

Abstract
NAD(P)H:quinone oxidoreductase 1 (NQO1) is a flavoenzyme that is important in maintaining the cellular redox state and regulating protein degradation. The NQO1 polymorphism C609T has been associated with increased susceptibility to various age-related pathologies. We show here that NQO1 protein level is regulated by the E3 ligase STUB1/CHIP (C terminus of Hsc70-interacting protein). NQO1 binds STUB1 via the Hsc70-interacting domain (tetratricopeptide repeat domain) and undergoes ubiquitination and degradation. We demonstrate here that the product of the C609T polymorphism (P187S) is a stronger STUB1 interactor with increased susceptibility to ubiquitination by the E3 ligase STUB1. Furthermore, age-dependent decrease of STUB1 correlates with increased NQO1 accumulation. Remarkably, examination of hippocampi from Alzheimer disease patients revealed that in half of the cases examined the NQO1 protein level was undetectable due to C609T polymorphism, suggesting that the age-dependent accumulation of NQO1 is impaired in certain Alzheimer disease patients.
Keywords: Aging, Alzheimer Disease, E3 Ubiquitin Ligase, Genetic Polymorphism, Proteasome, Protein Degradation, C Terminus of HSC70-interacting Protein, C609T NQO1 Polymorphism, NQO1, STUB1
Introduction
NQO12 is a ubiquitous homodimeric flavoenzyme containing one molecule of FAD/subunit. It catalyzes two-electron reduction of various quinones and aromatic nitro compounds by utilizing NAD(P)H as an electron donor. NQO1 function has been shown to be cytoprotective with highly inducible protein levels as part of the cellular defense mechanism in response to electrophilic and/or oxidative stress generated due to exposure to chemicals or endogenous quinones (1–3). It was recently discovered that NQO1 also plays a regulatory role in the ubiquitin-independent 20S proteasomal degradation pathway. NQO1 was shown to be associated with the 20S proteasome preventing the degradation of various key regulatory proteins and enzymes such as p53, p73, p33, and c-Fos (4–6). The association of NQO1 with the 20S is conserved over evolution, and it was recently shown in yeast that Lot6, the NQO1 ortholog (7), is also associated with the 20S proteasome (8).
A common genetic polymorphism of NQO1, C609T, results in the substitution of a proline to serine at position 187. People carrying the polymorphic C609T NQO1 gene show increased sensitivity to benzene toxicity (9), are at an increased risk for development of different solid tumors and leukemia (10–12), and are also suggested to be more susceptible to neurodegenerative diseases such as Alzheimer disease (AD) (13). To date, NQO1 protein accumulation has been shown to be regulated mainly at the transcriptional level by Nrf1 and Nrf2 (14), and not much is known on the regulation of NQO1 at the posttranslational level. The NQO1 P187S encoded by the C609T variant has been reported to be an unstable protein that is rapidly ubiquitinated and subjected to proteasomal degradation (15, 16), but it is still unknown what E3 ligase mediates this rapid ubiquitination and proteasomal degradation.
STUB1/CHIP was originally described as an Hsc70 co-chaperone containing an E3 ligase activity. As such, it comprises three functional domains: a tetratricopeptide repeat (TPR) at the N terminus mediating the interaction with the Hsc70, a U-box domain at the C terminus with the E3 ubiquitin ligase activity, and a highly charged region (17). It is believed that STUB1/CHIP exerts biological effects by inducing chaperone client proteins for proteasomal degradation (18, 19). STUB1 plays a critical regulatory function in the protein quality control machinery and implicated in the pathology of several neurological disorders characterized by protein misfolding and aggregation. It has been shown to target various unfolded proteins for degradation including proteins that are associated with numerous pathologies in the brain, some also age-associated such as tau (20), ataxin-1 (21), CFTR (22), and LRRK2 (23).
Age-associated pathologies are speculated to arise as a result of impairment in both the cellular redox regulation and the protein degradation machinery (24). The ability of NQO1 to regulate these two processes led us to speculate that the function of NQO1 might be of value in aging. Furthermore, the phenotypes observed in NQO1 knock-out mice that included significantly lower levels of pyridine nucleotides with an increase in NAD(P)H/NAD(P)+ ratio and reduction of adipose tissue, were significant only in old mice (25). Interestingly, newly synthesized NQO1 interacts with Hsp70 (15), suggesting an interplay between NQO1 and Hsp70-binding protein such as STUB1.
EXPERIMENTAL PROCEDURES
Ethics Statement
The Weizmann Institute Animal Care and Use Committee approved all studies in mice, according to institutional guidelines and Israeli law. The IACUC approval number is 03410709-1.
Brain Fractions
Frozen blocks of human hippocampus were obtained from The Netherlands Brain Bank. Human hippocampal tissue was obtained through the rapid autopsy program of The Netherlands Brain Bank (Coordinator: Dr R. Ravid). The Netherlands Brain Bank abides by all local ethical legislation. All tissue was obtained with informed consent from the donor or next of kin to perform brain autopsy and the subsequent use of brain tissue for scientific purposes. Male C57BL mice of specified age were used in this study.
Cells and Transfections
Human embryonic kidney HEK293T cells were grown in DMEM supplemented with 10% FBS, 100 units/ml penicillin, and 100 mg/ml streptomycin and cultured at 37 °C in a humidified incubator with 5.6% CO2. Transfections were carried out by the calcium phosphate method. Cells were treated with 25 μm MG132 (Sigma) where specified.
