

Abstract
KATP channels incorporate a regulatory subunit of the ATP-binding cassette (ABC) transporter family, the sulfonylurea receptor (SUR), which defines their pharmacology. The therapeutically important K+ channel openers (e.g. pinacidil, cromakalim, nicorandil) act specifically on the SUR2 muscle isoforms but, except for diazoxide, remain ineffective on the SUR1 neuronal/pancreatic isoform. This SUR1/2 dichotomy underpinned a chimeric strategy designed to identify the structural determinants of opener action, which led to a minimal set of two residues within the last transmembrane helix of SUR. Transfer of either residue from SUR2A to SUR1 conferred opener sensitivity to SUR1, while the reverse operation abolished SUR2A sensitivity. It is therefore likely that these residues form part of the site of interaction of openers with the channel. Thus, openers would target a region that, in other ABC transporters, is known to be tightly involved with the binding of substrates and other ligands. This first glimpse of the site of action of pharmacological openers should permit rapid progress towards understanding the structural determinants of their affinity and specificity.
Keywords: ABC transporter/ATP-sensitive K channel/cromakalim/pinacidil/sulfonylurea receptor
Introduction
ATP-sensitive K+ (KATP) channels are widely distributed among most types of excitable cells, where their primary function is to couple membrane excitability to cellular energy levels (Ashcroft and Ashcroft, 1990). In pancreatic β-cells, they serve as metabolic sensors in the cascade linking insulin secretion to hyperglycemia, and in muscle and neuronal cells they are thought to serve a protective role against ischemic insults. KATP channels are the main targets of K+ channel openers, a chemically diverse group of molecules that includes cromakalim, pinacidil, nicorandil and diazoxide (Gopalakrishnan et al., 1993). Openers are of considerable therapeutic interest as they constitute efficient tools to adjust down cell excitability and they are beneficial in conditions such as hypertension and ischemia (Lawson, 1996). Clinical use of openers is impaired, however, by insufficient tissue specificity and somewhat low affinity, a situation unlikely to improve without a better understanding of the sites and mechanisms of action of these pharmacological agents.
The ultrastructure of the KATP channel is unique among ion channels as it is a complex of two proteins: the sulfonylurea receptor (SUR) (Aguilar-Bryan et al., 1995), which is a member of the ABC (ATP-binding cassette) transporter family, and a smaller protein Kir6.2 (Inagaki et al., 1995), which belongs to the inward rectifier K+ channel family. Four Kir6.2 subunits assemble to form a K+-selective pore, which is constitutively associated to four SUR subunits (Clement et al., 1997; Inagaki et al., 1997; Shyng and Nichols, 1997; Zerangue et al., 1999). The variable tissue-specific properties, particularly the response to openers, of KATP channels arise from the identity of the SUR isoform expressed in that tissue. Channels containing isoform SUR1, which is found in neuronal cells and pancreatic β-cells, are blocked with high affinity by sulfonylureas (Gribble et al., 1998) and can be activated by only one opener, diazoxide (D’hahan et al., 1999b). Coded by a distinct gene, the muscle isoforms SUR2, SUR2A predominant in cardiac and skeletal muscles and its splice variant SUR2B found in smooth muscle, impart activation by the full range of K+ channel openers (Inagaki et al., 1995, 1996; Isomoto et al., 1996; Babenko et al., 1998; D’hahan et al., 1999a).
Recently, several groups have taken advantage of the differential sensitivity to openers of the SUR1 and SUR2A isoforms, to analyze chimeric constructs in order to identify the site of action of openers on SUR. We first used this approach to restrict the cromakalim site to the third transmembrane domain (TMD2; see Figure 1A for terminology) of SUR (D’hahan et al., 1999a), a finding later confirmed by Babenko et al. (2000). Within this domain, two separate regions, a short 29-residue segment connecting helices H13 and H14 and a longer 103-residue segment encompassing H16, H17 and part of NBD2 (Figure 1A), were identified as essential for binding of the pinacidil analog P1075 (Uhde et al., 1999).
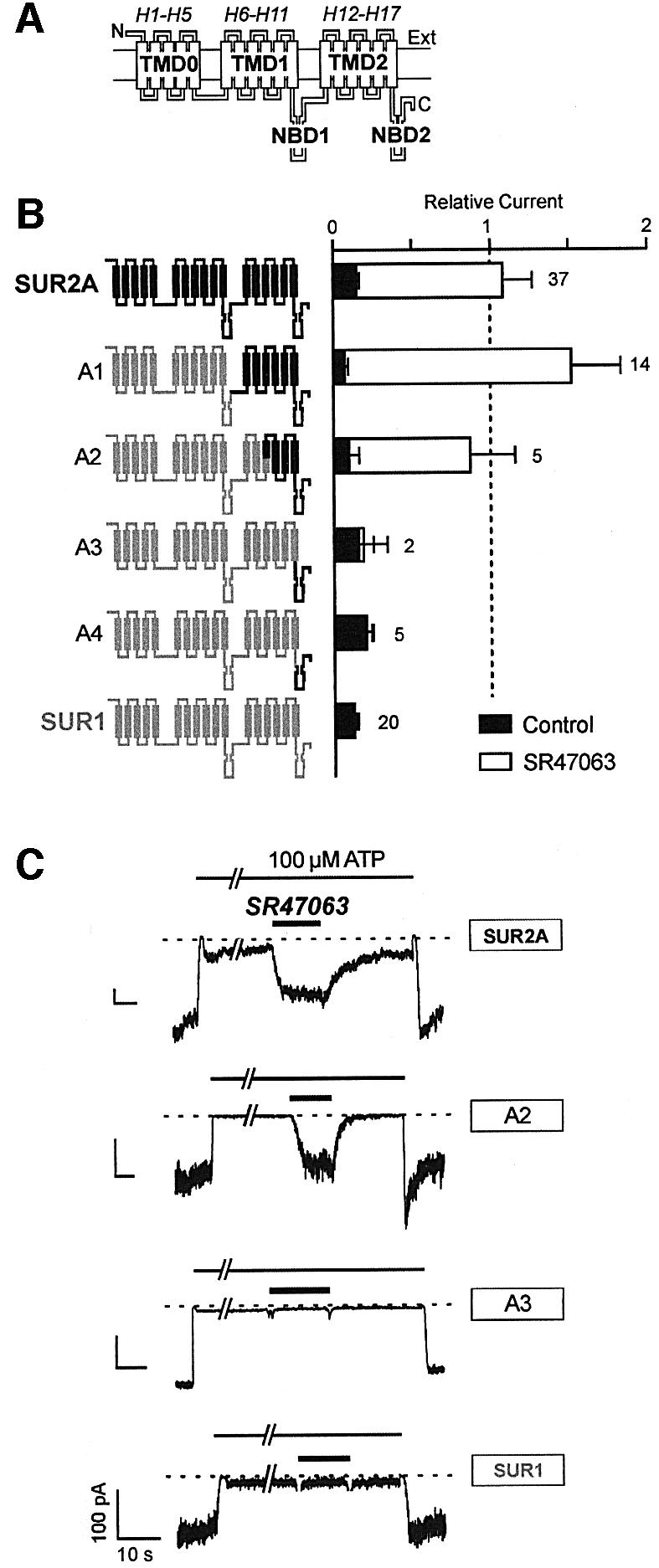
Fig. 1. The C-terminal half of the second transmembrane domain of SUR2A is an essential determinant of the response of chimeric KATP channels to the cromakalim analog SR47063. (A) Hypothetical topology of the SUR. SUR is represented as a modular transmembrane protein with an extracellular N-terminal and an intracellular C-terminal domain, three transmembrane domains, TMD0, TMD1 and TMD2, and two cytoplasmic nucleotide-binding domains, NBD1 and NBD2. Rectangular boxes stand for postulated transmembrane helices (Tusnády et al., 1997) designated here H1–H17. (B) Effects of SR47603 on KATP currents recorded in inside-out patches excised from oocytes co-expressing Kir6.2 and wild-type or chimeric SURs. Chimeras are schematized to the left with SUR1 and SUR2A sequence elements drawn in gray and black, respectively. Average currents in 100 µM ATP (relative to current measured in the absence of ATP) measured before (Control) and during application of 100 µM SR47063 are shown on the right. Numbers above bars indicate the number of patches included in the average. (C) Representative patch–clamp records illustrating the responses of specified wild-type and chimeric channels and the protocols used to test them.
