

Abstract
Gene-selective epigenetic reprogramming and shifts in cellular bioenergetics develop when Toll-like receptors (TLR) recognize and respond to systemic life-threatening infections. Using a human monocyte cell model of endotoxin tolerance and human leukocytes from acute systemic inflammation with sepsis, we report that energy sensor sirtuin 1 (SIRT1) coordinates the epigenetic and bioenergy shifts. After TLR4 signaling, SIRT1 rapidly accumulated at the promoters of TNF-α and IL-1β, but not IκBα; SIRT1 promoter binding was dependent on its co-factor, NAD+. During this initial process, SIRT1 deacetylated RelA/p65 lysine 310 and nucleosomal histone H4 lysine 16 to promote termination of NFκB-dependent transcription. SIRT1 then remained promoter bound and recruited de novo induced RelB, which directed assembly of the mature transcription repressor complex that generates endotoxin tolerance. SIRT1 also promoted de novo expression of RelB. During sustained endotoxin tolerance, nicotinamide phosphoribosyltransferase (Nampt), the rate-limiting enzyme for endogenous production of NAD+, and SIRT1 expression increased. The elevation of SIRT1 required protein stabilization and enhanced translation. To support the coordination of bioenergetics in human sepsis, we observed elevated NAD+ levels concomitant with SIRT1 and RelB accumulation at the TNF-α promoter of endotoxin tolerant sepsis blood leukocytes. We conclude that TLR4 stimulation and human sepsis activate pathways that couple NAD+ and its sensor SIRT1 with epigenetic reprogramming.
Keywords: Chromatin Immunoprecipitation (ChIP), Chromatin Remodeling, Inflammation, Innate Immunity, Lipopolysaccharide (LPS), Macrophage, NAD, Toll-like Receptors (TLR), SIRT1, Endotoxin Tolerance
Introduction
Two cellular processes predictably accompany TLR-mediated acute systemic inflammation caused by sepsis, a highly destructive and often lethal process. The first process occurs when epigenetic alterations reprogram distinct functional sets of genes to both activate and repress transcription of hundreds of genes (1, 2); this transcriptome reprogramming generates the phenomenon known as endotoxin tolerance (3). Endotoxin tolerance requires TLR3 receptor signaling of NFκB master regulator, which first induces and then represses rapid response and potentially autotoxic proinflammatory TNF-α and IL-1β. To control the initial recognition and response phases induced by TLR, gene-specific reprogramming selectively modifies chromatin structure and shifts nucleosomes on responsive euchromatin to form silent heterochromatin at acute proinflammatory genes; in contrast, genes encoding anti-inflammatory and antimicrobial mediators maintain responsive euchromatin (4, 5). This gene set-selective reprogramming generates a clinically relevant phenotypic transition from the hyperinflammatory to the hypoinflammatory endotoxin tolerant state, which may last hours, days, or weeks, depending on the strength of the initial TLR response (4, 5). The physiologic importance of endotoxin tolerance is still incompletely understood, but likely reflects an attempt to recover homeostasis (3).
Others and we (6–9) reported how temporal transitions in epigenetic programming alter the course of acute inflammation. NFκB master transcription switch directs a phase-shift between initiating acute inflammation and developing endotoxin tolerance. During this sequel, a RelA/p65-dependent feed-forward loop induces de novo production of NFκB factor RelB, which directly recruits G9a histone H3K9 methyltransferase and assembles a histone and DNA multicomponent transcription repressor complex (6, 10–12). The repressor complex converts structurally responsive euchromatin of acute proinflammatory genes to condense and reversibly silence facultative heterochromatin, which is maintained until sepsis is resolved. In contrast, the epigenetic shift generated by the RelB feed-forward loop also persistently activates euchromatin of genes encoding anti-inflammatory and antimicrobial mediators (13). Thus, RelB in innate immunity phagocytes acts as an inducible dual transcription regulator.
A second predictable feature of sepsis is a shift in cellular bioenergetics affecting many tissues and cell-types: phagocytes, hepatocytes, and muscle cells (14, 15). During this process, TLR-dependent signaling first increases ATP production by mitochondria to support the high energy requirements of phagocytosis. As a by-product, reactive oxygen species injure structural and functional cell constituents and activate cell signaling. Within hours after the initial TLR signaling, mitochondria are reprogrammed to uncouple oxidative phosphorylation, creating a state of “relative intracellular hypoxia” (16). If TLR responses are too exuberant, apoptosis kills many cells, and multiorgan failure occurs. If cells survive, the number of mitochondria and ATP levels drop, during which time NAD+/NADH ratios shift to favor NAD+-dependent deacetylation processes. During this time, increased glucose uptake provides ATP from glycolysis. These sequential shifts in bioenergetics occur in human and animal sepsis and have been linked to a prosurvival state of cellular “hibernation,” during which time autophagy becomes an alternate energy compartment and limits apoptosis (17). As sepsis evolves and adaptation continues, further shifts in gene expression induce mitochondrial biogenesis; ultimately, metabolic homeostasis returns. Recent data indicate that mitochondrial biogenesis in human muscle correlates with sepsis resolution and survival (18).
As a unifying concept, we hypothesized that NAD+-dependent bioenergetics and epigenetics may combine to influence the chromatin shifts that generate endotoxin tolerance during sepsis. To test this, we used the well established THP-1 (the human promonocytic cell) cell model of endotoxin tolerance and human sepsis blood leukocytes. Our findings support that redox sensor SIRT1 and NAD+ elevations controlled by nicotinamide phosphoribosyl transferase (Nampt) coordinate the epigenetic NFκB-dependent p65 and RelB feed-forward loop that regulates gene-selective changes during endotoxin tolerance.
EXPERIMENTAL PROCEDURES
Preparation of Human Blood Samples
Blood samples were collected from sepsis subjects with septic shock and multiorgan failure and healthy controls according to the IRB protocol approved by Wake Forest University. Leukocytes were separated by layering heparinized whole blood over Isolymph (Gallard-Schlesinger Industries, Carle Place, NY) and settling for 1 h. Cells were washed in phosphate-buffered saline, and residual red blood cells were removed by hypotonic lysis using three parts distilled H2O for 20 s followed by one part of 3.6% NaCl. Pelleted leukocytes were subjected to cell culture under the indicated conditions or NAD+ extraction. Cells were >95% viable, and because both neutrophils and mononuclear cells form silenced heterochromatin upon TLR stimulation (unpublished data), no attempt was made to assess cell-type specificity. Serum samples were prepared by clotting the blood for 30 min at room temperature followed by centrifugation for 10 min at 1000 rpm and were stored at −80 °C.