Plasmids and Antibodies
The NQO1 plasmids used were pEFIRES NQO1, FLAG-NQO1 or NQO1 C609T, FLAG-NQO1 C609T, and pcDNA3.1 NQO1-FLAG. The STUB1 plasmids used were pcDNA3.1 His-STUB1, His-ΔTPR, and His-ΔΕ4 were kindly provided by Prof. Patterson. CHIP siRNA and control siRNA were described previously (27) and were also kindly provided by Prof. Patterson. Antibodies used were goat anti-NQO1 C19 R20 (Santa Cruz Biotechnology, Santa Cruz, CA), rabbit anti-STUB1 (BETHYL), anti-Hsc-70 (26) (kindly provided by Prof. Ginzburg), mouse anti-actin, HA, FLAG (Sigma), AT8 (Innogenetics, Gent, Belgium).
Protein Extraction
For immunoblotting, protein extraction from mouse brains was performed by homogenization in Nonidet P-40 buffer (100 mm Tris, pH 7.5, 2 mm EDTA, 150 mm NaCl, 1% Nonidet P-40, 1 mm DTT, 1:100 protease inhibitors). The extract was subjected to ultracentrifugation (13,000× g, 15 min), and the supernatant was used as the protein extract. Protein concentration was determined by Bradford assay. Immunoblot analysis was carried out with indicated antibodies, and analysis and quantification were performed by ChemiDoc XRS (Bio-Rad) or by ImageQuantTM TL (General Electrics). Protein from frozen blocks of human hippocampus were obtained from The Netherlands Brain Bank as described previously (28). Proteins were extracted in buffer A (20 mm HEPES, pH 7.4, 0.32 m sucrose, 150 mm KCl, 3 mm MgCl2, 10 mm NaF, 20 mm β-glycerol phosphate). Fractions of the homogenate were centrifuged for 15 min at 4 °C at 13,000 × g. The supernatant was subjected to protein analysis.
Immunoprecipitation Assay
Cell extracts were prepared by lysis of PBS-washed cells in radioimmune precipitation assay lysis buffer (50 mm Tris-HCl, pH 8, 150 mm NaCl, 1% Nonidet P-40 (v/v), 0.5% deoxycholate (v/v), 0.1% SDS (v/v), 1 mm DTT, and Sigma protease inhibitor mixture (P8340) diluted 1:100). Equal amounts of protein were subjected to immunoprecipitation using anti-FLAG-agarose conjugate beads (Sigma). Proteins were identified and quantified by the indicated antibodies. The elution of the FLAG immunoprecipitated complexes was done with FLAG peptide (purchased from Sigma) according to the manufacturer's instructions.
Ubiquitination Assay
HEK293T cells were transiently transfected with mammalian expression plasmids for HA-ubiquitin, together with the indicated plasmid. At 24 h after transfection, cells were treated for 2 h with 25 μm MG132 (Sigma) before harvesting. Cell extracts were then subjected to immunoprecipitation using anti-FLAG-agarose conjugated beads (Sigma). Ubiquitin conjugates were detected using an anti-HA antibody (Biotest).
Pulse-Chase Experiments
Pulse-chase experiments were conducted as described previously with slight modifications (29). HEK293T cells were incubated with medium depleted of methionine (−M) for 20 min. [35S]Methionine (10 mCi/ml; Amersham Biosciences) was added reaching a final concentration of 0.2 mCi/μl for a pulse of 30 min. Cells were washed, and medium containing 2% unlabeled methionine was added for the indicated time (chase). Cells were collected, and protein extracts were subjected to immunoprecipitation as described above.
RNA Isolation and Quantitative RT-PCR
The RNA was purified from DNA and other contaminants by DNase I treatment and LiCl precipitation, respectively. 1 μg of RNA was reverse-transcribed using iScript cDNA kit (Bio-Rad). cDNA was amplified and quantified with a Roche Light Cycler 480 system using SYBR Green (KAPA Biosystems) and the ΔΔCt threshold cycle method. Gene expression levels were normalized to TBP (TATA-binding protein) mRNA and expressed relative to control. Primers used were NQO1: forward, AGGCGTCCTTCCTTATATGCTA and reverse, AGGATGGGAGGTACTCGAATC; STUB1: forward, CGGCAGCCCTGATAAGAGC and reverse, CACAAGTGGGTTCCGAGTGAT; TBP: forward, CCTATCACTCCTGCCACACCAGC and reverse, GTGCAATGGTCTTTAGGTCAAGTTTACAGCC.
Detection of WT and C609T NQO1 Alleles
Genomic DNA was purified using TRI reagent. WT or C609T mutated alleles were detected by the method described previously (30).
RESULTS
STUB1 Binds, Ubiquitinates, and Down-regulates NQO1 Protein Levels
STUB1/CHIP is an Hsc70-interacting E3 ligase that plays a significant role in the brain (31). NQO1 has been previously shown to interact with Hsp70, a paralog of Hsc70 (15). Therefore, we hypothesized that STUB1, a known Hsc70-binding E3 ligase, may induce NQO1 degradation. Overexpression of increasing amounts of STUB1 resulted in a significant decrease in the NQO1 protein levels (Fig. 1A). In a reciprocal experiment, specific siRNA knockdown of endogenous STUB1, but not a control siRNA, resulted in a decrease in the STUB1 protein levels and an increase in the NQO1 protein levels (Fig. 1B). These data suggest that the endogenous STUB1 regulates NQO1 level.
FIGURE 1.

