

Abstract
Prion diseases occur following the conversion of the cellular prion protein (PrPC) into a disease related, protease-resistant isoform (PrPSc). In these studies, a cell painting technique was used to introduce PrPC to prion-infected neuronal cell lines (ScGT1, ScN2a, or SMB cells). The addition of PrPC resulted in increased PrPSc formation that was preceded by an increase in the cholesterol content of cell membranes and increased activation of cytoplasmic phospholipase A2 (cPLA2). In contrast, although PrPC lacking one of the two acyl chains from its glycosylphosphatidylinositol (GPI) anchor (PrPC-G-lyso-PI) bound readily to cells, it did not alter the amount of cholesterol in cell membranes, was not found within detergent-resistant membranes (lipid rafts), and did not activate cPLA2. It remained within cells for longer than PrPC with a conventional GPI anchor and was not converted to PrPSc. Moreover, the addition of high amounts of PrPC-G-lyso-PI displaced cPLA2 from PrPSc-containing lipid rafts, reduced the activation of cPLA2, and reduced PrPSc formation in all three cell lines. In addition, ScGT1 cells treated with PrPC-G-lyso-PI did not transmit infection following intracerebral injection to mice. We propose that that the chemical composition of the GPI anchor attached to PrPC modified the local membrane microenvironments that control cell signaling, the fate of PrPC, and hence PrPSc formation. In addition, our observations raise the possibility that pharmacological modification of GPI anchors might constitute a novel therapeutic approach to prion diseases.
Keywords: Cholesterol, Glycosyl Phosphatidyl Inositol Anchors, Lipid Raft, Phospholipase A, Prions
Introduction
The transmissible spongiform encephalopathies, which are also known as prion diseases, are invariably fatal neurodegenerative disorders that include scrapie in sheep, bovine spongiform encephalopathy in cattle, and Creutzfeldt-Jakob disease in human(s). A key event in these diseases is the conversion of a normal host protein, designated PrPC,3 into a disease-associated isoform (PrPSc), which represents the major component of infectious scrapie prions (1). This process involves a portion of PrPC that is mostly α-helix being refolded into a β-pleated sheet in the PrPSc molecule (2). PrPSc acts as a template that facilitates the conversion of PrPC to PrPSc. The conversion of PrPC to PrPSc is accompanied by changes in biological and biochemical properties, including an increased resistance to proteases (3), resulting in the accumulation of PrPSc in affected brain areas (4).
Although the presence of PrPC is essential for prion replication (5, 6), it does not ensure prion replication, and it is thought that the targeting of PrPC to specific membranes is required for efficient PrPSc formation. For example, antibody studies suggest that PrPC conversion occurs at the cell surface (7, 8), and treatments that altered the intracellular trafficking of PrPC also affected PrPSc formation (9, 10). In addition, treatment with cholesterol synthesis inhibitors that altered the cellular location of PrPC also reduced PrPSc formation (11, 12). Collectively, these observations indicate that the factors that affect the intracellular trafficking of PrPC affect its conversion to PrPSc.
The majority of PrPC molecules are linked to membranes via a glycosylphosphatidylinositol (GPI) anchor (13). The role of the GPI anchor in prion disease is controversial in that although transgenic mice producing anchorless PrPC produced large amounts of extracellular PrPSc (14), a recent study showed that cells producing anchorless PrPC were resistant to scrapie infection (15). Moreover, these studies employed genetic methods that removed the entire GPI anchor rather than specific modifications of GPI anchors. The GPI anchor targets PrPC to detergent-resistant membrane microdomains that are commonly called lipid rafts and which are necessary for efficient PrPSc formation (12). Many GPI-anchored molecules transfer between cell membranes, a process called cell painting (16, 17). The transfer of PrPC between cells in vitro (18) showed that cell painting could be used to introduce PrPC with different GPI anchors to recipient cells. In this study, we report the effects of modification of the GPI anchor attached to PrPC on PrPSc formation using a combination of a cell painting technique and PrPC that had been digested by phospholipase A2 (PrPC-G-lyso-PI) or phosphatidylinositol-phospholipase C (PrPC-IPG) (Fig. 1).
FIGURE 1.

Phospholipase digestion of PrPC affects the GPI anchors. Shown is a cartoon displaying the putative GPI anchor attached to PrPC. Glycan residues shown include inositol (Inos), mannose (Man), sialic acid (SA), galactose (Gal), N-acetyl galactosamine (GalNAc), and glucosamine (GlcN) as well as phosphate (P). Also shown are the products of PrPC after digestion with either PLA2 (PrPC-G-lyso-PI) or phosphatidylinositol-phospholipase C (PrPC-IPG).
EXPERIMENTAL PROCEDURES
Cell Lines
Prion-infected ScN2a, ScGT1, and SMB cells were grown in Ham's F12 medium containing 2 mm glutamine, 2% FCS, 100 units/ml penicillin, and 100 μg/ml streptomycin. To determine the effect of PrPC preparations on PrPSc formation, ScN2a, SMB, or ScGT1 cells were plated in six-well plates at 105 cells/well and allowed to adhere overnight. Cells were then cultured in the presence or absence of test preparations, with daily changes of media, for 7 days. Cells were washed twice in PBS, and extracts were collected. Spent medium was collected and concentrated by centrifugation with a 10-kDa filter (Vivaspin, Sartorius) to examine whether PrPSc was released into culture supernatants. The survival of cells was determined by the addition of 25 μm thiazolyl blue tetrazolium for 3 h and reported as a percentage of untreated cells.
Evaluation of Infectivity
Treated ScGT1 cells were detached and counted, washed twice with PBS, frozen, and thawed. Membranes were centrifuged at 16,000 × g for 30 min, washed twice, and homogenized in sterile 0.9% (w/v) saline at 2.5 × 106 cell equivalents/ml. C57/BL mice under halothane anesthesia were injected intracerebrally with 30 μl (7.5 × 104 cell equivalents) of this homogenate. Mice were monitored for clinical signs of scrapie until reaching a predefined clinical end point. All animal work was conducted according to local and national guidelines.
