

Abstract
Type 2 diabetes results from an impairment of insulin action. The first demonstrable abnormality of insulin signaling is a decrease of insulin-dependent glucose disposal followed by an increase in hepatic glucose production. In an attempt to dissect the relative importance of these two changes in disease progression, we have employed genetic knock-outs/knock-ins of the insulin receptor. Previously, we demonstrated that insulin receptor knock-out mice (Insr−/−) could be rescued from diabetes by reconstitution of insulin signaling in liver, brain, and pancreatic β cells (L1 mice). In this study, we used a similar approach to reconstitute insulin signaling in tissues that display insulin-dependent glucose uptake. Using GLUT4-Cre mice, we restored InsR expression in muscle, fat, and brain of Insr−/− mice (GIRKI (Glut4-insulin receptor knock-in line 1) mice). Unlike L1 mice, GIRKI mice failed to thrive and developed diabetes, although their survival was modestly extended when compared with Insr−/−. The data underscore the role of developmental factors in the presentation of murine diabetes. The broader implication of our findings is that diabetes treatment should not necessarily target the same tissues that are responsible for disease pathogenesis.
Keywords: Brain Metabolism, Carbohydrate Metabolism, Cell Surface Receptor, Fatty Acid Metabolism, Gene Knock-out, Insulin Resistance, Insulin Secretion, Mouse Genetics
Introduction
Type 2 (non-insulin-dependent) diabetes arises as a consequence of complex changes in insulin action in different target tissues. Although the factors that favor disease progression are heterogeneous, evidence from prospective human studies and from animal models indicates that an impairment of insulin-dependent glucose uptake and utilization is an early event in the pathogenesis of diabetes (1).
To analyze the contribution of individual organs and cell types to the integrated physiology of insulin action, we and others have used gene targeting by homologous recombination to carry out gain- and loss-of-function experiments affecting insulin signaling (2). In view of its cardinal position in the insulin signaling cascade, the insulin receptor (InsR)3 represents an ideal target to manipulate insulin action and study the systemic consequences of insulin resistance in any given tissue (3).
We have previously shown that Insr−/− mice can be rescued from neonatal diabetic ketoacidosis by restoring InsR expression in liver, brain, and pancreatic β cells (4, 5). These findings are surprising as they ostensibly indicate that insulin action in tissues with insulin-dependent glucose uptake, such as muscle and fat, is dispensable for metabolic control. However, it can be argued that the restoration of insulin sensitivity in L1 mice is simply an effect of mass action rather than a specific effect of the triple tissue combination. In other words, it does not matter where insulin action is restored so long as a certain minimal number of insulin-sensitive cells are present. Although our mosaic analysis of InsR signaling fails to support this view (6), the possibility has not been formally excluded. Moreover, we have recently shown that InsR inactivation in Glut4-expressing cell types gives rise to insulin-resistant diabetes (47). For these reasons, we sought to determine the metabolic consequences of reconstituting InsR expression in Glut4-expressing cell types of Insr−/− mice. In this study, we generated InsR knock-in mice in which insulin action was selectively restored in muscle, adipocytes, and Glut4-expressing brain cells. The data show that restoring insulin signaling in these tissues is not sufficient to rescue Insr−/− from diabetes and that the phenotype of these mice does not differ from that of mice with brain-restricted InsR reactivation.
EXPERIMENTAL PROCEDURES
Animals
Insr−/− (7), βac/INSR knock-in allele encoding the INSR-B isoform containing exon 11 (4), Synapsin-Cre (8), and GLUT4-Cre mice (47) have been previously described. Insr+/−::βac/INSR mice were intercrossed with Insr+/−::Synapsin-Cre or Insr+/−::GLUT4-Cre mice to generate Insr−/−::βac/INSR::Synapsin-Cre (NIRKI) and Insr−/−:: βac/INSR::GLUT4-Cre (GIRKI lines 1 and 2) mice. WT (Insr+/+::βac/IR, Insr+/+::Synapsin-Cre, Insr+/+::GLUT4-Cre, Insr+/+::βac/INSR::Synapsin-Cre, or Insr+/+::βac/INSR:: GLUT4-Cre) littermates were used as controls. Animals were maintained on a mixed background derived from 129/Sv, C57BL/6, and FVB strains. Genotyping was performed as described previously (4, 9, 47). All animal procedures have been approved by the Columbia University Institutional Animal Care and Utilization Committee.
Metabolic Analyses
Blood glucose was measured by the OneTouch Ultra meter (LifeScan). Urine ketone bodies and glucose were measured by Ketostix and Diastix (Bayer). Free fatty acids and triglycerides were measured by NEFA-HR test reagents (Wako Chemicals) and a serum triglyceride determination kit (Sigma-Aldrich). Insulin was measured by ELISA; adiponectin, leptin, monocyte chemotactic protein-1 (MCP-1), and tumor necrosis factor-α (TNFα) were measured by Luminex (Linco Research). C-peptide was measured by the Hormone and Metabolite Core at the New York Obesity Research Center. Hepatic glycogen and triglycerides were measured as described previously (10).
Western Blotting
Detergent extracts were prepared in buffer containing 20 mm Tris (pH 7.6), 150 mm NaCl, 1 mm DTT, 10 mm EGTA, 1% Nonidet P-40, 2.5 mm Na4P2O7, 1 mm NaVO3, 1 mm β-glycerophosphate, and protease inhibitor mixture (Roche Applied Science). Protein concentration of extracts was determined by the BCA assay (Pierce). Equal amounts of protein (40–100 μg) were resolved on SDS-PAGE and transferred onto nitrocellulose membranes (Schleicher & Schuell). Membranes were probed with antibodies against InsR β (C-19) and β-actin (Santa Cruz Biotechnology).