Cell Culture
THP-1 cells were obtained from the American Type Culture Collection and were maintained in RPMI 1640 medium (Invitrogen) supplemented with 100 units/ml penicillin, 100 μg/ml streptomycin, 2 mm l-glutamine, and 10% fetal bovine serum (HyClone, Logan, UT) in a humidified incubator with 5% CO2 at 37 °C. For analysis of TNF-α mRNA, cells were pretreated for 1 h with either 1 mm nicotinamide (Sigma), 1 μm Ex527 (Tocris Bioscience), or 250 μm resveratrol (Sigma) followed by incubation for the indicated times with 1 μg/ml of Gram-negative bacteria LPS (Escherichia coli 0111:B4, Sigma). For determination of half-life of LPS-induced TNF-α mRNA, the transcription inhibitor actinomycin D (5 μg/ml) was added at the peak time of TNF-α transcription for indicated times. LPS-tolerated cells were prepared by incubation of THP-1 cells for 16 h with 1 μg/ml of LPS. In some experiments, cells were pretreated with 10 nm FK866 (Cayman Chemical) for 24 h (to deplete cellular NAD+) or pretreated with 1 μm cycloheximide (Sigma) for 15 min (to block protein synthesis).
Real-time RT-PCR
Levels of TNF-α, SIRT1, Nampt, and RelB mRNA were measured by quantitative real-time RT-PCR. Cellular RNA was isolated using the STAT-60 RNA extraction kit (Tel-Test, Friendswood, TX). One μg of RNA was reverse-transcribed to cDNA using murine leukemia reverse transcriptase (Applied Biosystems). The PCR analysis was performed in an ABI prism 7000 Sequence Detection System (Applied Biosystems) in a 25-μl reaction containing 12.5 μl of 2× TaqMan universal Master Mix, gene-specific predesigned TaqMan primer/probe sets, and 1 μl cDNA. GAPDH mRNA served as an internal control. Data are presented as fold change relative to unstimulated cells.
ChIP Assay
DNA-protein interactions at the promoters of TNF-α, IL-1β, RelB, and IκBα were analyzed by using a ChIP assay kit (Active Motif, Carlsbad, CA) as detailed previously (7). Chromatin was immunoprecipitated for overnight at 4 °C by incubation with 5 μg of antibody against SIRT1, NF-κB p65, RelB, linker histone H1, IκBα (Santa Cruz Biotechnology), acetylated p65 at lysine 310 (Abcam), acetylated histone H4 at lysine 16 (Millipore), or total histone H4 (Cell Signaling). Isotype-matched IgG served as a negative control.
For sequential ChIP assay, chromatin was first immunoprecipitated with the SIRT1 or H1 antibody. The primary immunoprecipitates were then eluted by incubation with 10 mm dithiothreitol at 37 °C for 30 min, diluted 40 times in immunoprecipitation buffer, and reimmunoprecipitated with the indicated secondary antibody.
The precipitated DNA was analyzed using promoter-specific PCR primer pairs (Integrated DNA Technologies) covering the κB binding sites at promoter regions. Primer sequences were as follows: TNF-α κB1 (at −598) forward, 5′-CCAAGACTGAAACCAGCAT-3′ and reverse, 5′-TAGCAGGGACAAGCCT-3′; TNF-α κB2 (at −216) forward, 5′-GAGGCAATAGGTTTTGAGG-3′ and reverse, 5′-AAGCATCAAGGATACCCCTC-3′; TNF-α κB3 (at −98) forward, 5′-TACCGCTTCCTCCAGATGAG-3′ and reverse, 5′-TGCTGGCTGGGTGTGCCAA-3′; RelB (forward), 5′-CAGAGCAATGGTCAGCGACG-3′ and reverse, 5′-CACAGT CTGGTGGACGATCG-3′ encircling κB1 (at −247) and κB2 (at −175) sites; IκBα (forward), 5′-AGCAGAGGACGAAGCCAGTTCT-3′ and reverse, 5′-GACTGCTGTGGGCTCTGCAG-3′ (surrounding κB1 site at −96). TNF-α gene-specific primers were obtained from Applied Biosystems (Hs00174128_m1). Five μl of immunoprecipitated DNA and 1 μl of input DNA were analyzed in a 25-μl reaction volume containing 1 μm of each primer, 2 mm MgCl2, 0.2 μm dNTP, and 0.04 units/μl AmpliTaq Gold DNA polymerase (Applied Biosystems). PCR conditions were set as follows; one cycle for 10 min at 94 °C, 30 cycles of 30-s each at 94 °C, 58 °C and 72 °C followed by one cycle at 72 °C for 5 min. Equal amounts of PCR products were run onto 1.8% agarose gel and scanned using a typhoon scanner (GE Healthcare). For SIRT1/p65 ChIP co-immunoprecipitations, DNA-protein complexes were immunoprecipitated with anti-SIRT1 antibody and immunoblotted with anti-p65 antibody.
Cellular NAD+ Extraction and Colorimetric NAD+ Assay
Cellular NAD+ extraction and evaluation was performed using EnzyChrom NAD+/NADH Assay kit (BioAssay System) according to the manufacturer's instructions. For cellular NAD+ extraction, cells were incubated with NAD+ extraction buffer for 5 min at 60 °C. After neutralization by adding NADH extraction buffer and assay buffer, the NAD+ containing supernatants were obtained by spinning the samples down at 14,000 rpm for 5 min. Forty microliters of NAD+ standard and samples were mixed with 80 μl of working reagent in duplicate in a 96-well plate, and the optical density was read immediately at 565 nm (OD0). After a 15-min incubation at room temperature, the plate was read again at 565 nm (OD15). The OD0 values were subtracted from OD15 for concentration analysis. Cellular NAD+ levels of unknown samples were calculated from the standard curve and analyzed by Prism software (GraphPad Prism, version 4.0, GraphPad Software, San Diego, CA). NAD+ values were normalized against protein levels and are presented as % of control or μm/mg proteins.
Transfection
for RNA interference, 60 pmol of a pool of three target-specific siRNA (Santa Cruz Biotechnology) were electronically transfected into responsive or LPS-tolerated cells using Amaxa Nucleofector kit V and Amaxa nucleofector II device (Lonza, Inc.). Twenty four hours after transfection, cells were stimulated for the indicated times with 1 μg/ml LPS before harvest. A pool of scrambled siRNAs was transfected as a negative control.
RelB plasmid transfection was performed as detailed previously (12). In brief, 0.5 μg of either pcDNA3-HA vector DNA or HA-RelB plasmid DNA were electronically transfected into THP-1 cells. Forty-eight hours after transfection, cells were stimulated for 1 h with 1 μg/ml LPS in the absence or presence of 1 μm Ex527. Cell lysates were subjected to ChIP analysis of RelB at TNF-α promoter.
Pulse Chase Assay
LPS-responsive or tolerated cells were starved for 30 min in methionine-free RPMI medium followed by pulse labeling for 1 h in 5% dialyzed FBS RPMI medium containing [35S]methionine. After washes, cells were chased in complete RPMI medium for 0–4 h. The radiolabeled SIRT1 protein was immunoprecipitated with SIRT1 antibody and resolved onto 9% SDS-PAGE.