STUB1 ubiquitinates and down-regulates NQO1. A, NQO1 levels were detected in HEK293T cells co-transfected with NQO1 in the absence or the presence of increasing concentrations of STUB1. IB, immunoblotting. B, NQO1 protein levels were examined following the knockdown of endogenous STUB1 by a specific STUB1 siRNA versus a control siRNA in HEK293T cells. C, NQO1-f (NQO1 with a FLAG antigen at its C terminus) ubiquitination was detected in the presence of increasing concentrations of STUB1 (as described under “Experimental Procedures”). Ubiquitination of NQO1 by STUB1 was determined by overexpressing FLAG-NQO1 in HEK293T cells with HA-ubiquitin and in the presence or absence of increasing concentrations of STUB1. Following treatment with 25 μm MG132 for 2 h, NQO1 was immunoprecipitated with FLAG beads (IP). Total input (Total) and the level of ubiquitination were detected by probing with anti-NQO1 and ant-HA antibodies, respectively. D, half-life of NQO1 in the presence or absence of STUB1 was determined by analyzing the NQO1 protein level following cycloheximide (CHX) treatment (50 μg/ml) of HEK293T cells overexpressing NQO1 in the presence or absence of STUB1.
Next, to demonstrate that STUB1 polyubiquitinates NQO1 we co-transfected HA-tagged ubiquitin together with FLAG-tagged NQO1 and increasing amounts of STUB1. The tagged NQO1 was immunoprecipitated, and the level of ubiquitination was monitored by anti HA antibody. We revealed that the immunoprecipitated NQO1 was shifted to high molecular weight, indicating that it was heavily ubiquitinated (Fig. 1C). Additionally, the level of ubiquitination was STUB1 dose-dependent. Furthermore, upon STUB1 overexpression, the half-life of the endogenous NQO1 was dramatically decreased (Fig. 1D), as measured by inhibiting translation with cycloheximide and examining the degradation of NQO1 in a time-kinetic manner. These results suggest that STUB1 is an NQO1 E3 ligase.
NQO1 Interaction with STUB1 Requires the STUB1 TPR Domain
STUB1/CHIP was originally described as an Hsc70 co-chaperone containing an E3 ligase activity and a significant player in the cellular quality control machinery. The TPR domain of STUB1 mediates the interaction with the Hsc70 whereas the U-box domain contains the E3 ubiquitin ligase activity (17). To map the STUB1 domain that mediates NQO1 interaction, we co-transfected NQO1 in the presence of various STUB1 truncation mutants. These experiments revealed that NQO1 was co-immunoprecipitated with the full-length STUB1 and delta U-box domain (ΔE4) but not with the ΔTPR STUB1 mutant (Fig. 2). The latter mutant lacks the domain that binds Hsc70. These data suggest that the NQO1 binding to STUB1 is either direct or Hsc70-mediated.
FIGURE 2.
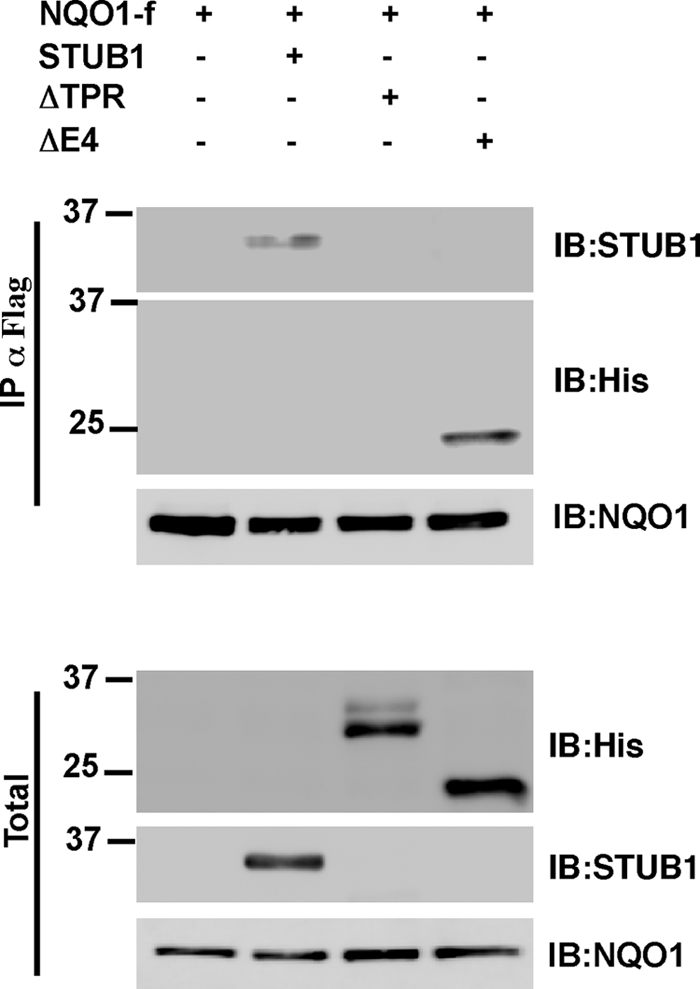
TPR domain of STUB1 is necessary for NQO1 binding. WT STUB1 and the ΔTPR and ΔE4 STUB1 mutants were co-transfected with NQO1-f in HEK293T cells. Following treatment with 25 μm MG132 for 2 h NQO1-f was immunoprecipitated (IP) with FLAG beads. The immunoprecipitated complexes were released with FLAG peptide elution, and the total input (Total) and the co-immunoprecipitation of the various forms of STUB1 were detected. IB, immunoblotting.
