of which is a C-terminal PXVXL consensus sequence. A docking model of the complex formed by β4c and the CSD dimer (referred to as CSD throughout) is presented, and the possible mechanisms of gene regulation are discussed. Our results open up new and interesting research avenues that may be especially important for our understanding of the function of the vestibular system in health and disease.
EXPERIMENTAL PROCEDURES
Polymerase Chain Reaction, Subcloning, and Mutagenesis
PCR primers designed based on genomic human β4 sequence were used to amplify exon 9 splice variants. The four primers used for PCR reactions were as follows: exon 8 forward (5′-TATCATATGGCCACCCCTTACGATGTTGTACCGTCAAT-3′); exon 12 reverse (5′-CTGCTCGAGCTATTAATCAACCGCTGTAAAAC-3′); β4a forward (5′-TCACATATGATGTATGACAATTTGTACCTGCATGG-3′); and β4c reverse (5′-CTGCTCGAGTCAGCTGTCACTCTCGTTATTGAAATCCTCG-3′). NdeI and XhoI restriction sequences were added to the 5′-end of primers for ligation into the vector pET-15b. Human brain first-strand cDNA was purchased from Biochain (Hayward, CA), and 10 ng of cDNA was used as templates for PCR amplification. The thermal cycling program was as follows: 1 cycle of 98 °C for 30 s and 35 cycles of 98 °C for 10 s, 58 °C for 25 s, and 72 °C for 30 s. A final step of 72 °C for 5 min was used for the final extension. All PCR products were separated on 1% agarose gel containing ethidium bromide. For protein expression, PCR fragments were excised and purified from the gel, digested with NdeI and XhoI restriction enzymes, and ligated into the similarly digested hexahistidine pET-15b (Novagen) vector. Human β4c 38–184 (β4cΔ184), β4c 38–199 (β4cΔ199), and the CSD of human HP1γ (residues 113–183) were constructed in this manner. Site-directed mutagenesis was performed with the QuikChange (Stratagene) mutagenesis kit according to manufacturer's instructions. All constructs were verified by DNA sequencing.
Mouse Cerebellum/Brainstem Membranes
Snap-frozen cerebellum and brainstem from adult (>2 months) C57BL/6 mice were thawed and homogenized (10% w/v) in ice-cold 5 mm HEPES, pH 7.4, 0.32 m sucrose, 1 mm PMSF, 10 mm benzamidine, containing 1% protease inhibitor mixture (Calbiochem). The crude cerebellar/brainstem homogenate was centrifuged for 30 min at 900 × g to obtain the nuclear pellet. The resulting supernatant was centrifuged for 30 min at 25,000 × g to obtain the crude membrane pellet (P2), which was then resuspended in the same buffer. The resulting suspension was layered on a solution of homogenization buffer containing 1.6 m sucrose and centrifuged for 2 h at 60,000 × g to obtain the synaptosomal fraction (floating layer). This fraction was centrifuged for 1 h at 60,000 × g through 0.8 m sucrose in homogenization buffer to obtain the P4, synaptosome pellet.
Western Blot Analysis
Proteins from cerebellar/brainstem, nuclear, and synaptosome fractions were separated by SDS-PAGE and transferred at 30 V to Immun-BlotTM PVDF membranes (Bio-Rad) overnight at 4 °C. Membranes were blocked with TBST plus 5% milk (Blotto) and incubated with rabbit polyclonal primary antibody (anti-β4aA; 1:5000) overnight at 4 °C. Membranes were washed three times with Blotto prior to a 2-h incubation with alkaline phosphatase-conjugated goat anti-rabbit IgG (1:2000) at 26 °C. Membranes were washed four times with Blotto and two times with 150 mm NaCl, 50 mm Tris, pH 7.5, prior to development with 5-bromo-4-chloro-3-indolyl phosphate/nitro blue tetrazolium (Novagen) at 26 °C.
Immunocytochemistry
Adult (>2 months) C57BL/6 mice anesthetized with xylazine (4 mg/kg) and ketamine (21 mg/kg) were transcardially perfused with 10 ml (5 ml/min) of heparanized saline (0.9%), followed by 25 ml of 4% paraformaldehyde in 0.1 m phosphate buffer, pH 7.5. For light microscopy, brains were removed and post-fixed overnight in the same fixative at 4 °C. Brains were washed repeatedly in 0.1 m phosphate buffer, pH 7.5. Cerebellum with attached brainstem was isolated, embedded in 5% agarose, sectioned at 50 μm with a vibratome (Leica VT1000S, E. Licht, Denver, CO), and collected into phosphate-buffered saline (PBS), pH 7.5. Sections were subjected to the following antigen retrieval procedures prior to antibody treatments: a 30-min soak in 0.1 m glycine in PBS, followed by three washes in PBS at 4 °C; a 15-min soak in 0.5% sodium borohydride in PBS, followed by four washes in PBS at 4 °C; a 60-min soak in 0.05 sodium citrate, pH 8.6, at 26 °C; and a 30-min soak in 0.05 sodium citrate, pH 8.6, at 80 °C, followed by a 30-min slow cool to 24 °C and three washes in PBS. Cerebellar sections were blocked with PBS containing 5% normal goat serum, 0.5% Triton X-100, and 1% hydrogen peroxide at 4 °C. Sections were incubated for 36 h at 4 °C with a 1:1000 dilution of primary affinity-purified rabbit anti-β4a polyclonal antibody. Following incubation with primary antibody, sections were washed four times with PBS and then incubated with biotin-conjugated donkey anti-rabbit or donkey anti-mouse (1:2500; Jackson ImmunoResearch, West Grove, PA) in 1% normal goat serum, 0.5% Triton X-100 for 2 h at 24 °C. Immunoreactions were detected using a Vectastain Elite kit (Vector Laboratories, Burlingame, CA). Some sections were lightly counterstained with 1% basic fuschin to visualize nuclei.