Immunoprecipitation
THP-1 cells were stimulated for 24 h with 1 μg/ml of LPS. Nuclear extracts were prepared by lysing cells in harvest buffer (10 mm HEPES (pH 7.9), 50 mm NaCl, 0.5 m sucrose, 0.1 mm EDTA, 0.5% Triton X-100, protease inhibitor mixture) for 5 min in ice followed by spin down for 5 min at 1000 rpm at 4 °C. The supernatants were collected as cytosol samples. The pellets were washed with wash buffer (10 mm HEPES (pH 7.9), 10 mm KCl, 0.1 mm EDTA, 0.1 mm EGTA, protease inhibitor mixture) and incubated for 15 min in ice with extraction buffer (20 mm HEPES (pH 7.9), 1 m NaCl, 0.2 mm EDTA, 0.2 mm EGTA, 0.2% IGEPAL, protease inhibitor mixture). Nuclear extracts were collected after centrifugation at 1000 rpm for 5 min and were incubated with anti-SIRT1 antibody for overnight at 4 °C. The immunocomplexes were then precipitated with protein A-Sepharose CL4B beads and analyzed by Western blot using anti-RelB antibody. IgG immunoprecipitation severs as negative control.
Immunoblotting
Equal amounts (50 μg) of cell lysates or nuclear extracts were separated by SDS-PAGE electrophoresis and transferred to a polyvinylidene difluoride membrane (PerkinElmer Life Sciences). Blots were blocked with 5% milk-PBS for 1 h at room temperature and probed for overnight at 4 °C with 0.4 μg/ml of primary antibody against SIRT1 or Nampt (Santa Cruz Biotechnology). β-Actin was used as a loading control and probed with 0.04 μg/ml anti-human β-actin monoclonal antibody (Sigma). Protein complexes were detected by incubation for 1 h at room temperature with secondary antibody conjugated to horseradish peroxidase (Sigma) diluted at 1:5000 in blocking buffer and then detected by Enhanced Chemiluminescence Plus (GE Healthcare).
Statistical Analysis
Data were analyzed with unpaired Student's t test (GraphPad Prism) and are expressed as mean ± S.E. p values of < 0.05 were considered significant.
RESULTS
TLR4 Stimulation Post-transcriptionally Induces SIRT1 during Endotoxin Tolerance
We first determined whether SIRT1 expression changes during endotoxin tolerance. Upon TLR stimulation, THP-1 cells rapidly produced high levels of TNF-α mRNA, which peaked at ∼1 h and quickly decreased to a background level as endotoxin tolerance developed (Fig. 1A), as reflected by repression of TNF-α gene expression in response to LPS restimulation (Fig. 1B). We then assessed SIRT1 mRNA and protein levels in whole cell extracts of LPS-treated THP-1 cells. We found that SIRT1 mRNA did not change for up to 24 h after TLR stimulation. In contrast, SIRT1 protein levels decreased transiently and then substantially increased between 8 and 24 h (Fig. 1, C and D). Next, we examined the mechanism responsible for SIRT1 protein increases. Pulse labeling showed increases in newly synthesized SIRT1 in tolerant cells over that of nontolerant normal cells (Fig. 2A, lane 0). We then tested whether SIRT1, which is degraded in the basal state in the proteasome (19), also is stabilized following TLR stimulation (Fig. 2A, right panel). To do this, THP-1 cells were treated with cycloheximide to block protein translation and then monitored for SIRT1 degradation after TLR stimulation. SIRT1 levels rapidly decreased in normal cells but remained relatively unchanged in TLR-stimulated tolerant cells (Fig. 2, B and C). However, the prolonged half-life of proteins in tolerant cells is not a general phenomenon of endotoxin tolerance. We have reported previously that the anti-inflammatory protein IL-1ra is stabilized, but the inflammatory protein cox2 is degraded rapidly during development of tolerance (20). NFκB factor RelB is also degraded rapidly in tolerance (21).
FIGURE 1.

SIRT1 increases during TLR4-induced endotoxin tolerance in THP1 cells. A, kinetics of TNF-α transcription during the course of endotoxin tolerance. TNF-α mRNA levels were measured using quantitative real-time RT-PCR. B, TNF-α gene transcription was repressed in endotoxin-tolerant cells in response to second LPS stimulation. THP-1 cells pretreated with 1 μg/ml LPS for indicated times were restimulated with LPS for another 1 h. C, SIRT1 protein expression during the course of endotoxin tolerance. Normal THP-1 cells were stimulated for the indicated times with 1 μg/ml LPS, and cell lysates were subjected to Western blot analysis for SIRT1 level. β-Actin serves as loading control. D, real-time PCR analysis of SIRT1 transcription and densitometry analysis of SIRT1 protein expression in C. med, medium.
FIGURE 2.
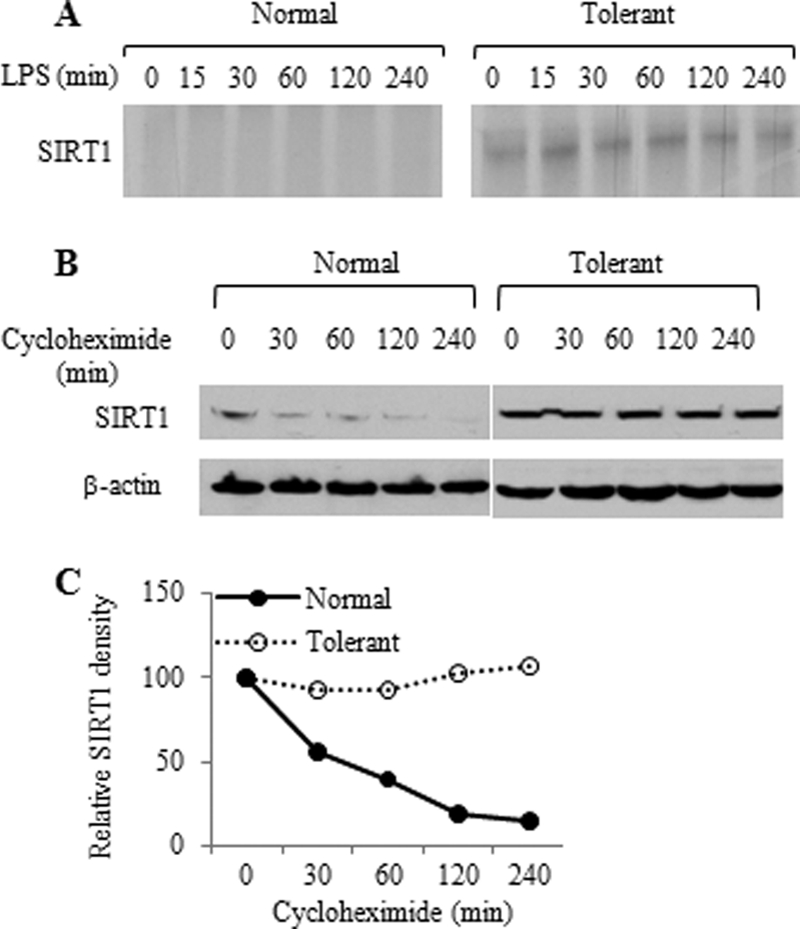
THP1 cells translate and stabilize SIRT1 following TLR4 stimulation. A, pulse chase analysis of SIRT1. Normal and tolerant THP-1 cells were pulsed for 1 h with [35S]methionine and chased for indicated times. The radiolabeled SIRT1 protein was immunoprecipitated with SIRT1 antibody and resolved onto 9% SDS-PAGE. B, SIRT1 protein degradation. Normal and tolerant cells were pretreated with 1 μm cycloheximide for 15 min followed by incubation with LPS for the indicated times. SIRT1 protein level in cell lysates was analyzed by Western blot analysis. C, densitometry analysis of the Western blot data shown in B.
