

Abstract
ATP-binding cassette transporter A1 (ABCA1) is a membrane-bound protein that regulates the efflux of cholesterol derived from internalized lipoproteins. Using a mouse macrophage cell line, this report studied the impact of low-density lipoproteins (LDL) on ABCA1 expression and the signaling pathway responsible for lipoprotein-induced ABCA1 expression. Our data demonstrated that treatment of macrophages with LDL increased ABCA1 mRNA and protein levels 4.3- and 3.5-fold, respectively. LDL also induced an ∼2-fold increase in macrophage surface expression of ABCA1 and a 14-fold-increase in apolipoprotein AI-mediated cholesterol efflux. In addition, LDL significantly increased the level of phosphorylated specificity protein 1 (Sp1) and the amount of Sp1 bound to the ABCA1 promoter without alteration in total Sp1 protein level. Mutation of the Sp1 binding site in the ABCA1 promoter and inhibition of Sp1 DNA binding with mithramycin A suppressed the ABCA1 promoter activity and reduced the ABCA1 expression level induced by LDL. LDL treatment also elevated protein kinase C-ζ (PKC-ζ) phosphorylation and induced PKC-ζ binding with Sp1. Inhibition of PKC-ζ with kinase inhibitors or overexpression of kinase-dead PKC-ζ attenuated Sp1 phosphorylation and ABCA1 expression induced by LDL. These results demonstrate for the first time that activation of the PKCζ-Sp1 signaling cascade is a mechanism for regulation of LDL-induced ABCA1 expression.
Keywords: ABC Transporter, Low Density Lipoprotein (LDL), Macrophage, Protein Kinase C (PKC), Sp1
Introduction
Low-density lipoproteins (LDL)3 could infiltrate from plasma into the arterial intima, where they could undergo oxidative modifications and therefore initiate atherogenic events (1). Under physiological conditions, arterial resident macrophages function as scavengers by endocytosis of the lipoprotein deposits (2). In this process, efficient efflux of lipoprotein-derived cholesterols is essential for prevention of macrophage cholesterol accumulation and atherosclerosis development (2). ATP-binding cassette transporter A1 (ABCA1) is one of the plasma membrane proteins that export excess cholesterol derived from internalized lipoproteins (3–6). Mutation of ABCA1 in humans has been shown to induce cholesterol accumulation in cells of the reticuloendothelial system and increase susceptibility to atherosclerosis (7). Specific disruption of the macrophage ABCA1 gene aggravates atherosclerosis development in LDL receptor knockout (LDLR−/−) mice (3), and macrophages obtained from ABCA1 knockout mice are deficient in cholesterol efflux (4). In contrast, overexpression of ABCA1 inhibits atherosclerosis in mouse models (5, 6). Likewise, macrophages obtained from transgenic mice that overexpress ABCA1 have an elevated cholesterol efflux compared with normal macrophages (5). These studies clearly indicate an antiatherogenic role for ABCA1, especially ABCA1 in the macrophages.
The cellular ABCA1 protein level is regulated by transcriptional activation and protein degradation (8). The transcription of ABCA1 is regulated by nuclear transcription factor liver X receptor (LXR) (9). Two isoforms of LXR have been identified in mammalian cells. LXRα is primarily expressed in liver, kidney, macrophages, and intestine, whereas LXRβ is expressed ubiquitously (10). Several oxysterols have been shown to function as ligands to bind and activate LXR (11). The oxysterol-activated LXRs form heterodimers with retinoid X receptors (RXR) and bind to the ABCA1 promoter, inducing transcription (9, 11). In addition to LXR and RXR, transcription factor specificity protein 1 (Sp1) has been suggested to be a regulator in ABCA1 expression. A recent study showed that treatment of cells with oxysterols induced physical interaction of Sp1 with the LXR and RXR heterodimer in the ABCA1 promoter (12). Inhibition of Sp1 DNA binding attenuated oxysterol-induced ABCA1 expression (12). These observations suggest that oxysterol-induced ABCA1 expression is modulated by Sp1. However, the mechanism of how Sp1 is involved in the regulation of ABCA1 expression is still not clear.
It is known that phosphorylation of Sp1 increases its DNA binding and transcription activity (12). Several protein kinases, including DNA-dependent protein kinase, protein kinase C-ζ (PKC-ζ), casein kinase II, ERK, and cyclin-dependent kinase 2 (Cdk2), have been shown to mediate Sp1 phosphorylation (12). Currently, the effect of Sp1 phosphorylation on ABCA1 expression remains unclear.
Uptake of native LDL has been shown to increase ABCA1 expression (13, 14). However, the mechanism responsible for native LDL-induced ABCA1 expression has not been defined. This report demonstrates for the first time that native LDL-induced ABCA1 expression is associated with an increased phosphorylation of Sp1 and PKC-ζ. Inhibition of PKC-ζ with kinase inhibitor or overexpression of kinase-dead PKC-ζ attenuates LDL-induced Sp1 phosphorylation and ABCA1 expression in macrophages. Taken together, our data suggest a novel pathway, except for LXRα, in regulating macrophage ABCA1 expression.