Protein Expression and Purification
All proteins were expressed in Escherichia coli strain BL21 Rosetta (DE3) pLysS. A single colony of freshly transformed E. coli was inoculated into 20 ml of LB medium containing 50 μg/ml ampicillin. The overnight culture was further inoculated into 1 liter of LB or isotope (15N or 15N/13C)-containing medium (Cambridge Isotope Laboratories) supplemented with 50 μg/ml ampicillin. The cells were grown at 37 °C until the A600 reached 0.6 absorbance units. Isopropyl 1-thio-β-d-galactopyranoside was added to a final concentration of 0.2 mm, and protein expression was induced for 3 h (for unlabeled protein) or 4 h (for labeled protein) before the cells were harvested by centrifugation at 4500 rpm for 20 min. The harvested cells were resuspended in a binding buffer (50 mm Tris-HCl, 0.3 m NaCl, 1 mm DTT, pH 8.0) and lysed by sonication. The lysate was incubated with 50 units of DNase I (Bio-Rad) and 12.5 mm MgCl2 for 30 min and further centrifuged at 17,000 RPM for 30 min, and the supernatant was filtered and collected. The supernatant was then loaded onto a 15-ml Ni2+ resin column (Bio-Rad) pre-equilibrated with 100 ml of binding buffer. After washing with eight column volumes of binding buffer, the desired protein was eluted with increasing concentrations of imidazole (up to 300 mm). For Δ199 or Δ184 β4c proteins used for pull-down experiments, fractions eluted from the Ni2+ column were further purified by gel filtration using a Superdex 75HR column or a Sephacryl S-200 HR column on an AKTA FPLC (Amersham Biosciences) in 500 mm NaCl, 50 mm sodium phosphate, 1 mm DTT, pH 7.0. For CSD and β4c proteins used for other experiments, after the Ni2+ column, the His6 tag was removed by overnight thrombin cleavage (Amersham Biosciences) at 4 °C. The His6 tag-cleaved proteins were further purified by gel filtration using the same protocols. Protein purity was analyzed on SDS-polyacrylamide gels, and the protein samples were concentrated via centrifugal filtration. Protein concentrations were determined by the UV absorbance at 280 nm using predicted extinction coefficients.
Pull-down Assays
Purified His6-tagged β4cΔ199 was incubated for 1 h at 4 °C with fresh Ni2+ resin beads (Bio-Rad) and centrifuged at 2500 rpm for 5 min. The supernatant was discarded, and beads were washed with binding buffer (50 mm Tris, 0.3 m NaCl, pH 8.0). β4cΔ199-bound beads were incubated for 1 h in loading buffer (50 mm Tris-HCl, 0.3 m NaCl, 1 mm DTT, pH 8.0) with 20 μm untagged CSD in a 1.5-ml Eppendorf tube and subsequently centrifuged at 2500 rpm for 5 min. The supernatant was discarded, and beads were washed with 1 ml of binding buffer five times. Elution buffer (500 mm imidazole, 0.3 m NaCl, 50 mm Tris, pH 8.0) was added, and the sample was centrifuged at 2500 rpm for 5 min. Contents of the supernatant were analyzed using SDS-PAGE.
Isothermal Titration Calorimetry
Isothermal titration calorimetry (ITC) measurements were carried out at 298 K with a MicroCal (Northampton, MA) VP-ITC microcalorimeter. All proteins were dialyzed against the same buffer (50 mm sodium phosphate, 150 mm NaCl, pH 7.0, 2 mm DTT), and all buffers were degassed before experiments. For all measurements, 28 injections of 10 μl of CSD dimer were titrated into 1.4-ml solutions of either β4cΔ199 or β4cΔ184 proteins (or, for base-line correction, buffer alone) to a stoichiometric ratio of 2.1:1. Data were processed and analyzed with MicroCal Origin 7.0.
NMR Spectroscopy and Titrations
All NMR experiments were conducted at 298 K with a Varian Inova 500-MHz spectrometer equipped with a triple-resonance, z-gradient cryogenic probe. For the first set of titration experiments, NMR protein samples, 15N-labeled β4cΔ199 or β4cΔ184 and unlabeled CSD, were dissolved in buffer containing 50 mm HEPES, 100 mm NaCl, 50 mm Glu/Arg, 10% D2O, pH 7.0. Aliquots of 2 mm CSD dimer were titrated into 300 μm 15N β4cΔ199 or β4cΔ184 to a final molar ratio of 1.25; 1H-15N HSQC spectra were recorded following each aliquot. For CSD resonance assignments, 15N,13C-double-labeled CSD dimer was dissolved in 25 mm sodium phosphate, 50 mm NaCl, 10 mm DTT, 10% D2O, pH 7.4, to a final concentration of 0.8 mm. The 15N HSQC backbone amide assignments of the CSD were completed with standard three-dimensional heteronuclear NMR experiments (15), including CBCA(CO)NH, HNCACB, HNCA, and HN(CO)CA, and confirmed by a three-dimensional 15N HSQC nuclear Overhauser effect spectroscopy experiment. An HSQC of 15N-labeled CSD in complex with unlabeled β4cΔ199 in excess was also recorded to confirm the complex formation. For peptide binding experiments, a 17-mer peptide, PSMRPVVLVGPSLKGYE (>95% purity), derived from the C-terminal sequence of β4cΔ199 was synthesized by Invitrogen. The peptide was dissolved in sodium phosphate NMR buffer and used in NMR titrations without further purification (concentration was determined by an extinction coefficient, ϵ280 = 1490 m−1 cm−1). In peptide titration experiments, 1H-15N HSQC spectra were recorded with samples of 0.8 mm 15N-labeled CSD dimer into which 5-μl aliquots of unlabeled 17-mer peptide were titrated. The final molar ratio of peptide over CSD dimer was 2:1. The backbone amide assignments of 15N,13C-labeled CSD in complex with the β4c peptide were finished via the connectivity of Cα chemical shifts with a pair of three-dimensional experiments, HNCA and HN(CO)CA. Protein NMR data processing was performed using NMRPipe (16), and the spectra were analyzed in CCPNMR (17) and Sparky (18).
Structural Modeling and Docking
The structure coordinates of β4cΔ199 were modeled (Modeler 7.0) (19) based on the structure of the β4 core protein (Protein Data Bank entry 1VYV). Using default parameters of the program, the structure of human HP1γ CSD dimer was modeled based on mouse HP1β CSD in a complex with CAF1 peptide (Protein Data Bank entry 1S4Z) (20). HADDOCK (high ambiguity-driven docking) (21) was used for the docking of β4cΔ199 with CSD dimer. Based on homology modeling using the structure of the complex CAF1·CSD (Protein Data Bank entry 1S4Z) as a template, a set of distance restraints describing the contact between β4cΔ199 residues Pro187–Val191 and C-terminal residues of the CSD dimer were applied. Another group of CSD residues (Gly128, Ala129, and Trp173 for each monomer of the dimer) with large chemical shift changes in the peptide titration experiments were used for ambiguous restraints. These residues are surface-exposed in the free proteins and thought to be at the interface of the complex, thus showing large chemical shift changes upon complex formation. These residues can be restrained within 6–8 Å of any residue (ambiguous) on the interacting partner during docking (21). During the first cycle docking calculation, 2000 structures were generated, followed by a refinement step for 200 structures. These 200 structures were subsequently subjected to a cycle of simulated annealing during which the C-terminal of CSD and the HOOK region and residues Pro183–Arg186 of β4cΔ199 were allowed to be flexible to optimize the contact. Finally, all the structures were scored and clustered using a root mean square deviation value of 2.0 Å. The first model from the highest scored cluster is used to represent the structure model of the complex β4cΔ199·CSD.