Using SUR1–SUR2A chimeras and point mutants expressed and characterized in Xenopus oocytes by patch–clamp techniques, we narrowed the search down to two amino acid residues (L1249/T1253 in SUR2A and T1286/M1290 in SUR1) within the last helix of TMD2. Our results, which imply a close association between these residues and the cromakalim binding site, provide the first close-up view of the molecular mechanism of action of K+ channel openers.
A preliminary account of this work has been published in abstract form (Jacquet et al., 2000).
Results
In order to identify the elements of the primary sequence of the SUR that are responsible for the activation of KATP channels by K+ channel openers, we have pursued an approach based on the construction of chimeras between SUR isoforms having easily distinguishable opener sensitivity. We used the pancreatic isoform SUR1 and the cardiac muscle isoform SUR2A with SR47063, an analog of cromakalim, as a test substance. Although SUR1 and SUR2A display a high degree of homology in their primary sequence with 67% identical amino acids, they possess ∼300 non-matching amino acids distributed throughout their sequences. Our strategy aimed at identifying which of these mismatches were responsible for the observed phenotypic differences in terms of opener pharmacology.
Our standard assay, illustrated in Figure 1C, consisted of co-expressing wild-type or modified SURs with Kir6.2 in Xenopus oocytes, monitoring the activity of the resulting KATP channels by the patch–clamp technique in the inside-out configuration, and measuring the change in activity produced by a saturating dose (100 µM) of SR47063. In the presence of a concentration of ATP (100 µM) sufficient in most patches to produce an 80% channel inhibition, such a dose of opener robustly activated SUR2A/Kir6.2 channels but had no discernible effects on SUR1/Kir6.2 channels. As demonstrated in the following, the same rapid and simple single-dose assay proved appropriate and sufficient, since practically all of our constructs gave clear-cut responses to this simple assay and could be unambiguously classified as having a SUR2A phenotype (≥3-fold increase in current) or a SUR1 phenotype (<20% increase in current).
Initially, we replaced progressively longer portions of the N-terminus of SUR2A with the corresponding segments of SUR1. The first chimera, A1, which contains only the last transmembrane domain (TMD2) and nucleotide binding domain (NBD2) of SUR2A (see Figure 1A for terminology), was found to retain full sensitivity to SR47063 (Figure 1B). While the lack of response of chimeras A3 and A4 dismisses NBD2 as non-essential, the strong response of chimera A2 reduces the candidate region to a 149-residue fragment of TMD2 going from SUR2A residues L1119 to D1267.
New chimeras consisting of SUR1 containing only fragments of domain TMD2 from SUR2A were constructed and tested (Figure 2). Chimera B1, which possesses the entire domain TMD2 from SUR2A, was responsive to opener as expected. Chimeras C1, C2 and C3 showed no significant activation by SR47063, confirming that the N-terminal half of TMD2 is not an essential determinant of opener sensitivity. Chimeras B3, B4, B6 and B7 demonstrate that, within the C-terminal half of TMD2, the last helix H17 (Figure 1A) is crucial. When H17 from SUR2A is transplanted into SUR1, the resulting construct B7 acquires the opener responsiveness of SUR2A.
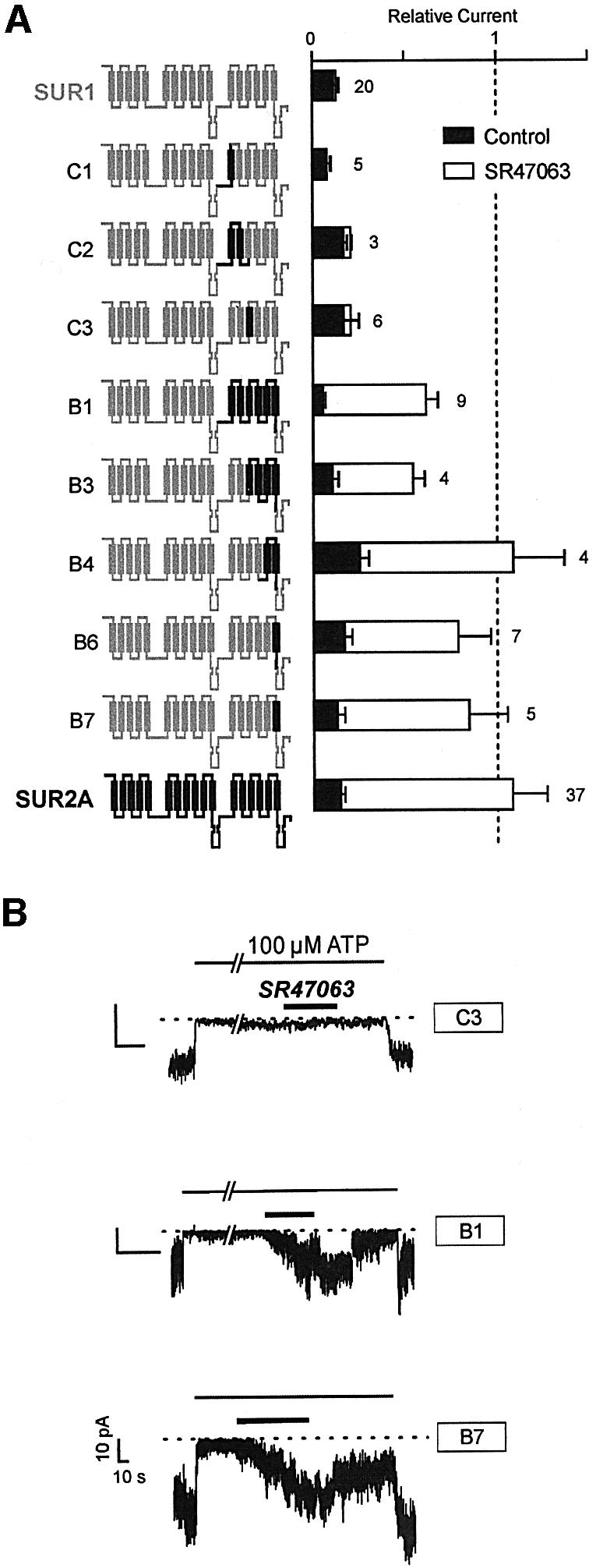
Fig. 2. Central role of the last helix of SUR (H17) in KATP channel activation by SR47063. (A) Effects of SR47603 on KATP currents recorded from oocytes co-expressing Kir6.2 and wild-type or SUR1-based chimeras incorporating progressively shorter fragments of domain TMD2 of SUR2A. (B) Representative current records showing the effects of SR47063 on the indicated chimeras.