MATERIALS AND METHODS
Chemicals and Reagents
DMEM, FBS, and penicillin/streptomycin for cell culture were purchased from Invitrogen. Restriction enzymes and modifying enzymes (T4 DNA ligase and calf intestinal alkaline phosphatase, λ-protein phosphatase) were purchased from New England Biolabs (Beverly, MA). The luciferase assay system was purchased from Promega (Madison, WI). The Galacto-Startm β-galactosidase reporter gene assay system was purchased from Applied Biosystems (Foster City, CA). Pfu Ultra DNA polymerase was purchased from Stratagene (La Jolla, CA). Glutathione-Sepharose 4B and protein G-Sepharose were purchased from GE Healthcare-Amersham Biosciences. PI3K inhibitor 2-(4-morpholinyl)-8-phenyl-4H-1-benzopyran-4-one (LY294002), MEK inhibitor (U0126), and PKC inhibitor bisindolylmaleimide I hydrochloride (109203X) were purchased from Cell Signaling Technology (Boston, MA). The M-PER mammalian protein extraction reagent and BCA protein assay kit were purchased from Thermo Scientific. Protein A/G PLUS-agarose beads, rabbit IgG, antibodies against p-PKC-ζ, Sp1, ABCA1 (H-220), and β-actin, as well as horseradish peroxidase-conjugated secondary antibodies were purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). [1,2-3H(N)]cholesterol was obtained from PerkinElmer (Waltham, MA).
Animals and LDL Preparation
Wild-type C57BL/6 and LDL receptor-deficient (LDLR−/−) mice at 3–4 months of age were obtained from the Jackson Laboratory (Bar Harbor, ME) and used for collection of blood samples. To minimize oxidation, the collected blood was immediately mixed with 50 μm butylated hydroxytoluene and 2 mm EDTA and cooled on ice. All procedures were approved by the Institutional Animal Care and Use Committee at Meharry Medical College and were performed in accordance with the national law. Mouse plasma was overlaid with a potassium bromide gradient solution (d, 1.063) and centrifuged at 120,000 rpm for 2 h. LDL was collected, dialyzed in PBS (pH 7.4) containing 10 mm EDTA for 48 h at 4 °C, and filtered through a 0.45-μm filter (15).
Recombinant Plasmid Construction
For functional analysis of the ABCA1 promoter, four 5′-deletion and three mutant ABCA1 promoter fragments were generated by PCR using Pfu Ultra DNA polymerase. For generation of the deletion fragments, wild-type C57BL/6J mouse genomic DNA was used as a template. The PCR reverse primer for these fragments starts at nucleotide 205 downstream from the transcription start site of the ABCA1 gene. The forward primers for ABC-1094, ABC-257, ABC-125, and ABC-42 start at nucleotides −1094, −257, −125, and −42 upstream from the transcription start site of the ABCA1 gene, respectively. The ABC-125 fragment was used as a template to generate three mutant ABCA1 promoter fragments; i.e. an Sp1 binding site mutation, an LXRα binding site mutation, and an Sp1- and LXRα binding site double mutation. The PCR products were cloned into the KpnI-XhoI sites of the pGL2 luciferase vector (Promega).
For determination of the regulatory role of PKC-ζ on LDL-induced ABCA1 expression, rat PKC-ζ expression plasmid vector (FLAG.PKC-ζ) was purchased from Addgene, Inc. (Cambridge, MA). The PKC-ζ kinase-dead expression vector was generated by site-directed mutagenesis using the FLAG.PKC-ζ plasmid as a temperate and using the oligo-nucleotide cagatttacgccatgtgggtggtgaagaaggag to substitute PKC-ζ lysine 281 with tryptophan (16).
Luciferase and β-Galactosidase Assays
Luciferase and β-galactosidase assays were performed as described previously (17). Briefly, RAW264.7 cells were transfected with a β-galactosidase expression plasmid and the ABCA1 promoter-reporter constructs or the empty pGL-2 vector. The transfected cells were treated with 20 μg/ml LDL for 4 h and lysed with 100 μl of lysis buffer provided by the luciferase assay kit or the galactosidase assay kit (Galacto-Startm β-galactosidase reporter gene assay system). A 10-μl aliquot of lysate was incubated in a 96-well plate at room temperature for 2 min with 100 μl of luciferase assay reagent (Promega) or for 60 min with 100 μl of galactosidase assay reagent. Luminescence was measured using the BL10000 LumiCount (PerkinElmer, Meriden, CT). Luciferase activity was expressed as the luminosity ratio of the luciferase assay to the β-galactosidase assay.