RESULTS
Identification of a Truncated Splice Variant of the Human Ca2+ Channel β4 Subunit
A truncated Ca2+ channel β4 subunit spice variant, termed β4c, was previously shown to be expressed in chicken cochlea (14). This variant results from splicing out exon 9 from β4a subtype mRNA (Fig. 1). To determine whether the same alternative splicing event occurs in human RNA, we performed PCR with exon 9-flanking primers (exon 8F and exon 12R) using whole brain and cerebellum cDNA as templates. Two PCR products could clearly be identified (Fig. 1A, right). DNA sequencing confirmed the identities of both fragments: the bright 433 bp upper band is the amplification product of full-length β4; the lower 374 bp band is the product of an alternatively spliced β4 that lacks exon 9. We previously reported that alternative splicing of human β4 N-terminal exons creates β4a and β4b subtypes (11). Because the published chicken β4c sequence contained the β4a N terminus, we chose to amplify human full-length β4c using a forward primer that contained the start codon of β4a (in exon 2a) and a reverse primer that bridged exon 8 and exon 10 (Fig. 1B). We used cDNA from a variety of brain regions as template and found an especially high level of amplification from pons (Fig. 1B, right). The resulting 639-bp PCR product was ligated into pET-15b vector, sequenced, and subsequently expressed as protein in bacteria. The deduced amino acid sequence indicates that skipping exon 9 in human β4 RNA causes a frameshift and premature stop codon that creates a truncated β4c protein with 207 amino acids (∼23 kDa). Sequence alignment of human and chicken β4c shows that these two proteins are nearly identical (Fig. 1C). Both proteins contain the β4a N terminus, the Src homology 3 domain and HOOK sequence, a truncated GK domain, and additional C-terminal sequence resulting from the frameshift.
FIGURE 1.

Identification of a truncated splice variant of the human Ca2+ channel β4 subunit. A, diagram of exon structure and corresponding protein domains of the full-length human β4 subunit (486 amino acids). The arrows indicate two primer sites used to detect the alternative splicing (skipping) of exon 9 (left). SH3, Src homology 3 domain. Right, results of PCR amplification using brain and cerebellum cDNA as template. The larger amplified fragment (433 bp) corresponds to a β4 product containing exon 9, whereas the smaller fragment (374 bp) corresponds to a product without exon 9. B, diagram of splice variant mRNA and resulting truncated β4c protein (left). The arrows indicate primer sites used for amplification and cloning of the coding region of the truncated splicing variant, β4c (left). Note that the downstream primer spans exons 8 and 10. The deduced domains of the human β4c protein are also shown (207 amino acids). Right, a single 639 bp band was PCR-amplified using pons cDNA as template. The cDNA codes for full-length β4c. C, sequence alignment of human and chicken β4c proteins. Tubes above the sequence indicate α-helical secondary structure. Arrows, β-strands. *, divergent amino acids. Underlined sequence delineates the β4cΔ199 construct. ∨, β4cΔ199 truncation site used to create β4cΔ184.
Mammalian β4c Protein Is Located in the Nucleus
To determine whether or not β4c is found in the nucleus, we used a well characterized antibody directed toward the N terminus of β4a (22) in Western blot and immunohistochemistry experiments (Fig. 2). Fig. 2A shows that the β4a antibody labels two bands in crude cerebellar/brainstem membranes (CB), one corresponding to a 52-kDa protein and the other to a ∼20-kDa protein. The 52-kDa protein co-purifies with synaptosomes (Fig. 2, S) and represents full-length β4a; the 20 kDa band co-purifies with nuclei (N) and is probably β4c. The β4c protein is a highly acidic protein (pI = 5.16) and therefore highly negatively charged at the pH (8.3) of the SDS running buffer. This can account for the mobility shift and size anomaly (predicted size is 23 kDa) compared with the neutral charged standards used for SDS-PAGE. It is also possible that the protein undergoes post-translational modification. Fig. 2B shows comparisons of antibody and fuschin-stained cross-sections of mouse cerebellar cortex (molecular layer (ML), Purkinje cell layer (P), and granule cell layer (GCL)), dorsal cochlear nucleus (DCN), and medial vestibular nucleus (MVN). The fuschin staining provides an approximate count of the total number of nuclei present in each section. Interestingly, there is no antibody staining in the granule cell layer of the cerebellar cortex, which contains the highest number of cells, and no staining in Purkinje cells (Fig. 2B, a). The staining in the molecular layer corresponds to full-length β4a localized to synapses (22). Interestingly, there is sparse staining within the dorsal cochlear nucleus (Fig. 2B, c), where β4c was reported to be the only β4 subunit splice variant expressed in chicken (14). The medial vestibular nucleus contains the most antibody staining, where nearly half of all nuclei are labeled (the slightly higher number of fuschin-stained cells may be due to higher stain penetrability compared with the antibody), and the structures identified appear to be variably labeled nuclei (Fig. 2B, e). A closer view (e, inset) shows a staining pattern that is nearly identical to that revealed for HP1 in other cells (23) and suggests that β4c may be located along with HP1 in heterochromatin foci. Taken together, our results suggest the intriguing possibility that β4c plays an epigenetic role in regulating nuclear transcription in specific neuronal cell types. This hypothesis is supported by the fact that we saw β4a antibody staining in the nuclei of deep cerebellar neurons and that we could not amplify β4c from human cerebral cortex, hippocampus, amygdala, or thalamus (not shown).
FIGURE 2.

Two isoforms of the β4a subunit in mouse cerebellum and brainstem. A, Western blot showing that an affinity-purified β4a-specific polyclonal antibody labels a 52–55-kDa protein in a crude mouse cerebellar/brainstem homogenate (CB) as well as in sucrose step gradient-purified synaptosomes (S). The same antibody labels an 18–20-kDa protein in a slow speed pellet containing nuclei (N). Numbers to the left indicate molecular mass in kDa. B, distribution of β4a isoforms in cerebellar cortex and brainstem. Antibody labeling of 50-μm-thick sections was assessed by light microscopy (a, c, and e). Sections were counterstained with basic fuschin to label nuclei throughout the section (b, d, and f). Nuclear staining varies from none (a) to sparse (c) to significant (e), depending on the region of the cerebellum or brainstem. Punctate labeling of the β4a subunit can be seen in the molecular layer (ML) of the cerebellar cortex, whereas there is no specific labeling in the Purkinje (P) or granular cell (GC) layers (a). Fuschin staining confirms that the granular cell layer is rich in cell nuclei (b). c and d, sparse labeling of nuclei in the dorsal cochlear nucleus (DCN) relative to the number of fuschin-stained nuclei present. e and f, a significant number of nuclei labeled in the medial vestibular nucleus (MVN) relative to the number of fuschin-stained nuclei present. Staining of nuclei in the medial vestibular nucleus is quite variable. The predominant pattern within nuclei (inset) is suggestive of staining in heterochromatin foci.