Helix H17 is rather well conserved between SUR1 and SUR2A, and possesses only five non-identical residues among 20: these residues are T1286, M1290, V1291, S1292 and M1298 in SUR1, and L1249, T1253, I1254, T1255 and V1261 in SUR2A. The respective ability of these residues to support opener activation was investigated by site-directed mutagenesis.
As shown in Figure 3, chimera B7 is equivalent to SUR1 with the five mutations T1286L, M1290T, V1291I, S1292T and M1298V. The two conservative mutations V1291I and S1292T appeared non-essential since chimera B12, which retains only the three non-conservative mutations T1286L, M1290T and M1298V, was still vigorously activated by the opener. When these mutations were examined individually, only mutation M1290T was able to confer to SUR1 the ability to be activated by SR47063 as efficiently as SUR2A. Mutation T1286L rendered SUR1 sensitive to SR47063, but activation was noticeably weaker. Mutation M1298V was the least effective and yielded only marginal or no increases in current when probed with opener.
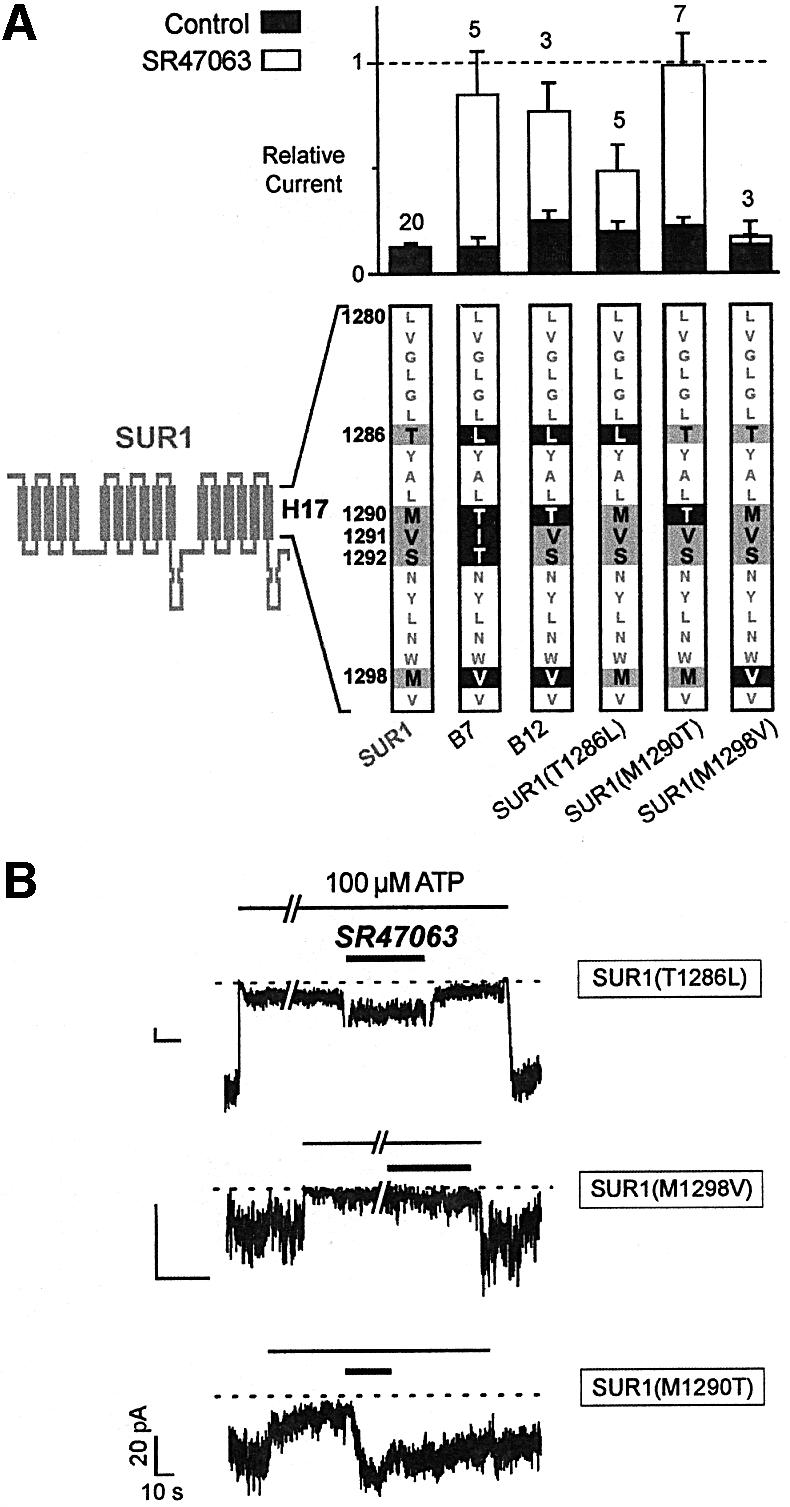
Fig. 3. Single-residue substitutions within helix H17 confer SR47063 sensitivity to SUR1. (A) Effects of SR47603 on KATP currents recorded from oocytes co-expressing Kir6.2 and SUR1 subunits carrying mutations within helix H17. The sequence of helix H17 is shown at the bottom with residues common to SUR1 and SUR2A shown as gray characters on a white background, residues specific to SUR1 as black on gray, and residues mutated to their SUR2A counterparts as white on black. (B) Representative current records showing the effects of SR47063 on single-point mutants.
These results suggest that SUR2A residues T1253 and, to a lesser extent, L1249 are sufficient to confer opener sensitivity to SUR1. Whether these residues are necessary was addressed by the experiments described in Figure 4 where, logically, we examined the effects of their substitutions in the context of SUR2A. Construct D1, which is SUR2A with most of helix H17 from SUR1, did not respond to opener SR47063, confirming the major role of this helix. Within helix H17, L1249 and T1253 were most closely linked to opener activation since mutant SUR2A(L1249T,T1253M) became almost immune to activation, but mutant SUR2A(I1254V,T1255S) kept a wild-type phenotype. The behavior of the SUR2A mutants is therefore fully consistent with that of the converse SUR1 mutants.