NQO1 C609T Is Highly Sensitized to STUB1-mediated Ubiquitination and Degradation
NQO1 C609T polymorphism is highly susceptible to ubiquitination and proteasomal degradation (15, 16). To validate this observation in our experimental system we determined the half-life of the WT NQO1 and polymorphic form NQO1 P187S by conducting a metabolic 35S-labeled pulse-chase experiment. The results are in good agreement with the fact that NQO1 P187S is a highly labile protein degraded with much higher kinetics than the WT NQO1 (Fig. 3A). Unlike the WT NQO1, NQO1 P187S polymorphic form does not bind Hsp70. It was suggested that the defect in Hsp70 binding is responsible for its instability (15). Hsc70, the STUB1-interacting protein, is a member of the Hsp70 family bearing ∼85% homology in sequence and constitutively expressed, whereas Hsp70 is an inducible gene responding to heat or other stressors. Recently, the differential cellular roles of Hsc70 and Hsp70 have been demonstrated (32), leading us to hypothesize that the two might also have distinct roles in the NQO1 P187S regulation. To examine whether NQO1 P187S (NQO1 C609T gene) can bind STUB1, we overexpressed FLAG-tagged WT or NQO1 P187S either alone or in the presence of STUB1 in HEK293T cells. Cell extracts were subjected to immunoprecipitation with anti-FLAG antibody. Surprisingly, we found that STUB1 was co-immunoprecipitated with NQO1 P187S with a much higher efficiency than with the WT NQO1 (Fig. 3B). Furthermore, the endogenous Hsc70, the mediator of the interaction with STUB1, was also co-immunoprecipitated with NQO1 P187S to a much higher extent than with the WT NQO1. These data hint toward the possibility that NQO1 P187S is a better STUB1 substrate. To challenge this prediction, we overexpressed STUB1 and HA-ubiquitin to detect ubiquitinated transfected NQO1. Remarkably, under this condition much higher levels of NQO1 P187S were polyubiquitinated compared with the WT NQO1 (Fig. 3C). Note that under this low exposure the ubiquitinated NQO1 is barely seen. To examine whether STUB1 sensitizes NQO1 P187S to degradation we conducted three independent pulse-chase experiments of NQO1 P187S in the presence or absence of STUB1 (Fig. 3D). STUB1 markedly increased the susceptibility of NQO1 P187S to degradation to reach a much shorter half-life. These results suggest that NQO1 P187S is under tight regulation of the Hsc70 chaperone-mediated STUB1-dependent ubiquitination and degradation.
FIGURE 3.

P187S NQO1 is highly susceptible to STUB1-mediated ubiquitination and degradation. A, WT NQO1 and P187S NQO1 were overexpressed in HEK293T cells, and their half-life was analyzed by pulse-chase experiment. WT and P187S NQO1 were immunoprecipitated (IP), and the decay of the radiolabeled NQO1 (35S) was detected by autoradiography. Immunoprecipitated NQO1 was detected by immunoblotting (IB) (NQO1). B, binding of STUB1 to NQO1 and P187S NQO1 was analyzed by overexpression of either WT FLAG-NQO1 or FLAG-P187S NQO1 in HEK293T cells in the presence or absence of STUB1. Following treatment with 25 μm MG132 for 2 h, NQO1 and P187S NQO1 were immunoprecipitated, and the level of total input protein (Total) and the co-immunoprecipitated STUB1 and endogenous Hsc70 were analyzed. C, ubiquitination of WT and P187S NQO1 by STUB1 was determined by overexpressing WT FLAG-NQO1 or FLAG-P187S NQO1 in HEK293T cells with HA-ubiquitin and in the presence or absence of increasing concentrations of STUB1. Following treatment with 25 μm MG132 for 2 h NQO1 and P187S NQO1 were immunoprecipitated (αFLAG). Total input (Total) and the level of ubiquitination were detected by probing with anti-HA antibody (IP). D, half-life of P187S NQO1 in the presence or absence of STUB1 was determined by conducting pulse-chase experiment (as described under “Experimental Procedures”). The decay of overexpressed P187S NQO1 in the presence or absence of STUB1 was quantified from three experiments, and the calculated averages with standard deviations (as the error bars) were plotted.
Increase in NQO1 Protein Level Observed in Aged Brain Correlates with STUB1 Decrease
It has been suggested that the STUB1 level is down-regulated with aging (33). Indeed, when examining STUB1 protein levels in the brain of mice of different ages (1, 12, and 36 weeks old) a decrease in STUB1 protein levels was observed with aging whereas the level of its interacting proteins Hsc70 remained unchanged (Fig. 4A). The decrease in the STUB1 levels from 1 to 12 weeks and 1 to 36 weeks was analyzed and shown to be statistically significant (paired t test, p = 0.0053 and 0.023, respectively) (Fig. 4B). Interestingly, STUB1 mRNA did not decrease with aging (Fig. 4C), suggesting that STUB1 undergoes posttranscriptional regulation by a yet unknown mechanism.
FIGURE 4.