RNA Isolation and RT-PCR Analyses
Total tissue RNA was extracted using RNeasy mini kit and RNase-free DNase set (Qiagen). RNA was reverse-transcribed using oligo(dT) and an AffinityScript cDNA synthesis kit (Stratagene). Quantitative PCR reactions were performed in triplicate using a DNA Engine Opticon 3 system (Bio-Rad) and DyNAmo HS SYBR green quantitative PCR kit (New England Biolabs). See supplemental Table 1 for primer sequences. Relative mRNA levels were calculated using standard curves, with the PCR product for each primer set normalized to 36B4 for muscle and liver and Rps3 for fat.
Pancreas Immunohistochemistry
Pancreata were fixed in formalin overnight and embedded in paraffin, and consecutive sections 5 μm in thickness were mounted on slides. Sections were stained with antibodies against insulin, glucagon, Ki-67, active caspase-3 (Sigma-Aldrich), and FoxO1 (11). The β cell area and islet cell proliferation were determined using three sections spanning 200 μm/pancreas as described previously (4, 12).
Statistical Methods
All data represent means ± S.E. Data sets were analyzed for statistical significance with one-way analysis of variance followed by post hoc Bonferroni test using the SPSS software.
RESULTS
Generation of GIRKI and NIRKI Mice
To analyze whether reconstitution of insulin action in Glut4-expressing tissues sufficed to prevent diabetes in Insr−/− mice, we used a genetic knock-in approach to reactivate InsR expression (4). Insr+/− mice bearing a β-actin locus modified to encode a loxP-stop-LoxP INSR cassette were bred with two different lines of GLUT4-Cre transgenic mice. The knock-in allele encodes INSR-B, the predominant isoform in muscle and fat that includes exon 11 of the human INSR gene (13, 14). Line GLUT4-Cre 546 exhibits recombination in muscle, fat, and brain, whereas line GLUT4-Cre 535 differs from line 546 by the absence of recombination in fat (47). Thus, we generated mice in which InsR expression was restricted to muscle, fat, and brain (Glut4-insulin receptor knock-in line 1, henceforth GIRKI-1) or to skeletal muscle and brain (henceforth, GIRKI-2).
Western blot analyses showed reconstitution of InsR expression in skeletal muscle of GIRKI-1 and GIRKI-2 mice (levels were estimated at 134 ± 23 and 113 ± 15% of WT, respectively), whereas only GIRKI-1 mice displayed InsR reconstitution in white adipose tissue (WAT) (levels were 60 ± 8% of WT) (Fig. 1A). InsR expression was not detected in brown adipose tissue (BAT), heart, and liver of GIRKI-1 and GIRKI-2 mice (Fig. 1A). We also detected InsR expression in total brain lysates of GIRKI-1 and GIRKI-2 mice (Fig. 1A). More detailed analyses indicated that InsR was expressed in cortex, cerebellum, brainstem, hypothalamus, hippocampus, and the olfactory bulb (Fig. 1B and data not shown), consistent with our previous findings that Glut4-Cre induces recombination in selected neuronal subpopulations (47) overlapping with endogenous Glut4 expression (15–17).
FIGURE 1.
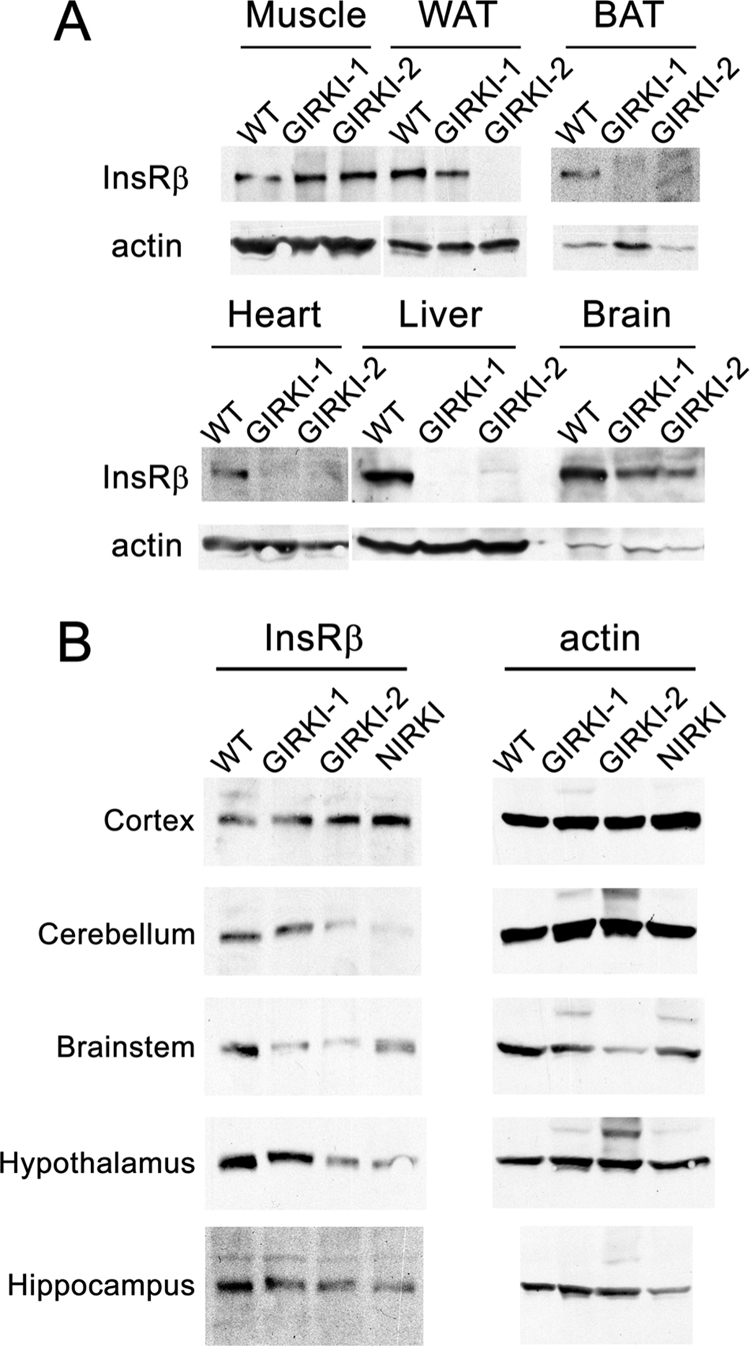
Tissue-specific reconstitution of InsR expression in Insr−/− mice. A, representative immunoblots of InsR β subunit and actin in hind limb muscle, subcutaneous WAT, BAT, heart, liver, and whole brain lysates of 1–2-week-old GIRKI-1, GIRKI-2, and WT littermates. B, representative immunoblots of InsR β subunit and actin in different brain regions of 1–2-week-old GIRKI-1, GIRKI-2, NIRKI, and WT littermates.