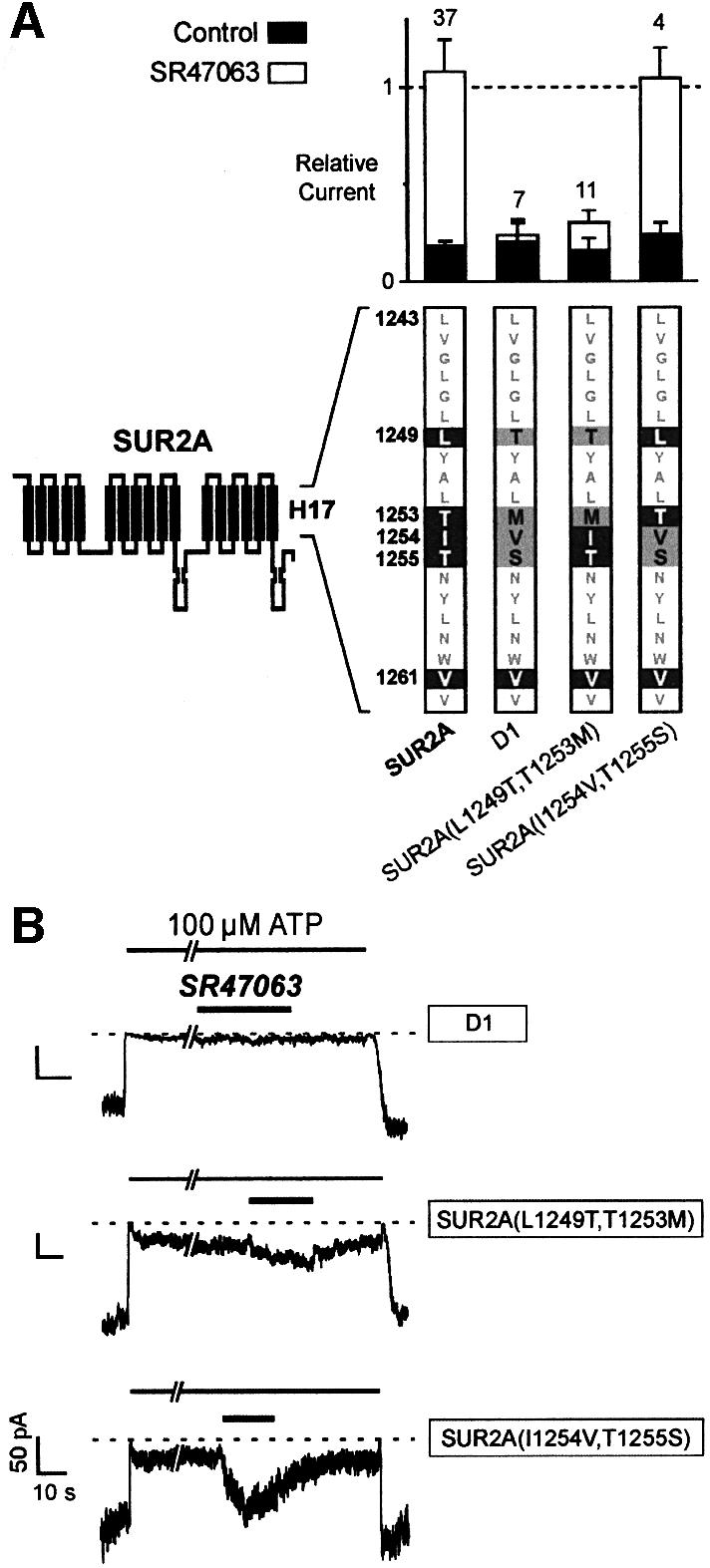
Fig. 4. Single-residue substitutions within helix H17 abolish the effects of SR47063 on SUR2A. (A) Effects of SR47603 on KATP currents recorded from oocytes co-expressing Kir6.2 and SUR2A subunits carrying mutations within helix H17. The sequence of helix H17 is shown at the bottom with residues common to SUR1 and SUR2A shown as gray characters on a white background, residues specific to SUR2A as white on black, and residues mutated to their SUR1 counterparts as black on gray. (B) Representative current records illustrating the effects of SR47063 on the mutants of (A).
Experiments described so far applied to the single opener SR47063 and results would not necessarily apply to other openers of different chemical structures. We conducted experiments to assess the role of T1253 and L1249 in the response to two other openers: the pinacidil analog P1075 and rilmakalim (HOE234), a bulkier derivative of cromakalim possessing an added phenylsulfonyl group (Terzic et al., 1994). As shown in Figure 5, transfer of T1253 from SUR2A to SUR1 to yield mutant SUR1(M1290T) conferred sensitivity not only to SR47063 but also to rilmakalim and P1075. This was not true of mutation T1286L, which did not make SUR1 any more sensitive to rilmakalim and P1075.

Fig. 5. Comparative effects of different classes of K+ channel openers on single-point SUR1 mutants. (A) Representative current trace illustrating the effects in the same membrane patch of diazoxide (300 µM), SR47063 (100 µM), rilmakalim (HOE234; 10 µM) and the pinacidil analog P1075 (100 µM) on channels formed of SUR1(T1286L) and Kir6.2. (B) The same as (A) for SUR1(M1290T). (C) Average effects of ADP (100 µM), diazoxide (300 µM), SR47063 (100 µM), rilmakalim (10 µM) and the pinacidil analog P1075 (100 µM) on KATP currents recorded in 100 µM ATP from oocytes co-expressing Kir6.2 and wild-type or mutant SURs.
These data concur to designate the Thr residue at position 1253 of SUR2A as a most critical determinant of KATP channel activation. We therefore proceeded to investigate the role of this residue in the response of KATP channels to two other singular activators: ADP, the physiological opener of KATP channels (Nichols et al., 1996), and diazoxide, the only synthetic opener acting on SUR1 (Figure 5C).
ADP, which activates SUR1 and SUR2A channels to the same extent, also activated both mutants. Although ADP responses are notably variable from patch to patch and even in the same patch (in our hands, they tend to fade away with time after patch excision), it appears that mutant SUR1(T1286L) was more responsive than wild-type SUR and the SUR1(M1290T) mutant.
Diazoxide is able to activate SUR2A channels provided sufficient internal ADP is present (D’hahan et al., 1999b). In our conditions, with no added ADP, only SUR1 channels are strongly activated by diazoxide, whereas SUR2A channels are not. Mutations T1286L and M1290T had little effect on the diazoxide responsiveness of SUR1, in sharp contrast to the dramatic effects of these mutations on the responsiveness of SUR1 to other openers. This observation agrees with our previous conclusion that separate regions mediate the effects of diazoxide and that of other openers (D’hahan et al., 1999a). Nonetheless, mutation M1290T caused a reduction in the activation by diazoxide while mutation T1286L caused a slight increase.
Discussion
This study has identified two SUR residues that are intimately involved with the activation of KATP channels by K+ channel openers and could possibly form part of a binding pocket able to accommodate openers of different chemical classes. These residues, which we shall call I and II, are located within the last transmembrane helix of SUR at positions 1249 and 1253 of SUR2A and at the aligned positions 1286 and 1290 of SUR1. Residues I and II are Leu and Thr in opener-sensitive isoforms SUR2A and SUR2B and Thr and Met in opener-insensitive SUR1, respectively.