Primary Cortical Neurons
Cortical neurons were prepared from the brains of mouse embryos (day 15.5) derived from PrP null mice as described (11) and plated at 106 cells/well in six-well plates precoated with poly-l-lysine. Neurons were grown in neurobasal medium containing B27 components (PAA) for 10 days. Neurons were incubated with PrPC preparations for different time periods and washed three times with PBS, and extracts were prepared. In some assays, cells were pulsed with PrPC for 2 h, washed three times with PBS, and incubated in fresh culture medium for between 24 and 96 h. The amount of PrPC expressed at the cell surface was determined by treating cells with 0.2 units of phosphatidylinositol-phospholipase C/106 cells for 1 h at 37 °C, and the amount of PrPC released into the supernatant was measured by ELISA.
Cell Membrane Extracts
Treated cells were homogenized in an extraction buffer (10 mm Tris-HCl, pH 7.4, 100 mm NaCl, 10 mm EDTA, 0.5% Nonidet P-40, 0.5% sodium deoxycholate, and 0.2% SDS) at 106 cells/ml, and nuclei and large fragments were removed by centrifugation (300 × g for 5 min). Mixed protease inhibitors (4-(2-aminoethyl)benzenesulfonyl fluoride hydrochloride, aprotinin, leupeptin, bestatin, pepstatin A, and E-46) (Sigma) and a phosphatase inhibitor mixture including PP1, PP2A, microcystin LR, cantharidin, and p-bromotetramisole (Sigma) were added to extracts where appropriate. To determine the amount of PrPSc produced, cell extracts/supernatants were digested with 1 μg/ml proteinase K for 1 h at 37 °C. Samples were heated to 95 °C for 5 min and tested in a PrP ELISA.
Isolation of Detergent-resistant Membranes
Cells were homogenized in an ice-cold buffer containing 1% Triton X-100, 10 mm Tris-HCl, pH 7.2, 100 mm NaCl, 10 mm EDTA, and mixed protease inhibitors at 106 cells/ml. Nuclei and large fragments were removed by centrifugation (500 × g for 5 min). The postnuclear supernatant was incubated on ice for 60 min and centrifuged (16,000 × g for 30 min at 4 °C). The soluble material contained the normal cell membrane (detergent-resistant membranes). Pellets were homogenized in 10 mm Tris-HCl, pH 7.4, 10 mm NaCl, 10 mm EDTA, 0.5% Nonidet P-40, 0.5% sodium deoxycholate, and 0.2% SDS and mixed protease inhibitors, centrifuged again (16,000 × g for 10 min), and the supernatant containing the lipid raft constituents was collected.
Sucrose Density Gradients
Cultured neurons were harvested with a Teflon scraper and homogenized in 250 mm sucrose, 10 mm Tris-HCl, pH 7.2, 1 mm EDTA, and 1 mm dithiothreitol at 106 cells/ml. Nuclei and membrane fragments were removed by centrifugation (1000 × g for 5 min). Membranes were washed by centrifugation at 16,000 × g for 10 min at 4 °C and suspended in an ice-cold buffer containing 1% Triton X-100, 10 mm Tris-HCl, pH 7.4, 150 mm NaCl, 10 mm EDTA, and protease and phosphatase inhibitors (as above). 5–40% sucrose solutions were prepared and layered to produce a gradient. Membranes were added and centrifuged (50,000 × g for 4 h at 4 °C). Fractions were collected from the bottom of gradients.
Isolation of PrPC
PrPC was isolated from GT1 neuronal cell membranes that had been homogenized in a buffer containing 10 mm Tris-HCl, pH 7.4, 100 mm NaCl, 10 mm EDTA, 0.5% Nonidet P-40, 0.5% sodium deoxycholate, and mixed protease inhibitors using an affinity column loaded with mAb ICSM35 (d.gen, Inc.). Bound PrPC was digested with 0.2 units/ml phosphatidylinositol-phospholipase C extracted from Bacillus cereus (Sigma) to generate PrPC-IPG, or 100 units/ml bee venom phospholipase A2 (PLA2) (Sigma) to generate PrPC-G-lyso-PI at 37 °C for 1 h. Digested PrPC preparations were eluted using glycine-HCl, neutralized with 1 m Tris, and isolated via reverse phase chromatography on C18 columns (Waters). Proteins were eluted using a gradient of acetonitrile in water and 0.1% TFA as shown in supplemental Fig. 1A. PrP containing fractions were pooled, desalted, and concentrated. For bioassays, samples were diluted in culture medium and solubilized by sonication.
Isolation of GPI Anchors
GPIs were isolated from PrPC by digestion with 100 μg/ml proteinase K at 37 °C for 24 h. The released GPIs were extracted with water-saturated butan-1-ol, washed with water, lyophilized, and stored in ethanol at 20 μm. GPIs were examined by high performance thin-layer chromatography on silica gel 60 plates and probed with mAb 5AB3-11 that binds to phosphatidylinositol as described (19).
PrP ELISA
The amount of PrP in samples was measured by ELISA as described (20). Maxisorb Immunoplates (Nunc) were coated with mAb ICSM18 (d.gen, Inc.) and blocked with 5% milk powder. Samples were applied and detected with biotinylated mAb ICSM35 (d.gen, Inc.), followed by extravidin-alkaline phosphatase and 1 mg/ml 4-nitrophenyl phosphate (Sigma). Absorbance was measured on a microplate reader at 405 nm, and the amount of PrP in samples was calculated by reference to a standard curve of recombinant murine PrP (Prionics).
Activated cPLA2 ELISA
The activation of cPLA2 is accompanied by phosphorylation of the serine 505 residue, which creates a unique epitope, and the amount of activated cPLA2 in samples was measured by ELISA as described (20). Maxisorb immunoplates were coated with 0.5 μg/ml of mouse mAb anti-cPLA2 (clone CH-7 (Upstate)). Samples were incubated for 1 h, and the amount of activated cPLA2 was detected using a rabbit polyclonal antiphospho-cPLA2 (Cell Signaling Technology) followed by a biotinylated anti-rabbit IgG (Dako), extravidin-alkaline phosphatase, and 1 mg/ml 4-nitrophenyl phosphate. Samples were expressed as “units of activated cPLA2,” where 100 units was the amount of activated cPLA2 in extracts from 106 untreated cells. To measure cPLA2 protein immunoplates were coated with mAb CH-7, and samples were applied as above. Bound cPLA2 was detected using a goat polyclonal anti-cPLA2 (Santa Cruz Biotechnology) followed by biotinylated anti-goat IgG, extravidin-alkaline phosphatase, and 1 mg/ml 4-nitrophenyl phosphate.