ChIP
Quiescent RAW264.7 cells were treated with or without 20 μg/ml LDL for 4 h. Sp1 binding to the ABCA1 promoter region was determined by ChIP as described previously (17). Briefly, cells were crosslinked with 1% formaldehyde at room temperature and then lysed in 500 μl of cell lysis buffer (5 mm PIPES, 85 mm KCl, 0.5% Nonidet P-40, 1 mm PMSF, protease inhibitor mixture (pH 8.0)). Nuclei were isolated and homogenized in 300 μl of nuclear lysis buffer (50 mm Tris-HCl, 1% SDS, 10 mm EDTA, protease inhibitor mixture (pH 8.1)). The resulting nuclear lysate was sonicated until crosslinked chromatin was sheared to an average length of 0.3–1.0 kb. A 5-μl aliquot of supernatant was used as an input control. The remaining lysate was diluted 10-fold with ChIP dilution buffer (16.7 mm Tris-HCl, 167 mm NaCl, 0.01% SDS, 1.1% Triton X-100, 1.2 mm EDTA, protease inhibitor mixture (pH 8.1)) and precleaned with salmon sperm DNA/protein A-agarose (Santa Cruz Biotechnology). The precleaned sample was incubated with Sp1 antibody followed by salmon sperm DNA/protein A-agarose. Bound protein-DNA complexes were eluted with a solution containing 0.1 m NaHCO3 and 1% SDS. Following reversion of the protein-DNA crosslinks, DNA fragments in the eluate and input controls were purified using the QIAquick PCR purification kit (Qiagen, Valencia, CA). PCR amplification of a 220-bp fragment containing the Sp1 binding site sequence of the ABCA1 promoter region was performed with the following primers: forward, 5′-dTTCCCGTTTCCCGAAGGCTA-3′; and reverse, 5′-dAACGCTGTTCTCTCCCTCTT-3′. The amount of DNA-bound Sp1 was expressed as a ratio of the PCR product amplified from the anti-Sp1 immunoprecipitants versus that from the input controls.
Quantitative Real-time RT-PCR Assay
Raw264.7 cells grown to confluence in six-well plates were made quiescent in serum-free DMEM for 12 h and then treated with 20 μg/ml LDL or medium alone as a control for 4 h. Total RNA was extracted using TRIzol reagent (Invitrogen) and subjected to reverse transcription using the High Capacity cDNA reverse transcription kit (Applied Biosystems). The resulting cDNAs were subjected to quantitative real-time PCR with an iCycler system (Bio-Bad) using primers synthesized by Qiagen for amplification of ABCA1 and GAPDH (18). The expression levels of the ABCA1 mRNAs were normalized to GAPDH mRNA.
Western Blot Analysis
Quiescent Raw264.7 cells in serum-free DMEM were treated with 20 μg/ml LDL or medium alone as a control for 4 h and then lysed in M-PER mammalian protein extraction reagent (Thermo Scientific, Rockford, IL). Samples containing 40 μg of protein were resolved on 6% (for separation of phosphorylated and non-phosphorylated Sp1) or 10% SDS-PAGE gels (for separation of other proteins). Proteins were transferred to a PVDF membrane (Millipore, Billerica, MA). After blocking with 5% fat-free milk, the membranes were incubated with antibodies as indicated (18). Immunoreactive bands were visualized using the ECL-plus chemiluminescence reagent (GE Healthcare-Amersham Biosciences) and analyzed with a GS-700 imaging densitometer (Bio-Rad).
ELISA
The expression of ABCA1 on the macrophage surface was determined with ELISA as described previously (19). Raw 264.7 cells grown in 96-well plates at confluence were treated with 20 μg/ml LDL or medium alone as a control for 4 h. After fixing with 0.1% glutaraldehyde, the cells were incubated with 100 μl of ABCA1 antibody or nonspecific IgG as a control for 1 h, followed by incubation with peroxidase-conjugated secondary antibody for 1 h. The ELISA was developed using 100 μl of 0.1 mg/ml 3,3′,5,5′-tetramethylbenzidine (Sigma) with 0.003% H2O2 for ∼30 min at room temperature. The reaction was stopped by the addition of 25 μl of 8 n sulfuric acid. The absorbance reading was recorded at 450 nm using a microplate reader (Dynex Technologies, Chantilly, VA). The optical densities of the wells incubated with nonspecific IgG were subtracted from those of the wells incubated with ABCA1 antibody.
Cholesterol Efflux Assay
Raw 264.7 cells grown in 24-well plates were incubated for 24 h with 5 μCi/ml of [1,2-3H(N)]cholesterol in the presence or absence of 20 μg/ml LDL (20). After washing with PBS, the cells were further incubated with 20 μg/ml human ApoAI (Sigma-Aldrich) or culture medium. After a 2-h incubation, the culture medium was collected. Cells were lysed with 0.5 m NaOH. The lysate and medium were mixed with scintillation fluid for a radioactivity assay using a Tri-Carb 2300TR liquid scintillation analyzer (PerkinElmer). Cholesterol efflux was expressed as the percentage of radioactivity in medium to total radioactivity (cells plus medium). ApoAI-mediated cholesterol efflux was calculated as the difference between the values obtained in the presence or absence of ApoAI in the medium.
Statistical Analysis
For experiments using the microplate reader, the mean value for each experiment was averaged from triplicate wells in the same plate. Data are reported as the mean ± S.E. Differences between treatments and controls were analyzed by analysis of variance and Student's unpaired t test. Statistical significance was considered when p was less than 0.05. Statistix software (Statistix, Tallahassee, FL) was used for statistical analysis.
RESULTS
LDL Induces Macrophage ABCA1 Expression and Cholesterol Efflux
Because of a small amount of LDL obtained from wild-type mice, this report used LDL from LDLR−/− mice. The results on ABCA1 expression and cholesterol efflux were confirmed using LDL from wild-type mice. We observed that the effect of LDL from wild-type and LDLR−/− mice on ABCA1 expression and cholesterol efflux was comparable. Fig. 1 shows the average data generated using LDL obtained from these two lines of mice. Data in Fig. 1, A–C show that treatment of macrophages with 20 μg/ml LDL for 4 h elevates the ABCA1 mRNA level by more than 4.3-fold, which is associated with a 3.5-fold increase in ABCA1 protein level. Correspondingly, LDL induced an ∼2-fold increase in ABCA1 expression on the surface of macrophages (Fig. 1D).