As a control for the specificity of the findings in GIRKI-1 and -2 mice, we also generated knock-in mice expressing InsR in neurons only (NIRKI) using Synapsin-Cre mice (4, 8). As shown previously, we detected widespread InsR reconstitution in the brain but not in peripheral tissues (Fig. 1B) (4).
Survival and Growth Phenotypes in GIRKI-1, GIRKI-2, and NIRKI Mice
GIRKI-1, GIRKI-2, and NIRKI mice were born in Mendelian ratios without apparent abnormalities. When compared with Insr−/− mice, whose median lifespan is 4 days (7, 10, 18), GIRKI-1, GIRKI-2, and NIRKI mice displayed significant lifespan extensions (Fig. 2A). The median lifespan of the three groups was similar (11–12 days), with the GIRKI-2 group attaining the longest mean lifespan (15 versus 10.6 and 11 days in GIRKI-1 and NIRKI mice, respectively). A small fraction of GIRKI-1, GIRKI-2, and NIRKI mice (3, 16, and 7%, respectively) survived longer than 3 weeks, and few GIRKI-2 and NIRKI mice survived up to 4 months. GIRKI-1, GIRKI-2, and NIRKI mice were stunted, attaining 47–69% of WT body weight at postnatal day 9 and ∼30% of WT body weight at 4 weeks of age (Fig. 2, B and C), similar to InsR mosaic mice (6). This was correlated with 7–10-fold increases in hepatic insulin-like growth factor-binding protein-1 (Igfbp1) mRNA levels when compared with WT (Fig. 2D) (10).
FIGURE 2.

Survival and growth phenotypes. A, Kaplan-Meier survival curves of GIRKI-1, GIRKI-2, and NIRKI mice. Data for Insr−/− mice represent historical controls based on previous publications (7, 10, 18). B, growth curves of GIRKI-1, GIRKI-2, NIRKI, and WT littermates. n = 4–20. *, p < 0.05, ***, p < 0.001 WT versus all three knock-in groups. C, representative images of GIRKI-1 and NIRKI mice and age-matched WT littermates. D, hepatic Igfbp1 mRNA expression in 1–2-week-old GIRKI-1, GIRKI-2, NIRKI, and WT littermates. Data are normalized against 36B4 mRNA. n = 7–10. *, p < 0.05, **, p < 0.01, ***, p < 0.001 versus WT.
Diabetes, Lipodystrophy, and Dyslipidemia Are Common Features of GIRKI and NIRKI Mice
More than 80% of GIRKI-1 and GIRKI-2 mice became hyperglycemic within 2 weeks of birth (Fig. 3A), with attendant glycosuria (Table 1) and 30–40-fold increases in plasma insulin levels (Fig. 3A). The increases in plasma C-peptide levels (Table 1) were more modest (2.2–2.8-fold), resulting in >20-fold increases in the insulin/C-peptide ratios (Table 1) and indicating that increased insulin secretion and defective insulin clearance contribute to hyperinsulinemia in these mice. 1–2-week-old NIRKI mice also displayed hyperglycemia and hyperinsulinemia similar to those observed in GIRKI-1 and GIRKI-2 mice (Fig. 3A). In addition, 50–80% of 1–2-week-old GIRKI-1, GIRKI-2, and NIRKI mice showed ketonuria (Table 1), indicating that ketoacidosis is a likely cause of death.
FIGURE 3.

Metabolic parameters. A, scatter plots of blood glucose and serum insulin levels in ad libitum fed 1–2- and 3–5-week-old GIRKI-1, GIRKI-2, NIRKI, and WT littermates. B, blood glucose and serum insulin levels in ad libitum fed 1–2-week-old versus 3–5-week-old GIRKI-2 and NIRKI mice. n = 5–29. *, p < 0.05, **, p < 0.01.
TABLE 1.