The β4c C Terminus Contains a Chromo Shadow Domain Consensus Binding Motif
The CSD is highly conserved among all HP1 isoforms (Fig. 3A) and is the binding partner for a variety of proteins involved in transcription. These proteins have recently been classified into four groups according to their binding profiles (24). The majority of HP1-interacting proteins fall into group I and contain a consensus PXVXL motif. Phage display and NMR studies have revealed that the PXVXL pentapeptide motif contained in these proteins binds specifically to a surface formed by a CSD dimer (25). In the study by Hibino et al. (14), the yeast two-hybrid method was used to identify the region of β4c responsible for interacting with the HP1γ CSD. The results demonstrated that the 170–199 sequence of β4c contains critical residues for the interaction with CSD. On further examination of the sequence of β4c, we found a candidate CSD binding motif, PVVLV, located at residues 187–191 of the C terminus (Fig. 3B). Although the flanking sequences of PVVLV do not show high homology to other PXVXL motif-containing proteins, such as transcriptional intermediary factor 1 (TIF1α) and chromatin assembly factor 1 (CAF1), the PVVLV sequence itself contains the three most important hydrophobic residues essential for the CSD dimer interaction. The finding of a PXVXL motif at positions 187–191 is consistent with the general findings of Hibino et al. (14) that the interaction occurs in the last 29 amino acids of the β4c sequence. The discovery of the PXVXL sequence prompted us to design further experiments to characterize the physical interaction of β4c with CSD. We hypothesized that the C terminus of β4c binds to the CSD dimer in a manner similar to the CAF1 peptide (Fig. 3C).
FIGURE 3.

The β4c protein contains a PXVXL consensus HP1 binding motif. A, chromo shadow domains from different HP1 proteins. The proteins are as follows (from top to bottom): human HP1γ, mouse HP1γ, mouse HP1β, and chicken HP1γ. CSD residues shown previously to be involved in the interaction with PXVXL-containing proteins are boxed. B, a PXVXL motif is located in the C terminus of the β4c subunit. Alignment with other PXVXL motifs from other proteins suggests its role in binding to CSD. The PXVXL-containing proteins are human Ca2+ channel β4c (hβ4c_199), human transcriptional intermediary factor 1α (hTIF1α), mouse chromatin assembly factor 1 (mCAF1), and chicken Ca2+ channel β4c (cβ4c_199). C, NMR structure of a complex (Protein Data Bank entry 1S4Z) formed by mCAF1 peptide (shown as sticks) and CSD of mouse HP1β (shown as ribbons) (20, 39). SH3, Src homology 3 domain.
β4cΔ199 Interacts with CSD
Guided by the structure of the β4a A domain (26), we created an N-terminal truncation construct, β4c 38–199 (β4cΔ199; see underlined sequence in Fig. 1C), to characterize further the interaction of β4c with CSD. (This construct produced fewer interfering resonances in the NMR HSQC experiments shown below). For our first set of experiments, we used the pull-down method employing His6-β4cΔ199 (with PVVLV motif) and His6-Δ184 (without PVVLV motif) proteins bound to nickel resin to test for interactions with soluble CSD. We surmised based on sequence examination that truncation of 15 C-terminal β4cΔ199 residues was unlikely to have an overall effect on the core structure. This was confirmed by NMR experiments that show similar and well dispersed resonances for both β4cΔ184 and β4cΔ199 (see Fig. 5). The proteins used in the pull-down experiments are shown in the first three lanes in Fig. 4A (Input), whereas lanes 4 and 5 show, respectively, that soluble CSD does not bind to β4cΔ184 beads but does bind to β4cΔ199 beads. This indicates that the N terminus is not crucial for the interaction but that the C terminus is critical. The specific interaction between β4cΔ199 and CSD was further confirmed and characterized by ITC measurements. Fig. 4B (top) shows the raw data for titration of β4cΔ199 against soluble CSD and indicates that the interaction is exothermic. Fig. 4B (bottom) reveals that the binding stoichiometry between β4cΔ199 and the CSD dimer is 1:1 (n = 1.04), and the binding affinity of the interaction is 700 nm under our experimental conditions. Furthermore, no detectable interaction between Δ184 and CSD was found by ITC, indicating that the 15 C-terminal residues of β4cΔ199 do contain specific determinants for the interaction of β4c with the CSD dimer.
FIGURE 5.

NMR evidence for a specific interaction between β4cΔ199 and CSD. A, two identical overlaid HSQC spectra of 15N-labeled Δ184 in the absence (black, below) and presence (red, above) of unlabeled CSD. This is indicative of no interaction. B, two overlaid HSQC spectra of 15N-labeled β4cΔ199 in the absence (black, below) and presence (red, above) of unlabeled CSD. Note that the spectra are not identical and that many of the cross-peaks have undergone broadening or chemical shift changes that are indicative of complex formation. Residues Val189 (V189), Leu190 (L190), Gly192 (G192), and Gly197 (G197) are labeled as examples of resonances undergoing these changes.
FIGURE 4.

A CSD dimer interacts specifically with β4cΔ199. A, SDS-polyacrylamide gel showing results of pull-down experiments. Lanes 1–3 contain individual proteins used in the experiments (Input): His6-β4cΔ184; His6-β4cΔ199; and untagged CSD, respectively. Lane 4 shows that when combined with β4cΔ184 beads, CSD is not present in the pellet. Lane 5 shows that when combined with β4cΔ199 beads, soluble CSD does spin down with the pellet. B, isothermal titration calorimetry. Top, raw data after base-line correction shows saturating binding of CSD when titrated into β4cΔ199 solution. Bottom, integrated data corrected for the heat of dilution of the CSD dimer. Open and filled rectangles, β4cΔ184 and β4cΔ199 data, respectively. The solid line in the bottom panel represents the best fit to a one-site binding model of the interaction of the CSD dimer with β4cΔ199.