Increase in NQO1 protein levels correlated with STUB1 decrease in the aged brain. A, equal amounts of proteins from mouse brains of different ages (1, 12, and 36 weeks) were analyzed for NQO1, STUB1, and Hsc70 protein levels by immunoblot analysis (IB) with goat anti-NQO1, rabbit anti-STUB1, and rabbit anti-Hsc70. B and D, protein level and STUB1/Hsc70 (B) and NQO1/Hsc70 (D) protein ratio were determined and quantified with ImageQuantTM TL. The average ± S.D. (error bars) of each age group are presented. *, p < 0.05; **, p < 0.01 compared with the 1 week group. C and E, real-time PCR analysis of STUB1 (C) and NQO1 (E) transcripts were conducted with RNA isolated from mouse brains of different ages (1, 12, and 36 weeks) as indicated.
Given the decrease in the level of STUB1 we predicted that the level of NQO1 has to increase. To this end the NQO1 protein level in the very same age groups was examined. Remarkably, the NQO1 protein level significantly increased in aged mouse brains, exhibiting up to an ∼10-fold increase from 1 week to 36 weeks (Fig. 4A). The increase in the NQO1 levels from 1 week to 12 weeks and 1 week to 36 weeks was statistically significant (paired t test, p = 0.006 and 0.002, respectively) (Fig. 4D). However, it is well documented that the NQO1 level is mostly regulated at the level of transcription, and not much is known on NQO1 accumulation. The inverse correlation between the level of STUB1 and NQO1 hints toward a posttranscriptional mechanism of NQO1 accumulation. To challenge this prediction, mRNA was extracted from these mice and quantified. Remarkably, real-time RT-PCR analysis revealed that mRNA levels, although exhibiting only a limited increase, cannot explain the observed increase at the level of the protein in the aged brain samples (Fig. 4E), suggesting that age-dependent NQO1 accumulation is in part a posttranscriptional process. Our finding of specific inverse correlation between the levels of STUB1 and NQO1 in the brain might offer an explanation for the age-dependent increase of NQO1.
NQO1 Protein Levels Are Undetectable in Hippocampus of Some AD Patients
Many have linked AD pathologies to oxidative stress (34, 35). NQO1 as a regulator of cellular redox state has also been connected to AD (36, 37). Furthermore, STUB1 has been also suggested to be important in preventing the onset of AD (38). Given the high levels of NQO1 protein in the aged brain and low levels of STUB1, we examined NQO1 levels in the hippocampus of AD patients and compared them with those in nondemented controls. Protein samples were prepared, and the NQO1 levels were examined by Western blot analysis with different antibodies. As expected, phosphorylated tau (AT8) was abundant in AD samples (Fig. 5A). Surprisingly, in half of the tested AD samples, the NQO1 protein level was too low to be detected (Fig. 5A), although there was no apparent difference in the STUB1 protein levels (data not shown). We next examined whether the poor expression of NQO1 in the AD samples is because these patients carry the polymorphic C609T form of NQO1. Remarkably, the lack of NQO1 expression was correlated one to one with the presence of the double allelic C609T NQO1 polymorphism (Fig. 5B). The product of the NQO1 C609T polymorphic gene is an efficient STUB1 substrate and therefore highly labile. This may explain why NQO1 protein levels could not be detected by immunoblotting in samples containing only the polymorphic form of NQO1. Furthermore, although the number of samples tested here (six of each group) is relatively small, these results suggest that NQO1 inactivation might be correlated with AD pathology onset.
FIGURE 5.

NQO1 protein levels are undetectable in some AD patient hippocampus. A, NQO1 protein levels in the hippocampus of six examined AD patients are shown and compared with six examined nondemented controls (see “Experimental Procedures”). Immunoblot analysis (IB) was carried out with goat anti-NQO1 and mouse anti-actin and mouse anti-AT8. B, genotype pattern of NQO1 was analyzed by PCR and digestion by HinfI. WT NQO1 and polymorphic C609T plasmids were used as controls.
DISCUSSION
In this study we show that STUB1 is a novel E3 ligase for NQO1 and to the best of our knowledge the first E3 ligase to be described for NQO1. A number of findings support the possibility that STUB1 is a likely NQO1 E3 ligase. First, we found that STUB1 binds NQO1 via the TPR domain that is involved in targeting client proteins. Second, we demonstrated that in the presence of STUB1, NQO1 undergoes polyubiquitination. Finally, we showed that in the presence of STUB1 NQO1 stability is sharply reduced. Furthermore, the polymorphic NQO1 P187S binds to STUB1 with higher affinity, resulting in hyperubiquitination and shorter half-life, suggesting that STUB1 is responsible for the instability of the NQO1 P187S. We further show that in the brain STUB1 level is decreased whereas that of NQO1 markedly increased with aging. Consistent with our model, we show that the increase at the level of NQO1 is at least in part has to do with protein accumulation. Surprisingly, in half of the examined hippocampi of AD patients there were undetectable levels of NQO1 due to C609T polymorphism. Thus, our demonstration of the regulation of NQO1 on the protein level by the E3 ligase STUB1 might be of significance in age-associated pathologies in the brain such as AD.