FIGURE 1.

LDL induces ABCA1 expression and cholesterol efflux. To study the effect of LDL on ABCA1 expression, mouse macrophages were incubated with 20 μg/ml LDL or culture medium alone. A and B, the level of ABCA1 protein was determined by Western blot analysis and quantitated relative to β-actin. C, the level of ABCA1 mRNA was determined by quantitative real-time RT-PCR and normalized to GAPDH mRNA. D, the cell surface level of ABCA1 was determined by ELISA. E, to study the effect of LDL on cholesterol efflux, mouse macrophages were incubated with [3H]cholesterol in the presence or absence of 20 μg/ml LDL. ApoAI-mediated cholesterol efflux was measured as described under “Materials and Methods.” Values represent the mean ± S.E. of six independent experiments. *, p < 0.05 compared with control.
It has been suggested that ABCA1 transports cholesterol from the cell membrane to lipid-free ApoAI (21). We therefore studied the effect of LDL on ApoAI-mediated cholesterol efflux. In the absence of LDL, efflux of cholesterol from macrophages to ApoAI is relatively low (Fig. 1E). Treatment of macrophages with LDL induced an ∼14-fold increase in ApoAI-mediated cholesterol efflux (Fig. 1E). Taken together, these results suggest that native LDL is able to activate the cholesterol efflux system that exports excess cholesterol from macrophages.
LDL Induces ABCA1 Promoter Activity
To study the mechanism underlying the increased ABCA1 expression in response to LDL treatment, we examined the ABCA1 promoter activity by transfection of macrophages with a series of ABCA1 promoter-luciferase reporter chimeric constructs (Fig. 2A). The data in Fig. 2A show that the basal luciferase activity in the cells transfected with the ABC-1094, ABC-257, and ABC-125 constructs is comparable and that LDL treatment induces an approximately 3.2-fold elevation in luciferase activity in all these three cell lines. The luciferase activity in cells transfected with the ABC-42 construct is relatively low. In addition, LDL treatment does not significantly induce luciferase activity in these cells (Fig. 2A). It therefore appears that the regulatory elements responsible for the basal and LDL-induced ABCA1 transcription are located between 125 and 42 nucleotides upstream of the transcription start site of the ABCA1 promoter.
FIGURE 2.

Functional analysis of the mouse ABCA1 promoter. A, four constructs containing different lengths of the ABCA1 promoter region were subcloned into a luciferase (Luc) reporter plasmid (pGL-2). Numbers in the schematic diagram indicate the relative positions to the transcription start site of the ABCA1 promoter. B, the wild-type (WT) ABC-125 construct was used as a template to generate the Sp1-mutated (Mut Sp1), LXR-mutated (Mut LXR), or the Sp1/LXR-mutated (Mut Sp1/LXR) ABCA1 promoter constructs. The mutated nucleotides are shown in the sequence graphic. Mouse macrophages were transfected with a β-galactosidase expression plasmid and the ABCA1 promoter-reporter constructs or the empty pGL-2 vector. The transfected macrophages were treated with 20 μg/ml LDL or culture medium alone as a control for 4 h. Luciferase activity was normalized to the β-galactosidase activity and expressed relative to that of the pGL2 basic vector. Values represent the mean ± S.E. of three independent experiments. *, p < 0.05 compared with WT; †, p < 0.05 compared with Mut Sp1 or Mut LXR alone.
It has been demonstrated that LXRα and Sp1 are transcription factors that control ABCA1 expression (9, 12). Computer analysis of the ABCA1 promoter shows an LXRα binding site and an Sp1 binding site, respectively, located 67 and 99 nucleotides upstream of the transcription start site. The impact of these DNA motifs on ABCA1 transcription was assessed using mutated promoter-reporter constructs (Fig. 2B). Data in Fig. 2B show that mutation of the LXRα or the Sp1 binding site alone, or combined mutation of the LXRα and the Sp1 binding sites, significantly reduces the expression of the reporter gene induced by lipoproteins and that the inhibitory effect induced by combinational mutation of the LXRα and the Sp1 binding sites was greater than those induced by mutation of either the LXRα and the Sp1 binding site alone. Specifically, mutation of the Sp1 or the LXRα motif alone reduces the luciferase activity by about 59 and 63%, respectively, when compared with that derived from the wild-type ABCA1 promoter counterpart, ABC-125 (Fig. 2B). Mutation of both LXRα and Sp1 motifs almost completely blocks LDL-induced luciferase activity (Fig. 2B). Taken together, both the Sp1 and LXRα motifs are essential for LDL-induced ABCA1 promoter activity.
LDL Increases Sp1 binding to the ABCA1 Promoter
To confirm the regulatory role of Sp1 in LDL-induced ABCA1 promoter activity, we determined the effect of LDL on the amount of Sp1 bound to the ABCA1 promoter with a ChIP assay. The typical image in Fig. 3A shows a faint PCR band in the untreated cells, suggesting few Sp1 proteins bound to the ABCA1 promoter under the control conditions. In contrast, a clear PCR band is visualized in LDL-treated cells. Quantitative data indicate that the amount of Sp1 proteins bound to the ABCA1 promoter is more than 2.5-fold higher in lipoprotein-treated cells than in control untreated cells (Fig. 3A). These results suggest that LDL increases Sp1 binding to its cognate regulatory element in the ABCA1 promoter region.