Characterization of the β4c-CSD Interaction by NMR Titration
NMR provides a highly sensitive tool for studying protein-protein interactions. To further confirm that CSD binding is specific for β4cΔ199, we prepared uniformly 15N-labeled β4cΔ184 and β4cΔ199 proteins for NMR titration experiments. The large chemical shift dispersion of the 1H dimension in the 1H-15N HSQC spectra of both β4cΔ184 and β4cΔ199 indicates that the two proteins are well folded (Fig. 5). Comparison of β4cΔ184 sequence (see Fig. 1C) with the solved structure of the β4 core suggests that β4cΔ184 consists of a long N-terminal helix and a split Src homology 3 domain in which a HOOK domain is located between the second helix and the fifth β-strand of Src homology 3. To confirm the β4c-CSD ITC data, CSD protein was titrated into solutions containing either 15N-labeled β4cΔ184 or 15N-labeled β4cΔ199. Fig. 5A shows that neither chemical shift changes nor line broadening was observed for 15N-labeled Δ184 HSQC resonances when unlabeled CSD was titrated up to a 1:1 ratio. This confirms by a very sensitive methodology that β4cΔ184 does not interact with CSD. The addition of CSD protein to 15N-labeled β4cΔ199, however, led to the broadening of many resonances along with chemical shift changes for a number of cross-peaks in the HSQC spectrum (residues Val189, Leu190, Gly192, and Gly197 are identified as examples in Fig. 5B). This result supports the general conclusion that the two proteins associate to form a complex. For residues showing chemical shift changes, only two sets of resonances are visible during the titration, suggesting that the interaction occurs by slow exchange on the chemical shift scale. Consistent with the ITC results, the NMR titration experiments confirmed that the stoichiometry of the β4cΔ199-CSD dimer interaction is 1:1. Moreover, strong resonances of the additional C-terminal backbone amides of β4cΔ199 suggest that this region is likely to form a random coil conformation in the free form. It is these resonances that experience either large shifts or line broadening upon complex formation. Thus, a comparative NMR study further identifies the β4cΔ199 C terminus as the site containing critical residues for the specific interaction with CSD.
Site-directed Mutagenesis Confirms the PVVLV Binding Motif
To pinpoint the critical residues involved in the β4c-CSD interaction, we next turned to site-directed mutagenesis of the PVVLV motif. As mentioned previously, a number of studies indicate that CSD-interacting pentapeptides contain a predominant proline at position 1, a valine at position 3, and two hydrophobic residues at positions 4 and 5. Mutations of the highly conserved residues at positions 1 and 3 should therefore compromise the interaction of the peptide with CSD. To test this, we created two additional His6-β4cΔ199 constructs: one with a single V189A mutation and the other with double P187A and V189A mutations. The proteins used in the pull-down experiments are shown in the first three lanes in Fig. 6A (Input), whereas lanes 4 and 5 show, respectively, that soluble CSD does bind to β4cΔ199 beads but does not bind to β4cΔ199-P187A/V189A beads. More quantitative ITC measurements (Fig. 6B) indicate that the single mutation V189A reduces the binding affinity of β4cΔ199 for CSD 5-fold to 3.9 μm. Moreover, as with the pull-down experiments, there was no evidence that the P189A/V189A double mutant interacted with CSD (flat isotherm in Fig. 6B, bottom).
FIGURE 6.
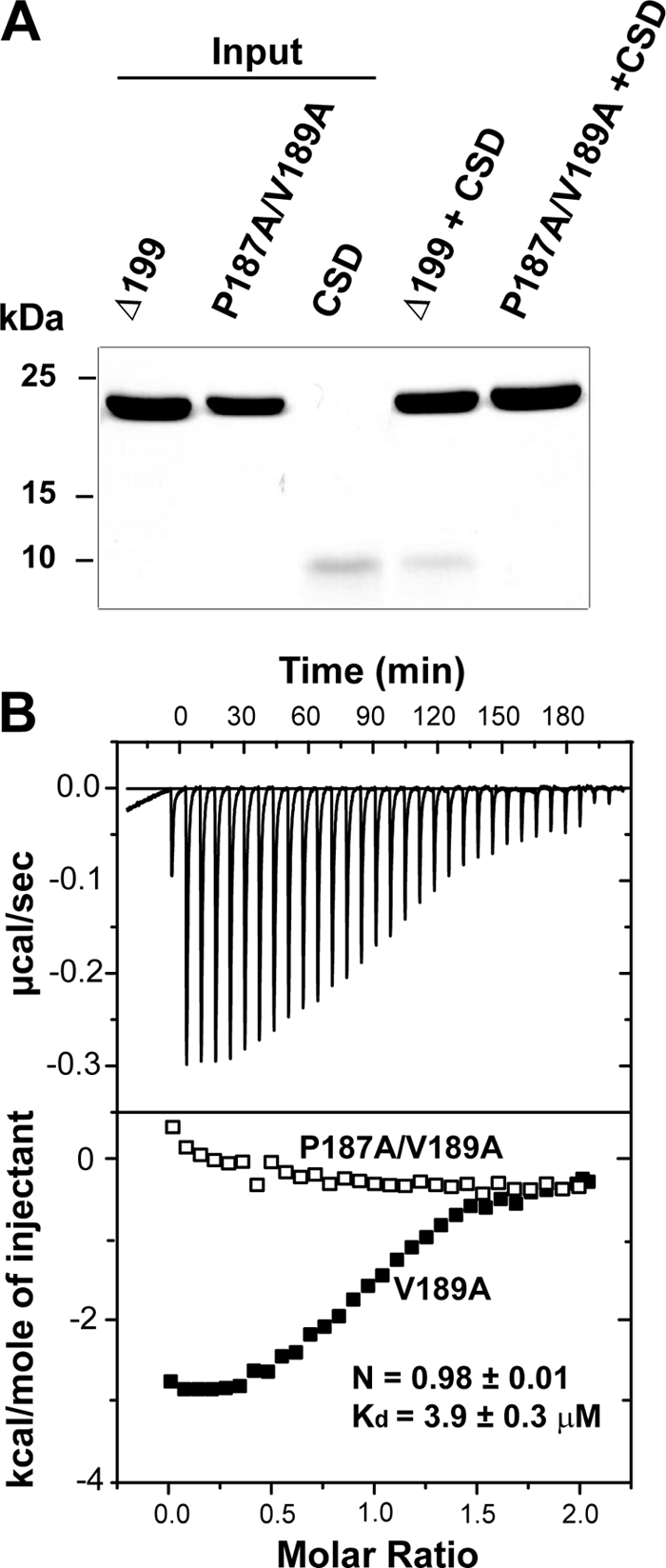
Mutations in the PXVXL motif eliminate β4cΔ199-CSD dimer interaction. A, SDS-polyacrylamide gel showing results of pull-down experiments. Lanes 1–3 contain individual proteins used in the experiments: His6- β4cΔ199; His6-β4cΔ199/P187A/V189A; and untagged CSD, respectively (Input). Lane 4 shows that when combined with β4cΔ199 beads, CSD is present in the pellet. Lane 5 shows that when combined with β4cΔ199/P187A/V189A beads, soluble CSD does not spin down with the pellet. B, isothermal titration calorimetry. Top, raw data after base-line correction shows saturating binding of CSD when titrated into β4cΔ199/V189A solution. Bottom, integrated data corrected for the heat of dilution of the CSD dimer. Open and filled rectangles, β4cΔ199/P187A/V189A and β4cΔ199/V189A data, respectively. Binding data were derived from the best fit to a one-site binding model of the interaction of the CSD dimer with β4cΔ199/V189.