Immunoprecipitations
Treated cells were solubilized in ice cold 1% Triton X-100, 10 mm Tris-HCl, pH 7.2, 100 mm NaCl, 10 mm EDTA 106 cells/ml for 1 h at 4 °C. Cell debris was removed by centrifugation (500 × g for 5 min), and the supernatant was incubated with mAbs to PrP (ICSM35), cPLA2 (CH-7), or isotype controls for 30 min at 4 °C on rollers. Magnetic microbeads containing protein G were added (10 μl/ml) (Miltenyi Biotech) for 30 min, and protein G bound antibody complexes were isolated using a μMACS magnetic system (Miltenyi Biotech.) at 4 °C. To determine PrPSc levels, isolated beads were predigested with 1 μg/ml proteinase K.
Western Analysis
Samples were mixed with Laemmli buffer containing β-mercaptoethanol, boiled, and separated by PAGE on a 15% gel. Proteins were transferred onto a Hybond-P PVDF membrane (Amersham Biosciences) by semi-dry blotting. Membranes were blocked using 10% milk powder. PrP was detected by incubation with mAb ICSM18 (d.gen, Inc.), β-actin by mAb clone AC-74 (Sigma), and cPLA2 by mAb CH-7, followed by a secondary anti-mouse IgG conjugated to peroxidase. Bound antibody was visualized using enhanced chemiluminescence.
Cholesterol and Protein Content
Protein concentrations were measured using a micro-BCA protein assay kit (Pierce), and the amount of cholesterol was measured using the Amplex Red cholesterol assay kit (Invitrogen). The assay was performed after digestion with cholesterol esterase to determine the amount of esterified cholesterol within samples.
Statistical Analysis
Comparison of treatment effects was carried out using one-way and two-way analysis of variance. For all statistical tests, significance was set at the 1% level.
RESULTS
Addition of PrPC Increased PrPSc Formation in Prion-infected Cells
Control and phosphatidylinositol-phospholipase C- or PLA2-digested PrPC preparations were isolated by reverse phase chromatography on C18 columns. Mock-treated PrPC containing an intact GPI anchor (PrPC-GPI) was eluted between 68 and 74% acetonitrile, PrPC-G-lyso-PI was eluted between 30 and 36% acetonitrile, whereas PrPC-IPG did not bind (supplemental Fig. 1A). Immunoblots showed that digestion with PLA2 or phosphatidylinositol-phospholipase C did not affect the protein or glycan components of PrPC (supplemental Fig. 1B). However, the GPI anchor isolated from PrPC-G-lyso-PI and PrPC-IPG had differed to that isolated from PrPC-GPI (supplemental Fig. 1C).
The daily addition of PrPC-GPI increased the amount of PrPSc in ScN2a cells in a dose-dependent manner (Fig. 2A). The increased PrPSc formation did not affect the survival of ScN2a cells, for example the addition of 10 ng PrPC-GPI increased PrPSc formation without affecting cell survival (97% cell survival ± 9 compared with 100% ± 7, n = 12, p = 0.7). Immunoblots confirmed ELISA data and showed that the addition of PrPC-GPI increased the PrPSc content of ScN2a cells without affecting the amount of β-actin (Fig. 2B). This effect of PrPC-GPI was not cell-line specific; the daily addition of 10 ng of PrPC-GPI also increased the PrPSc content of ScGT1 and SMB cells (Table 1). The amount of PrPSc in these cells was not altered by the addition of 10 ng PrPC-IPG and was reduced following the addition of 10 ng PrPC-G-lyso-PI. Supernatants were collected to determine whether extracellular PrPSc had been formed during these experiments. The daily addition of 10 ng PrPC-GPI increased the amount of extracellular PrPSc released from ScN2a cells (1.8 ng of PrPSc ± 0.67 compared with 0.38 ng ± 0.29, n = 28, p < 0.05). The addition of 10 ng PrPC-IPG had no affect (0.41 ng of PrPSc ± 0.34 compared with 0.38 ng ± 0.29, n = 28, p = 0.7), and no PrPSc was detected in the supernatants of ScN2a cells incubated with 10 ng PrPC-G-lyso-PI (<0.05 ng of PrPSc).
FIGURE 2.
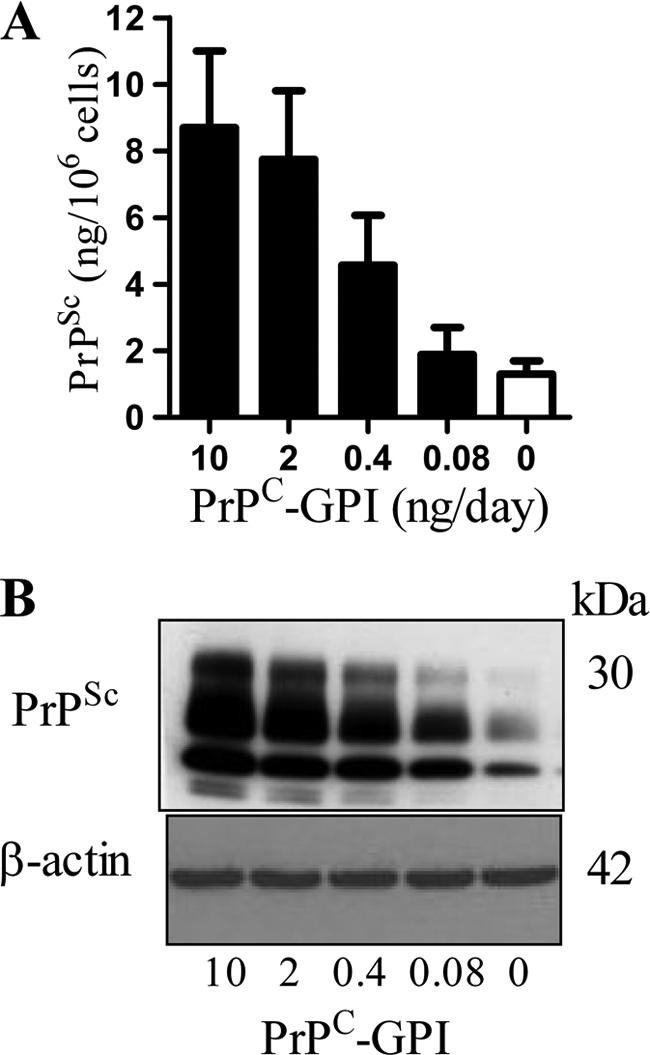
The addition of PrPC-GPI increased the PrPSc content of ScN2a cells. A, The amount of PrPSc in ScN2a cells treated with different concentrations of PrPC-GPI as shown for 7 days. Values shown are the mean amount of PrPSc (ng/106 cells) ± S.D. from triplicate experiments performed 10 times (n = 30). B, ScN2a cells were incubated for 7 days with different concentrations of PrPC-GPI as shown, and cell extracts prepared (± proteinase K digestion) and separated by PAGE and transferred to membranes. β-Actin in cell extracts was detected by immunoblot with clone AC-74, and PrPSc was detected by immunoblot with mAb ICSM18.