FIGURE 3.

Sp1-binding to the ABCA1 promoter is indispensible for LDL-induced ABCA1 expression. A, LDL increases Sp1-binding to the ABCA1 promoter. Mouse macrophages were incubated with 20 μg/ml LDL or medium alone as a control for 4 h. The amount of Sp1 bound to the ABCA1 promoter was detected using ChIP analysis and expressed as the ratio of input controls. Values represent the mean ± S.E. of five separate experiments. *, p < 0.05 compared with medium alone (control). B, mithramycin A inhibits ABCA1 gene induction by LDL. Macrophages were treated with 20 μg/ml LDL or culture medium in the absence or presence of 100 mm mithramycin A (mmA) for 10 h. The ABCA1 protein level was determined by Western blot analysis and expressed relative to the immunoblot intensity of β-actin. Values represent the mean ± S.E. of five separate experiments. *, p < 0.05 compared with medium with or without mmA; †, p < 0.05 compared with LDL treatment alone.
Mithramycin A Inhibits ABCA1 Expression
To address if Sp1 binding to the ABCA1 promoter is necessary for LDL-induced expression of ABCA1, we studied the effect of mithramycin A on ABCA1 expression. Mithramycin A is a chemotherapeutic drug that binds to GC-rich DNA sequences, thereby blocking the binding of transcription factors such as Sp1 to GC-specific regions of DNA (22). A wide range of concentrations of mithramycin A (10 nm-1 μm) have been used to inhibit the transcriptional factor activity of Sp1 in various types of cells, including human bronchial epithelial cells, multiple myeloma cells (22, 23), and mouse endothelial cells (17). As the data in Fig. 3B show, LDL treatment increases the protein level of ABCA1 in macrophages. Addition of 100 nm mithramycin A inhibits the lipoprotein-induced ABCA1 protein expression by about 43%. These data suggest that binding of the Sp1 protein to the ABCA1 promoter is crucial to LDL-induced ABCA1 expression.
LDL Induces Sp1 Phosphorylation
To uncover the mechanism underlying the increased Sp1 binding to the ABCA1 promoter, we examined the effect of LDL on the Sp1 protein level in macrophages. As the typical Western blot images in Fig. 4 illustrate, two Sp1 immunoreactive bands are detectable, indicating covalent modifications in Sp1 proteins. The immunoblotting images in Fig. 4 also show that LDL treatment does not significantly alter the total Sp1 protein level. However, a band shift is visualized in cells treated with lipoproteins (Fig. 4). Specifically, LDL treatment induces part of the Sp1 proteins to shift to the top band. To determine whether the slowly migrating forms of Sp1 are generated by phosphorylation, we incubated the cell lysates with λ-phosphatase before Western blot analysis. As shown in Fig. 4, λ-phosphatase abolishes the slowly migrating form of Sp1 induced by lipoproteins. These observations thus provide direct evidence that the LDL-induced shift of the Sp1 immunoreactive band is due to Sp1 phosphorylation.
FIGURE 4.
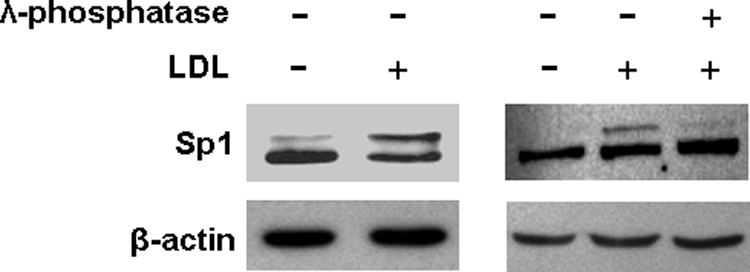
LDL induces Sp1 phosphorylation. Mouse macrophages were treated with 20 μg/ml LDL or medium alone as a control for 4 h. The lysates obtained from LDL-treated cells were incubated with or without λ-phosphatase. LDL-induced Sp1 phosphorylation was detected by Sp1 immunoblot band shift induced by the LDL and λ-phosphatase treatments.
Activation of PKC-ζ Is a Mechanism for LDL-induced Sp1 Phosphorylation and ABCA1 Expression
Having established the regulatory role of LDL on ABCA1 expression and Sp1 phosphorylation, we next identified the kinases responsible for LDL-induced Sp1 phosphorylation. Data in Fig. 5A show that treatment of macrophages with a MAP kinase inhibitor U0126 (24) does not affect Sp1 phosphorylation and ABCA1 protein expression. In contrast, 109203X, a chemical that has been shown to inhibit PKC-ζ activity (25), significantly reduces the level of ABCA1 protein and phosphorylated Sp1 in cells treated with LDL (Fig. 5A). In addition, 109203X also reduces ABCA1 promoter activity by about 48% (Fig. 5D). It has been reported that PI3K is a kinase that phosphorylates PKC-ζ (26). Data in Fig. 5A show that PI3K inhibitor LY294002 (27) attenuates Sp1 phosphorylation and ABCA1 protein expression induced by LDL. We also observed that inhibition of PI3K suppressed LDL-induced PKC-ζ phosphorylation (data not shown). These results suggest that activation of the PI3K-PKC-ζ cascade is critical for Sp1 phosphorylation and ABCA1 expression induced by LDL.