NQO1 is an antioxidant enzyme with several protective roles that might be especially important for the aged brain. Oxidative stress that overcomes the antioxidant defenses can cause cell death and neurodegenerative diseases (34, 35). NQO1 plays a role in the maintenance of the cellular redox state by the reduction of various quinones and prevention of reactive oxygen species accumulation (39, 40). NQO1 also protects p53 from proteasomal degradation by default as a protective mechanism (5, 41, 42). In addition, NQO1 can also alter the NADH/NAD+ balance in the cell (25), a balance that regulates life span and neuroprotective processes (43, 44). Therefore, the observed age-dependent elevation of NQO1 might be an important cellular protection mechanism. Following elevation of reactive oxygen species, NQO1 transcription is induced by the transcription factor Nrf2 (14), but this is not the case in the aged organs. We show here that the age-dependent NQO1 accumulation is a transcription-independent process. Recently, a reported microarray analysis of the aged brain showed no significant alteration in NQO1 expression levels in the aged brain (45), which is in agreement with our results. The basal level of NQO1 mRNA varies between different tissues and is significantly higher in the liver than in the brain (14), suggesting that the NQO1 promoter responds to certain tissue-specific transcription factors. However, so far no evidence for transcription-independent NQO1 accumulation was documented, and little is known about mechanisms that control NQO1 posttranslational regulation.
The NQO1 C609T polymorphic allele is present in the heterozygous form in up to 45% and in the homozygous form in up to 7% of the population (46, 47) and is associated with the susceptibility to various cancers (10, 11, 48). It is therefore important to understand the mechanism that renders the NQO1 P187S protein (encoded by NQO1 C609T polymorphic allele) unstable. It was shown previously that unlike WT NQO1, the P187S polymorphic protein does not bind Hsp70 (15). It was suggested that newly synthesized NQO1 is stabilized via binding to Hsp70, and the lack of Hsp70 binding by NQO1 P187S may explain its rapid degradation. We show here that NQO1 P187S can bind the constitutively expressed form of Hsp70, namely Hsc70, to a higher extent than WT NQO1. Hsc70 and Hsp70 are two distinct proteins playing unique roles while sharing high sequence homology (32). It seems that in the case of NQO1 there is also a differential regulation by these two Hsp70 family members; the inducible Hsp70 stabilizes only the WT NQO1 whereas the constitutively expressed Hsc70 mediates ubiquitination of both WT and NQO1 P187S by STUB1. It would be interesting to explore further whether this dual role of the Hsp70 family members is a more common phenomenon.
STUB1 appears to play important roles in the aged brain. Several of the STUB1 substrates such as mutant keratins (49), tau (38), and α-synuclein (50) form aggregates in numerous neurological pathologies, therefore STUB1 could be a regulator of neurodegeneration onset (31). Furthermore, STUB1 has been implicated in cancer. Recently, it was shown that STUB1 can induce the ubiquitination and degradation of both WT p53 (51) and mutant forms of p53 (52). As NQO1 stabilizes p53 by protecting its ubiquitin-independent degradation (5), a high level of STUB1 may facilitate ubiquitin-independent p53 degradation as well by decreasing NQO1 levels. The ability of STUB1 to down-regulate some oncogenic proteins led to the suggestion that STUB1 is an upstream regulator of oncogenic pathways, and it was indeed shown that STUB1 can inhibit tumor progression (53). Thus, the interplay between STUB1-Hsc70 and NQO1 could be of significance in cancer pathologies as well.
The high levels of NQO1 in the brain could suggest that NQO1 has an important role in neuronal setting. It has been shown that NQO1 levels increase due to calorie restriction in aged brains, suggesting that high levels of NQO1 could be indeed neuroprotective (54). In age-associated pathologies such as AD, NQO1 protein level is reported to be high in some histological analysis (36, 37). On the other hand, studies on correlating AD with NQO1 C609T polymorphism proved inconclusive (13, 55). Furthermore, genetic screening did not find NQO1 gene expression to be associated with the onset of AD (56). In this work, we show that NQO1 protein levels are high in the aged brain, and surprisingly the polymorphic C609T NQO1 that is highly unstable was present in 50% of our tested AD sample. Although our sample is not big enough to draw a valid statistical conclusion there seems to be a strong indication that polymorphic C609T NQO1 might be correlated with AD pathology.
In many aspects, NQO1 can be considered neuroprotective. NQO1 maintains the cellular redox state by reducing various possibly toxic quinones (1, 39), by regulating protein degradation (4, 5, 57), and increasing the cellular NAD+/NADH ratio (25). Therefore, inactivation of NQO1 by pharmacological interventions or by genetic mutations could well correlate with the onset of age-associated pathologies in the brain such as AD and Parkinson disease. It would be further interesting to explore whether such a correlation exists in human samples and to look further into the upstream effectors of NQO1 stability, such as STUB1 in relation to the onset of AD and possibly other neurodegenerative disorders.
Acknowledgments
We thank Prof. Cam Patterson for the STUB1 plasmids and siRNA, Dr. Nina Reuven for critical reading of the manuscript, and Ayelet Fohrer and Michal Elbaz for technical assistance.
This work was supported by Grant 1040/03-18.2 from the Israel Science Foundation and a grant from the Minerva Foundation with funding from the Federal German Ministry for Education and Research.
- NQO1
- NAD(P)H:quinone oxidoreductase 1
- AD
- Alzheimer disease
- E4
- U-box domain
- NQO1-f
- NQO1 with a FLAG antigen at its C terminus) STUB1/CHIP, C terminus of Hsc70-interacting protein
- TPR
- tetratricopeptide repeat.