TABLE 1.
PrPC-G-lyso-PI inhibited the conversion of PrPC to PrPSc
Prion-infected neuronal cell lines were treated daily with 10 ng of PrPC-GPI, PrPC-IPG, or PrPC-G-lyso-PI as shown. After 7 days, cells were collected, and the amount of PrPSc was measured by ELISA. Values shown are the mean amount of PrPSc (ng/106 cells) ± S.D. in ScN2a cells (n = 28), ScGT1 cells (n = 22), or SMB cells (n = 18).
| Treatment | PrPSc (ng/106 cells) |
||
|---|---|---|---|
| ScN2a cells | ScGT1 cells | SMB cells | |
| None | 1.3 ± 0.4 | 9.6 ± 1.4 | 5.8 ± 1.2 |
| PrPC-GPI | 8.7 ± 2.3 | 19.4 ± 3.1 | 15.4 ± 3.3 |
| PrPC-IPG | 1.1 ± 0.4 | 8.6 ± 2.2 | 5.2 ± 1.3 |
| PrPC-G-lyso-PI | 0.6 ± 0.3 | 3.8 ± 1.6 | 1.6 ± 1.1 |
PrPC-G-lyso-PI reduced PrPSc Formation
The inhibitory effect of PrPC-G-lyso-PI on PrPSc formation was studied further. We report that PrPC-G-lyso-PI reduced the PrPSc content of ScN2a cells (Fig. 3A), SMB cells (Fig. 3B), and ScGT1 cells in a dose-dependent manner (Fig. 3C). Immunoblots confirmed that PrPC-G-lyso-PI reduced the PrPSc content of ScGT1 cells without affecting the amount of β-actin (Fig. 3D). The addition of 25 ng PrPC-G-lyso-PI did not affect the survival of ScGT1 cells (101% cell survival ± 9 compared with 100 ± 7, n = 12, p = 0.6). PrPC-G-lyso-PI also reduced the infectivity of ScGT1 cells. Groups of mice were injected intra-cerebrally with homogenates from ScGT1 cells treated for 7 days with 25 ng of PrPC-G-lyso-PI or with a vehicle control. The mean incubation period of mice given the control homogenate was 164 days ± 4 (incubation period ± S.D., n = 13). None of the six mice inoculated with homogenates from cells treated with PrPC-G-lyso-PI had died by day 600, indicating that treatment with PrPC-G-lyso-PI reduced infectivity.
FIGURE 3.

The addition of PrPC-G-lyso-PI reduced the PrPSc content of ScN2a cells. A, the amount of PrPSc in ScN2a cells treated with PrPC-G-lyso-PI as shown for 7 days. Values shown are the mean amount of PrPSc (ng/106 cells) ± S.D. from triplicate experiments performed 10 times (n = 30). B, the amount of PrPSc in SMB cells treated with PrPC-G-lyso-PI as shown for 7 days. Values shown are the mean amount of PrPSc (ng/106 cells) ± S.D. from triplicate experiments performed 8 times (n = 24). C, the amount of PrPSc in ScGT1 cells treated with PrPC-G-lyso-PI as shown for 7 days. Values shown are the mean amount of PrPSc (ng/106 cells) ± S.D. from triplicate experiments performed 8 times (n = 24). D, ScGT1 cells were incubated for 7 days with different concentrations of PrPC-G-lyso-PI as shown, cell extracts prepared (± proteinase K digestion) and separated by PAGE and transferred to membranes. β-Actin in cell extracts was detected by immunoblot with clone AC-74, and PrPSc was detected by immunoblot with mAb ICSM18.
PrPC-GPI Increased Amount of Cholesterol in Cells
The amount of cellular cholesterol affects the formation of PrPSc (11, 12). Here, we show that the addition of 10 ng PrPC-GPI, but not PrPC-G-lyso-PI, for 12 h increased the amount of cholesterol in ScN2a cell membranes (Fig. 4A) and reduced the amount of cholesterol esters (Fig. 4B) suggesting that the increase in cholesterol was partially derived from the hydrolysis of cholesterol esters. Pretreatment of cells with 100 μm diethylumbelliferyl phosphate, which inhibits the hydrolysis of cholesterol esters (21), reduced both the PrPC-GPI-induced increase in cholesterol (613 ng/106 cells ± 38 compared with 727 ± 51, n = 12, p < 0.01), and prevented the reduction in cholesterol esters (Fig. 4B). The addition of 100 μm diethylumbelliferyl phosphate also reduced PrPSc production in ScN2a cells incubated with 10 ng of PrPC-GPI (3.8 ng PrPSc ± 1.4 compared with 8.7 ± 2.3, n = 12, p < 0.01), indicating that the hydrolysis of cholesterol esters provides cholesterol to facilitate PrPSc formation.
FIGURE 4.