Taken together, these data indicate that both translation and protein stabilization contribute to the SIRT1 protein increments during the endotoxin tolerance. This prompted our determining SIRT1 function during endotoxin tolerance.
SIRT1 Influences Proinflammatory Cytokine Gene Transcription during Endotoxin Tolerance
We observed that the LPS-induced peak TNF-α mRNA levels were substantially increased in the presence of the SIRT1-specific inhibitor Ex527 (Fig. 3A). We then confirmed this effect using RNA interference to knockdown SIRT1 expression before LPS stimulation (Fig. 3B). In further support of SIRT1 inhibitory function, pretreatment of cells with resveratrol, an activator of SIRT1 deacetylase activity, markedly depressed TNF-α mRNA synthesis after LPS stimulation (Fig. 3C).
FIGURE 3.

SIRT1 represses TLR4-induced TNF-α transcription in normal and endotoxin-tolerant cells. A, effect of SIRT1-specific inhibitor Ex527 on TNF-α transcription. Normal THP-1 cells were pretreated with 1 μm Ex527 for 1 h followed by stimulation with 1 μg/ml LPS. TNF-α mRNA was quantified using real-time PCR. B, SIRT1 knockdown augments LPS-induced TNF-α transcription. Normal THP-1 cells were transfected with SIRT1-specific siRNA for 24 h as described under “Experimental Procedures.” Cells were then stimulated for 1 h with LPS. The insert shows Western blot analysis of SIRT1 protein levels. C, SIRT1 activator resveratrol inhibits LPS-induced TNF-α transcription. Normal THP-1 cells were pretreated for 1 h with 250 μm resveratrol followed by stimulation for 1 h with 1 μg/ml LPS. D, degradation of LPS-induced TNF-α mRNA. THP-1 cells were stimulated with 1 μg/ml LPS for 1 h in the presence or absence of Ex527 to induce maximal TNF-α mRNA. Cellular gene transcription was then inhibited by incubation of cells with 5 μg/ml of actinomycin D for indicated times. TNF-α mRNA levels are quantified using real-time PCR analysis and are presented as percentage of the maximum TNF-α mRNA. One of two similar experiment results is shown. SIRT1 inhibitor Ex527 (E) and SIRT1 knockdown (F) partially restore TNF-α transcription of tolerant THP-1 cells in response to second LPS stimulation. LPS-tolerant cells were treated as in A (Ex527) or as in B (SIRT1 knockdown) before LPS stimulation for 1 h. Data in A, B, C, E, and F are presented as percentage of control TNF-α mRNA and shown as mean ± S.E. Ctrl, control; KD, knockdown; Ex, Ex527; Res, resveratrol; ETOH, ethyl alcohol.
To exclude the possibility that repression of TNF-α transcription by SIRT1 is due to the increased degradation of TNF-α mRNA, we compared the half-life of LPS-induced TNF-α mRNA in the presence or absence of the SIRT1 specific inhibitor Ex527. Inhibition of SIRT1 activity did not change TNF-α mRNA degradation rate (Fig. 3D), suggesting that SIRT1 regulates TNF-α transcription, instead of its degradation.
Because these data addressed only the initial wave of TLR4-induced transcription, we examined whether SIRT1 also contributed to the hyporesponsive status of fully developed endotoxin-tolerance, as assessed at 16 h after the initial TLR4 stimulus. We found that Ex527 treatment of tolerant cells partially (∼50%) restored TNF-α gene transcription after a second TLR stimulation with LPS (Fig. 3E). Similar results occurred after knockdown of SIRT1 in tolerant cells (Fig. 3F). Together, these data suggested that SIRT1 might participate in both the initiating and sustaining phases of endotoxin tolerance.
SIRT1 Deacetylates RelA/p65 at Lysine 310 at Promoters to Limit Gene Transcription of Proinflammatory Cytokine Genes during Initiation of Endotoxin Tolerance
We next reasoned that SIRT1 might promote the initial shift toward endotoxin tolerance by inactivating NFκB p65 transcription factor, which is required to generate the feed-forward loop that generates the tolerant phenotype (6, 10–12). To test this, we used ChIP assays. There are three NFκB binding sites at the TNF-α promoter area: the distal κB1 and κB2 sites and proximal κB3 site. Although κB1 and κB2 sites play a minor role in TNF-α transcription, we reported that the proximal site (κB3) plays a crucial role in TNF-α transcription silencing (7). Hence, we performed ChIP assay using primers covering κB3 site. We found no SIRT1 bound at the proximal TNF-α promoter in resting cells, but SIRT1 rapidly accumulated at proximal promoter after LPS stimulation (Fig. 4A). Further ChIP analysis using a TNF-α gene specific primer shows SIRT1 exclusively binds to TNF-α promoter but not to the TNF-α gene coding sequence (data not shown). As predicted, RelA/p65, a crucial initiator of TNF-α gene transcription, also rapidly accumulated at proximal TNF-α promoter, reached its peak after 30 min of LPS stimulation and dissociated from promoter after 4 h. SIRT1 also bound to the proximal promoter of acute proinflammatory gene, IL-1β (data not shown), but did not accumulate at the IκB α promoter (data not shown). These ChIP standard PCR data were confirmed by quantitative real-time PCR analysis (data not shown).
FIGURE 4.

SIRT1 deacetylates RelA/p65 at lysine 310 at the TNF-α promoter in the initiation phase of TLR4 response. A, kinetics of SIRT1 and RelA/p65 association to the proximal TNF-α promoter. THP-1 cells were stimulated for the indicated times with 1 μg/ml LPS. Cell lysates were subjected to ChIP analysis using antibodies against SIRT1 or RelA/p65 as described under “Experimental Procedures.” B, SIRT1 interacts with RelA/p65 at promoters. THP-1 cells were stimulated for the indicated times, and chromatin-bound SIRT1 was immunoprecipitated using anti-SIRT1 antibody. SIRT1 immunoprecipitates were probed using anti-RelA/p65 antibody. C, LPS-induced dynamic changes of RelA/p65 protein. THP-1 cells were stimulated for indicated times, cytosol and nuclear extracts were prepared as described under “Experimental Procedures” and were subjected to Western blot analysis using anti-RelA/p65 antibody. D, SIRT1-specific inhibitor Ex527 accumulates acetylated RelA/p65 at lysine 310 (p65k310Ac) at proximal TNF-α promoter. THP-1 cells were cultured for 1 h in the absence or presence of 1 μg/ml LPS, 1 μm Ex527, or LPS plus Ex527, respectively. Cell lysates were subjected to ChIP assay using the indicated antibodies. IB, immunoblotting; EX, Ex527; HC, heavy chain.