FIGURE 5.

Activation of PKC-ζ is a mechanism for LDL-induced Sp1 phosphorylation and ABCA1 expression. Macrophages were treated with 20 μg/ml LDL or medium alone as a control. The activity of protein kinases was inhibited with inhibitors as indicated (A and D) or transfected with kinase-dead PKC-ζ (PKC-ζ KD) (C and E). In the studies where PKC-ζ KD was used, macrophages were transfected with wild-type PKC-ζ (PKC-ζ WT) or an empty pEGFP vector as controls (C and E). The protein levels of ABCA1, Sp1, and total and phosphorylated PKC-ζ were determined by Western blot analysis (A–C). In the studies where the ABCA1 promoter activity was determined (D and E), macrophages were transfected with an ABCA1 promoter construct (ABC-125) and a β-galactosidase expression plasmid. Luciferase activity was measured using a luminescence assay and expressed relative to the luminosity of the β-galactosidase assay. Values represent the mean ± S.E. of three separate experiments. *, p < 0.05 compared with medium without LDL treatment; †, p < 0.05 compared with LDL treatment alone (D). *, p < 0.05 compared with cells transfected with empty vector and treated with LDL; †, p < 0.05 compared with cells transfected with PKC-ζ WT and treated with LDL (E).
This report further confirmed the regulatory role of PKC-ζ on Sp1 phosphorylation and ABCA1 expression. Data in Fig. 5B show that LDL significantly increases PKC-ζ phosphorylation but does not alter the total protein level of PKC-ζ. Transfection of macrophages with kinase-dead PKC-ζ attenuates Sp1 phosphorylation induced by LDL (Fig. 5C). In addition, kinase-dead PKC-ζ reduces ABCA1 promoter activity by 43% in cells treated with LDL (Fig. 5E). These observations provide direct evidence that activation of PKC-ζ is at least partially responsible for the increased Sp1 phosphorylation induced by LDL. It is highly likely that LDL activates PKC-ζ, which in turn phosphorylates Sp1, increasing its binding to the ABCA1 promoter and up-regulating ABCA1 transcription.
LDL Induces PKC-ζ and Sp1 Physical Interaction
To determine the physical interaction of PKC-ζ and Sp1, binding was tested in cells transfected with a FLAG.PKC-ζ expression vector. The images in Fig. 6 show that immunoprecipitation of the FLAG.PKC-ζ barely co-precipitates Sp1 under the control conditions. In contrast, a substantial amount of Sp1 is co-precipitated with the FLAG.PKC-ζ in cells treated with LDL, as shown by anti-Sp1 Western blotting (Fig. 6). These data suggest that LDL treatment induces PKC-ζ binding to Sp1, a reaction necessary for PKC-ζ to phosphorylate Sp1.
FIGURE 6.
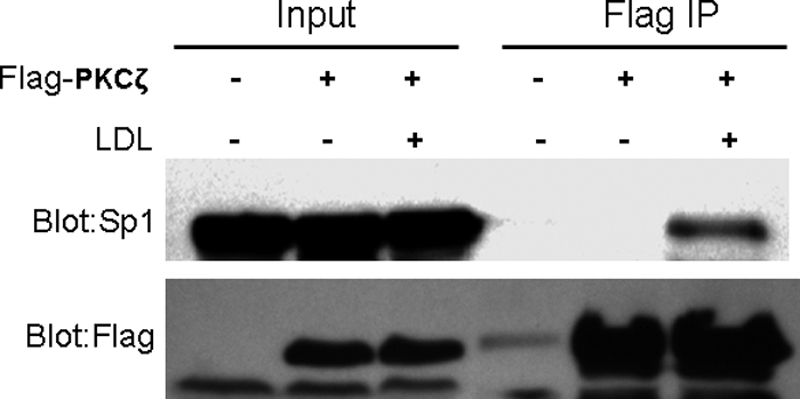
PKC-ζ physically interacts with Sp1. Macrophages were transfected with empty vector or FLAG-PKC-ζ expression vector for 48 h. The transfected cells were treated with 20 μg/ml LDL or medium alone for 4 h. The FLAG-tagged PKC-ζ protein was precipitated using FLAG-conjugated agarose beads. The precipitants were analyzed by Western blots with antibodies against Sp1 and FLAG.
DISCUSSION
In agreement with previous reports (13, 14), data from this study clearly indicate that native LDL increases macrophage ABCA1 expression and induces ApoAI-mediated cholesterol efflux. Additionally, our data demonstrate for the first time that an increase in Sp1 DNA binding activity is a mechanism underlying the increased ABCA1 expression in macrophages in response to native LDL treatment. It is known that Sp1 regulates basal and inducible transcription of many genes via binding to GC boxes (GGGCGG), GT motifs (GGGTGTGGC), or CT elements (CCTCCTCCTCCTCGGCCTCCTCCCC) in the promoter region of target genes (28). The mouse ABCA1 promoter contains one GC box located 99 nucleotides upstream the transcription start site. Our data demonstrated that LDL increased Sp1 binding to the ABCA1 promoter region and that mutation of the Sp1 binding motif attenuated the transcription activity of ABCA1 promoter induced by LDL. In addition, we observed that inhibition of Sp1 DNA binding by mithramycin A suppressed LDL-induced ABCA1 expression.