Metabolic characteristics of experimental animals (7–12 days old)
Ketonuria, liver steatosis, lipemia, and glycosuria were scored as positive or negative and were presented as fractions of total animals examined. *, p < 0.05, **, p < 0.01 vs. WT. ND, not determined. †, MCP-1 was below detection limit (25 pg/ml) in all WT (n = 8) and NIRKI (n = 3) mice examined. Two out of seven GIRKI-1 mice examined and four out of six GIRKI-2 mice examined had MCP-1 levels below detection limit and were all included in the group means as 25 pg/ml. FFA, free fatty acids.
| Genotype | WT | GIRKI-1 | GIRKI-2 | NIRKI |
|---|---|---|---|---|
| Ketonuria | None | 9/11 | 5/10 | 8/11 |
| Liver steatosis | None | 16/22 | 15/22 | 15/18 |
| Lipemia | None | 6/22 | 6/14 | 7/20 |
| Glycosuria | None | 10/10 | 21/22 | 13/13 |
| FFA (meq/liter) | 0.78 ± 0.10 | 1.28 ± 0.14* | 1.23 ± 0.17* | 1.38 ± 0.18* |
| Adiponectin (mg/ml) | 8.23 ± 1.63 | 4.37 ± 0.17* | 4.37 ± 0.16* | 4.77 ± 0.85* |
| Leptin (ng/ml) | 2.12 ± 0.39 | 0.59 ± 0.31* | 1.16 ± 0.56 | 0.24 ± 0.08** |
| MCP-1 (pg/ml) | ≤25† | ≤156 ± 44* | ≤113 ± 85 | ≤25 |
| TNFα (pg/ml) | 62.2 ± 4.5 | 94.6 ± 13.8* | 63.1 ± 2.2 | 55.3 ± 6.2 |
| C-peptide (nm) | 1.15 ± 0.17 | 2.55 ± 0.49* | 3.20 ± 0.67** | ND |
| Insulin/C-peptide (arbitrary units) | 1.0 ± 0.2 | 22.5 ± 7.3* | 38.6 ± 15.5* | ND |
The rare GIRKI-2 and NIRKI survivors at 3–5 weeks (the GIRKI-1 group did not have sufficient survivors for statistical analysis) displayed a further 40% increase in glycemia accompanied by a 60–70% decrease in insulin levels (Fig. 3B) when compared with 1–2-week-old mice, indicating progressive exhaustion of the pancreatic β cell compensatory capacity. These data indicate that InsR reconstitution in brain (NIRKI), in brain/muscle (GIRKI-2), and in brain/muscle/WAT (GIRKI-1) prolongs survival of Insr−/− mice but fails to prevent diabetes and premature death. The similarities in survival and metabolic phenotypes between NIRKI and GIRKI-1 or GIRKI-2 mice suggest that the predominant effect in these mice is due to restored CNS insulin signaling.
Histological analyses of skeletal muscle in 1–2-week-old GIRKI-1 and GIRKI-2 mice showed normal fiber composition (Fig. 4A and data not shown). This is consistent with restoration of InsR signaling to FoxO1, which can disrupt myogenesis and reduce slow-twitch fibers (19). Moreover, mRNA expression of PPARγ (peroxisome proliferator-activated receptor γ) coactivator 1α (Ppargc1a), a transcriptional coactivator promoting the formation of slow-twitch fibers (20), was modestly increased in GIRKI-1 and tended to be increased in GIRKI-2 muscles (Fig. 4B), thus restoring the formation of slow-twitch fibers.
FIGURE 4.

Histological and gene expression analyses of skeletal muscle. A, slow myosin heavy chain (MyHC) immunohistochemistry (brown) of soleus muscle from 9–11-day-old WT, GIRKI-1, and GIRKI-2 mice. Representative images are shown. B, hind limb skeletal muscle mRNA expression of Ppargc1a in 7–12-day-old WT, GIRKI-1, and GIRKI-2 mice. n = 5–9. **, p < 0.01.
Histological analyses in 1-week-old GIRKI-1, GIRKI-2, and NIRKI pups revealed interscapular BAT with normal morphology (Fig. 5A) but a substantial reduction of subcutaneous WAT and absent visceral WAT (21). Cellular infiltration, occasionally of granulomatous appearance, and fibrosis were present in the residual fat pads (Fig. 5, B and C), consistent with lipodystrophy (22, 23). Circulating leptin levels were significantly decreased in GIRKI-1 and NIRKI mice, as were adiponectin levels in all three groups of knock-in mice when compared with WT controls (Table 1) (9). 27–43% of 1–2-week-old GIRKI-1, GIRKI-2, and NIRKI mice were hyperlipidemic (Table 1), and serum free fatty acids were increased by 57–76% in all three groups when compared with WT (Table 1), consistent with increased lipolysis. Expression of Pparg, the master adipogenic regulator, was normal in subcutaneous WAT of GIRKI-1 and GIRKI-2 mice, and fatty acid synthase (Fasn) expression was reduced in GIRKI-1 mice (Fig. 5D). MCP-1 and TNFα, both pro-inflammatory cytokines, were significantly increased in GIRKI-1 mice but not in GIRKI-2 or NIRKI mice (Table 1). Expression of F4/80, a macrophage marker, and Cd3e, a T cell marker, was not affected in WAT of GIRKI-1 mice (Fig. 5D), suggesting that adipocytes are the source of increased cytokines as a consequence of restored InsR signaling and excess nutrient flux. In addition, glucose-regulated protein 78 (Grp78) and CCAAT/enhancer-binding protein homologous protein (Chop), key mediators of the unfolded protein response in the endoplasmic reticulum, were reduced in GIRKI-1 mice, as was uncoupling protein 1 (Ucp1) (Fig. 5D).
FIGURE 5.