As SIRT1 contains no DNA-binding domain, it must associate with partner proteins to affect transcription. One of these, RelA/p65, can directly bind SIRT1 (22). Accordingly, we tested whether SIRT1 partners with RelA/p65 during the initiation phase of endotoxin tolerance. Using ChIP co-immunoprecipitation, we observed that SIRT1 and RelA/p65 formed a complex at the promoter, peaking at 1 h and decreasing by 4 h (Fig. 4B). The kinetics of both p65 promoter binding (Fig. 4A) and SIRT1-p65 interaction at the promoter (Fig. 4B) parallel the dynamic changes of nuclear p65 protein, which increases upon LPS stimulation and decreases after 4 h of LPS stimulation (Fig. 4C).
Lysine 310 on RelA/p65 is a key residue that controls RelA/p65 transactivation by p300 acetyl transferase; deacetylation at lysine 310 by SIRT1 can disrupt transactivation by RelA/p65 (22). Accordingly, we tested whether SIRT1 might limit transactivation by RelA/p65 by deacetylating lysine 310, while these two proteins are co-bound in vivo. Although we could not detect acetylation of RelA/p65 lysine 310 at TNF-α promoter 1 h after TLR stimulation, inhibition of SIRT1 activity by Ex527 significantly increased RelA/p65 lysine 310 acetylation (Fig. 4D). We obtained similar results using nicotinamide, a SIRT1-nonspecific inhibitor (data not shown). Together, these data supported that SIRT1 deacetylase activity participated in shifting activated to repressed transcription as endotoxin tolerance develops.
SIRT1 Remains Bound to TNF-α Promoter during Endotoxin Tolerance and Recruits NFκB Factor RelB
Others and we (3, 5 and Fig. 1B) have shown that phagocytes become tolerant to a second stimulation with LPS within 3–6 h after the initial TLR stimulus. Our finding that either SIRT1 inhibitor Ex527 or SIRT1-gene specific knockdown in tolerant cells partially reversed tolerance supported that SIRT1 may facilitate the shift from responsive euchromatin to repressed facultative heterochromatin. In support of this, we found that SIRT1 remained bound to the TNF-α promoter as tolerance developed (Fig. 5A).
FIGURE 5.

SIRT1 accumulates with RelB and H1 at the TNF-α promoter. A, ChIP analysis of TNF-α promoter-bound SIRT1, RelA/p65, and RelB in tolerant cells. LPS-tolerant cells were restimulated for indicated times with 1 μg/ml LPS. Cell lysates were subjected to ChIP analysis using indicated antibodies as detailed under “Experimental Procedures.” B, SIRT1 interacts with H1 and RelB at the TNF-α promoter of tolerant cells. Normal and tolerant cells were stimulated for 1 h by LPS, SIRT1/H1, and RelB sequential ChIP analysis was performed as detailed under “Experiment Procedures.” C, H1/SIRT1 and RelB sequential ChIP analysis at the TNF-α promoter after SIRT1 knockdown. Cells were stimulated with LPS for 12 h followed by SIRT1 knockdown for 24 h. Chromatin was immunoprecipitated with H1 antibody and reimmunoprecipitated with SIRT1 or RelB antibody. D, SIRT1 interacts with RelB. THP-1 cells were stimulated for 24 h with 1 μg/ml LPS, nuclear extracts were immunoprecipitated with SIRT1 antibody. Immunoprecipitates were subjected to Western blot analysis using anti-RelB antibody. E, overexpression of RelB in THP-1 cells. Normal THP-1 cells were transfected for 48 h with 0.5 μg of either pcDNA3-HA vector plasmid DNA or HA-RelB plasmid DNA. Total cell lysates were subjected to Western blot analysis of RelB expression using anti-RelB antibody. F, SIRT1 facilitates RelB loading onto the TNF-α promoter. RelB-transfected THP-1 cells as detailed in E were stimulated for 1 h with LPS in the presence or absence of Ex527. ChIP analysis of TNF-α promoter bound RelB was performed as described under “Experimental Procedures.” IP, immunoprecipitation; IB, immunoblotting; Ctrl, control; KD, knockdown; Med, medium; EX, EX527.
As RelA/p65 is replaced at the TNF-α promoter in tolerant THP-1 cells by dimer exchange with RelB (23), we asked whether SIRT1 interacts with the multicomponent repressor complex proteins RelB and heterochromatin linker histone H1 (8, 11, 24, 25). Our previous work showed that RelB promoter recruitment directs facultative heterochromatin formation by first binding to G9a methyltransferase, which methylates H3K9; other members of chromatin modifiers include histone linker H1, heterochromatin protein 1, and DNA CpG methyltransferases DNMT 3a/3b (11, 13, 25). Using sequential ChIP analysis of SIRT1 followed by histone H1 and RelB re-ChIP, we showed that SIRT1, RelB, and H1 co-accumulate at the tolerant TNF-α promoter (Fig. 5B). Furthermore, knockdown of SIRT1 reduced promoter binding of RelB, but not histone H1 (Fig. 5C). This supports that SIRT1 binding preceded that of RelB and that H1 linker histone binding may be proximal to both SIRT1 and RelB.
Our previous data indicated the RelB is both necessary and sufficient for promoting transcription repression during endotoxin tolerances (12). Our observation here that removal of SIRT1 decreased RelB promoter binding suggested that SIRT1 might bind directly or indirectly to RelB to facilitate generation of the multicomponent repressor complex. Using immunoprecipitation analysis, we further substantiated that SIRT1 and RelB interacts (Fig. 5D).
We also have found that RelB promoter binding requires TLR signaling. Overexpressed RelB in normal THP-1 cells does not bind to promoter without TLR signaling.4 To investigate whether SIRT1 is required for RelB loading onto promoter, we overexpressed RelB in normal THP-1 cells for 48 h (Fig. 5F). Cells were then stimulated with LPS for 1 h in the presence or absence of the SIRT1-specific inhibitor Ex527. ChIP analysis shows that the LPS-stimulated promoter bound RelB is blocked by inhibition of SIRT1 activity (Fig. 5G), supporting SIRT1 facilitates RelB loading onto promoter. To exclude the possibility that RelB may bind to the two distal κB sites at the TNF-α promoter without SIRT1 support, we performed RelB ChIP analysis using primers covering κB1 and κB2 sites and found no RelB binds to these two distal kB sites (data not shown).
SIRT1 Promotes Transcription and Repression of Select Genes
RelB acts as both a repressor and activator of transcription of specific genes during endotoxin tolerance and acute systemic inflammation (5, 10, 13). Reports indicate that SIRT1 also can activate or repress transcription by deacetylating distinct transcription mediators (26, 27). Accordingly, we asked whether SIRT1 might promote transcription and de novo synthesis of RelB. In support of this, we found that SIRT1 accumulated at the RelB promoter after LPS stimulation (Fig. 6A) and that inhibition of SIRT1 deacetylase activity with Ex527 reduced RelB gene transcription (Fig. 6B) and protein expression (Fig. 6C). These data support a dual and cooperative function between SIRT1 and RelB during endotoxin tolerance.