REFERENCES
- 1. Dinkova-Kostova A. T., Talalay P. (2010) Arch. Biochem. Biophys. 501, 116–123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ross D., Siegel D. (2004) Methods Enzymol. 382, 115–144 [DOI] [PubMed] [Google Scholar]
- 3. Talalay P., Dinkova-Kostova A. T. (2004) Methods Enzymol. 382, 355–364 [DOI] [PubMed] [Google Scholar]
- 4. Adler J., Reuven N., Kahana C., Shaul Y. (2010) Mol. Cell. Biol. 30, 3767–3778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Asher G., Tsvetkov P., Kahana C., Shaul Y. (2005) Genes Dev. 19, 316–321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Garate M., Wong R. P., Campos E. I., Wang Y., Li G. (2008) EMBO Rep. 9, 576–581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sollner S., Nebauer R., Ehammer H., Prem A., Deller S., Palfey B. A., Daum G., Macheroux P. (2007) FEBS J. 274, 1328–1339 [DOI] [PubMed] [Google Scholar]
- 8. Sollner S., Schober M., Wagner A., Prem A., Lorkova L., Palfey B. A., Groll M., Macheroux P. (2009) EMBO Rep. 10, 65–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Moran J. L., Siegel D., Ross D. (1999) Proc. Natl. Acad. Sci. U.S.A. 96, 8150–8155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lafuente M. J., Casterad X., Trias M., Ascaso C., Molina R., Ballesta A., Zheng S., Wiencke J. K., Lafuente A. (2000) Carcinogenesis 21, 1813–1819 [DOI] [PubMed] [Google Scholar]
- 11. Smith M. T., Wang Y., Kane E., Rollinson S., Wiemels J. L., Roman E., Roddam P., Cartwright R., Morgan G. (2001) Blood 97, 1422–1426 [DOI] [PubMed] [Google Scholar]
- 12. Wiemels J. L., Pagnamenta A., Taylor G. M., Eden O. B., Alexander F. E., Greaves M. F. (1999) Cancer Res. 59, 4095–4099 [PubMed] [Google Scholar]
- 13. Bian J. T., Zhao H. L., Zhang Z. X., Bi X. H., Zhang J. W. (2008) J. Mol. Neurosci. 34, 235–240 [DOI] [PubMed] [Google Scholar]
- 14. Jaiswal A. K. (2000) Free Radic. Biol. Med. 29, 254–262 [DOI] [PubMed] [Google Scholar]
- 15. Anwar A., Siegel D., Kepa J. K., Ross D. (2002) J. Biol. Chem. 277, 14060–14067 [DOI] [PubMed] [Google Scholar]
- 16. Siegel D., Anwar A., Winski S. L., Kepa J. K., Zolman K. L., Ross D. (2001) Mol. Pharmacol. 59, 263–268 [DOI] [PubMed] [Google Scholar]
- 17. Ballinger C. A., Connell P., Wu Y., Hu Z., Thompson L. J., Yin L. Y., Patterson C. (1999) Mol. Cell. Biol. 19, 4535–4545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Qian S. B., McDonough H., Boellmann F., Cyr D. M., Patterson C. (2006) Nature 440, 551–555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Connell P., Ballinger C. A., Jiang J., Wu Y., Thompson L. J., Höhfeld J., Patterson C. (2001) Nat. Cell Biol. 3, 93–96 [DOI] [PubMed] [Google Scholar]
- 20. Dickey C. A., Kamal A., Lundgren K., Klosak N., Bailey R. M., Dunmore J., Ash P., Shoraka S., Zlatkovic J., Eckman C. B., Patterson C., Dickson D. W., Nahman N. S., Jr., Hutton M., Burrows F., Petrucelli L. (2007) J. Clin. Invest. 117, 648–658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Al-Ramahi I., Lam Y. C., Chen H. K., de Gouyon B., Zhang M., Pérez A. M., Branco J., de Haro M., Patterson C., Zoghbi H. Y., Botas J. (2006) J. Biol. Chem. 281, 26714–26724 [DOI] [PubMed] [Google Scholar]
- 22. Meacham G. C., Patterson C., Zhang W., Younger J. M., Cyr D. M. (2001) Nat. Cell Biol. 3, 100–105 [DOI] [PubMed] [Google Scholar]
- 23. Ko H. S., Bailey R., Smith W. W., Liu Z., Shin J. H., Lee Y. I., Zhang Y. J., Jiang H., Ross C. A., Moore D. J., Patterson C., Petrucelli L., Dawson T. M., Dawson V. L. (2009) Proc. Natl. Acad. Sci. U.S.A. 106, 2897–2902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Friguet B. (2006) FEBS Lett. 580, 2910–2916 [DOI] [PubMed] [Google Scholar]
- 25. Gaikwad A., Long D. J., 2nd, Stringer J. L., Jaiswal A. K. (2001) J. Biol. Chem. 276, 22559–22564 [DOI] [PubMed] [Google Scholar]
- 26. Elliott E., Tsvetkov P., Ginzburg I. (2007) J. Biol. Chem. 282, 37276–37284 [DOI] [PubMed] [Google Scholar]
- 27. Hwang J. R., Zhang C., Patterson C. (2005) Cell Stress Chaperones 10, 147–156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Elliott E., Laufer O., Ginzburg I. (2009) J. Neurochem. 109, 1168–1178 [DOI] [PubMed] [Google Scholar]