Histological and gene expression analyses of adipose tissue. A and B, hematoxylin and eosin staining of BAT (A) and subcutaneous WAT (B) in 9–11-day-old WT, GIRKI-1, GIRKI-2, and NIRKI mice. Representative images are shown. C, subcutaneous WAT of a 4-week-old GIRKI-1 mouse shows extensive fibrosis and granulomatous infiltrates. D, subcutaneous WAT mRNA expression in 7–12-day-old WT, GIRKI-1, and GIRKI-2 mice. n = 6–9. Data are normalized against Rps3 mRNA. *, p < 0.05, **, p < 0.01 versus WT. #, p < 0.05, ##, p < 0.01 between groups. AU, arbitrary units.
A majority of 1-week-old GIRKI-1, GIRKI-2, and NIRKI mice had pale livers (Table 1); Oil Red O staining revealed extensive accumulation of intracellular lipid droplets (Fig. 6A) and a 5–14-fold increase in hepatic triglyceride (TG) content when compared with WT (Fig. 6C). Conversely, all three groups of mice showed significant decreases in liver glycogen content (Fig. 6, B and D). Interestingly, lipid accumulation was accompanied by significant decreases in hepatic mRNA expression of sterol regulatory element binding transcription factor 1 (Srebf1) and its target genes Fasn and acetyl-CoA carboxylase α (Acaca) (Fig. 7, A–C) in GIRKI-1 and GIRKI-2 mice, consistent with decreased triglyceride synthesis. Although Ppara mRNA was modestly decreased in GIRKI-1 mice (Fig. 7D), carnitine palmitoyltransferase 1a (Cpt1a) expression was normal (Fig. 7E), and acyl-CoA oxidase-1 (Acox1) was increased 2-fold in both GIRKI-1 and GIRKI-2 mice when compared with WT (Fig. 7F), indicative of increased free fatty acid oxidation. These data suggest that lipid accumulation in GIRKI and NIRKI mice primarily results from increased free fatty acids and lipoprotein flux to the liver. The decrease of lipogenic gene expression is likely to result from hepatic InsR deficiency (24–26). Furthermore, hepatic mRNA levels of F4/80 and Tnfa were decreased in GIRKI-1 and GIRKI-2 mice (Fig. 7, G and H), indicating that intrahepatic inflammation does not contribute to insulin resistance in these mice.
FIGURE 6.

Hepatic steatosis and glycogen content. A and B, Oil Red O (A) staining and periodic acid-Schiff (PAS) staining (B) in liver of 9–11-day-old WT, GIRKI-1, GIRKI-2, and NIRKI mice. Representative images are shown. C and D, triglyceride (TG) contents (C) and glycogen contents (D) in liver of 9–15-day-old WT, GIRKI-1, and NIRKI mice. n = 4–8. *, p < 0.05 versus WT.
FIGURE 7.

Hepatic gene expression profile. A–H, hepatic mRNA expression in ad libitum fed 1–2-week-old WT, GIRKI-1, and GIRKI-2 mice. n = 7–10. Data are normalized against 36B4 mRNA. *, p < 0.05, **, p < 0.01 versus WT. AU, arbitrary units.
β Cell Compensation in GIRKI and NIRKI Mice
We next analyzed pancreatic islet morphology. 9–11-day-old NIRKI, GIRKI-1, and GIRKI-2 mice displayed enlarged islets with normal architecture (Fig. 8, A and B). The pancreatic β cell area was increased 2–3-fold in all three groups when compared with WT (Fig. 8C). By 4 weeks of age, islets displayed reduced insulin immunoreactivity and decreased size in all genotypes (Fig. 8, D and E), consistent with the relative decrease in plasma insulin levels at this age (Fig. 3B). In addition, cellular infiltration and islet loss were apparent in a subset of all three groups of knock-in mice (Fig. 8, F and G and data not shown). The number of Ki-67-positive cells detected by immunohistochemistry increased in all mutant mice, indicating that the increase of the β cell area likely results from hyperproliferation (Fig. 9, A and D). These data indicate that InsR signaling in β cells is dispensable for β cell hyperproliferation in response to insulin resistance, as shown previously (27–29). In addition, all three groups of knock-in mice showed increased intra-islet apoptosis detected by active caspase-3 immunohistochemistry (Fig. 9E and data not shown), consistent with increased loss of β cells. FoxO1 plays an important role in β cell compensation to metabolic stress by promoting β cell diapause (30, 31) and premature senescence (11) and preventing β cell replication (12). Immunohistochemistry showed normal FoxO1 expression in β cells of 1–2-week-old NIRKI and GIRKI-1 mice (Fig. 9B and data not shown) but virtually no expression in islets of 5-week-old NIRKI mice (Fig. 9C), consistent with FoxO1 degradation due to cellular stress and prolonged hyperglycemia (11).
FIGURE 8.

Histological analyses of pancreatic islets. A and B, insulin (brown) (A) and glucagon (brown) (B) immunohistochemistry of pancreata from 9–11-day-old (P9∼P11) WT, NIRKI, GIRKI-1, and GIRKI-2 mice. Representative images are shown. C, the β cell area was quantified as the ratio of insulin-positive area relative to total pancreatic area. n = 6–22. *, p < 0.05, **, p < 0.01 versus WT. D and E, insulin (brown) (D) and glucagon (brown) (E) immunohistochemistry of pancreata from 4-week-old WT, NIRKI, GIRKI-1, and GIRKI-2 mice. Representative images are shown. F and G, cellular infiltration in islets of 4-week-old (F) and 4-month-old (G) GIRKI-2 mice.
FIGURE 9.