FIGURE 6.

SIRT1 regulates RelB expression. A, SIRT1 binds to RelB promoter. THP-1 cells were stimulated with 1 μg/ml of LPS for 0, 1, or 16 h, and chromatin was immunoprecipitated with SIRT1 antibody. The SIRT1-DNA complex was analyzed by standard PCR using TNF-α promoter-specific or RelB promoter-specific primers. IgG-immunoprecipitated samples served as negative control. B and C, inhibition of SIRT1 deacetylase activity decreases LPS-induced RelB gene transcription and protein expression during the development of endotoxin tolerance. THP-1 cells were pretreated for 1 h with 1 μm Ex527 followed by stimulation with 1 μg/ml of LPS for indicated times. RelB mRNA was quantified by real-time PCR and RelB protein levels were analyzed using Western blot. RelB mRNA levels are presented as fold changes relative to unstimulated control. Ctrl, control; med, medium.
NAMPT Generates NAD+ Accumulation during Endotoxin Tolerance
Because SIRT1 activity is NAD+-dependent, and because redox states change during acute inflammation, we tested whether NAD+ levels increase following TLR stimulation. We detected NAD+ at basal levels in nonstimulated THP-1 cells and observed that TLR stimulation decreased cellular NAD+ to ∼20% of its basal level by 15 min to 1 h. Thereafter, NAD+ gradually and substantially increased for up to 24 h (Fig. 7A), which correlated with elevations in SIRT1 protein (Fig. 1, C and D).
FIGURE 7.

Nampt expression and cellular NAD+ increase during TLR4-induced endotoxin tolerance. A, changes in cellular NAD+ levels during the course of endotoxin tolerance. Cells were cultured for different times in the presence of 1 μg/ml LPS. Intracellular NAD+ was extracted and evaluated using a commercial kit as detailed under “Experimental Procedures”. Results are reported as mean ± S.E. of 3 independent experiments. B: Western blot analysis of Nampt expression at the indicated times after 1 μg/ml of LPS stimulation. C, Real-time PCR analysis of Nampt transcription after LPS stimulation and densitometry analysis of Nampt protein levels in B. D, Nampt inhibitor FK866 inhibits LPS-induced NAD+ biosynthesis and depletes cellular NAD+. THP-1 cells were cultured for the indicated times with 1 μg/ml LPS or LPS plus 10 nm FK866. Intracellular NAD+ was extracted and analyzed as in A.
Cellular NAD+ homeostasis is partially sustained by recycling NAD+ degradation products by the rate-limiting enzyme Nampt (28). Because Nampt has an NF-κB consensus binding domain in its proximal promoter, we hypothesized that TLR signaling might increase cellular NAD+ by inducing Nampt expression, which could thereby provide substrate for SIRT1 deacetylase activity. Western blot analysis showed increased Nampt expression after 4 h, which peaked at 8 h, and then decreased by 16 h (Fig. 7B). The increased protein expression was preceded by induction of Nampt mRNA (Fig. 7C). As predicted, inhibition of Nampt activity by FK866, a Nampt-specific inhibitor (29, 30), markedly decreases in total cellular NAD+ (Fig. 7D).
Nampt-dependent Up-regulation of NAD+ Supports SIRT1 Activation and Epigenetic Gene Reprogramming during Endotoxin Tolerance
Given that SIRT1 deacetylase activity is NAD+-dependent, we tested whether Nampt activity promotes endotoxin tolerance. To do this, we employed the Nampt-specific inhibitor FK866 to diminish cellular NAD+ before LPS stimulation (Fig. 8A). We found that predepletion of cellular NAD+ by FK866 treatment enhanced LPS-induced TNF-α mRNA (Fig. 8B). We then demonstrated that FK866 prevented NAD+ accumulation and disrupted SIRT1 binding to the TNF-α promoter (Fig. 8C); FK866 also increased acetylation of histone H4 on lysine 16 at the TNF-α promoter (Fig. 8D). Together, these results supported that TLR-dependent up-regulation of Nampt expression positively regulates cellular NAD+ biosynthesis, SIRT1 activation, and SIRT1 promoter binding during the development of endotoxin tolerance.
FIGURE 8.

Cellular NAD+ is required for SIRT1 promoter binding and repression of TNF-α transcription. A, cellular NAD+ is diminished by the Nampt-specific inhibitor FK866. THP-1 cells were pretreated for 24 h with 10 nm FK866. Cells were washed and stimulated for indicated times with 1 μg/ml LPS in the presence of 10 nm FK866. B, depletion of cellular NAD+ by FK866 augments LPS-induced TNF-α transcription. THP-1 cells were pretreated for overnight with 10 nm FK866 followed by stimulation for 1 h with 1 μg/ml LPS. TNF-α mRNA was quantified using real-time PCR. C, depletion of cellular NAD+ inhibits SIRT1 binding to the TNF-α promoter. THP-1 cells were treated as in A. Cell lysates were subjected to SIRT1 ChIP assay at the TNF-α proximal promoter. D, depleting NAD+ by FK866 simultaneously with inhibiting SIRT1 activity enhances accumulation of acetylated histone H4 at lysine 16. Cells were treated for 16 h with LPS in the presence or absence of 10 nm FK866 and 1 mm nicotinamide followed by ChIP analysis with H4K16Ac antibody. Med, medium; Nic, nicotinamide.
Cellular NAD+ Increases and SIRT1 and RelB Epigenetic Regulators Accumulate at TNF-α Promoter of Blood Leukocytes during Human Sepsis
We previously demonstrated that RelB is induced in sepsis blood leukocytes during endotoxin tolerance, concomitant with the formation of facultative heterochromatin (10, 12). To test whether our unifying concept of SIRT1 incorporating bio-energy and epigenetics might translate to human acute systemic inflammation, we isolated peripheral blood leukocytes from healthy volunteers and patients with sepsis and multiorgan failure and then assessed the binding of SIRT1 and RelB at the TNF-α promoter. As expected, leukocytes from sepsis patients were endotoxin-tolerant with repressed TLR4-induced transcription of TNF-α (Fig. 9A). Coincident with this phenotype, we found SIRT1 and RelB accumulated at the TNF-α promoter (Fig. 9B). Two more sepsis participants showed increased SIRT1 accumulated at both TNF-α and IL-1β promoters as determined by real-time RT-PCR analysis (data not shown). We also detected that cellular NAD+ in sepsis blood leukocytes and extracellular Nampt in sepsis serum were increased (Fig. 10, C and D).
FIGURE 9.