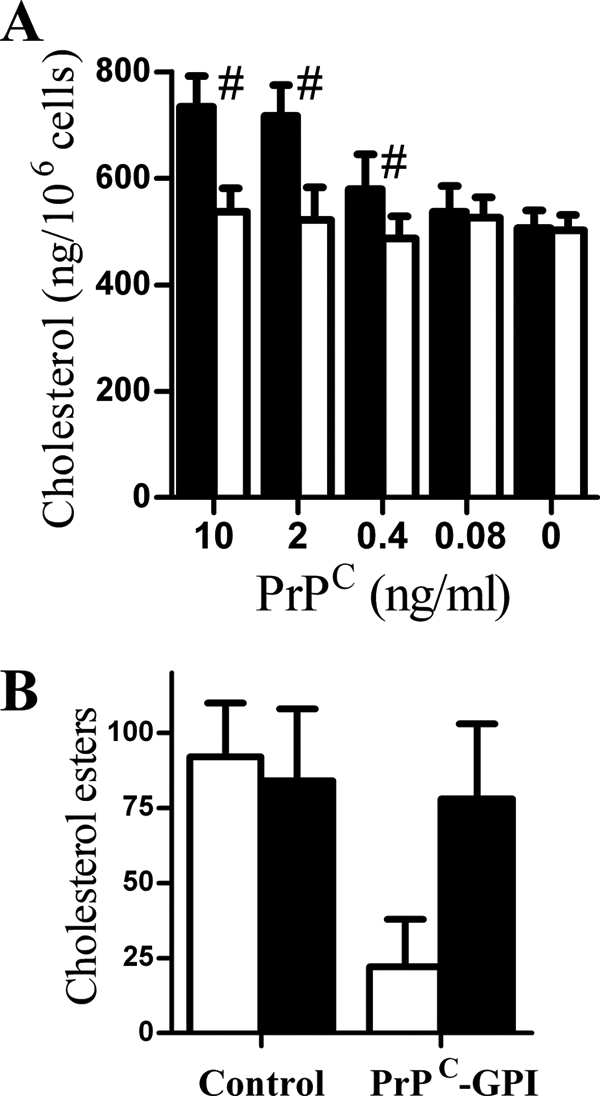
PrPC-GPI increased the cholesterol content of cell membranes. A, the amount of cholesterol in extracts from ScN2a cells that had been treated for 12 h with different concentrations of PrPC-GPI (■) or PrPC-G-lyso-PI (□) as shown. #, amount of cholesterol in cell extracts significantly higher than that of untreated ScN2a cells. Values shown are the mean amount of cholesterol (ng/106 cells) ± S.D. from triplicate experiments performed four times (n = 12). B, the amount of cholesterol esters in extracts from ScN2a cells treated for 12 h with control medium or 10 ng of PrPC-GPI as shown in the absence (□) or presence (■) of 100 μm diethylumbelliferyl phosphate. Values shown are the mean amount of cholesterol esters (ng/106 cells) ± S.D. from triplicate experiments performed three times (n = 9).
Acylation of GPI Anchor Altered Membrane Targeting of PrPC
Cortical neurons derived from PrP null mice were incubated with 10 ng of PrPC to determine the effect of the GPI anchor on the targeting of PrPC. PrPC-GPI and PrPC-G-lyso-PI bound rapidly to cortical neurons, whereas PrPC-IPG did not (Table 2). Although PrPC-GPI was mostly found within detergent-resistant membranes, PrPC-G-lyso-PI was detergent soluble (Table 2). Acylation of the GPI anchor also affected the expression of PrPC at the cell surface; the amount of PrPC-G-lyso-PI at the surface of these cells after 2 h was higher than the amount of PrPC-GPI (8.3 ng PrPC ± 0.8 compared with 3.7 ng ± 0.5, n = 12, p < 0.01). Cortical neurons from PrP null mice were incubated for 2 h with 10 ng of PrPC preparations, and membrane constituents were separated on sucrose density gradients. Although most of the PrPC-GPI was found in low density membranes, PrPC-G-lyso-PI was found in different fractions (Fig. 5A). The targeting of proteins to lipid rafts can affect their trafficking within cells and hence their fate. After PrP null neurons were pulsed with 10 ng PrPC-GPI or PrPC-G-lyso-PI for 2 h PrPC-GPI was rapidly cleared from neurons and was absent after 48 h, whereas PrPC-G-lyso-PI remained in neurons for up to 96 h (Fig. 5B).
TABLE 2.
Acylation of the GPI anchor altered the targeting of PrPC to specific membranes
Cortical neurons derived from PrP null mice were incubated with 10 ng of PrPC-GPI, PrPC-IPG, or PrPC-G-lyso-PI for 2 h. The amounts of PrPC in whole cell extracts (Total), lipid rafts (detergent-resistant membrane (DRM)), detergent-soluble membranes (DSM), or expressed at the surface of cells were measured by ELISA. Values shown are the mean amount of PrPC ± S.D. in total cell extracts (n = 18), detergent-resistant membranes, or detergent-soluble membranes (n = 18) and cell surface PrPC (n = 21).
| PrPC (ng/106 cells) |
|||
|---|---|---|---|
| PrPC-GPI | PrPC-IPG | PrPC-G-lyso-PI | |
| Total | 9.4 ± 0.9 | <0.05 | 9.2 ± 1.3 |
| DRM | 8.2 ± 0.9 | <0.05 | 2.3 ± 0.4 |
| DSM | 1.1 ± 0.2 | <0.05 | 7.4 ± 1.1 |
| Cell surface | 3.7 ± 0.5 | <0.05 | 8.3 ± 0.8 |
FIGURE 5.