Cell proliferation and apoptosis in pancreatic islets. A, Ki-67 immunohistochemistry (brown) in pancreata of 4-week-old WT, GIRKI-1, GIRKI-2, and NIRKI mice. B and C, glucagon (green) and FoxO1 (red) immunohistochemistry in 9-day-old (B) and 5-week-old (C) WT and NIRKI mice. Representative images are shown. D, percentages of Ki-67-positive cells in all pancreatic islet cells in 4-week-old WT, GIRKI-1, GIRKI-2, and NIRKI mice. n = 1 for GIRKI-1, n = 3–7 for WT, GIRKI-2, and NIRKI. **, p < 0.01, ***, p < 0.001 versus WT. # p < 0.05 GIRKI-2 versus NIRKI. E, cleaved (active) caspase-3 immunohistochemistry (brown) in pancreata of 9-day-old WT and GIRKI-1 and 4–5-week-old WT and GIRKI-2 mice.
DISCUSSION
Perhaps the most intriguing question about insulin-resistant diabetes is about the site of onset of the disease. That skeletal muscle is an important site of insulin resistance in humans, leading to adaptive changes in nutrient utilization from carbohydrates to lipids and to compensatory β cell hyperplasia, is beyond dispute (32). However, targeted mutagenesis of genes required for insulin-dependent glucose disposal in muscle and/or fat of mice has limited metabolic consequences (33–36). In addition, gain-of-function experiments show that diabetes due to InsR knock-out can be “cured” by restoration of InsR function in liver, brain, and β cells despite the absence of InsR in muscle and fat (4, 5). These data indicate that mice can overcome the insulin resistance of muscle and adipose tissue as long as hepatic insulin action is preserved and there are no additional strains on the β cell. The truism that mice are different from humans and that this set of observations amounts to little more than a quirk of comparative physiology brings little solace to the urgency of developing effective diabetes treatments and does not change the fact that only rodent models combine the ability to modify genes with advanced phenotyping capabilities and a detailed understanding of integrated physiology.
The lesson learned from the failure of muscle- and fat-specific InsR knock-outs to develop either insulin resistance or diabetes is that additional insulin target tissues must be involved in the primary defect of insulin action. A prime, if overlooked, candidate in that regard is the CNS as a reemerging body of work indicates that insulin signaling in hypothalamic neurons accounts for its indirect effects on hepatic glucose production (5, 37–39). Interestingly, although the nature of the effects of insulin on these neurons remains unclear, the fact that both insulin and glucose regulate their electrical activity would appear to link insulin signaling to glucose sensing (39, 40).
Based on evidence demonstrating that the insulin-sensitive glucose transporter Glut4 is scattered throughout the CNS (41), we developed transgenic mice in which Cre-mediated DNA excision occurred under the transcriptional control of the GLUT4 promoter in muscle, fat, and Glut4-expressing CNS neurons. Consistent with the prediction, we found that Glut4-InsR knock-out mice (GIRKO) recapitulated all aspects of insulin resistance and developed diabetes with high frequency (47). These data indicate that insulin resistance can singlehandedly cause diabetes, regardless of primary defects of β cell function, when it involves Glut4-expressing cells in CNS, muscle, and fat.
However, are the sites of onset of insulin resistance necessarily the same sites that should be targeted to treat diabetes? The question has more than academic interest as prospective data indicate that the contribution of different tissues to hyperglycemia changes as a function of disease progression and control (42). To address this question, we asked whether restoring InsR function in InsR-deficient mice would suffice to rescue them from diabetes. In other words, the loss-of-function experiment explored the pathophysiology of insulin-resistant diabetes, whereas the gain-of-function experiment addressed its treatment.
The finding that the muscle/fat/GLUT4 neuron combination is insufficient to rescue Insr−/− mice from diabetes provides several useful lessons. First, comparison of the phenotype of GIRKI-1 and -2 mice with those of NIRKI mice suggests that the admittedly modest metabolic amelioration seen in the former, when compared with Insr−/− mice, is likely to reflect the contribution of the brain (4). Second, the absence of InsR in pancreatic β cells accelerates their failure and is accompanied by the loss of FoxO1 expression, consistent with a protective role of FoxO1 against β cell failure and the role of hyperglycemia to promote FoxO1 degradation (11). These data add to a body of evidence indicating that InsR function in β cells promotes compensatory hyperplasia and survival (12, 27, 43, 44).
Why is the outcome of the knock-out different from that of the knock-in? We show that the levels of InsR expression in GIRKI-1 and -2 mice are similar to WT controls, indicating that the experiment worked from a technical standpoint. INSR-B, the isoform used in this study, is the predominant splicing isoform in muscle and fat, which also express the 12-amino acid-shorter isoform INSR-A at lower levels (14). It remains formally possible that certain INSR-A-specific functions may not have been rescued by re-expression of INSR-B. However, unlike its specific prenatal role in response to high affinity binding of IGF-II (45), it is unclear that the postnatal metabolic role of INSR-A is sufficiently different from INSR-B to engender the differences seen in this study. Therefore, we propose that the differences between the knock-out and the knock-in have a developmental basis. Newborn mice depend on liver for conversion of triglycerides into ketones for brain growth, whereas insulin-dependent glucose uptake is less important in the neonatal period (46). In fact, adipose tissue begins to form in earnest only after birth, whereas skeletal muscle can compensate for the lack of InsR through contraction-stimulated glucose uptake (1). Thus, Insr−/−mice can survive if their hepatic insulin action is restored, but they are relatively unresponsive to restoration of insulin action in muscle and fat. Conversely, in adult mice, muscle and fat have achieved their target mass and account for a significant fraction of glucose disposal, and thus impairments of their insulin sensitivity are bound to have metabolic consequences. In addition, the CNS can affect hepatic glucose production; hence, the combination of impaired glucose uptake and excessive glucose production is the likely cause of diabetes in the GIRKO mice (47).