Identification of residues responsible for the differential sensitivity of SUR1 and SUR2A to SR47063
The critical role of residues I and II was discovered by following a systematic chimeric strategy destined to isolate the elements of the primary sequence of SUR2A capable of conferring opener sensitivity when substituted in SUR1. Matched chimeras incorporating progressively shorter fragments of SUR2A in a SUR1 background led to the remarkable finding that the opener phenotype of SUR2A could be carried over to SUR1 through single-residue substitutions. Thus, single-point SUR1 mutants with residues I or II from SUR2A acquired a responsiveness to the cromakalim analog, SR47063, which was totally absent from their SUR1 parents. Conversely, the transfer of both residues I and II from SUR1 in SUR2A did not completely obliterate the responses to SR47063, suggesting that other determinants remained, but the residual effect was extremely weak, implying that these determinants were of minor importance compared with residues I and II. While we are aware of the pitfalls of overinterpreting mutagenesis results, the combination of experiments showing both gain-of-function in the SUR1 context and loss-of-function in the SUR2A context argues strongly in favor of a direct involvement of residues I and II in the binding and effect of cromakalim.
Structural implications: binding and specificity of openers
Since our testing assay is purely functional, residues I and II could intervene either at the binding step or at the transducing step linking binding to channel activation. The second hypothesis is highly improbable given that biochemical binding studies indicate that wild-type SUR1 does not bind levcromakalim [the active isomer of cromakalim resembling SR47063 in its structure and efficacy (Forestier et al., 1996)] to any measurable extent at the concentrations we have used (Schwanstecher et al., 1998). If substitutions of residues I or II alter the binding of openers, one can not jump directly to the conclusion that they line the binding site. These mutations could uncover a pre-existing binding site through long-range allosteric effects. Such a possibility can not be formally discarded either with our functional experiments or with ligand binding experiments (Colquhoun, 1998). Nonetheless, we believe that its probability is low here for several reasons: (i) it would entail a complex structural rearrangement of SUR, whereas residues I and II are located in the middle of a stable structure, a predicted transmembrane helix that is highly conserved in SUR1 and SUR2; (ii) it would require that SUR1 and SUR2A exist in well defined, distinct conformations with residues I or II acting as switches. If this were the case, it would have been extremely difficult to reach the consistency of our results with numerous chimeric constructs of unpredictable intermediate conformations; and (iii) finally, an allosteric effect of mutations could not easily account for the facts that substitution of residue II conferred sensitivity to three distinct openers to SUR1 but substitution of residue I made SUR1 sensitive solely to SR47063 (Figure 5).
We therefore feel that the most appropriate interpretation of our results is that residues I and II form part of the binding site for openers. These residues are one α-helix turn away from each other and are therefore positioned next to each other on the same hydrophilic face of amphipathic helix H17 (Figure 6). The three other residues of H17 that differ between SUR1 and SUR2A (I254, T1255 and V1261 of SUR2A, and V1291, S1292 and M1298 of SUR1) are predicted to lie on the opposite face of the helix and, fittingly, their modifications did not perturb the opener phenotypes of either SUR1 or SUR2A.
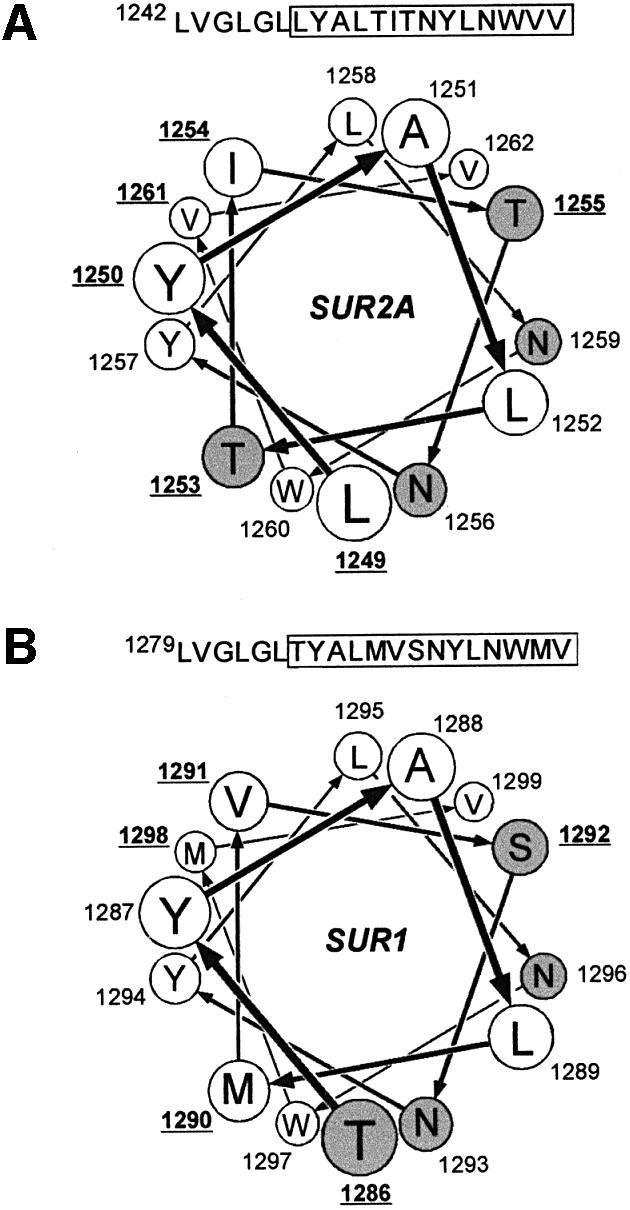
Fig. 6. Helical wheel projections of the last transmembrane helix H17 of SUR2A (A) and SUR1 (B). The helix is viewed from the extracellular side or the N-terminal end. Hydrophobic residues are represented as white circles and polar residues are shaded in gray. Numbers indicate residue positions in the sequence of the protein. Underlined numbers point out residues that are different in SUR1 and SUR2A. The N-terminal hydrophobic residues LVGLGL, identical in SUR1 and SUR2A, have been omitted for clarity.
If transfer of residue I of SUR2A to SUR1 (i.e. T1286L) conferred only weak sensitivity to SR47063 and none to P1075 and rilmakalim, transfer of residue II (M1290T) gave SUR1 the complete opener phenotype of SUR2A. These observations indicate that openers of different chemical structures do target the same site. They also suggest that Thr1253 has a central role in the SUR2A–opener interaction and that the other residues crucial to that interaction are shared by the two SUR isoforms. These other residues could very well include any of the three adjacent aromatic residues, Tyr1250, Tyr1257 and Trp1260 in SUR2A (Figure 6), which could interact favorably with the hydrophobic rings present in all K+ channel openers. Such weak interactions could contribute to the broad opener specificity of SUR2A (Pawagi et al., 1994). Further substitutions of residue II will have to be evaluated to establish whether hydrogen-bonding capacity or compactness is the property of Thr that makes it superior to Met in favoring ligand binding, and whether the ligand interacts physically with this residue.