NMR Mapping of CSD Binding Site
A 17-mer peptide (PSMRPVVLVGPSLKGYE) derived from β4cΔ199 C-terminal sequence was used to map the CSD binding region using NMR. Fig. 7A shows that titration of the unlabeled peptide into a solution containing uniformly 15N-labeled CSD dimer resulted in chemical shift perturbations of many residues relative to their positions in the absence of the peptide. At least 20 CSD residues show asymmetrical shifts in the HSQC spectrum, resulting in two cross-peaks for each of those residues in the final protein·peptide complex. This is consistent with what is expected if a single peptide binds to an interface formed by two subunits of a dimer and subsequently induces asymmetry within the binding region of each monomer. Interestingly, many of the residues that show two cross-peaks in the peptide-bound state display HSQC resonances of different intensities; one of them is more broadened than the other (see, for example, Gly128, Asp131, and Thr130 in Fig. 7A). This is indicative of the same residue in each of the CSD monomers having different dynamic behavior within the peptide-bound complex. Also interesting, the side chain indole amide of the two ϵTrp174 residues shows three cross-peaks in the complex state, one of which has a stronger intensity than the other two. This also suggests that in the final complex, the side chain of Trp174 for one monomer may exist in a single conformation, whereas the side chain of the other monomer adopts two conformations in a slow exchange. Fig. 7B shows a detailed comparison of two HP1 CSD dimer peptide-binding maps, one for the β4c peptide (left; HP1γ) and the other for mouse CAF1 (right; HP1β from Ref. 20). In both cases, the peptide-binding region on CSD is composed of CSD C-terminal residues, the end of the second helix, and the first β-strand (Fig. 7B). Although a few residues of the second β-strands also show chemical shift changes, these effects are probably from a secondary binding effect.
FIGURE 7.

Chemical shift mapping of β4c peptide-binding region on HP1γ CSD. A, two overlaid HSQC spectra of 15N HP1γ CSD in the absence (black, above) and presence (red, below) of a 17-mer PXVXL-containing peptide derived from β4c. Residues experiencing large chemical shifts are labeled with blue lines. B, comparison of β4c binding to HP1γ CSD and CAF1 binding to HP1β CSD dimers. Residues experiencing large chemical shift changes in the complex are colored in red. Residues without assignment are shown in gray. The remainder are green. Left, mapping of the β4c peptide-binding region on the modeled structure of human HP1γ CSD dimer (see “Experimental Procedures”). The CSD dimer structure is represented in ribbons. Right, previously published mapping of mouse CAF1 peptide-binding region on the CSD dimer of mouse HP1β (39).
Structural Model of β4cΔ199 Binding to CSD Dimer
Our combined results from pull-down, ITC, NMR, and mutagenesis experiments indicate that the β4c C-terminal PXVXL motif binds to CSD in a manner similar to other CSD-interacting proteins. Thus, we used combined homology modeling and docking methods to define the complex structure of β4cΔ199·CSD. The β4cΔ199 structure was based on the crystal structure of the β4 subunit core domains (Protein Data Bank entry 1VYV) (7); the large disordered loop of the HOOK domain was added in the modeled structure. The human CSD dimer was modeled based on Protein Data Bank entry 1S4Z (20). A set of distance restraints (see “Experimental Procedures”) was used to define the specific interaction between residues on the PVVLV sequence and residues of the CSD dimer. Another set of ambiguous distance restraints were derived from the residues showing large chemical shift perturbations shown in Fig. 7A. In the final model of the complex, the two proteins make contact primarily through the PVVLV sequence (Fig. 8A). Apart from this region, the model also predicts that C-terminal residues of one monomer in the CSD dimer make contact with a region of the β4cΔ199 HOOK domain. This additional contact is shown clearly by the chemical shift perturbation experiments in which unlabeled β4cΔ199 was titrated into 15N-labeled CSD. Upon β4cΔ199 binding, the C-terminal residues 179E-183Q of one monomer in the CSD dimer show significant chemical shift changes, whereas the same five residues of the other monomer show minimum shifts relative to their HSQC positions in the absence of β4cΔ199 (Fig. 8B). These interactions must be of very low affinity given that they are not detected in ITC and NMR experiments using β4cΔ184 as the CSD binding partner.
FIGURE 8.

Two sites of β4cΔ199 interaction with CSD based on modeling and NMR. A, HADDOCK (high ambiguity-driven docking) (21) model of the complex of β4cΔ199 (purple ribbon) with the CSD dimer (green ribbons). The β4cΔ199 C terminus with the PXVXL binding motif is shown in orange. B, view of part of the overlaid spectra of 15N-labeled CSD in the absence (black, below) and in the presence (red, above) of β4cΔ199. The highlighted CSD residues undergoing chemical shifts (Glu179–Gln183; 179E–183Q) are from one monomer of the CSD dimer in complex with β4cΔ199.
DISCUSSION
Alternative Splicing of the Ca2+ Channel β Subunit
Alternative splicing is an adaptive mechanism that evolved to enhance protein diversity from a limited number of eukaryotic genes (27). Only four genes code for voltage-gated Ca2+ channel β subunits; however, multiple splice variants exist that allow for broad functional diversity. Splicing leads to heterogeneity in β subunit structure, subcellular localization, and its ability to regulate electrophysiological properties (12). This heterogeneity is well correlated with the finely tuned Ca2+ channel gating properties found in specialized synapses and cell types (11). Although alternatively spliced N-terminal, HOOK, and C-terminal domains predominate, recent studies have also shown that the highly conserved GK domain can undergo alternative splicing (14, 28). Skipping of GK exon 9 creates a premature stop codon and a truncated β subunit that can no longer bind to α1 subunits. This form of splicing was first recognized in chicken cochlear RNA, and the resulting protein, β4c, was found to regulate gene expression in vitro (14). Our current report confirms and significantly extends the preliminary findings of Hibino et al. (14), and we consider three of their results highly relevant to our study: 1) truncated β4c, but not full-length β4a, dramatically attenuates the gene-silencing activity of HP1γ in a chloramphenicol transferase assay; 2) β4c co-localizes with HP1γ in isolated nuclei from cochlear hair cells; and 3) binding to HP1γ is required for β4c to enter the nucleus. We have confirmed in intact tissue that β4c is found in vestibular nuclei and used NMR spectroscopy to identify the structural determinants for β4c binding to HP1γ. Interestingly, a nuclear localization sequence was recently identified within the N terminus of full-length β4b that was shown to be essential for nuclear targeting; alternatively spliced full-length β4a lacking the sequence was not able to enter the nucleus (5).