In future studies, it will be important to address whether, in addition to the established mechanisms of insulin resistance, there are developmental circuits involving brain, liver, and β cells, whose disruption by the congenital deficiency of InsR may prevent restoration of insulin sensitivity in GIRKI mice. In view of the growing interest in the role of the uterine environment on adult metabolism, such studies are likely to have wide-ranging implications in the fight against metabolic disorders.
Supplementary Material
Acknowledgments
We thank K. Aizawa and Q. Xu for technical assistance and members of the Accili laboratory for helpful discussions and critical reading of the manuscript.
This work was supported, in whole or in part, by National Institutes of Health Grants DK58282 and DK57539 (to D. A.). This work was also supported by the Berrie Fellowship Award (to H. V. L.) and by Grant DK63608 from the Columbia University Diabetes and Endocrinology Research Center.

The on-line version of this article (available at http://www.jbc.org) contains supplemental Table 1.
- InsR
- insulin receptor
- WAT
- white adipose tissue
- BAT
- brown adipose tissue.
REFERENCES
- 1. Accili D. (2004) Diabetes 53, 1633–1642 [DOI] [PubMed] [Google Scholar]
- 2. Nandi A., Kitamura Y., Kahn C. R., Accili D. (2004) Physiol. Rev. 84, 623–647 [DOI] [PubMed] [Google Scholar]
- 3. Okamoto H., Accili D. (2003) J. Biol. Chem. 278, 28359–28362 [DOI] [PubMed] [Google Scholar]
- 4. Okamoto H., Nakae J., Kitamura T., Park B. C., Dragatsis I., Accili D. (2004) J. Clin. Invest. 114, 214–223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Okamoto H., Obici S., Accili D., Rossetti L. (2005) J. Clin. Invest. 115, 1314–1322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kitamura T., Kitamura Y., Nakae J., Giordano A., Cinti S., Kahn C. R., Efstratiadis A., Accili D. (2004) J. Clin. Invest. 113, 209–219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Accili D., Drago J., Lee E. J., Johnson M. D., Cool M. H., Salvatore P., Asico L. D., José P. A., Taylor S. I., Westphal H. (1996) Nat. Genet. 12, 106–109 [DOI] [PubMed] [Google Scholar]
- 8. Zhu Y., Romero M. I., Ghosh P., Ye Z., Charnay P., Rushing E. J., Marth J. D., Parada L. F. (2001) Genes Dev. 15, 859–876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lin H. V., Kim J. Y., Pocai A., Rossetti L., Shapiro L., Scherer P. E., Accili D. (2007) Diabetes 56, 1969–1976 [DOI] [PubMed] [Google Scholar]
- 10. Matsumoto M., Pocai A., Rossetti L., Depinho R. A., Accili D. (2007) Cell Metab. 6, 208–216 [DOI] [PubMed] [Google Scholar]
- 11. Kitamura Y. I., Kitamura T., Kruse J. P., Raum J. C., Stein R., Gu W., Accili D. (2005) Cell Metab. 2, 153–163 [DOI] [PubMed] [Google Scholar]
- 12. Okamoto H., Hribal M. L., Lin H. V., Bennett W. R., Ward A., Accili D. (2006) J. Clin. Invest. 116, 775–782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Seino S., Bell G. I. (1989) Biochem. Biophys. Res. Commun. 159, 312–316 [DOI] [PubMed] [Google Scholar]
- 14. Moller D. E., Yokota A., Caro J. F., Flier J. S. (1989) Mol. Endocrinol. 3, 1263–1269 [DOI] [PubMed] [Google Scholar]
- 15. Vannucci S. J., Koehler-Stec E. M., Li K., Reynolds T. H., Clark R., Simpson I. A. (1998) Brain Res. 797, 1–11 [DOI] [PubMed] [Google Scholar]
- 16. Sankar R., Thamotharan S., Shin D., Moley K. H., Devaskar S. U. (2002) Brain Res. Mol. Brain Res. 107, 157–165 [DOI] [PubMed] [Google Scholar]
- 17. Komori T., Morikawa Y., Tamura S., Doi A., Nanjo K., Senba E. (2005) Brain Res. 1049, 34–42 [DOI] [PubMed] [Google Scholar]
- 18. Joshi R. L., Lamothe B., Cordonnier N., Mesbah K., Monthioux E., Jami J., Bucchini D. (1996) EMBO J. 15, 1542–1547 [PMC free article] [PubMed] [Google Scholar]
- 19. Kitamura T., Kitamura Y. I., Funahashi Y., Shawber C. J., Castrillon D. H., Kollipara R., DePinho R. A., Kitajewski J., Accili D. (2007) J. Clin. Invest. 117, 2477–2485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lin J., Wu H., Tarr P. T., Zhang C. Y., Wu Z., Boss O., Michael L. F., Puigserver P., Isotani E., Olson E. N., Lowell B. B., Bassel-Duby R., Spiegelman B. M. (2002) Nature 418, 797–801 [DOI] [PubMed] [Google Scholar]
- 21. Cinti S., Eberbach S., Castellucci M., Accili D. (1998) Diabetologia 41, 171–177 [DOI] [PubMed] [Google Scholar]