Interactions with other regions of SUR
Our experiments were conducted with the cardiac isoform of SUR2: SUR2A. The smooth muscle isoform, SUR2B, responds to the same openers but with a significantly higher affinity (Schwanstecher et al., 1998; Hambrock et al., 1999). Since SUR2B is identical to SUR2A except for 28 residues at the C-terminal end (Isomoto et al., 1996), one could envisage that opener affinity is modulated by direct interactions between the C-terminal extremity and the neighboring helix, H17. Our results could serve as a seed for experiments designed to address this possibility and further our knowledge of the topology of SUR and other similar ABC transporters. Another region to investigate is the loop connecting helices H13 and H14 (T1059–L1087) in TMD2 of SUR2. Although we did not analyze this region in detail with our simple assay, its modulatory role was uncovered by Uhde et al. (1999), who found it to be critical for maintaining high-affinity binding of P1075 to SUR2B.
We did not address here the role of nucleotides in modulating opener action. ATP and ADP, acting primarily on NBD1 and NBD2, respectively, are important regulators of opener binding and effect (Dickinson et al., 1997; Gribble et al., 1997; Shyng et al., 1997; Schwanstecher et al., 1998; D’hahan et al., 1999b) and are themselves regulated by openers (Bienengraeber et al., 2000). It has been proposed that the different opener phenotypes of SUR isoforms could be due to their different nucleotide-binding properties as measured by photoaffinity labeling (Matsuo et al., 2000). That the responses to openers of SUR2A and SUR1(M1290T) were similar proves that, within the limits of our coarse examination, opener responsiveness is not critically associated with the specific properties of the nucleotide-binding domains. A more detailed study is warranted, however, and the availability of SUR1 single-point mutants sensitive to openers could be a useful tool in further investigations of this aspect of SUR pharmacology.
Openers as ABC transporter substrates
Recent work by Bienengraeber et al. (2000) has provided evidence that SUR possesses detectable intrinsic ATPase activity and that openers increase that activity. If and how this relates to activation of KATP channels remain to be formally established. However, this observation suggests that openers display the behavior expected of ABC transporter substrates. In that respect, the discovery of helix H17 as part of the opener site is significant since in the most studied ABC transporter, P-glycoprotein, the matching helix H12 also appears to be crucial to substrate binding. By analyzing cross-linking experiments with a thiol-reactive substrate, Loo and Clarke (1997) identified several residues of H12 of P-glycoprotein within reach of the bound substrate. In spite of poor homology between P-glycoprotein and SUR, primary sequence alignment (ClustalW1.7 program; Thompson et al., 1994) places these residues within one position or one helix turn of residues I and II of SUR. This underscores the relevance of this helix in ligand binding and raises the hypothesis that openers could act by occupying the substrate site of SUR, a site that could be an evolutionary remnant or the target of a hitherto unknown endogenous substance. Considering such a hypothesis, it would be tempting to speculate that the same mechanisms underlie the ability of SUR to recognize opener molecules of varied structures and the broad substrate specificity of the multidrug resistance transporters, P-glycoprotein (Ueda et al., 1997) and the SUR homolog MRP (Borst et al., 1999).
To our knowledge, the only other ABC transporter shown to interact with K+ channel openers is CFTR (cystic fibrosis transmembrane regulator). CFTR possesses intrinsic chloride channel activity, which is blocked by cromakalim (Sheppard and Welsh, 1992). Given the nearly 30% overall amino acid identity and 50% homology over helix H17 between SUR and CFTR, it might be worth looking at this last helix as a pharmacologically relevant region of CFTR.
Conclusions
We have demonstrated here that the differential responsiveness of KATP channel isoforms to openers arises from amino acid differences at two positions in the last transmembrane helix of SUR. Evidence suggests that these residues belong to a site accessible to openers of distinct chemical structures, a site that could resemble the substrate-binding site of other ABC transporters. Although the mechanism linking binding to channel opening remains to be elucidated, the discovery of the first piece of the puzzle provides a starting point toward assembling the full picture.
Materials and methods
Molecular biology
Mouse Kir6.2 (Inagaki et al., 1995; DDBJ/EMBL/GenBank accession No. D50581), hamster SUR1 (Aguilar-Bryan et al., 1995; DDBJ/EMBL/GenBank accession No. L40623) and rat SUR2A (Inagaki et al., 1996; DDBJ/EMBL/GenBank accession No. D83598) were subcloned in the Xenopus oocyte expression vectors derived from pGEMHE (Liman et al., 1992). Chimeric cDNA constructs were produced using the Splicing by Overlap Extension (SOE) PCR technique (Horton et al., 1989) as previously described (D’hahan et al., 1999a). Hamster SUR1 and rat SUR2A were used as templates for making all the chimeras tested. To minimize errors, PCRs were performed for 25 cycles using the high-fidelity Vent DNA polymerase (New England Biolabs). Site-directed mutagenesis was carried out by PCR amplification of both DNA strands of full-length plasmids with complementary primers mutated to produce the desired amino acid change (QuikChange Site-Directed Mutagenesis Kit; Stratagene, La Jolla, CA).
The exact amino acid composition of the SUR1–SUR2A chimeric constructs was: A1 = SUR1(M1-V850) + SUR2A(V839-K1545); A2 = SUR1(M1-C1150) + SUR2A(L1118-K1545); A3 = SUR1(M1-D1304) + SUR2A(L1268-K1545); A4 = SUR1(M1-V1361) + SUR2A(L1325-K1545); B1 = SUR1(M1-V850) + SUR2A(V839-V1324) + SUR1(L1362-K1582); B3 = SUR1(M1-I1105) + SUR2A(I1073-V1324) + SUR1(L1362-K1582); B4 = SUR1(M1-I1214) + SUR2A(R1182-V1324) + SUR1(L1362-K1582); B6 = SUR1 (M1-A1278) + SUR2A(G1242-V1324) + SUR1(L1362-K1582); B7 = SUR1(M1-A1278) + SUR2A(G1242-V1261) + SUR1(V1299-K1582); B12 = SUR1 with mutations T1286L, M1290T and M1298V; C1 = SUR1(M1-V850) + SUR2A(V839-L1011) + SUR1(A1035-K1582); C2 = SUR1(M1-V850) + SUR2A(V839-I1072) + SUR1(I1106-K1582); C3 = SUR1(M1-I1105) + SUR2A(I1073-R1091) + SUR1(F1125-K1582); D1 = SUR2A with mutations T1286L, M1290T, V1291L and S1292T. Other single- and double-point mutants are designated by the name of the wild-type parent with the mutations indicated in parentheses.