Increases in cellular NAD+ and extracellular Nampt and accumulation of SIRT1 and RelB at TNF-α promoter in septic leukocytes. A, TNF-α transcription in blood leukocytes in response to LPS stimulation. Blood leukocytes were isolated from normal and septic subjects and stimulated with LPS for 1 h. RNA was isolated and analyzed for TNF-α mRNA by real-time PCR. B, ChIP analysis of TNF-α promoter-bound SIRT1 and RelB in normal and septic leukocytes. C, intracellular NAD+ levels in normal (n = 3) and septic blood leukocytes (n = 12). D, extracellular Nampt levels in normal (n = 3) and septic serum (n = 12). Data in C and D are shown as mean ± S.E. N, normal control; P, septic patient.
FIGURE 10.

A model for SIRT1 in bridging bioenergetics and epigenetics during endotoxin tolerance and sepsis. TLR induces rapid promoter binding of constitutive SIRT1, which uses available nuclear NAD+ to support its promoter binding and deactivate RelA/p65 through lysine 310 deacetylation, thus limiting transcription of acute proinflammatory genes. SIRT1 then remains promoter bound and TLR-dependent responses increase the expression of Nampt, which sustains NAD+ elevation by recycling nicotinamide. Increases in Nampt are accompanied by elevations in SIRT1 by stabilizing the protein and enhancing translation. The combined availability of nuclear NAD+ and its effect on SIRT1 promoter accumulation stimulates RelB expression and supports recruitment of RelB to direct formation of locus-specific facultative heterochromatin to silence acute proinflammatory genes or its activating gene sets with distinct functions.
DISCUSSION
This study shows that endotoxin tolerance, which reflects the epigenome of acute systemic inflammation associated with sepsis (3), involves interplay between energy sensor SIRT1 and the NFκB feed-forward loop that shifts TLR4-responsive euchromatin state of the TNF-α and IL-1β promoters to silent facultative heterochromatin. This coordinated process uses distinct pathways (redox sensor SIRT1, Nampt-dependent generation of NAD+, transcription regulators NF-κB p65 and RelB, and modifiers of chromatin structure) to shift from inflammation initiation to the tolerant or adaptive phase of TLR responses. Fig. 10 models this temporal coupling of energy and epigenetics. First, TLR induces rapid promoter binding of constitutive SIRT1, which uses available nuclear NAD+ to support its promoter binding and deactivate RelA/p65 through lysine 310 deacetylation, thus limiting transcription of acute proinflammatory genes. SIRT1 then remains promoter bound, and TLR-dependent responses increase the expression of Nampt, which sustains NAD+ elevation by recycling nicotinamide. Increases in Nampt are accompanied by elevations in SIRT1 by stabilizing the protein and enhancing translation. The combined availability of nuclear NAD+ and its effect on SIRT1 promoter accumulation, supports recruitment of RelB and its directly facilitating formation of locus-specific facultative heterochromatin to silence acute proinflammatory genes, or activate other genes with distinct physiologic effects. Unexpectedly, we also observed that SIRT1 also participates in inducing RelB transcription of RelB, which later becomes a binding partner. Thus, there is an interplay between feed-forward loops that involve transcription, translation, and post-translation programming. A product of this sequel is the development of endotoxin tolerance through epigenetic reprogramming.
We (in humans) and others (in mice) (7–11) showed that epigenetic reprogramming regulates inflammation by both repressing acute proinflammatory genes and activating anti-inflammatory, antimicrobial, and other functionally distinct gene sets, including those that regulate cellular energy and mitochondrial biogenesis. In this study, we further find, like RelB (13), that SIRT1 becomes a gene-specific dual regulator of epigenetic reprogramming, because it does not bind to the anti-inflammatory IkBα promoter, it represses transcription of TNF-α and IL-1β, and it activates RelB transcription. Thus, SIRT1 is part of the master switch that coordinates epigenetic reprogramming and shifts functional phenotypes during acute TLR4 responses and sepsis inflammation.
Although the THP-1 cell model of endotoxin tolerance has been faithful in predicting what happens in human and animal sepsis (3, 5), interpretations of this model alone require caution. To support application of the cell model to the epigenetics of human acute systemic inflammation, we previously demonstrated gene-specific formation of facultative heterochromatin at the TNF-α and IL-1β promoters in both THP-1 cells and in human blood leukocytes from human sepsis (5). Others and we (9) have shown gene-specific heterochromatin formation in murine macrophages stimulated with endotoxin ex vivo and in peritoneal macrophages and splenocytes from murine sepsis.5 Here, we provide support of concept for energy and epigenetic coordination during human sepsis by showing elevated cellular NAD+ and concomitant SIRT1 and RelB accumulation at the TNF-α promoter of blood leukocytes obtained during the endotoxin tolerant state of septic humans. This leukocyte population is mixed and contains TLR4-expressing neutrophils and macrophages, both of which develop the endotoxin-tolerant phenotype (13, 25). A recent study supports the concept that SIRT1 may regulate inflammation after endotoxin administration to humans and mice (31). Further investigations are needed to determine whether this paradigm extends to other cells or compartments, such as skeletal muscle, liver, lung, and heart.
The sirtuin family, whose founding member Sir2 was discovered as a chromatin modifier in yeast (32), is intimately involved in many cellular functions, including stem cell development, cell intermediary metabolism, and cell senescence (33). Although this study focused on Sir2 homologue SIRT1, other NAD+-dependent sirtuin family members might also coordinate energy transitions in many cells: in immunocytes or other cell types affected by sepsis. For example, SIRT3 localizes to the mitochondria in hepatocytes and promotes fatty acid oxidation as an energy source, although the functions of SIRT3 in innate immunity cells are unclear (34). SIRT6, like SIRT1, can bind to and inactivate RelA/p65, which modifies development of newborn mice (35). SIRT6 also promotes translation of TNF-α and deacetylates histone H3 lysine 9 to augment heterochromatin formation in some tissues (36). We have demonstrated that SIRT6 also represses TNF-α transcription in our THP-1 cell model of acute inflammation6 and thus may have redundant functions with SIRT1. Finally, SIRT1 itself has diverse non-nuclear functions that may impact inflammation regulation (37, 38). It enhances autophagy as an alternate energy compartment, modifies circadian rhythm, enhances antioxidant production, and promotes mitochondrial biogenesis, all of which are prominent features of endotoxin tolerance (3). Taken together, this study and others support that the sirtuin family may link energy and inflammation by distinct spatial and temporal processes among diverse cell types.
In summary, we report that redox-dependent cellular energy and gene-specific epigenetic programming converge through SIRT1 to influence the course of TLR-induced endotoxin tolerance as an indicator of the epigenome of acute systemic inflammatory responses such as sepsis. When this occurs, four highly conserved biologic processes interact: TLR sensing, NAD+-directed deacetylation, NF-κB, and epigenetic modifications of germ line DNA. Our findings may inform new ways to analyze and treat acute inflammatory diseases such as sepsis.
Acknowledgments
We thank Sue Cousart, Jean Hu, and Ashley Church for technical assistance and Drs. Richard Loeser and Douglas Lyles for critical discussion.