- 22. Shimomura I., Hammer R. E., Richardson J. A., Ikemoto S., Bashmakov Y., Goldstein J. L., Brown M. S. (1998) Genes Dev. 12, 3182–3194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Pajvani U. B., Trujillo M. E., Combs T. P., Iyengar P., Jelicks L., Roth K. A., Kitsis R. N., Scherer P. E. (2005) Nat. Med. 11, 797–803 [DOI] [PubMed] [Google Scholar]
- 24. Shimomura I., Matsuda M., Hammer R. E., Bashmakov Y., Brown M. S., Goldstein J. L. (2000) Mol. Cell. 6, 77–86 [PubMed] [Google Scholar]
- 25. Matsumoto M., Han S., Kitamura T., Accili D. (2006) J. Clin. Invest. 116, 2464–2472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Biddinger S. B., Hernandez-Ono A., Rask-Madsen C., Haas J. T., Alemán J. O., Suzuki R., Scapa E. F., Agarwal C., Carey M. C., Stephanopoulos G., Cohen D. E., King G. L., Ginsberg H. N., Kahn C. R. (2008) Cell Metab. 7, 125–134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Okada T., Liew C. W., Hu J., Hinault C., Michael M. D., Krtzfeldt J., Yin C., Holzenberger M., Stoffel M., Kulkarni R. N. (2007) Proc. Natl. Acad. Sci. U.S.A. 104, 8977–8982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Xuan S., Szabolcs M., Cinti F., Perincheri S., Accili D., Efstratiadis A. (2010) J. Biol. Chem. 285, 41044–41050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kido Y., Nakae J., Hribal M. L., Xuan S., Efstratiadis A., Accili D. (2002) J. Biol. Chem. 277, 36740–36747 [DOI] [PubMed] [Google Scholar]
- 30. Buteau J., Shlien A., Foisy S., Accili D. (2007) J. Biol. Chem. 282, 287–293 [DOI] [PubMed] [Google Scholar]
- 31. Kawamori D., Kaneto H., Nakatani Y., Matsuoka T. A., Matsuhisa M., Hori M., Yamasaki Y. (2006) J. Biol. Chem. 281, 1091–1098 [DOI] [PubMed] [Google Scholar]
- 32. Petersen K. F., Dufour S., Savage D. B., Bilz S., Solomon G., Yonemitsu S., Cline G. W., Befroy D., Zemany L., Kahn B. B., Papademetris X., Rothman D. L., Shulman G. I. (2007) Proc. Natl. Acad. Sci. U.S.A. 104, 12587–12594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lauro D., Kido Y., Castle A. L., Zarnowski M. J., Hayashi H., Ebina Y., Accili D. (1998) Nat. Genet. 20, 294–298 [DOI] [PubMed] [Google Scholar]
- 34. Brüning J. C., Michael M. D., Winnay J. N., Hayashi T., Hörsch D., Accili D., Goodyear L. J., Kahn C. R. (1998) Mol. Cell. 2, 559–569 [DOI] [PubMed] [Google Scholar]
- 35. Blüher M., Michael M. D., Peroni O. D., Ueki K., Carter N., Kahn B. B., Kahn C. R. (2002) Dev. Cell. 3, 25–38 [DOI] [PubMed] [Google Scholar]
- 36. Kotani K., Peroni O. D., Minokoshi Y., Boss O., Kahn B. B. (2004) J. Clin. Invest. 114, 1666–1675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Obici S., Feng Z., Karkanias G., Baskin D. G., Rossetti L. (2002) Nat. Neurosci. 5, 566–572 [DOI] [PubMed] [Google Scholar]
- 38. Lin H. V., Plum L., Ono H., Gutiérrez-Juárez R., Shanabrough M., Borok E., Horvath T. L., Rossetti L., Accili D. (2010) Diabetes 59, 337–346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Könner A. C., Janoschek R., Plum L., Jordan S. D., Rother E., Ma X., Xu C., Enriori P., Hampel B., Barsh G. S., Kahn C. R., Cowley M. A., Ashcroft F. M., Brüning J. C. (2007) Cell Metab. 5, 438–449 [DOI] [PubMed] [Google Scholar]
- 40. Pocai A., Lam T. K., Gutierrez-Juarez R., Obici S., Schwartz G. J., Bryan J., Aguilar-Bryan L., Rossetti L. (2005) Nature 434, 1026–1031 [DOI] [PubMed] [Google Scholar]
- 41. Maher F., Vannucci S. J., Simpson I. A. (1994) FASEB J. 8, 1003–1011 [DOI] [PubMed] [Google Scholar]
- 42. Monnier L., Colette C., Dunseath G. J., Owens D. R. (2007) Diabetes Care. 30, 263–269 [DOI] [PubMed] [Google Scholar]
- 43. Kulkarni R. N., Brüning J. C., Winnay J. N., Postic C., Magnuson M. A., Kahn C. R. (1999) Cell 96, 329–339 [DOI] [PubMed] [Google Scholar]
- 44. Ueki K., Okada T., Hu J., Liew C. W., Assmann A., Dahlgren G. M., Peters J. L., Shackman J. G., Zhang M., Artner I., Satin L. S., Stein R., Holzenberger M., Kennedy R. T., Kahn C. R., Kulkarni R. N. (2006) Nat. Genet. 38, 583–588 [DOI] [PubMed] [Google Scholar]
- 45. Nakae J., Kido Y., Accili D. (2001) Endocr. Rev. 22, 818–835 [DOI] [PubMed] [Google Scholar]
- 46. Girard J., Ferré P., Pégorier J. P., Duée P. H. (1992) Physiol. Rev. 72, 507–562 [DOI] [PubMed] [Google Scholar]
- 47. Lin H. V., Ren H., Samuel V. T., Lee H. Y., Lu T. Y., Shulman G. I., Accili D. (2011) Diabetes, in press [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.