Plasmid DNAs were amplified, confirmed by restriction analysis and sequencing of the PCR-derived regions, linearized and transcribed in vitro using the T7 mMessage mMachine kit (Ambion). cRNAs were electrophoresed on formaldehyde gels and concentrations were estimated from two dilutions using RNA marker as a standard.
cRNAs coding Kir6.2 (∼2 ng) and wild-type or modified SURs (∼6 ng) were co-injected into defoliculated Xenopus laevis oocytes. Injected oocytes were stored at 19°C in Barth’s solution [in mM: 1 KCl, 0.82 MgSO4, 88 NaCl, 2.4 NaHCO3, 0.41 CaCl2, 0.3 Ca(NO3)2, 16 HEPES pH 7.4] with 100 U/ml penicillin, 100 µg/ml streptomycin and 100 µg/ml gentamycin.
Electrophysiology
Two to 15 days after injection, oocytes were devitellinized and recombinant KATP channels were characterized by the patch–clamp technique in the excised inside-out configuration (Hamill et al., 1981). Patch pipettes (2–10 MΩ) contained (in mM) 154 K+, 146 Cl–, 5 Mg2+ and 10 PIPES pH 7.1. The cytoplasmic face of the patch was bathed in solutions that all contained (in mM) 174 K+, 40 Cl–, 1 EGTA, 1 Mg2+, 10 PIPES pH 7.1 and methanesulfonate– as the remaining anions. ATP and ADP (potassium salt; Sigma), SR47063 [20 mM stock in dimethylsulfoxide (DMSO); Sanofi Recherche, Montpellier, France], diazoxide (100 mM stock in DMSO; Sigma), P1075 (20 mM stock in DMSO; Leo Pharmaceutical Products, Copenhagen, Denmark) and rilmakalim (a.k.a. HOE 234; 100 mM stock in DMSO; Hoechst, Frankfurt, Germany) were added as specified. The membrane potential was maintained at –50 mV. Experiments were conducted at room temperature (22–24°C).
Applications of the various solutions to the intracellular face of the patch were performed using an RSC-100 rapid-solution-changer (Bio-Logic, Claix, France) controlled by in-house software Perf 2.10. Analog signals were filtered at 300 Hz and sampled at 1 kHz. Slow fluctuations of the no-channel-open baseline of the signal were removed by interactive fitting of the baseline with a spline curve and subtraction of this fit from the signal. Acquisition, analysis and presentation were performed with in-house software Erwin 3.2. The tracings shown in the figures represent continuous records with sporadic solution-switching artifacts blanked out and occasional short segments of superfluous data removed for display purposes. Since the same Kir6.2 subunit is implicitly present in all the reconstituted channels of this study, for the sake of simplicity the text often incorrectly designates a given channel by the name of its SUR subunit. Results are displayed as mean ± SEM.
Acknowledgments
Acknowledgements
We thank Mylène Robert for technical assistance with mutant constructs and Dr Patrice Catty for many helpful discussions and advice. We are grateful to Dr J.Bryan (Houston, TX) for hamster SUR1, Dr S.Seino (Chiba, Japan) for mouse Kir6.2 and rat SUR2A, Dr P.Gautier (Sanofi Recherche, Montpellier, France) for SR 47063, Dr L.Billerup (Leo Pharmaceutical Products, Copenhagen, Denmark) for P1075 and Dr A.Terzic (Mayo Clinic, Rochester, MN) for rilmakalim. This work was made possible by grants from the A.F.M. (Association Francaise contre les Myopathies), A.F.L.M. (Association Francaise de Lutte contre la Mucoviscidose), the Rhône-Alpes Région with additional support provided by C.E.A. (Commissariat à l’Energie Atomique) and the C.N.R.S. (Centre National de la Recherche Scientifique). C.M., H.J., A.-L.P. and N.D. were supported by fellowships from La Ligue contre le Cancer, ARC (Association pour la Recherche contre le Cancer), C.E.A. and La Société des Amis des Sciences, respectively.
References
- Aguilar-Bryan L. et al. (1995) Cloning of the β cell high-affinity sulfonylurea receptor: a regulator of insulin secretion. Science, 268, 423–426. [DOI] [PubMed] [Google Scholar]
- Ashcroft S.J.H. and Ashcroft,F.M. (1990) Properties and functions of ATP-sensitive K-channels. Cell Signal., 2, 197–214. [DOI] [PubMed] [Google Scholar]
- Babenko A.P., Gonzalez,G., Aguilar-Bryan,L. and Bryan,J. (1998) Reconstituted human cardiac K-ATP channels—functional identity with the native channels from the sarcolemma of human ventricular cells. Circ. Res., 83, 1132–1143. [DOI] [PubMed] [Google Scholar]
- Babenko A.P., Gonzalez,G. and Bryan,J. (2000) Pharmaco-topology of sulfonylurea receptors—separate domains of the regulatory subunits of K-ATP channel isoforms are required for selective interaction with K+ channel openers. J. Biol. Chem., 275, 717–720. [DOI] [PubMed] [Google Scholar]
- Bienengraeber M., Alekseev,A.E., Abraham,M.R., Carrasco,A.J., Moreau,C., Vivaudou,M., Dzeja,P.P. and Terzic,A. (2000) ATPase activity of SUR2A: a catalytic function for the KATP channel complex. FASEB J., 14, 1943–1952. [DOI] [PubMed] [Google Scholar]
- Borst P., Evers,R., Kool,M. and Wijnholds,J. (1999) The multidrug resistance protein family. Biochim. Biophys. Acta, 1461, 347–357. [DOI] [PubMed] [Google Scholar]
- Clement J.P., Kunjilwar,K., Gonzalez,G., Schwanstecher,M., Panten,U., Aguilar-Bryan,L. and Bryan,J. (1997) Association and stoichiometry of K-ATP channel subunits. Neuron, 18, 827–838. [DOI] [PubMed] [Google Scholar]
- Colquhoun D. (1998) Binding, gating, affinity and efficacy: the interpretation of structure–activity relationships for agonists and of the effects of mutating receptors. Br. J. Pharmacol., 125, 924–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’hahan N., Jacquet,H., Moreau,C., Catty,P. and Vivaudou,M. (1999a) A transmembrane domain of the sulfonylurea receptor mediates activation of ATP-sensitive K+ channels by K+ channel openers. Mol. Pharmacol., 56, 308–315. [DOI] [PubMed] [Google Scholar]
- D’hahan N., Moreau,C., Prost,A.L., Jacquet,H., Alekseev,A.E., Terzic,A. and Vivaudou,M. (1999b) Pharmacological plasticity of cardiac ATP-sensitive potassium channels toward diazoxide revealed by ADP. Proc. Natl Acad. Sci. USA, 96, 12162–12167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickinson K.E.J., Bryson,C.C., Cohen,R.B., Rogers,L., Green,D.W. and Atwal,K.S. (1997) Nucleotide regulation and characteristics of potassium channel opener binding to skeletal muscle membranes. Mol. Pharmacol., 52, 473–481. [DOI] [PubMed] [Google Scholar]