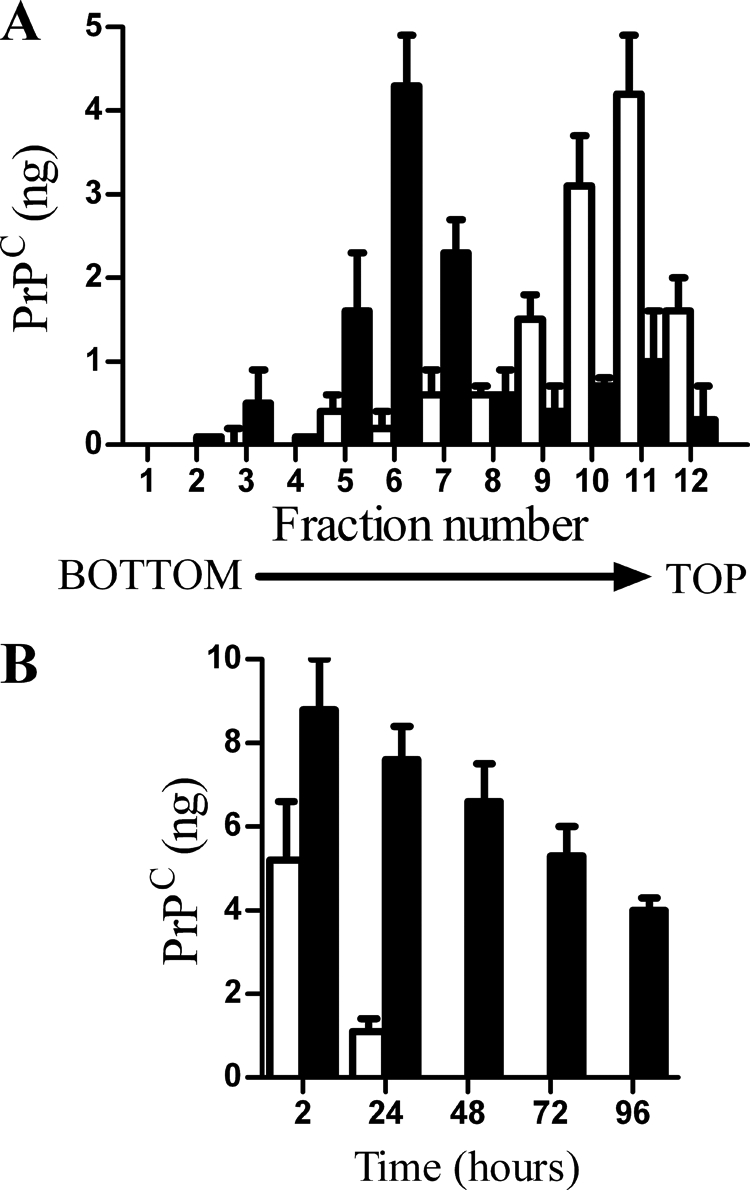
Acylation of GPI anchors affected the targeting of PrPC to lipid rafts. A, PrP null cortical neurons were incubated with 10 ng of PrPC-GPI (□) or PrPC-G-lyso-PI (■) for 2 h. Cell extracts were prepared and separated by centrifugation on a sucrose density gradient, and the amount of PrPC in each fraction was determined by ELISA. Values shown are the mean amount of PrPC (ng) ± S.D. from triplicate experiments performed three times (n = 9). B, PrP null cortical neurons were pulsed with 10 ng of PrPC-GPI (□) or PrPC-G-lyso-PI (■) for 2 h. The amount of PrPC in cells was determined at time points thereafter as shown. Values shown are the mean amount of PrPC (ng/106 cells) ± S.D. from triplicate experiments performed four times (n = 12).
PrPC-G-lyso-PI reduced activation of cPLA2 in Prion-infected Cells
As PLA2 is involved in prion formation (22), the effects of PrPC-G-lyso-PI on cPLA2 were studied. The addition of 25 ng of PrPC-G-lyso-PI for 3 h reduced the amount of activated cPLA2 in ScN2a cells (55 units ± 16 compared with 100 ± 12, n = 12, p < 0.01), ScGT1 cells (38 units ± 14 compared with 100 ± 12, n = 12, p < 0.01), or SMB cells (48 units ± 15 compared with 100 ± 12, n = 15, p < 0.01). Upon activation, cPLA2 migrates to specific membranes utilizing a Ca2+-dependent lipid binding domain (23). Sucrose density gradients showed that 3 h after the addition of 25 ng of PrPC-G-lyso-PI to ScN2a cells, a proportion of cPLA2 had relocated (Fig. 6A).
FIGURE 6.

PrPC-G-lyso-PI reduced the co-precipitation of cPLA2 with PrPSc. A, membrane extracts from ScN2a cells that had been treated for 3 h with control medium (■) or 25 ng of PrPC-G-lyso-PI (□) were separated by centrifugation on a sucrose density gradient, and the amount of cPLA2 in each fraction was determined by ELISA. Values shown are the mean amount of cPLA2 (units) ± S.D. from an experiment run in triplicate. Cell extracts were prepared from control ScN2a cells (fraction 1) or ScN2a cells that had been pretreated with 25 ng of PrPC-G-lyso-PI (fraction 2). Membranes were isolated and incubated with either mAb ICSM35 (anti-PrP) or mAb CH-7 (anti-cPLA2) and protein G magnetic beads. Immunoprecipitates were prepared (± proteinase K digestion), separated by PAGE, and transferred to membranes. The presence of PrPSc was detected by mAb ICSM18, and cPLA2 was detected by mAb CH-7. Immunoblots show the amount of PrPSc (B) and cPLA2 (C) precipitated by mAb ICSM35 (anti-PrP) or the amount of PrPSc (D) and cPLA2 (E) precipitated by mAb CH-7 (anti-cPLA2).
Prior studies showed that cPLA2 was found within PrPSc-containing lipid rafts (20). We report that pretreatment of ScN2a cells with 25 ng of PrPC-G-lyso-PI for 3 h did not affect the amount of PrPSc precipitated by ICSM35 (Fig. 6B) but reduced the amount of co-precipitated cPLA2 (Fig. 6C), indicating that PrPC-G-lyso-PI caused the dissociation of some cPLA2 from PrP-containing membranes. Conversely, an mAb to cPLA2 (CH-7) precipitated similar amounts of cPLA2 from extracts from control cells and cells incubated with 25 ng of PrPC-G-lyso-PI (Fig. 6E). However, it precipitated less PrPSc from ScN2a cells treated with 25 ng of PrPC-G-lyso-PI than from control cells (1.6 ng of PrPSc ± 1.2 compared with 5.2 ng ± 1.4, n = 9, p < 0.01) and (Fig. 6D). These studies were conducted on cells 3 h after the addition of PrPC-G-lyso-PI and before any differences in the PrPSc content of cells were observed.
DISCUSSION
A cell painting technique was used to demonstrate the effect of the GPI anchor on the targeting of PrPC to specific membranes, cell activation, and on the conversion of PrPC to PrPSc. While treating prion-infected cells with PrPC increased PrPSc formation, this effect was lost after the removal of acyl chains from the GPI anchor attached to PrPC. Moreover, the addition of high amounts of PrPC-G-lyso-PI altered membrane domains surrounding PrPSc, reducing the activation of cPLA2, PrPSc formation, and infectivity.
The addition of PrPC-IPG did not affect PrPSc formation in cells, a surprising observation considering of a report that transgenic mice producing anchorless PrPC produced large amounts of PrPSc (14). However, the PrPSc produced in those mice was extracellular, and cells producing anchorless PrPC are resistant to scrapie infection (15). It should also be noted that PrPC-IPG contains the glycan component of the GPI anchor, whereas the transgenic mice produced PrPC lacking the whole GPI anchor. Although PrPC-G-lyso-PI was readily taken up by cells, we found no evidence that it was converted to PrPSc. Once incorporated into recipient cells, the effects and fate of PrPC-GPI and PrPC-G-lyso-PI were different. Prion formation is dependent upon the amount of cholesterol in cell membranes and whereas the addition of PrPC-GPI increased the amount of cholesterol in membranes, consistent with reports that GPI anchors contain mostly saturated fatty acids, which help to solubilize cholesterol (24), the addition of PrPC-G-lyso-PI had no effect. The increase in cholesterol following the addition of PrPC-GPI was reduced by an inhibitor of cholesterol esterase, showing that it was partly derived from the hydrolysis of cholesterol esters. This release of this cholesterol was also necessary for the PrPC-GPI induced PrPSc formation.