This work was supported, in whole or in part, by National Institutes of Health Grants RO1 AI-065791 (to C. E. M.), R01 AI-079144 (to C. E. M.), and MO-1RR 007122 (Wake Forest University General Clinical Research Center). The research on human participants was endorsed by Wake Forest University IRB Protocol BG174.
B. K. Yoza and C. E. McCall, unpublished data.
M. El Gazzar and C. E. McCall, unpublished data.
T. F. Liu and C. E. McCall, unpublished data.
- TLR
- Toll-like receptor(s)
- Nampt
- nicotinamide phosphoribosyltransferase.
REFERENCES
- 1. Schreiber J., Jenner R. G., Murray H. L., Gerber G. K., Gifford D. K., Young R. A. (2006) Proc. Natl. Acad. Sci. U.S.A. 103, 5899–5904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Medzhitov R., Horng T. (2009) Nat. Rev. Immunol. 9, 692–703 [DOI] [PubMed] [Google Scholar]
- 3. McCall C. E., Yoza B., Liu T., El Gazzar M. (2010) J. Innate Immun. 2, 395–405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hotchkiss R. S., Karl I. E. (2003) N. Engl. J. Med. 348, 138–150 [DOI] [PubMed] [Google Scholar]
- 5. McCall C. E., Yoza B. K. (2007) Am. J. Respir. Crit. Care Med. 175, 763–767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chan C., Li L., McCall C. E., Yoza B. K. (2005) J. Immunol. 175, 461–468 [DOI] [PubMed] [Google Scholar]
- 7. El Gazzar M., Yoza B. K., Hu J. Y., Cousart S. L., McCall C. E. (2007) J. Biol. Chem. 282, 26857–26864 [DOI] [PubMed] [Google Scholar]
- 8. El Gazzar M., Liu T., Yoza B. K., McCall C. E. (2010) J. Biol. Chem. 285, 1259–1271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Foster S. L., Hargreaves D. C., Medzhitov R. (2007) Nature 447, 972–978 [DOI] [PubMed] [Google Scholar]
- 10. Chen X., El Gazzar M., Yoza B. K., McCall C. E. (2009) J. Biol. Chem. 284, 27857–27865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. El Gazzar M., Yoza B. K., Chen X., Hu J., Hawkins G. A., McCall C. E. (2008) J. Biol. Chem. 283, 32198–32208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Yoza B. K., Hu J. Y., Cousart S. L., Forrest L. M., McCall C. E. (2006) J. Immunol. 177, 4080–4085 [DOI] [PubMed] [Google Scholar]
- 13. Chen X., Yoza B. K., El Gazzar M., Hu J. Y., Cousart S. L., McCall C. E. (2009) Clin. Vaccine Immunol. 16, 104–110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Suliman H. B., Welty-Wolf K. E., Carraway M. S., Schwartz D. A., Hollingsworth J. W., Piantadosi C. A. (2005) FASEB J. 19, 1531–1533 [DOI] [PubMed] [Google Scholar]
- 15. Suliman H. B., Carraway M. S., Welty-Wolf K. E., Whorton A. R., Piantadosi C. A. (2003) J. Biol. Chem. 278, 41510–41518 [DOI] [PubMed] [Google Scholar]
- 16. Haden D. W., Suliman H. B., Carraway M. S., Welty-Wolf K. E., Ali A. S., Shitara H., Yonekawa H., Piantadosi C. A. (2007) Am. J. Respir. Crit. Care Med. 176, 768–777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Singer M. (2008) Clin. Chest Med. 29, 655–660 [DOI] [PubMed] [Google Scholar]
- 18. Carré J. E., Orban J. C., Re L., Felsmann K., Iffert W., Bauer M., Suliman H. B., Piantadosi C. A., Mayhew T. M., Breen P., Stotz M., Singer M. (2010) Am. J. Respir. Crit. Care Med. 182, 745–751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sasaki T., Kim H. J., Kobayashi M., Kitamura Y. I., Yokota-Hashimoto H., Shiuchi T., Minokoshi Y., Kitamura T. (2010) Endocrinology 151, 2556–2566 [DOI] [PubMed] [Google Scholar]
- 20. Learn C. A., Mizel S. B., McCall C. E. (2000) J. Biol. Chem. 275, 12185–12193 [DOI] [PubMed] [Google Scholar]
- 21. Yoza B. K., McCall C. E. (2011) Cytokine 53, 145–152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Yeung F., Hoberg J. E., Ramsey C. S., Keller M. D., Jones D. R., Frye R. A., Mayo M. W. (2004) EMBO J. 23, 2369–2380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Saccani S., Pantano S., Natoli G. (2003) Mol. Cell 11, 1563–1574 [DOI] [PubMed] [Google Scholar]
- 24. Agwu D. E., McPhail L. C., Sozzani S., Bass D. A., McCall C. E. (1991) J. Clin. Invest. 88, 531–539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. El, Gazzar M., Yoza B. K., Chen X., Garcia B. A., Young N. L., McCall C. E. (2009) Mol. Cell. Biol. 29, 1959–1971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Blander G., Guarente L. (2004) Annu. Rev. Biochem. 73, 417–435 [DOI] [PubMed] [Google Scholar]
- 27. Guarente L. (2007) Cold Spring Harb. Symp. Quant. Biol. 72, 483–488 [DOI] [PubMed] [Google Scholar]
- 28. Garten A., Petzold S., Körner A., Imai S., Kiess W. (2009) Trends Endocrinol. Metab. 20, 130–138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Nakahata Y., Sahar S., Astarita G., Kaluzova M., Sassone-Corsi P. (2009) Science 324, 654–657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hasmann M., Schemainda I. (2003) Cancer Res. 63, 7436–7442 [PubMed] [Google Scholar]
- 31. Zhang Z., Lowry S. F., Guarente L., Haimovich B. (2010) J. Biol. Chem. 285, 41391–41401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Irizar A., Yu Y., Reed S. H., Louis E. J., Waters R. (2010) Nucleic Acids Res. 38, 4675–4686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Finkel T., Deng C. X., Mostoslavsky R. (2009) Nature 460, 587–591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Scher M. B., Vaquero A., Reinberg D. (2007) Genes Dev. 21, 920–928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kawahara T. L., Michishita E., Adler A. S., Damian M., Berber E., Lin M., McCord R. A., Ongaigui K. C., Boxer L. D., Chang H. Y., Chua K. F. (2009) Cell 136, 62–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Van Gool F., Gallí M., Gueydan C., Kruys V., Prevot P. P., Bedalov A., Mostoslavsky R., Alt F. W., De Smedt T., Leo O. (2009) Nat. Med. 15, 206–210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Imai S., Johnson F. B., Marciniak R. A., McVey M., Park P. U., Guarente L. (2000) Cold Spring Harb. Symp. Quant. Biol. 65, 297–302 [DOI] [PubMed] [Google Scholar]
- 38. Imai S. (2009) Cell Biochem. Biophys. 53, 65–74 [DOI] [PMC free article] [PubMed] [Google Scholar]