β4c Interacts with HP1γ via a PXVXL Consensus Sequence
In examining the β4c C-terminal sequence, we found a candidate PXVXL consensus site that we hypothesized would contain the key residues for binding. The PXVXL pentapeptide sequence was first identified in a phage display study and was subsequently found to be present in many HP1-associated proteins (25). Of residues in the PXVXL sequence, the first proline and the third valine were found to be the most highly conserved (this is why we chose to mutate these residues in confirming the function of the β4c PXVXL motif); the fifth hydrophobic residue could be isoleucine or leucine but not valine (25). NMR structural studies defined a unique mode of sequence recognition by the CSD dimer of HP1 in which the PXVXL peptide binds to a groove formed by the CSD dimer and forms a sandwiched β-sheet (20). In a more recent x-ray crystallographic study, a broader definition of PXVXL consensus was extended to ΦX(V/P)X(L/M/V) (where Φ represents a hydrophobic residue) (29). We found the sequence, PVVLV, to be conserved in all GK domains of four β subunits; however, in full-length β subunits, PVVLV is part of a β-strand that is buried in the GK domain. It is therefore unavailable for interacting with other proteins. Alternative splicing of exon 9 creates the shorter variant, β4c, which lacks a complete GK domain and makes the PVVLV sequence available for interaction with CSD. This explains why we found that neither expression rate nor gating parameters were affected after co-injecting β4c199 cRNA with α1A and α2δ in Xenopus oocytes (data not shown) because the truncation completely removes the α1 subunit interaction domain (7, 8). Interestingly, Hibino et al. (14) found that β4c could not compete with TIF1β for binding to CSD. This is surprising, given the fact that TIF1β binds to CSD via the same mechanism. It could be that TIF1β binds to CSD with a higher affinity than the Kd of 0.7 μm we found for β4c; however, to our knowledge this information has not been published. Moreover, most HP1 binding studies have been performed with peptides; therefore, direct comparisons to other studies are difficult. We believe that our experiments with β4cΔ199 protein are the first to show binding to an additional site on CSD outside the PXVXL binding groove.
Possible Functional Consequences of β4c Binding to HP1 Proteins
Heterochromatin is a ubiquitous and dynamic component of the eukaryotic chromosome that functions in epigenetic gene repression and silencing (30). Members of the HP1 family serve as adaptor proteins and are critical to the process of heterochromatin formation (31). Their chromo and chromo shadow domains allow them to serve dual roles as ligands (HPs bind to methylated histone H3) and receptors (for β4c and other effectors), respectively. Although most studies of HP1s have focused primarily on their role in heterochromatin formation and long term gene silencing, it is becoming increasingly more evident that they also play critical roles in telomere maintenance and capping, centromere organization, and even positive expression of euchromatic genes. Thus, it is likely that β4c binding to HP1 might have several outcomes. One exciting possibility is that β4c may interact with HP1 to turn on a set of genes (perhaps involved in Ca2+ regulation) located at specific loci of euchromatin. This could happen in one of two ways; the interaction could attenuate the repressive effect of HP1 on specific genes, or alternatively, the interaction could promote the HP1 binding-specific region of euchromatin, where it has positive effects on gene expression. A detailed comparative study using transcriptional profiling will be required to answer these questions. Perhaps the most interesting finding of our present study is that β4c appears to localize to heterochromatin structures in nuclei of vestibular and deep cerebellar neurons but not in other regions of the brain, such as the granular layer of the cerebellar cortex. It will be especially interesting to determine what specific function is served by the β4c-HP1 interaction in the context of the vestibular system in whole animals.
Possible Roles for β4c in the Vestibular Nucleus
Vestibular nucleus neurons play a central role in the circuitry that mediates the vestibulo-ocular reflex, a relatively simple motor reflex of early phylogenetic origin (reviewed in Ref. 32). The circuit ensures that when the head moves in one direction, the eyes move in the opposite direction. This keeps visual images centered on the retina and avoids blurred vision, an adaptation of obvious evolutionary significance. Vestibular neurons are also important for long term vestibulo-ocular reflex memory and display a unique mechanism for intrinsic plasticity that ultimately results in firing rate potentiation (reviewed in Ref. 33). Head movements activate cerebellar granule cells whose parallel fibers form synapses on Purkinje cell dendrites. Interestingly, Ca2+ currents at these excitatory synapses are largely under the control of full-length β4 subunits (22). Purkinje cell axons descend to the brainstem, where, when activated, they have inhibitory effects on vestibular synapses. Inhibition decreases CAMKII activity and induces firing rate potentiation of vestibular neurons, in part through mechanisms that regulate voltage-dependent Ca2+ channels and Ca2+-activated potassium channels (34). Thus, as with other types of long term potentiation, firing rate potentiation is highly dependent on cytosolic Ca2+ concentrations. These effects are long lasting and are important not only for motor memory but for recovery from vestibular damage (35). An intriguing possibility might be that β4c, a splice variant of a Ca2+ channel β subunit, by interacting with HP1, contributes to firing rate potentiation and motor memory in the vestibular nuclei by activating genes that are involved in regulating Ca2+ homeostasis. Moreover, the importance of the β4c-HP1 interaction in normal motor/vestibular function may be embodied in the lethargic mouse (lh) that expresses a β4 truncation mutant that terminates 45 amino acids upstream from the PVVLV sequence (36). The motor syndrome in these mice consists of base-line mild ataxia with intermittent attacks of severe dyskinesia that arise subcortically (37). This syndrome resembles an inheritable form of paroxysmal kinesigenic dyskinesia that occurs at a rate of 1:150,000 in people; the underlying pathophysiology of the neurological disorder is not known (37, 38).
Acknowledgments
We thank Dr. Robert Weiss for advice on the manuscript, Dr. Stuart Tobet for the use of immunohistochemistry equipment and protocols, and Andy Hoyong Yi for outstanding technical assistance.
This work was supported, in whole or in part, by National Institutes of Health Grant NS42600 (to W. A. H.). This work was also supported by National Science Foundation Grant IOS 0719242 (to W. A. H.).
- GK
- guanylate kinase
- CSD
- chromo shadow domain
- ITC
- isothermal titration calorimetry
- HSQC
- heteronuclear single quantum correlation.