The amount of cholesterol in cell membranes is important for the formation and function of lipid rafts (25). Although both detergent resistance assays and sucrose density gradients showed that PrPC-GPI was found within lipid rafts, most of the PrPC-G-lyso-PI molecules were found within non-raft membranes. The presence of a GPI anchor targets PrPC to cholesterol-dense lipid rafts (12, 26), which is essential for PrPC to PrPSc conversion (11, 12). Thus, the targeting of PrPC-G-lyso-PI to non-raft membranes suggested that it limited interactions between raft-associated PrPSc and PrPC-G-lyso-PI.
PrPC interacts with other membrane molecules, including glycosaminoglycans (27), the laminin receptor precursor (28), the low density lipoprotein receptor related protein-1 (29), or glypican-1 (30). The targeting of PrPC-G-lyso-PI to the normal cell membrane may reduce interactions with these proteins, or facilitate interactions with other proteins that alter its trafficking. Differences in the composition of the GPI anchor affect the trafficking of proteins (31). Thus, greater amounts of PrPC-G-lyso-PI than PrPC-GPI were expressed at the cell surface, and although PrPC-GPI was rapidly removed from these cells, PrPC-G-lyso-PI remained in neurons for longer. One explanation of these results is that PrPC-G-lyso-PI targets a different membrane domain to PrPC-GPI and consequently most of the PrPC-G-lyso-PI molecules traffic via a pathway that is physically segregated from PrPSc.
Although these theories explain why PrPC-G-lyso-PI was not readily converted to PrPSc, a more refined hypothesis is required to explain why the addition of high amounts of PrPC-G-lyso-PI reduced PrPSc production and infectivity. One possibility is that PrPC-G-lyso-PI competed with endogenous PrPC for endocytic partner proteins and consequently altered the trafficking of endogenous PrPC and hence PrPSc formation. Another possibility is that PrPC binds to PrPSc and modifies lipid rafts. The composition and hence function of lipid rafts is dynamic and controlled by an induced fit model (32). Because PrPC-G-lyso-PI did not sequester cholesterol, the membrane surrounding a complex between PrPSc and PrPC-G-lyso-PI would contain less cholesterol than membranes formed following the interaction between PrPSc and PrPC-GPI. Thus, the binding of PrPC-G-lyso-PI to PrPSc may reduce the cholesterol content of local membranes to a level below that required for the conversion of PrPC to PrPSc. This hypothesis is consistent with observations that formation of PrPSc was affected by the lipid composition of membranes (33) and that lipids were essential co-factors in prion formation (34). Finally, it is possible that PrPC-G-lyso-PI is converted to PrPSc-G-lyso-PI, which in turn acts as an inefficient template for PrPC to PrPSc conversion (35).
Lipid rafts are enriched with signaling molecules, which suggests that they act as domains in which the GPI anchors attached to PrPC interact with cell signaling pathways (36). The activation of PLA2 is required for PrPSc formation (22), and in prion-infected cells, cPLA2 co-localized with PrPSc-containing lipid rafts (20). Such observations suggest that PrPC-GPI binds to PrPSc in cholesterol-dense lipid rafts, where it activates cPLA2 and facilitates the conversion of PrPC to PrPSc. The addition of PrPC-G-lyso-PI reduced activation of cPLA2 in prion-infected cell lines. Notably, the addition of PrPC-G-lyso-PI reduced the amount of cPLA2 within PrPSc-containing lipid rafts. We propose that the binding of PrPC-G-lyso-PI to PrPSc changed the composition of the underlying membrane so that it no longer captured cPLA2. Upon activation, cPLA2 migrated to intracellular membranes utilizing a Ca2+-dependent lipid binding domain (23). It is thought that the targeting of cPLA2 to membranes containing their endogenous substrates can regulate cell signaling. Thus, the targeting of cPLA2 to specific membranes is essential for the formation of second messengers such as platelet-activating factor that facilitate PrPSc formation (22). Here, we showed that the addition of PrPC-G-lyso-PI to prion-infected cells altered the location of cPLA2. More specifically, it caused the dissociation of cPLA2 from PrPSc, reduced activation of cPLA2 and hindered the conversion of PrPC to PrPSc. The activation of cPLA2 is essential to the maintenance of the Golgi network (37), which is involved in the trafficking of a GFP-tagged PrPC (38). Thus, altering the GPI anchor attached to PrPC may reduce the activation of cPLA2 and alter the trafficking of PrPC away from specific sites conducive to prion formation.
In conclusion, we showed that the addition of PrPC-G-lyso-PI reduced the activation of cPLA2 and PrPSc formation in prion-infected cells. We propose that the chemical composition of the GPI anchor is a factor that targets PrPC to sites conducive to conversion to PrPSc. Moreover, these results raise the possibility that drugs, which alter the structure of the PrPC-GPI anchor, may provide novel treatments for prion diseases.
Supplementary Material
This work was supported by a grant from the European Commission FP6 “Neuroprion” Network of Excellence.

The on-line version of this article (available at http://www.jbc.org) contains supplemental Fig. 1.
- PrPC
- cellular prion protein
- cPLA2
- cytoplasmic phospholipase A2
- PI
- phosphatidylinositol
- GPI
- glycosylphosphatidylinositol
- IPG
- inositolphosphoglycan.