Another important finding from this report is that LDL increased Sp1 phosphorylation in macrophages. It has been demonstrated that an increase in Sp1 phosphorylation augments its DNA binding and transcription factor activity (12). In addition, our data demonstrate that activation of PKC-ζ is at least partially responsible for LDL-induced Sp1 phosphorylation. Specifically, treatment of macrophages with LDL increased PKC-ζ phosphorylation and induced physical interaction of PKC-ζ and Sp1. Transfection of macrophages with a kinase-dead PKC-ζ or treatment of the cells with a PKC inhibitor attenuated LDL-induced Sp1 phosphorylation and ABCA1 promoter activity. Moreover, we observed that LDL-induced PKC-ζ phosphorylation was reduced by inhibition of PI3K, which is a protein kinase that mediates PKC-ζ phosphorylation. It appears that activation of the PI3K-PKC-ζ cascade is critical for LDL-induced Sp1 phosphorylation and ABCA1 expression. It is highly likely that LDL sequentially activates PI3K and PKC-ζ, which in turn phosphorylate Sp1, increasing its binding to the ABCA1 promoter and up-regulating ABCA1 transcription.
Activation of LXR by internalized oxysterols has been suggested to be a mechanism for oxidized LDL-induced ABCA1 expression (29). Data from this report demonstrated that mutation of the LXR binding site diminished native LDL-induced ABCA1 promoter activity, suggesting an involvement of this regulatory motif in ABCA1 expression elicited by native LDL. It has been shown that Sp1 physically binds with LXR/RXR heterodimers in the ABCA1 promoter and that genetic deficiency of Sp1 or chemical inhibition of Sp1 DNA binding suppresses ligand-induced recruitment of the LXR/RXR heterodimer to the ABCA1 promoter and reduces ABCA1 expression (12). These observations suggest that Sp1 is able to regulate the recruitment of LXR/RXR heterodimer to the ABCA1 promoter. It is highly possible that LDL increases Sp1 binding to its cognate response element and therefore promotes the recruitment of LXR/RXR heterodimers to the ABCA1 promoter. We observed that combinational mutation of the Sp1 and LXR binding sites almost completely blocked LDL-induced ABCA1 promoter activity, supporting the view that a coordinate work of Sp1 and the LXR/RXR heterodimer is essential for a full induction of ABCA1 expression elicited by LDL.
In summary, data from this report clearly indicate that native LDL is able to induce ABCA1 expression and cholesterol efflux and that the increase in Sp1 phosphorylation is an underlying mechanism for LDL-induced ABCA1 expression. We also demonstrate that sequential activation of PI3K and PKC-ζ is at least partially responsible for LDL-induced Sp1 phosphorylation. It is highly likely that uptake of native LDL activates the PI3K-PKC-ζ-Sp1 signaling cascade, which in turn up-regulates ABCA1 expression and therefore enhances cholesterol efflux. Further study is needed to explore the specific mechanism through which native LDL activates the PI3K-PKC-ζ-Sp1 signaling cascade.
This study was supported, in whole or in part, by National Institutes of Health Grants R01ES014471 and R01HL089382 (to Z. G.) and SC1HL101431 and U54RR026140 (to H. Y.).
- LDL
- low-density lipoprotein(s)
- LXR
- liver X receptor
- RXR
- retinoid X receptor
- PKC-ζ
- protein kinase C-ζ
- PIPES
- 1,4-piperazinediethanesulfonic acid.
REFERENCES
- 1. Chisolm G. M., 3rd, Penn M. S. (1996) in Atherosclerosis and Coronary Artery Disease (Fuster V., Ross R., Topol E. J. eds) pp. 129–149, Lippincott-Raven, Philadelphia [Google Scholar]
- 2. Linton M. F., Fazio S. (2003) Int. J. Obes. Relat. Metab. Disord. 27, S35–S40 [DOI] [PubMed] [Google Scholar]
- 3. Van Eck M., Bos I. S., Kaminski W. E., Orsó E., Rothe G., Twisk J., Böttcher A., Van Amersfoort E. S., Christiansen-Weber T. A., Fung-Leung W. P., Van Berkel T. J., Schmitz G. (2002) Proc. Natl. Acad. Sci. U.S.A. 99, 6298–6303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Orsó E., Broccardo C., Kaminski W. E., Böttcher A., Liebisch G., Drobnik W., Götz A., Chambenoit O., Diederich W., Langmann T., Spruss T., Luciani M. F., Rothe G., Lackner K. J., Chimini G., Schmitz G. (2000) Nat. Genet. 24, 192–196 [DOI] [PubMed] [Google Scholar]