- 29. Tsvetkov P., Reuven N., Prives C., Shaul Y. (2009) J. Biol. Chem. 284, 26234–26242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Chen H., Lum A., Seifried A., Wilkens L. R., Le Marchand L. (1999) Cancer Res. 59, 3045–3048 [PubMed] [Google Scholar]
- 31. Dickey C. A., Patterson C., Dickson D., Petrucelli L. (2007) Trends Mol. Med. 13, 32–38 [DOI] [PubMed] [Google Scholar]
- 32. Goldfarb S. B., Kashlan O. B., Watkins J. N., Suaud L., Yan W., Kleyman T. R., Rubenstein R. C. (2006) Proc. Natl. Acad. Sci. U.S.A. 103, 5817–5822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Cheng X. R., Zhou W. X., Zhang Y. X., Zhou D. S., Yang R. F., Chen L. F. (2007) Neurobiol. Aging 28, 497–506 [DOI] [PubMed] [Google Scholar]
- 34. Markesbery W. R. (1997) Free Radic. Biol. Med. 23, 134–147 [DOI] [PubMed] [Google Scholar]
- 35. Varadarajan S., Yatin S., Aksenova M., Butterfield D. A. (2000) J. Struct. Biol. 130, 184–208 [DOI] [PubMed] [Google Scholar]
- 36. Raina A. K., Templeton D. J., Deak J. C., Perry G., Smith M. A. (1999) Redox Rep. 4, 23–27 [DOI] [PubMed] [Google Scholar]
- 37. SantaCruz K. S., Yazlovitskaya E., Collins J., Johnson J., DeCarli C. (2004) Neurobiol. Aging 25, 63–69 [DOI] [PubMed] [Google Scholar]
- 38. Shimura H., Schwartz D., Gygi S. P., Kosik K. S. (2004) J. Biol. Chem. 279, 4869–4876 [DOI] [PubMed] [Google Scholar]
- 39. Dinkova-Kostova A. T., Talalay P. (2000) Free Radic. Biol. Med. 29, 231–240 [DOI] [PubMed] [Google Scholar]
- 40. Radjendirane V., Joseph P., Lee Y. H., Kimura S., Klein-Szanto A. J., Gonzalez F. J., Jaiswal A. K. (1998) J. Biol. Chem. 273, 7382–7389 [DOI] [PubMed] [Google Scholar]
- 41. Asher G., Lotem J., Sachs L., Kahana C., Shaul Y. (2002) Proc. Natl. Acad. Sci. U.S.A. 99, 13125–13130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Tsvetkov P., Asher G., Reiss V., Shaul Y., Sachs L., Lotem J. (2005) Proc. Natl. Acad. Sci. U.S.A. 102, 5535–5540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Beal M. F. (2005) Ann. Neurol. 58, 495–505 [DOI] [PubMed] [Google Scholar]
- 44. St-Pierre J., Drori S., Uldry M., Silvaggi J. M., Rhee J., Jäger S., Handschin C., Zheng K., Lin J., Yang W., Simon D. K., Bachoo R., Spiegelman B. M. (2006) Cell 127, 397–408 [DOI] [PubMed] [Google Scholar]
- 45. Lu T., Pan Y., Kao S. Y., Li C., Kohane I., Chan J., Yankner B. A. (2004) Nature 429, 883–891 [DOI] [PubMed] [Google Scholar]
- 46. Gaedigk A., Tyndale R. F., Jurima-Romet M., Sellers E. M., Grant D. M., Leeder J. S. (1998) Pharmacogenetics 8, 305–313 [DOI] [PubMed] [Google Scholar]
- 47. Traver R. D., Siegel D., Beall H. D., Phillips R. M., Gibson N. W., Franklin W. A., Ross D. (1997) Br. J. Cancer 75, 69–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Menzel H. J., Sarmanova J., Soucek P., Berberich R., Grünewald K., Haun M., Kraft H. G. (2004) Br. J. Cancer 90, 1989–1994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Löffek S., Wöll S., Höhfeld J., Leube R. E., Has C., Bruckner-Tuderman L., Magin T. M. (2010) Hum. Mutat. 31, 466–476 [DOI] [PubMed] [Google Scholar]
- 50. Shin Y., Klucken J., Patterson C., Hyman B. T., McLean P. J. (2005) J. Biol. Chem. 280, 23727–23734 [DOI] [PubMed] [Google Scholar]
- 51. Esser C., Scheffner M., Höhfeld J. (2005) J. Biol. Chem. 280, 27443–27448 [DOI] [PubMed] [Google Scholar]
- 52. Muller P., Hrstka R., Coomber D., Lane D. P., Vojtesek B. (2008) Oncogene 27, 3371–3383 [DOI] [PubMed] [Google Scholar]
- 53. Kajiro M., Hirota R., Nakajima Y., Kawanowa K., So-ma K., Ito I., Yamaguchi Y., Ohie S. H., Kobayashi Y., Seino Y., Kawano M., Kawabe Y., Takei H., Hayashi S., Kurosumi M., Murayama A., Kimura K., Yanagisawa J. (2009) Nat. Cell Biol. 11, 312–319 [DOI] [PubMed] [Google Scholar]
- 54. Hyun D. H., Emerson S. S., Jo D. G., Mattson M. P., de Cabo R. (2006) Proc. Natl. Acad. Sci. U.S.A. 103, 19908–19912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Wang B., Jin F., Xie Y., Tang Y., Kan R., Zheng C., Yang Z., Wang L. (2006) Neurosci. Lett 409, 179–181 [DOI] [PubMed] [Google Scholar]
- 56. Blalock E. M., Geddes J. W., Chen K. C., Porter N. M., Markesbery W. R., Landfield P. W. (2004) Proc. Natl. Acad. Sci. U.S.A. 101, 2173–2178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Tsvetkov P., Reuven N., Shaul Y. (2010) Cell Death Differ. 17, 103–108 [DOI] [PubMed] [Google Scholar]