REFERENCES
- 1. Prusiner S. B. (1998) Proc. Natl. Acad. Sci. U.S.A. 95, 13363–13383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Pan K. M., Baldwin M., Nguyen J., Gasset M., Serban A., Groth D., Mehlhorn I., Huang Z., Fletterick R. J., Cohen F. E. (1993) Proc. Natl. Acad. Sci. U.S.A. 90, 10962–10966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Prusiner S. B., McKinley M. P., Bowman K. A., Bolton D. C., Bendheim P. E., Groth D. F., Glenner G. G. (1983) Cell 35, 349–358 [DOI] [PubMed] [Google Scholar]
- 4. Jeffrey M., Halliday W. G., Bell J., Johnston A. R., MacLeod N. K., Ingham C., Sayers A. R., Brown D. A., Fraser J. R. (2000) Neuropathol. Appl. Neurobiol. 26, 41–54 [DOI] [PubMed] [Google Scholar]
- 5. Büeler H., Aguzzi A., Sailer A., Greiner R. A., Autenried P., Aguet M., Weissmann C. (1993) Cell 73, 1339–1347 [DOI] [PubMed] [Google Scholar]
- 6. Mallucci G., Dickinson A., Linehan J., Klöhn P. C., Brandner S., Collinge J. (2003) Science 302, 871–874 [DOI] [PubMed] [Google Scholar]
- 7. Enari M., Flechsig E., Weissmann C. (2001) Proc. Natl. Acad. Sci. U.S.A. 98, 9295–9299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Peretz D., Williamson R. A., Kaneko K., Vergara J., Leclerc E., Schmitt-Ulms G., Mehlhorn I. R., Legname G., Wormald M. R., Rudd P. M., Dwek R. A., Burton D. R., Prusiner S. B. (2001) Nature 412, 739–743 [DOI] [PubMed] [Google Scholar]
- 9. Gilch S., Winklhofer K. F., Groschup M. H., Nunziante M., Lucassen R., Spielhaupter C., Muranyi W., Riesner D., Tatzelt J., Schätzl H. M. (2001) EMBO J. 20, 3957–3966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Béranger F., Mangé A., Goud B., Lehmann S. (2002) J. Biol. Chem. 277, 38972–38977 [DOI] [PubMed] [Google Scholar]
- 11. Bate C., Salmona M., Diomede L., Williams A. (2004) J. Biol. Chem. 279, 14983–14990 [DOI] [PubMed] [Google Scholar]
- 12. Taraboulos A., Scott M., Semenov A., Avrahami D., Laszlo L., Prusiner S. B., Avraham D. (1995) J. Cell Biol. 129, 121–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Stahl N., Baldwin M. A., Hecker R., Pan K. M., Burlingame A. L., Prusiner S. B. (1992) Biochemistry 31, 5043–5053 [DOI] [PubMed] [Google Scholar]
- 14. Chesebro B., Trifilo M., Race R., Meade-White K., Teng C., LaCasse R., Raymond L., Favara C., Baron G., Priola S., Caughey B., Masliah E., Oldstone M. (2005) Science 308, 1435–1439 [DOI] [PubMed] [Google Scholar]
- 15. McNally K. L., Ward A. E., Priola S. A. (2009) J. Virol. 83, 4469–4475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Legler D. F., Doucey M. A., Schneider P., Chapatte L., Bender F. C., Bron C. (2005) FASEB J. 19, 73–75 [DOI] [PubMed] [Google Scholar]
- 17. Medof M. E., Nagarajan S., Tykocinski M. L. (1996) FASEB J. 10, 574–586 [DOI] [PubMed] [Google Scholar]
- 18. Liu T., Li R., Pan T., Liu D., Petersen R. B., Wong B. S., Gambetti P., Sy M. S. (2002) J. Biol. Chem. 277, 47671–47678 [DOI] [PubMed] [Google Scholar]
- 19. Bate C., Tayebi M., Williams A. (2010) J. Biol. Chem. 285, 22017–22026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bate C., Tayebi M., Williams A. (2008) BMC Biol. 6, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gocze P. M., Freeman D. A. (1992) Endocrinology 131, 2972–2978 [DOI] [PubMed] [Google Scholar]
- 22. Bate C., Reid S., Williams A. (2004) J. Biol. Chem. 279, 36405–36411 [DOI] [PubMed] [Google Scholar]
- 23. Nalefski E. A., Sultzman L. A., Martin D. M., Kriz R. W., Towler P. S., Knopf J. L., Clark J. D. (1994) J. Biol. Chem. 269, 18239–18249 [PubMed] [Google Scholar]
- 24. Schroeder R., London E., Brown D. (1994) Proc. Natl. Acad. Sci. U.S.A. 91, 12130–12134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Brown D. A., London E. (2000) J. Biol. Chem. 275, 17221–17224 [DOI] [PubMed] [Google Scholar]
- 26. Naslavsky N., Stein R., Yanai A., Friedlander G., Taraboulos A. (1997) J. Biol. Chem. 272, 6324–6331 [DOI] [PubMed] [Google Scholar]
- 27. Warner R. G., Hundt C., Weiss S., Turnbull J. E. (2002) J. Biol. Chem. 277, 18421–18430 [DOI] [PubMed] [Google Scholar]
- 28. Rieger R., Edenhofer F., Lasmézas C. I., Weiss S. (1997) Nat. Med. 3, 1383–1388 [DOI] [PubMed] [Google Scholar]
- 29. Parkyn C. J., Vermeulen E. G., Mootoosamy R. C., Sunyach C., Jacobsen C., Oxvig C., Moestrup S., Liu Q., Bu G., Jen A., Morris R. J. (2008) J. Cell Sci. 121, 773–783 [DOI] [PubMed] [Google Scholar]
- 30. Taylor D. R., Whitehouse I. J., Hooper N. M. (2009) PLoS. Pathog. 5, e1000666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Rajendran L., Simons K. (2005) J. Cell Sci. 118, 1099–1102 [DOI] [PubMed] [Google Scholar]
- 32. Pike L. J. (2004) Biochem. J. 378, 281–292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Graham J. F., Agarwal S., Kurian D., Kirby L., Pinheiro T. J., Gill A. C. (2010) J. Biol. Chem. 285, 9868–9880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wang F., Wang X., Yuan C. G., Ma J. (2010) Science 327, 1132–1135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bate C., Tayebi M., Williams A. (2010) Biochem. J. 428, 95–101 [DOI] [PubMed] [Google Scholar]
- 36. Simons K., Toomre D. (2000) Nat. Rev. Mol. Cell Biol. 1, 31–39 [DOI] [PubMed] [Google Scholar]
- 37. de Figueiredo P., Drecktrah D., Katzenellenbogen J. A., Strang M., Brown W. J. (1998) Proc. Natl. Acad. Sci. U.S.A. 95, 8642–8647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Magalhães A. C., Silva J. A., Lee K. S., Martins V. R., Prado V. F., Ferguson S. S., Gomez M. V., Brentani R. R., Prado M. A. (2002) J. Biol. Chem. 277, 33311–33318 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.