- 5. Van Eck M., Singaraja R. R., Ye D., Hildebrand R. B., James E. R., Hayden M. R., Van Berkel T. J. (2006) Arterioscler. Thromb. Vasc. Biol. 26, 929–934 [DOI] [PubMed] [Google Scholar]
- 6. Singaraja R. R., Fievet C., Castro G., James E. R., Hennuyer N., Clee S. M., Bissada N., Choy J. C., Fruchart J. C., McManus B. M., Staels B., Hayden M. R. (2002) J. Clin. Invest. 110, 35–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Brooks-Wilson A., Marcil M., Clee S. M., Zhang L. H., Roomp K., van Dam M., Yu L., Brewer C., Collins J. A., Molhuizen H. O., Loubser O., Ouelette B. F., Fichter K., Ashbourne-Excoffon K. J., Sensen C. W., Scherer S., Mott S., Denis M., Martindale D., Frohlich J., Morgan K., Koop B., Pimstone S., Kastelein J. J., Genest J., Jr., Hayden M. R. (1999) Nat. Genet. 22, 336–345 [DOI] [PubMed] [Google Scholar]
- 8. Wang N., Tall A. R. (2003) Arterioscler. Thromb. Vasc. Biol. 23, 1178–1184 [DOI] [PubMed] [Google Scholar]
- 9. Costet P., Luo Y., Wang N., Tall A. R. (2000) J. Biol. Chem. 275, 28240–28245 [DOI] [PubMed] [Google Scholar]
- 10. Repa J. J., Mangelsdorf D. J. (2002) Nat. Med. 8, 1243–1248 [DOI] [PubMed] [Google Scholar]
- 11. Janowski B. A., Grogan M. J., Jones S. A., Wisely G. B., Kliewer S. A., Corey E. J., Mangelsdorf D. J. (1999) Proc. Natl. Acad. Sci. U.S.A. 96, 266–271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Thymiakou E., Zannis V. I., Kardassis D. (2007) Biochemistry 46, 11473–11483 [DOI] [PubMed] [Google Scholar]
- 13. Chawla A., Boisvert W. A., Lee C. H., Laffitte B. A., Barak Y., Joseph S. B., Liao D., Nagy L., Edwards P. A., Curtiss L. K., Evans R. M., Tontonoz P. (2001) Mol. Cell 7, 161–171 [DOI] [PubMed] [Google Scholar]
- 14. Zhu Y., Liao H., Xie X., Yuan Y., Lee T. S., Wang N., Wang X., Shyy J. Y., Stemerman M. B. (2005) Cardiovasc. Res. 68, 425–432 [DOI] [PubMed] [Google Scholar]
- 15. Wu D., Sharan C., Yang H., Goodwin J. S., Zhou L., Grabowski G. A., Du H., Guo Z. (2007) J. Lipid Res. 48, 2571–2578 [DOI] [PubMed] [Google Scholar]
- 16. Chou M. M., Hou W., Johnson J., Graham L. K., Lee M. H., Chen C. S., Newton A. C., Schaffhausen B. S., Toker A. (1998) Curr. Biol. 8, 1069–1077 [DOI] [PubMed] [Google Scholar]
- 17. Tang T., Lin X., Yang H., Zhou L., Wang Z., Shan G., Guo Z. (2010) Free Radic. Biol. Med. 49, 487–492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wang Z., Yang H., Ramesh A., Roberts L. J., 2nd, Zhou L., Lin X., Zhao Y., Guo Z. (2009) Free Radic. Biol. Med. 47, 1221–1229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yang H., Shi M., Richardson A., Vijg J., Guo Z. (2003) Free Radic. Biol. Med. 35, 266–276 [DOI] [PubMed] [Google Scholar]
- 20. Dove D. E., Su Y. R., Zhang W., Jerome W. G., Swift L. L., Linton M. F., Fazio S. (2005) Arterioscler. Thromb. Vasc. Biol. 25, 128–134 [DOI] [PubMed] [Google Scholar]
- 21. Chen W., Sun Y., Welch C., Gorelik A., Leventhal A. R., Tabas I., Tall A. R. (2001) J. Biol. Chem. 276, 43564–43569 [DOI] [PubMed] [Google Scholar]
- 22. Blume S. W., Snyder R. C., Ray R., Thomas S., Koller C. A., Miller D. M. (1991) J. Clin. Invest. 88, 1613–1621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Reamon-Buettner S. M., Borlak J. (2008) Cancer Res. 68, 7587–7596 [DOI] [PubMed] [Google Scholar]
- 24. Marampon F., Bossi G., Ciccarelli C., Di Rocco A., Sacchi A., Pestell R. G., Zani B. M. (2009) Mol. Cancer Ther. 8, 543–551 [DOI] [PubMed] [Google Scholar]
- 25. Toullec D., Pianetti P., Coste H., Bellevergue P., Grand-Perret T., Ajakane M., Baudet V., Boissin P., Boursier E., Loriolle F. (1991) J. Biol. Chem. 266, 15771–15781 [PubMed] [Google Scholar]
- 26. Shenoy N. G., Gleich G. J., Thomas L. L. (2003) J. Immunol. 171, 3734–3741 [DOI] [PubMed] [Google Scholar]
- 27. Guo M., Joiakim A., Reiners J. J., Jr. (2000) Biochem. Pharmacol. 60, 635–642 [DOI] [PubMed] [Google Scholar]
- 28. Yu J. H., Schwartzbauer G., Kazlman A., Menon R. K. (1999) J. Biol. Chem. 274, 34327–34336 [DOI] [PubMed] [Google Scholar]
- 29. Geeraert B., De Keyzer D., Davey P. C., Crombé F., Benhabilès N., Holvoet P. (2007) J. Thromb. Haemost. 5, 2529–2536 [DOI] [PubMed] [Google Scholar]