REFERENCES
- 1. Dolphin A. C. (2003) J. Bioenerg. Biomembr. 35, 599–620 [DOI] [PubMed] [Google Scholar]
- 2. Dolphin A. C. (2009) Curr. Opin. Neurobiol. 19, 237–244 [DOI] [PubMed] [Google Scholar]
- 3. De Waard M., Pragnell M., Campbell K. P. (1994) Neuron 13, 495–503 [DOI] [PubMed] [Google Scholar]
- 4. Ebert A. M., McAnelly C. A., Srinivasan A., Linker J. L., Horne W. A., Garrity D. M. (2008) Proc. Natl. Acad. Sci. U.S.A. 105, 198–203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Subramanyam P., Obermair G. J., Baumgartner S., Gebhart M., Striessnig J., Kaufmann W. A., Geley S., Flucher B. E. (2009) Channels 3, 343–355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hanlon M. R., Berrow N. S., Dolphin A. C., Wallace B. A. (1999) FEBS Lett. 445, 366–370 [DOI] [PubMed] [Google Scholar]
- 7. Chen Y. H., Li M. H., Zhang Y., He L. L., Yamada Y., Fitzmaurice A., Shen Y., Zhang H., Tong L., Yang J. (2004) Nature 429, 675–680 [DOI] [PubMed] [Google Scholar]
- 8. Opatowsky Y., Chen C. C., Campbell K. P., Hirsch J. A. (2004) Neuron 42, 387–399 [DOI] [PubMed] [Google Scholar]
- 9. Van Petegem F., Clark K. A., Chatelain F. C., Minor D. L., Jr. (2004) Nature 429, 671–675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Van Petegem F., Duderstadt K. E., Clark K. A., Wang M., Minor D. L., Jr. (2008) Structure 16, 280–294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Helton T. D., Horne W. A. (2002) J. Neurosci. 22, 1573–1582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Foell J. D., Balijepalli R. C., Delisle B. P., Yunker A. M., Robia S. L., Walker J. W., McEnery M. W., January C. T., Kamp T. J. (2004) Physiol. Genomics 17, 183–200 [DOI] [PubMed] [Google Scholar]
- 13. Ebert A. M., McAnelly C. A., Handschy A. V., Mueller R. L., Horne W. A., Garrity D. M. (2008) Physiol. Genomics 35, 133–144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hibino H., Pironkova R., Onwumere O., Rousset M., Charnet P., Hudspeth A. J., Lesage F. (2003) Proc. Natl. Acad. Sci. U.S.A. 100, 307–312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Cavanagh J., Fairbrother W. J., Palmer A. G., III, Skelton N. J. (2006) Protein NMR Spectroscopy: Principles and Practice, 2nd Ed., pp. 613–654, Academic Press, Inc., New York [Google Scholar]
- 16. Delaglio F., Grzesiek S., Vuister G. W., Zhu G., Pfeifer J., Bax A. (1995) J. Biomol. NMR 6, 277–293 [DOI] [PubMed] [Google Scholar]
- 17. Vranken W. F., Boucher W., Stevens T. J., Fogh R. H., Pajon A., Llinas M., Ulrich E. L., Markley J. L., Ionides J., Laue E. D. (2005) Proteins 59, 687–696 [DOI] [PubMed] [Google Scholar]
- 18. Kneller D. G., Kuntz I. D. (1993) J. Cell. Biochem. 53, 254 [Google Scholar]
- 19. Eswar N., John B., Mirkovic N., Fiser A., Ilyin V. A., Pieper U., Stuart A. C., Marti-Renom M. A., Madhusudhan M. S., Yerkovich B., Sali A. (2003) Nucleic Acids Res. 31, 3375–3380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Thiru A., Nietlispach D., Mott H. R., Okuwaki M., Lyon D., Nielsen P. R., Hirshberg M., Verreault A., Murzina N. V., Laue E. D. (2004) EMBO J. 23, 489–499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Dominguez C., Boelens R., Bonvin A. M. J. J. (2003) J. Am. Chem. Soc. 125, 1731–1737 [DOI] [PubMed] [Google Scholar]
- 22. Vendel A. C., Terry M. D., Striegel A. R., Iverson N. M., Leuranguer V., Rithner C. D., Lyons B. A., Pickard G. E., Tobet S. A., Horne W. A. (2006) J. Neurosci. 26, 2635–2644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Festenstein R., Pagakis S. N., Hiragami K., Lyon D., Verreault A., Sekkali B., Kioussis D. (2003) Science 299, 719–721 [DOI] [PubMed] [Google Scholar]
- 24. Nozawa R. S., Nagao K., Masuda H. T., Iwasaki O., Hirota T., Nozaki N., Kimura H., Obuse C. (2010) Nat. Cell Biol. 12, 719–727 [DOI] [PubMed] [Google Scholar]
- 25. Smothers J. F., Henikoff S. (2000) Curr. Biol. 10, 27–30 [DOI] [PubMed] [Google Scholar]
- 26. Vendel A. C., Rithner C. D., Lyons B. A., Horne W. A. (2006) Protein Sci. 15, 378–383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gray A. C., Raingo J., Lipscombe D. (2007) Cell Calcium 42, 409–417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kobayashi T., Yamada Y., Fukao M., Shiratori K., Tsutsuura M., Tanimoto K., Tohse N. (2007) Biochem. Biophys. Res. Commun. 360, 679–683 [DOI] [PubMed] [Google Scholar]
- 29. Huang Y., Myers M. P., Xu R. M. (2006) Structure 14, 703–712 [DOI] [PubMed] [Google Scholar]
- 30. Grewal S. I., Jia S. T. (2007) Nat. Rev. Genet. 8, 35–46 [DOI] [PubMed] [Google Scholar]
- 31. Fanti L., Pimpinelli S. (2008) Curr. Opin. Genet. Dev. 18, 169–174 [DOI] [PubMed] [Google Scholar]
- 32. Broussard D. M., Kassardjian C. D. (2004) Learning Memory 11, 127–136 [DOI] [PubMed] [Google Scholar]
- 33. Gittis A. H., du Lac S. (2006) Curr. Opin. Neurobiol. 16, 385–390 [DOI] [PubMed] [Google Scholar]
- 34. Nelson A. B., Gittis A. H., du Lac S. (2005) Neuron 46, 623–631 [DOI] [PubMed] [Google Scholar]
- 35. Darlington C. L., Dutia M. B., Smith P. F. (2002) Eur. J. Neurosci. 15, 1719–1727 [DOI] [PubMed] [Google Scholar]
- 36. Burgess D. L., Jones J. M., Meisler M. H., Noebels J. L. (1997) Cell 88, 385–392 [DOI] [PubMed] [Google Scholar]
- 37. van Rootselaar A. F., van Westrum S. S., Velis D. N., Tijssen M. A. (2009) Pract. Neurol. 9, 102–109 [DOI] [PubMed] [Google Scholar]
- 38. Khan Z., Jinnah H. A. (2002) J. Neurosci. 22, 8193–8200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Brasher S. V., Smith B. O., Fogh R. H., Nietlispach D., Thiru A., Nielsen P. R., Broadhurst R. W., Ball L. J., Murzina N. V., Laue E. D. (2000) EMBO J. 19, 1587–1597 [DOI] [PMC free article] [PubMed] [Google Scholar]