- Forestier C., Pierrard,J. and Vivaudou,M. (1996) Mechanism of action of K channel openers on skeletal muscle K-ATP channels—interactions with nucleotides and protons. J. Gen. Physiol., 107, 489–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gopalakrishnan M., Janis,R.A. and Triggle,D.J. (1993) ATP-sensitive K+ channels—pharmacologic properties, regulation, and therapeutic potential. Drug Dev. Res., 28, 95–127. [Google Scholar]
- Gribble F.M., Tucker,S.J. and Ashcroft,F.M. (1997) The essential role of the Walker A motifs of SUR1 in K-ATP channel activation by Mg-ADP and diazoxide. EMBO J., 16, 1145–1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gribble F.M., Tucker,S.J., Seino,S. and Ashcroft,F.M. (1998) Tissue specificity of sulfonylureas: studies on cloned cardiac and β-cell K-ATP channels. Diabetes Care, 47, 1412–1418. [DOI] [PubMed] [Google Scholar]
- Hambrock A., Loffler-Walz,C., Kloor,D., Delabar,U., Horio,Y., Kurachi,Y. and Quast,U. (1999) ATP-sensitive K+ channel modulator binding to sulfonylurea receptors SUR2A and SUR2B: opposite effects of MgADP. Mol. Pharmacol., 55, 832–840. [PubMed] [Google Scholar]
- Hamill O.P., Marty,A., Neher,E., Sakmann,B. and Sigworth,F.J. (1981) Improved patch–clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflugers Arch. Eur. J. Physiol., 391, 85–100. [DOI] [PubMed] [Google Scholar]
- Horton R.M., Hunt,H.D., Ho,S.N., Pullen,J.K. and Pease,L.R. (1989) Engineering hybrid genes without the use of restriction enzymes: gene splicing by overlap extension. Gene, 77, 61–68. [DOI] [PubMed] [Google Scholar]
- Inagaki N., Gonoi,T., Clement,J.P., Namba,N., Inazawa,J., Gonzalez,G., Aguilar-Bryan,L., Seino,S. and Bryan,J. (1995) Reconstitution of I-KATP: an inward rectifier subunit plus the sulfonylurea receptor. Science, 270, 1166–1170. [DOI] [PubMed] [Google Scholar]
- Inagaki N., Gonoi,T., Clement,J.P., Wang,C.Z., Aguilar-Bryan,L., Bryan,J. and Seino,S. (1996) A family of sulfonylurea receptors determines the pharmacological properties of ATP-sensitive K+ channels. Neuron, 16, 1011–1017. [DOI] [PubMed] [Google Scholar]
- Inagaki N., Gonoi,T. and Seino,S. (1997) Subunit stoichiometry of the pancreatic β-cell ATP-sensitive K+ channel. FEBS Lett., 409, 232–236. [DOI] [PubMed] [Google Scholar]
- Isomoto S., Kondo,C., Yamada,M., Matsumoto,S., Higashiguchi,O., Horio,Y., Matsuzawa,Y. and Kurachi,Y. (1996) A novel sulfonylurea receptor forms with BIR (Kir6.2) a smooth muscle type ATP-sensitive K+ channel. J. Biol. Chem., 271, 24321–24324. [DOI] [PubMed] [Google Scholar]
- Jacquet H., Moreau,C., D’hahan,N., Prost,A.L. and Vivaudou,M. (2000) Identification of sulfonylurea receptor regions involved in K-ATP channel activation by cromakalim. Biophys. J., 78, 464A. [Google Scholar]
- Lawson K. (1996) Is there a therapeutic future for ‘potassium channel openers’? Clin. Sci., 91, 651–663. [DOI] [PubMed] [Google Scholar]
- Liman E.R., Tytgat,J. and Hess,P. (1992) Subunit stoichiometry of a mammalian K+ channel determined by construction of multimeric cDNAs. Neuron, 9, 861–871. [DOI] [PubMed] [Google Scholar]
- Loo T.W. and Clarke,D.M. (1997) Identification of residues in the drug-binding site of human P-glycoprotein using a thiol-reactive substrate. J. Biol. Chem., 272, 31945–31948. [DOI] [PubMed] [Google Scholar]
- Matsuo M., Tanabe,K., Kioka,N., Amachi,T. and Ueda,K. (2000) Different binding properties and affinities for ATP and ADP among sulfonylurea receptor subtypes, SUR1, SUR2A, and SUR2B. J. Biol. Chem., 275, 28757–28763. [DOI] [PubMed] [Google Scholar]
- Nichols C.G., Shyng,S.L., Nestorowicz,A., Glaser,B., Clement,J.P., Gonzalez,G., Aguilar-Bryan,L., Permutt,M.A. and Bryan,J. (1996) Adenosine diphosphate as an intracellular regulator of insulin secretion. Science, 272, 1785–1787. [DOI] [PubMed] [Google Scholar]
- Pawagi A.B., Wang,J., Silverman,M., Reithmeier,R.A. and Deber,C.M. (1994) Transmembrane aromatic amino acid distribution in P-glycoprotein. A functional role in broad substrate specificity. J. Mol. Biol., 235, 554–564. [DOI] [PubMed] [Google Scholar]
- Schwanstecher M., Sieverding,C., Dorschner,H., Gross,I., Aguilar-Bryan,L., Schwanstecher,C. and Bryan,J. (1998) Potassium channel openers require ATP to bind to and act through sulfonylurea receptors. EMBO J., 17, 5529–5535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheppard D.N. and Welsh,M.J. (1992) Effect of ATP-sensitive K+ channel regulators on cystic fibrosis transmembrane conductance regulator chloride currents. J. Gen. Physiol., 100, 573–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shyng S.L. and Nichols,C.G. (1997) Octameric stoichiometry of the K-ATP channel complex. J. Gen. Physiol., 110, 655–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shyng S.L., Ferrigni,T. and Nichols,C.G. (1997) Regulation of K-ATP channel activity by diazoxide and MgADP—distinct functions of the two nucleotide binding folds of the sulfonylurea receptor. J. Gen. Physiol., 110, 643–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terzic A., Jahangir,A. and Kurachi,Y. (1994) Hoe-234, a second generation K+ channel opener, antagonizes the ATP-dependent gating of cardiac ATP-sensitive K+ channels. J. Pharmacol. Exp. Ther., 268, 818–825. [PubMed] [Google Scholar]
- Thompson J.D., Higgins,D.G. and Gibson,T.J. (1994) CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res., 22, 4673–4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tusnády G.E., Bakos,E., Váradi,A. and Sarkadi,B. (1997) Membrane topology distinguishes a subfamily of the ATP-binding cassette (ABC) transporters. FEBS Lett., 402, 1–3. [DOI] [PubMed] [Google Scholar]
- Ueda K., Taguchi,Y. and Morishima,M. (1997) How does P-glycoprotein recognize its substrates? Semin. Cancer Biol., 8, 151–159. [DOI] [PubMed] [Google Scholar]
- Uhde I., Toman,A., Gross,I., Schwanstecher,C. and Schwanstecher,M. (1999) Identification of the potassium channel opener site on sulfonylurea receptors. J. Biol. Chem., 274, 28079–28082. [DOI] [PubMed] [Google Scholar]
- Zerangue N., Schwappach,B., Jan,Y.N. and Jan,L.Y. (1999) A new ER trafficking signal regulates the subunit stoichiometry of plasma membrane K-ATP channels. Neuron, 22, 537–548. [DOI] [PubMed] [Google Scholar]